

Abstract

Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
Keywords: GS-9350, cobicistat, pharmacoenhancer, PK enhancer, CYP3A inhibitor, ritonavir
The advent of highly active antiretroviral therapy (HAART) has transformed human immunodeficiency virus (HIV) infection to a manageable chronic disease in little over a decade. The use of a combination of antiretroviral drugs inhibiting different stages of the HIV life cycle has led to durable and full viral suppression, resulting in an increase in CD4 T cell counts, thus delaying disease progression and reducing the mortality rate.1,2 However, poor compliance with antiretroviral therapy increases the risk of incomplete viral suppression and the emergence of drug resistance. Adherence, driven by a low pill burden, convenient dosing schedule, and improved tolerability and safety profile, plays a critical role in achieving long-term efficacy of HAART and preventing the emergence of drug-resistant variants.3,4
Ritonavir (RTV, 1) (Scheme 1) has played a key role in enabling a better adherence to HAART regimens that contain a protease inhibitor (PI). As a class, HIV PIs are metabolized rapidly primarily by cytochrome P-450 enzymes of the 3A subfamily (CYP3A) in the liver and intestine, resulting in low systemic exposure and short half-lives after oral administration.5 RTV, although approved as an HIV PI (600 mg twice daily), is primarily used at a subtherapeutic dose (often 100−200 mg daily) in combination with other HIV PI drugs to “boost” their plasma concentrations. This widely accepted use of RTV as a pharmacokinetic (PK) enhancer (or pharmacoenhancer, booster) simplifies regimens and facilitates compliance by reducing the pill burden and dosing frequency.6−8 While a direct interaction between the RTV and the heme group of the CYP3A enzyme may play a role in the inhibition of CYP3A,9 several studies have shown that RTV predominantly exerts its boosting effect through mechanism-based inactivation of CYP3A.11−14 Using either recombinant CYP3A4 or hepatic microsomes, inactivation kinetic parameters kinact and KI ranged from 0.14 to 0.45 min−1 and 0.04 to 0.38 μM, respectively. This is consistent with the clinical observation that the pharmacoenhancement effect of RTV results from inhibition of CYP3A and that it persists beyond the time that the plasma concentrations of RTV are adequately above or close to Ki values conferring direct inhibition of the enzymes.16−18
Scheme 1. Synthesis of Desoxy-ritonavir.

Conditions: (i) 1,1′-Thiocarbonyldiimidazole (TCDI), 1,2-dichloroethane, 75 °C. (ii) 2,2′-Azobisisobutyronitrile (AIBN), Bu3SnH, toluene, 115 °C.
Besides affecting the PKs of HIV PI drugs, RTV has been shown in clinical studies to enhance the PKs of important antiviral drugs that are CYP3A substrates, including the HIV-1 integrase inhibitor elvitegravir,19 the CCR5 antagonist maraviroc,20,22 and the HCV PI narlaprevir.23 Elvitegravir is metabolized primarily by CYP3A4 in the liver and intestine. When dosed alone, elvitegravir needs to be administered 400 mg twice daily to achieve a plasma trough concentration that is adequately above its protein binding-adjusted EC95. When coadministered with 100 mg of RTV, elvitegravir can be dosed once daily to achieve this plasma level. However, the use of a subtherapeutic dose of RTV may have the potential to select PI-resistant virus in a non-PI-containing regimen. In addition, RTV has other disadvantages, including causing lipid disorders and triggering undesired drug interactions as an inducer of drug-metabolizing enzymes such as CYP, p-glycoprotein (Pgp), and UDP glucuronosyltransferases (UGT).
To enable a broader use of once-daily elvitegravir, we initiated a discovery effort to identify a new pharmacoenhancer with the following targeted properties: (1) no or minimal anti-HIV activity, (2) a mechanism of CYP inhibition similar to RTV, (3) higher aqueous solubility and better physicochemical properties, (4) reduced off-target drug interactions, and (5) improved tolerability with minimal side effects due to lipid disorder and gastrointestinal disturbance.
Because RTV (1) is a unique pharmacoenhancer that exerts sustained pharmacological effects with a record of long-term safety demonstrated in clinical settings, our strategy was to eliminate its anti-HIV activity while maintaining its potent inhibitory activity on CYP3A enzymes. Our initial attempts to eliminate the antiviral activity of RTV were focused on the removal of the key hydroxyl group that mimics the transition state of amