

Abstract
Objective
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease with incompletely understood etiology. Very little is known about the role of epigenetic dysregulation in the pathogenesis of pSS.
Methods
We performed a genome-wide DNA methylation study in naïve CD4+ T cells in eleven pSS patients compared to age-, sex-, and ethnicity-matched healthy controls. Cytosine methylation was quantified using the Illumina Infinium HumanMethylation450 BeadChip array and validated using bisulfite sequencing.
Results
We identified 553 hypomethylated and 200 hypermethylated CpG sites in naïve CD4+ T cells from pSS patients compared to healthy matched controls, representing 311 hypomethylated and 115 hypermethylated gene regions. Hypomethylated genes in pSS include LTA, coding for Lymphotoxin α. Other relevant genes such as CD247, TNFRSF25, PTPRC, GSTM1 and PDCD1 were also hypomethylated. The interferon signature pathway was represented by hypomethylation of STAT1, IFI44L, USP18 and IFITM1. A group of genes encoding for members of the solute carrier proteins were differentially methylated. In addition, the transcription factor RUNX1 was hypermethylated in patients, suggesting a possible connection to lymphoma predisposition. Gene ontology (GO) analysis of hypomethylated genes demonstrated enrichment of genes involved in lymphocyte activation and immune response. GO terms for hypermethylated genes included antigen processing and presentation.
Conclusion
This is the first epigenome-wide DNA methylation study in pSS. Our data highlight a role for DNA methylation in pSS and identify disease-associated DNA methylation changes in several genes and pathways in naïve CD4+ T cells in pSS that may be involved in the pathogenesis of this disease.
Introduction
Primary Sjögren’s syndrome (pSS) is a complex autoimmune disease characterized by production of autoantibodies against ribonucleoprotein (RNP) particles (Ro/SS-A and La/SS-B) and muscarinic acetylcholine receptor antigens, dysfunction of water transport processes, and lymphocytic infiltration of exocrine glands resulting in glandular atrophy and dysfunction (1, 2). Although, xerostomia (dry mouth) and xerophthalmia (dry eyes) are the main clinical features of pSS (3), the full spectrum of the disease encompasses involvement of different organ-systems and predisposition to lymphoproliferative disease (4). pSS predominantly affects women with a female to male ratio of 9:1 (5).
The etiology of pSS is incompletely understood, but there is growing body of evidence in diseases that often share certain clinical features with pSS, such as systemic lupus erythematosus, that epigenetic factors contribute to the pathogenesis of autoimmunity (6). Further, recent evidence suggests reduced global DNA methylation in salivary glands epithelial cells from patients with pSS (7). DNA methylation is considered the core epigenetic mechanism that regulates gene expression by altering transcriptional accessibility of regulatory regions within gene sequences (8). We have recently characterized DNA methylation changes in naïve CD4+ T cells from lupus patients, revealing DNA methylation changes prior to T cell differentiation and activation, and demonstrating that interferon-regulated genes in naïve CD4+ T cells from lupus patients are epigenetically poised for transcription (9). In this study, we performed a genome-wide DNA methylation study in naïve CD4+ T cells from pSS and control subjects. We next validated our results using bisulfite sequencing of selected differentially methylated loci. We demonstrated differential methylation in key genes and disease associated pathways pertinent to the pathogenesis of pSS.
Methods
Primary Sjögren’s syndrome patients and controls
We studied 11 patients with pSS and 11 healthy case controls. Patients and controls were matched for age (+/− 5 years), sex, and ethnicity. Demographic features of the study participants are shown in Table 1. Classification of pSS was based on the American-European Consensus Group 2002 revised criteria (10). The clinical features of the pSS patients included in this study are shown in Table 2. The cases were extensively characterized by a team of oral, ocular, and rheumatologic specialists for disease manifestations through the Oklahoma Medical Research Foundation (OMRF) Sjögren’s Research Clinic. Controls were recruited at OMRF and the University of Michigan. The institutional review boards at OMRF and the University of Michigan approved this study. All study participants signed a written informed consent prior to participation.
Table 1.
Demographic characteristics of the study participants. Mean age is 45.9 years and 45.54 years for primary Sjögren’s syndrome patients and controls, respectively (P value = 0.95).
| pSS cases
|
Controls
|
|||||||
|---|---|---|---|---|---|---|---|---|
| ID | Age | Ethnicity | Sex | Medication | ID | Age | Ethnicity | Sex |
| 1 | 50 | EA | Female | Methotrexate | 12 | 52 | EA | Female |
| 2 | 51 | EA | Female | Hydroxychloroquine | 13 | 47 | EA | Female |
| 3 | 30 | EA | Female | 14 | 34 | EA | Female | |
| 4 | 23 | EA | Female | Hydroxychloroquine | 15 | 23 | EA | Female |
| 5 | 62 | EA | Female | Azathioprin | 16 | 58 | EA | Female |
| 6 | 32 | EA | Female | 17 | 36 | EA | Female | |
| 7 | 35 | EA | Female | 18 | 36 | EA | Female | |
| 8 | 60 | EA | Female | 19 | 55 | EA | Female | |
| 9 | 58 | EA | Female | 20 | 58 | EA | Female | |
| 10 | 35 | EA | Female | Hydroxychloroquine | 21 | 38 | EA | Female |
| 11 | 69 | EA | Female | Hydroxychloroquine | 22 | 64 | EA | Female |
EA: European-American; pSS: primary Sjögren’s syndrome
Table 2.
Clinical characteristics of the primary Sjögren’s syndrome patients included in this study (n = 11).
| Patient number (%) | |
|---|---|
| Dry eyes | 11 (100) |
| Dry mouth | 11 (100) |
| Abnormal Schirmer’s test | 6 (55) |
| Average Schirmer’s response (mm) | 10.8 |
| Abnormal Lissamine green | 9 (82) |
| Abnormal WUSF | 5 (45) |
| Positive Anti SSA/Ro | 6 (55) |
| Positive Anti SSB/La | 6 (55) |
| MSGB consistent with pSS | 10 (91) |
| Average focus score | 4.5 |
Focus score: the number of mononuclear cell infiltrates containing at least 50 inflammatory cells in a 4 mm2 glandular section. MSGB: Minor salivary gland biopsy WUSF: Whole unstimulated salivary flow.
Naïve CD4+ T cell isolation and purity
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh blood samples obtained from patients and controls using density gradient centrifugation (Ficoll-Paque, GE Healthcare, Life Sciences, New Jersey, USA). Naïve CD4+ T cells were separated from PBMCs using either the naïve CD4+ T Cell Isolation Kit II (Miltenyi Biotec, Cambridge, MA) that allows for indirect isolation of naïve CD4+ T cells, or by sorting the CD3+CD4+CD45RA+ naïve T cell population on a FACSAria instrument (BD Biosciences, San Jose, CA). Naïve CD4+ T cell purity was confirmed by flow cytometry using fluorochrome-conjugated antibodies against CD4 and CD45RA. Isolated naïve CD4+ T cell purity was consistently > 95% regardless of the method used. DNA was isolated using the DNeasy Kit (Qiagen, Valencia, CA) as described in the manufacturer’s protocol.
DNA methylation studies and array validation
Genome-wide DNA methylation in naïve CD4+ T cells from pSS patients and controls included in this study was assessed using the Illumina Infinium HumanMethylation450 BeadChip array, which allows for the interrogation of over 485,000 methylation sites within the entire genome. This array covers 99% of RefSeq genes, with an average of 17 CpG sites per gene across the promoter region, 5′ untranslated region (5′-UTR), first exon, gene body, and 3′-UTR. It also covers 96% of CpG islands. Non CpG methylated sites recently identified in human stem cells are also covered as well as microRNA promoter regions.
Validation of the array data was performed using bisulfite DNA sequencing in selected hypermethylated and hypomethylated regions. The primers for HDAC4 were forward primer 5′-TGGTTTTATTTTTTGTAGTTAAAAA 3′ and reverse primer 5′ ATAAAACCTCTATACCTCACTCAAC 3′; for SLC38A4 were forward primer 5′ TTTGGATTTTTTAATTAAGTTGTTA3′, and reverse primer 5′ TCTACAATATTAATACTCCTACAAACC 3′; for DUSP22 were forward primer 5′TTATTTGTTTTTTTAGGGTAGGGAG 3′ and reverse primer 5′AATCTCCAAATCCCCCTTTAAC 3′; for GSTM1 were forward primer 5′ GTTAGGATTTGGTTGGTGTTTTAAG 3′ and reverse primer 5′ ATCCCAATACCCCAATATCATAAAC 3′; for RUFY1 were forward primer 5′ GTAGGAGAGGTTTTGAGTTGGATT 3′ and reverse primer 5′ TCCTCCATCATCTAACACTTAAAAA 3′; for NAPRT1 were forward primer 5′ TATGGTGGTTTGGTAGAGGTTAGTG 3′ and reverse primer 5′ ACTAATCTATCCTCCACCCTTTCC 3′; and for SLC9A were forward primer 5′ GTTTTTTTATTTAGAGAGGGGTAGG 3′ and reverse primer 5′ AACCAAAAAAAACTACAACTAAACC 3′. We used a Biorad T100 Thermocycler with the following protocol: 1 cycle at 94 °C for 5 minutes, followed by 40 cycles (94°C for 45 sec, annealing temp for each primer set for 45 sec, 72°C for 90 sec), and then 74°C for 10 minutes. The annealing temperatures for HDAC4, SLC38A, DUSP22, GSTM1, RUFY 1, NAPRT1, and SLC9A1 were 54°C, 57°C, 53°C, 54°C, 56°C, 56°C, 56°C and 53°C, respectively. We confirmed the presence of PCR product by using 1.6 % agarose gel electrophoresis. DNA was purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) as specified by the manufacturer. Bisulfite treated DNA was sequenced using Sanger sequencing. DNA methylation level on each CpG following bisulfite sequencing was quantified using the ESME software package (Epigenomics AG, Berlin).
Statistical and bioinformatics analysis
DNA methylation analysis was performed using the GenomeStudio methylation analysis package (Illumina) as previously described (8). The average level of DNA methylation (β) on each CpG site was compared between pSS patients and controls. To identify differentially methylated CpG sites between pSS cases and controls, we used three data filtering criteria: (i) CpG site with an average difference in DNA methylation level of at least 1.2-fold (ii) differential methylation score of ≥ 22 (P≤ 0.01) after adjusting for multiple testing using a false discovery rate of 5%, and (iii) exclusion of CpG sites assessed by probes with a genetic variant located within 10bp of the 3′ end of the probe.
To systematically highlight the most over-represented biological terms, out of the differentially methylated gene, we performed gene ontology, network, and pathway analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7(11). In pathway and gene ontology analysis, we used Expression Analysis Systematic Explorer (EASE) Score threshold of <0.1, for detection of gene enrichment analysis (EASE score represent a modified Fisher Exact P-Value, which is considered a measure to examine the significance of gene-term enrichment), in addition to fold enrichment of 1.5 and FDR for correction of multiple testing < 10% (12).
Results
We evaluated DNA methylation changes in naïve CD4+ T cells from pSS patients and age-, sex-, and ethnicity-matched controls. We identified 753 differentially methylated CpG sites in naïve CD4+ T cells from pSS patients (Table 3). Two hundred sites were hypermethylated and 553 were hypomethylated in patients with pSS compared to controls. A total of 426 differentially methylated unique genes were identified in naïve CD4+ T cells from pSS patients, with the majority (311 genes, 75%) being hypomethylated. Bisulfite DNA sequencing was used to validate the DNA methylation array results by studying a group of hypermethylated (4 genes, 7 loci) and hypomethylated (3 genes, 3 loci) CpG sites. The average Pearson’s correlation coefficient (r2) value between the Illumina Infinium HumanMethylation450 array data and bisulfite sequencing data was 0.803 (Supplementary Figure 1).
Table 3.
Summary of the differentially methylated CpG Sites and genes in naïve CD4+ T cells from primary Sjögren’s syndrome patients compared to healthy controls.
| Increased methylation | Decreased methylation | Total | |
|---|---|---|---|
| CpG sites | 200 | 553 | 753 |
| Fold change (Average) | 1.20–5.20 1.65 |
1.20–5.22 1.60 |
|
| Differential score (Average) | 22.0 to 342.0 (+95.6) | −22.0 to −339.8 (−66.1) | |
| Genes | 115 | 311 | 426 |
Despite consistently achieving naïve CD4+ T cell purity of >95%, to ensure that differential methylation patterns identified between patients and controls in our study were not influenced by potential differences in naïve CD4+T cell activation status induced by the isolation procedures used, we examined the DNA methylation levels of a region in the IL2 promoter-enhancer sequence that is known to readily demethylate upon T cell activation. Demethylation of the promoter-enhancer region of IL2 is a sensitive indicator of CD4+ T cell activation (13, 14) and is a prerequisite for IL2 transcription (15). We detected high DNA methylation levels in the IL2 promoter-enhancer region, as expected in naïve CD4+ T cells, and no difference between patients and controls (Supplementary Table 2). Further, we examined the DNA methylation levels in genetic loci known to demethylate in specific antigen experienced and regulatory CD4+ T cell subsets, including IFNG (Th1 cells), IL4, IL5 and IL13 (Th2 cells), IL17 (IL17A) and IL17F (Th17 cells) and FOXP3 (regulatory T cells). Again, we find consistent high DNA methylation levels across these genetic loci, and no difference between patients and controls (Supplementary Table 2).
Hypomethylated genes in naïve CD4+ T cells from pSS patients include LTA, encoding Lymphotoxin α, which is involved in the LTβ receptor signaling pathway, activation of follicular dendritic cells, and expression of interferon-α. We identified 5 differentially methylated CpG sites in LTA located at the 5′UTR, within 200 base pairs (bp) upstream of the transcriptional start site (abbreviated as TSS 200) and in the first exon (Figure 1). The average differential methylation score (Diff. score) of the 5 CpG sites was −35.4 (range −22.3 to −53.8; average fold change 1.82). Other relevant hypomethylated genes include CD247, which encodes for TCR-zeta chain (2 CpG sites located in the body of the gene; Diff. score −23.9 and −32.3; average fold change 1.96), TNFRSF25 (1 CpG site; TSS 1500; Diff. score −23.2; fold change 1.59), PTPRC (2 CpG sites; TSS 1500 and body of the gene; Diff. score −49.7 and −75.5; average fold change average 1.96), GSTM1 (2 CpG sites located in TSS 200; Diff. score −31.4 and −48.8; average fold change 1.40) and PDCD1 (1 CpG site; TSS 1500; Diff. score −46.9; fold change 2.63). The type-I interferon pathway, which plays a major role in the pathogenesis of pSS (16), was represented by hypomethylation of STAT1 (2 CpG sites; 5′UTR; Diff. score −23.9 and −79.2; average fold change 1.41), IFI44L (1 CpG site; 5′UTR; Diff. score −24.1; fold change 1.22), IFITM1 (1 CpG site; TSS 1500; Diff. score −35.8; average fold change 1.75), and USP18 (1 CpG site; 5′UTR; Diff. score −65.1; fold change 1.40).
Figure 1.
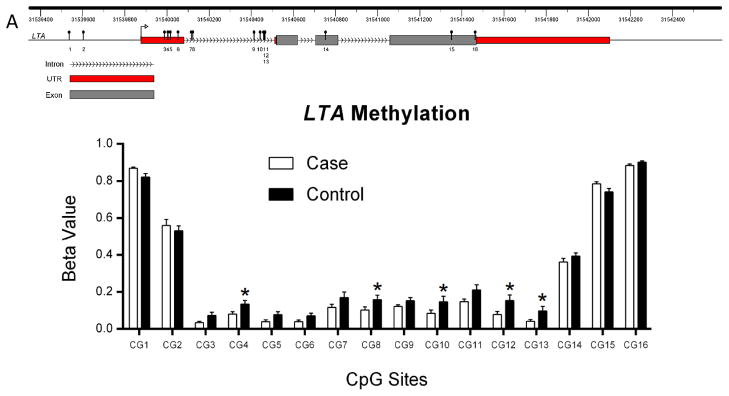
LTA gene DNA methylation in naïve CD4+ T cells from primary Sjögren’s syndrome patients and controls. A) LTA (chr6:31,539,814-–31,542,537; GRCh37/hg19) is shown depicting the location of CpG sites evaluated in this study. B) DNA methylation fractions (β value) across the CpG sites evaluated in LTA. We identified five hypomethylated CpG sites (*) in the LTA gene region in naïve CD4+ T cells from primary Sjögren’s syndrome patients compared to healthy controls.
A group of genes encoding members of the solute carrier proteins, which are membrane transport proteins that are important for maintenance of cell function were hypomethylated (SLC11A1, SLC11A2, SLC22A23, SLC25A25, ALC25A3, SLC25A33, SLC6A20), whereas, SLC9A1, which is important for the maintenance of pH homeostasis was hypermethylated in pSS patients compared to controls. In addition, the transcription factor RUNX1 was hypermethylated in patients with pSS. Supplementary Table 1 provides a summary of all CpG sites that were differentially methylated in patients with pSS in our study.
Next, we used DAVID software (11) to facilitate the systematic identification and grouping of differentially methylated genes into biological networks. Canonical pathway analysis identified type I diabetes mellitus (P= 4.59E-06), allograft rejection (P= 1.66E-04), viral myocarditis (P= 2.34E-04), graft-versus-host disease (P= 2.63E-04), autoimmune thyroid disease (P= 1.15E-03), antigen processing and presentation (P= 3.22E-03) and cell adhesion molecules (P= 3.73E-03) as the most significant pathways unifying the differentially methylated genes in naïve CD4+ T cells from pSS patients (Table 4).
Table 4.
Pathway analysis of the differentially methylated genes in naïve CD4+ T cells from primary Sjögren’s syndrome in comparison to healthy controls.
| Category | Term | Molecules | P Value | Fold Enrichment | FDR |
|---|---|---|---|---|---|
| KEGG_PATHWAY | Type I diabetes mellitus | PTPRN2; CD28; LTA; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB5 | 4.59E-06 | 9.005 | 0.005 |
| KEGG_PATHWAY | Allograft rejection | CD28; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB5 | 1.66E-04 | 8.171 | 0.191 |
| KEGG_PATHWAY | Viral myocarditis | MYH13; CD28; CASP9; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB5 | 2.34E-04 | 5.327 | 0.268 |
| KEGG_PATHWAY | Graft-versus-host disease | CD28; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB5 | 2.63E-04 | 7.543 | 0.301 |
| KEGG_PATHWAY | Autoimmune thyroid disease | CD28; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB-5 | 1.15E-03 | 5.768 | 1.315 |
| KEGG_PATHWAY | Antigen processing and presentation | LTA; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB-4; HLA-DRB5; TAP2 | 3.22E-03 | 4.051 | 3.637 |
| KEGG_PATHWAY | Cell adhesion molecules (CAMs) | NCAM1; SELL; PDCD1; CD28; HLA-A; HLA-B, HLA-C; HLA-F; HLA-DMA; HLA-DRB1, HLA-DRB4; HLA-DRB5 | 3.73E-03 | 3.184 | 4.201 |
FDR: False Discovery Rate; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Gene ontology (GO) analysis of hypomethylated genes demonstrated enrichment of genes involved in lymphocyte activation (P= 1.10E-04), leukocyte differentiation (P= 1.42E-03), immune response (P= 2.20E-03), chromatin organization (P= 3.90E-03), T cell differentiation (P= 4.20E-03), homophilic cell adhesion (P= 5.80E-03), and L-amino acid transport (P= 6.10E-03). GO terms for hypermethylated genes included antigen processing and presentation ontology (P= 6.89E-06) (Table 5).
Table 5.
Gene ontology (GO) analysis was performed on hypomethylated and hypermethylated genes in pSS. Statistically significant GO terms are shown.
| GO terms of Hypomethylated genes
| ||||||
|---|---|---|---|---|---|---|
| Category | Term | GO ID | Genes | P Value | Fold Enrichment | FDR |
| GOTERM_BP_FAT | lymphocyte activation | GO:0046649 | BCL11B; CD3G; CD28; CD7; CHD7; FOXP1; HDAC4; ITPKB; JMJD6; RHOH; SLC11A1; SKAP2; UNC13D | 1.10E-04 | 3.859 | 0.185 |
| GOTERM_BP_FAT | leukocyte differentiation | GO:0002521 | BCL11B; CEBPE; CD28; CHD7; FOXP1; HDAC4; ITPKB; JMJD6; RHOH | 1.42E-03 | 4.168 | 2.361 |
| GOTERM_BP_FAT | immune response | GO:0006955 | BTLA; CD28; CD300A; CD7; FAIM3; ST6GAL1; C4BPB; FOXP1; IFI44L; IL36G; IL16; LY86; LTA; HLA-A; HLA-B; HLA-H; HLA-DRB5; PDCD1; SLC11A1; TCF12; TCF7; UNC13D; ETS1 | 2.20E-03 | 1.969 | 3.646 |
| GOTERM_BP_FAT | chromatin organization | GO:0006325 | BCOR; CREBBP; DNMT3A; H2AFV; SATB1; SMARCB1; APBB1; CHD7; HDAC4; JMJD6; KDM2B; RBBP7; TSPY4; TSPY1; TSPY2; TSPY3; TSPY7P; TLK1 | 3.90E-03 | 2.344 | 6.356 |
| GOTERM_BP_FAT | T cell differentiation | GO:0030217 | BCL11B; CD28; CHD7; ITPKB; JMJD6; RHOH | 4.20E-03 | 5.453 | 6.855 |
| GOTERM_BP_FAT | homophilic cell adhesion | GO:0007156 | PCDHAC2; PCDHA4; PCDHA2; PCDHA3; PCDHA5; PCDHA7; PCDHA8; PCDHA9 | 5.80E-03 | 3.608 | 9.360 |
| GOTERM_BP_FAT | L-amino acid transport | GO:0015807 | CACNA1A; HTT; SLC11A1; SLC7A8 | 6.10E-03 | 10.274 | 9.687 |
|
| ||||||
| GO terms of Hypermethylated genes | ||||||
|
| ||||||
| GOTERM_BP_FAT | antigen processing and presentation | GO:0019882 | TAP2; HLA-B, HLA-C; HLA-F; HLA-H; HLA-DMA; HLA-DRB4; HLA-DRB5 | 6.89E-06 | 14.817 | 0.010 |
GOTERM_BP_FAT: Gene Ontology term biological process
Discussion
We performed an unbiased genome-wide DNA methylation study in naïve CD4+ T cells from pSS patients and identified differentially methylated genes and involved pathways compared to healthy matched controls.
The molecular basis of pSS is not well characterized; however, there is cumulative evidence that activation of the lymphotoxin-β receptor (LTβR) pathway plays an integral role in the pathogenesis of pSS (17). We have identified five differentially hypomethylated CpG sites in LTA gene in naïve CD4+ T cells from pSS patients compared to controls. Lymphotoxins (LTα, LTβ) and their receptors are part of the tumor necrosis factor (TNF) superfamily (18). Soluble LTα promotes production of IFNs and multiple chemokines that are important activating signals to the immune cells (19). Moreover, LTα forms a heterodimer with LTβ (LTα1β2), which in turn binds to LTβR (20). Activation of the LTβR pathway is crucial for the development, organization, and maintenance of lymphoid structures (21), and modulating expression of chemokines and adhesion molecules that aid in trafficking of lymphocytes and follicular dendritic cell activation (22). Further, murine models highlight an interesting role of LTα in the pathogenesis of Sjögren’s-like disease; LTα is overexpressed in salivary gland secretions and sera of a transgenic mouse model for SS (IL14αTG mouse) (23). The IL14αTG mouse reproduces the clinical and immunological changes characteristic of SS (24). Interestingly, IL14αTG mice with deletion of the LTA gene did not develop disease (23). In another murine model of Sjögren’s syndrome (Non-Obese Diabetic (NOD) mouse), Catumu and colleagues (22) showed that blocking the LTβR pathway results in ablation of the lymphoid organization in the NOD mouse salivary glands and an improvement in salivary gland function (22). In another study, blocking of LTβR reduced the size of leukocyte infiltrates in lacrimal glands, and improved tear production and corneal integrity (25). Additional evidence that LTα has a role in Sjögren’s syndrome comes from the observation that LTα is overexpressed in the salivary gland and sera of patients with Sjögren’s syndrome (23). Indeed, a clinical trial to evaluate the use of a LTβR fusion protein (Baminercept) in the treatment of pSS is currently in progress. Our results suggest that epigenetic factors may play a role in LTα overexpression in pSS, as cytosine demethylation predisposes to transcriptionally-permissive chromatin architecture.
Patients with pSS have an activated type I IFN response (16, 26), demonstrated by increased type I IFN activity and an “IFN signature” in peripheral blood mononuclear cells (27), saliva (28), and minor salivary gland biopsies (29, 30). Several hypomethylated genes that we identified in naïve CD4+ T in pSS patients were interferon-regulated genes (STAT1, IFI44L, IFITM, and USP18). The extent of hypomethylation in interferon-regulated genes in pSS seems to be less robust in comparison to SLE (9) where we have recently identified and reproduced 21 hypomethylated interferon-regulated genes in naïve CD4+ T cells.
We identified several differentially methylated genes that are important in activation of the immune system. CD247 encodes for TCR-zeta chain, is important for signal transduction upon antigen stimulation (31) and was hypomethylated in naïve CD4+ T cells in pSS. Other pertinent genes include PTPRC, which encodes for protein tyrosine phosphatase, receptor type C (also known as CD45 antigen or B220). PTPRC is a signaling molecule that is essential for T and B cell activation, cellular differentiation and oncogenic transformation (32). TNFRSF25, which plays a role in lymphocyte homeostasis and in apoptosis, was also hypomethylated in pSS. We demonstrated hypomethylation of Glutathione S-Transferase M1 (GSTM1), which encodes a cytoplasmic glutathione S-transferase that is important in detoxification of electrophilic compounds, including carcinogens and environmental toxins (33). Of particular interest, GSTM1 was proposed as a genetic risk locus for pSS in one study (34), which might suggest a role for genetic-epigenetic interaction in the pathogenesis of pSS. Indeed, we have previously reported the genetic association in pSS with variants in methyl-CpG-binding protein 2 (MECP2), a key transcriptional regulator with a critical role in DNA methylation (35). Among the transcription factors, RUNX-1 regulates the differentiation of hematopoietic stem cells into mature cells (36) and has been linked to cancer predisposition (37, 38). RUNX1 was hypermethylated in our study, suggesting a possible connection to lymphoma predisposition in pSS.
It is generally accepted that defects in membrane water channel proteins contribute to the exocrinopathy in pSS (39). Our study identified several differentially methylated genes encoding for members of the solute carrier proteins. The genes SLC11A1, SLC11A2, SLC22A23, SLC25A25, ALC25A3, SLC25A33, and SLC6A20 were hypomethylated, whereas SLC9A1, which is expressed in the kidneys, and plays an important role in the maintenance of pH homeostasis, was hypermethylated in pSS patients compared to controls. Mutation in solute carrier proteins have been previously linked to diseases associated with acid-base disturbances, like Bartter and Gitelman syndromes (40). We hypothesize that methylation changes in these proteins may explain some pathologic aspects of pSS like defects in molecular water transport, dysfunction of the exocrine glands, and perhaps distal renal tubular acidosis in some patients with pSS (41).
We used the DAVID database to facilitate the systematic identification and grouping of differentially methylated genes into biological networks. Of particular interest, there was enrichment of ontologies involved in both the adaptive and innate immune system, underscoring involvement of the two arms of immunity in the pathogenesis of pSS (Table 6).
Two cell separation methods were used to isolate naïve CD4+ T cells from patients and controls, albeit, with consistent and equal cell purity (>95%). We further confirmed the absence T cell activation or differentiation in the samples used in this study, using “epigenetic immunophenotyping” by examining the methylation status of IL2, IFNG, IL4, IL5, IL13, IL17, IL17F, and FOXP3. It is important to note that phenotypic cellular specificity is critical to accurately and reliably interpret differential methylation data in autoimmune diseases, while avoiding any perceived differences that could be related to altered cell constituents.
DNA methylation is tightly linked to chromatin accessibility, and gene expression requires both chromatin accessibility and appropriate transcription factors. Therefore, some but not all of the methylation difference we identified are expected to be associated with expression differences. Examining DNA methylation at a genome-wide level provides another level of discovering fundamental differences that might be pathogenic to the disease process at the chromatin level, which might or might not be linked directly to expression differences. For example, we recently reported wide-spread hypomethylation in interferon-regulated gene in lupus naïve CD4+ T cells, before an expression difference could be detected (9). Interestingly, there are no gene expression studies in naïve CD4+ T cells in pSS to our knowledge, so we do not know which genes are differentially expressed in naïve CD4+ T cells in this disease. Therefore, whether or not some of the methylation changes detected in this first DNA methylome study in pSS will reflect a difference in active (or potential) expression state remains to be seen.
The recent advances in epigenetics and the emergence of “epigenome-wide association studies” resulted in successfully identifying numerous intriguing associations between epigenomic perturbations and human disease. However, this field is still in its infancy and the issue of “causality” in the epigenetic changes detected will require careful experimental examination. The “causality” inference issue in pSS is even more complicated for several reasons: first, incomplete understanding of the genetic predisposition to pSS, which might influence epigenetic variations; second, the environmental factors involved in the pathogenesis of pSS remain by large uncharacterized; and third, the retrospective case-control design of our study. The alternatives are longitudinal studies of monozygotic twins discordant for pSS, or prospective studies to evaluate epigenetic perturbations prior to the onset of the disease, both study designs are cumbersome for obvious reasons, however, such studies are warranted in the future to move this field forward (42). Further, integrating emerging genomic data in pSS with epigenomic profiling might provide an avenue to discover mechanistic pathogenic pathways to this disease. For example, we suspect that some genetic susceptibility variants that will be discovered for pSS might influence some of the methylation differences observed. A simultaneous genomic-epigenomic analysis should therefore focus on allele-specific methylation changes induced by genetic variants associated with pSS. These approaches will help to identify novel mechanisms and therapeutic targets for this disease, and might enhance our understanding for the functional consequences of some of the genetic susceptibility loci in pSS.
In summary, we identified DNA methylation changes for the first time in naïve CD4+ T cells from pSS patients. These data indicate that abnormal DNA methylation exists in pSS CD4+ T cells even before activation and differentiation. Therefore, our findings emphasize the potential role of DNA methylation changes in the pathogenesis of pSS. Our study demonstrates differential methylation of LTA, type I interferon-regulated genes, and solute carrier proteins, in addition to other key genes and pathways involved in the pathogenesis of pSS. Future studies to replicate and determine the functional consequences of the methylation changes observed upon disease pathophysiology are warranted.
Supplementary Material
Supplementary Figure 1: Correlation in differentially methylated (DM) loci between the HumanMethylation450 BeadChip and bisulfite sequencing. DM loci of seven genes (3 hypomethylated: HDAC4, GSTM1, RUFY; 4 hypermethylated: DUSP22, NAPRT1, SLC38A4, SLC9A1) were validated using bisulfite sequencing. The X-axis depicts the percent methylated cytosines by bisulfite sequencing, while the Y-axis depicts the results from the methylation array. The average Pearson’s correlation coefficient (r2) value between the Illumina Infinium HumanMethylation450 array data and bisulfite sequencing data was 0.803.
Supplementary Table 2: The methylation status of CpG sites in the promoter region of genes known to demethylate upon T cell stimulation (IL2) and T cell differentiation (IL4, IL5, IL13, IFNG, IL17 (IL17A), IL17F, FOXP3). There was no significant difference in methylation status of these genes.
Acknowledgments
This work was supported by the National Institutes of Health through R01AI097134 from the National Institute of Allergy and Infectious Diseases and the Lupus Research Institute, and through P50AR060804 from the National Institute for Arthritis and Musculoskeletal and Skin Diseases. The authors are grateful to the clinicians and staff of the OMRF Sjögren’s Research Clinic (Kim Hefner, David Lewis and Glen Houston) and the expert technical assistance of Christina Lawrence
Footnotes
Conflict of interest: The authors have no relevant conflicts of interest to disclose.
References
- 1.Fox RI. Sjogren’s syndrome. Lancet. 2005;366(9482):321–31. doi: 10.1016/S0140-6736(05)66990-5. [DOI] [PubMed] [Google Scholar]
- 2.Delaleu N, Jonsson R, Koller MM. Sjogren’s syndrome. European journal of oral sciences. 2005;113(2):101–13. doi: 10.1111/j.1600-0722.2004.00183.x. [DOI] [PubMed] [Google Scholar]
- 3.Hughes GR, Whaley K. Sjogren’s syndrome. British medical journal. 1972;4(5839):533–6. doi: 10.1136/bmj.4.5839.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassan SS, Thomas TL, Moutsopoulos HM, Hoover R, Kimberly RP, Budman DR, et al. Increased risk of lymphoma in sicca syndrome. Annals of internal medicine. 1978;89(6):888–92. doi: 10.7326/0003-4819-89-6-888. [DOI] [PubMed] [Google Scholar]
- 5.Kassan SS, Moutsopoulos HM. Clinical manifestations and early diagnosis of Sjogren syndrome. Archives of internal medicine. 2004;164(12):1275–84. doi: 10.1001/archinte.164.12.1275. [DOI] [PubMed] [Google Scholar]
- 6.Altorok N, Sawalha AH. Epigenetics in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. 2013 doi: 10.1097/BOR.0b013e328364206f. [DOI] [PubMed] [Google Scholar]
- 7.Thabet Y, Le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, et al. Epigenetic dysregulation in salivary glands from patients with primary Sjogren’s syndrome may be ascribed to infiltrating B cells. J Autoimmun. 2013;41:175–81. doi: 10.1016/j.jaut.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Jeffries MA, Sawalha AH. Epigenetics in systemic lupus erythematosus: leading the way for specific therapeutic agents. International journal of clinical rheumatology. 2011;6(4):423–39. doi: 10.2217/ijr.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. Journal of autoimmunity. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Annals of the rheumatic diseases. 2002;61(6):554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4(5):P3. [PubMed] [Google Scholar]
- 12.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 13.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4(3):235–40. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 14.Bird A. Il2 transcription unleashed by active DNA demethylation. Nat Immunol. 2003;4(3):208–9. doi: 10.1038/ni0303-208. [DOI] [PubMed] [Google Scholar]
- 15.Murayama A, Sakura K, Nakama M, Yasuzawa-Tanaka K, Fujita E, Tateishi Y, et al. A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. Embo J. 2006;25(5):1081–92. doi: 10.1038/sj.emboj.7601012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mavragani CP, Crow MK. Activation of the type I interferon pathway in primary Sjogren’s syndrome. Journal of autoimmunity. 2010;35(3):225–31. doi: 10.1016/j.jaut.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9(1):71–9. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 18.Fava RA, Browning JL, Gatumu M, Skarstein K, Bolstad AI. LTBR-pathway in Sjogren’s syndrome: CXCL13 levels and B-cell-enriched ectopic lymphoid aggregates in NOD mouse lacrimal glands are dependent on LTBR. Advances in experimental medicine and biology. 2011;691:383–90. doi: 10.1007/978-1-4419-6612-4_39. [DOI] [PubMed] [Google Scholar]
- 19.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annual review of immunology. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 20.Browning JL, Dougas I, Ngam-ek A, Bourdon PR, Ehrenfels BN, Miatkowski K, et al. Characterization of surface lymphotoxin forms. Use of specific monoclonal antibodies and soluble receptors. J Immunol. 1995;154(1):33–46. [PubMed] [Google Scholar]
- 21.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annual review of immunology. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 22.Gatumu MK, Skarstein K, Papandile A, Browning JL, Fava RA, Bolstad AI. Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjogren’s syndrome in salivary glands of non-obese diabetic mice. Arthritis research & therapy. 2009;11(1):R24. doi: 10.1186/ar2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen L, Suresh L, Wu J, Xuan J, Li H, Zhang C, et al. A role for lymphotoxin in primary Sjogren’s disease. J Immunol. 2010;185(10):6355–63. doi: 10.4049/jimmunol.1001520. [DOI] [PubMed] [Google Scholar]
- 24.Shen L, Suresh L, Li H, Zhang C, Kumar V, Pankewycz O, et al. IL-14 alpha, the nexus for primary Sjogren’s disease in mice and humans. Clin Immunol. 2009;130(3):304–12. doi: 10.1016/j.clim.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Fava RA, Kennedy SM, Wood SG, Bolstad AI, Bienkowska J, Papandile A, et al. Lymphotoxin-beta receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjogren’s syndrome. Arthritis research & therapy. 2011;13(6):R182. doi: 10.1186/ar3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary Sjogren’s syndrome: a possible etiopathogenic mechanism. Arthritis and rheumatism. 2005;52(4):1185–95. doi: 10.1002/art.20998. [DOI] [PubMed] [Google Scholar]
- 27.Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, et al. Peripheral blood gene expression profiling in Sjogren’s syndrome. Genes and immunity. 2009;10(4):285–96. doi: 10.1038/gene.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu S, Wang J, Meijer J, Ieong S, Xie Y, Yu T, et al. Salivary proteomic and genomic biomarkers for primary Sjogren’s syndrome. Arthritis and rheumatism. 2007;56(11):3588–600. doi: 10.1002/art.22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis and rheumatism. 2005;52(5):1534–44. doi: 10.1002/art.21006. [DOI] [PubMed] [Google Scholar]
- 30.Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2770–5. doi: 10.1073/pnas.0510837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weissman AM, Hou D, Orloff DG, Modi WS, Seuanez H, O’Brien SJ, et al. Molecular cloning and chromosomal localization of the human T-cell receptor zeta chain: distinction from the molecular CD3 complex. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(24):9709–13. doi: 10.1073/pnas.85.24.9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark MC, Pang M, Hsu DK, Liu FT, de Vos S, Gascoyne RD, et al. Galectin-3 binds to CD45 on diffuse large B-cell lymphoma cells to regulate susceptibility to cell death. Blood. 2012;120(23):4635–44. doi: 10.1182/blood-2012-06-438234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun ZF, Zhang J, Xu HM, Wang GL, Dong P. Association between GSTM1 polymorphism and nasopharyngeal cancer susceptibility: a meta-analysis. Asian Pacific journal of cancer prevention : APJCP. 2012;13(11):5817–21. doi: 10.7314/apjcp.2012.13.11.5817. [DOI] [PubMed] [Google Scholar]
- 34.Morinobu A, Kanagawa S, Koshiba M, Sugai S, Kumagai S. Association of the glutathione S-transferase M1 homozygous null genotype with susceptibility to Sjogren’s syndrome in Japanese individuals. Arthritis and rheumatism. 1999;42(12):2612–5. doi: 10.1002/1529-0131(199912)42:12<2612::AID-ANR15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 35.Cobb BL, Fei Y, Jonsson R, Bolstad AI, Brun JG, Rischmueller M, et al. Genetic association between methyl-CpG binding protein 2 (MECP2) and primary Sjogren’s syndrome. Annals of the rheumatic diseases. 2010;69(9):1731–2. doi: 10.1136/ard.2009.122903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okuda T, Nishimura M, Nakao M, Fujita Y. RUNX1/AML1: a central player in hematopoiesis. International journal of hematology. 2001;74(3):252–7. doi: 10.1007/BF02982057. [DOI] [PubMed] [Google Scholar]
- 37.Asou N. The role of a Runt domain transcription factor AML1/RUNX1 in leukemogenesis and its clinical implications. Critical reviews in oncology/hematology. 2003;45(2):129–50. doi: 10.1016/s1040-8428(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 38.Kundu M, Compton S, Garrett-Beal L, Stacy T, Starost MF, Eckhaus M, et al. Runx1 deficiency predisposes mice to T-lymphoblastic lymphoma. Blood. 2005;106(10):3621–4. doi: 10.1182/blood-2005-04-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolov NP, Illei GG. Pathogenesis of Sjogren’s syndrome. Current opinion in rheumatology. 2009;21(5):465–70. doi: 10.1097/BOR.0b013e32832eba21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Archiv : European journal of physiology. 2004;447(5):465–8. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 41.Pertovaara M, Korpela M, Kouri T, Pasternack A. The occurrence of renal involvement in primary Sjogren’s syndrome: a study of 78 patients. Rheumatology (Oxford, England) 1999;38(11):1113–20. doi: 10.1093/rheumatology/38.11.1113. [DOI] [PubMed] [Google Scholar]
- 42.Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. International journal of epidemiology. 2012;41(1):161–76. doi: 10.1093/ije/dyr233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Correlation in differentially methylated (DM) loci between the HumanMethylation450 BeadChip and bisulfite sequencing. DM loci of seven genes (3 hypomethylated: HDAC4, GSTM1, RUFY; 4 hypermethylated: DUSP22, NAPRT1, SLC38A4, SLC9A1) were validated using bisulfite sequencing. The X-axis depicts the percent methylated cytosines by bisulfite sequencing, while the Y-axis depicts the results from the methylation array. The average Pearson’s correlation coefficient (r2) value between the Illumina Infinium HumanMethylation450 array data and bisulfite sequencing data was 0.803.
Supplementary Table 2: The methylation status of CpG sites in the promoter region of genes known to demethylate upon T cell stimulation (IL2) and T cell differentiation (IL4, IL5, IL13, IFNG, IL17 (IL17A), IL17F, FOXP3). There was no significant difference in methylation status of these genes.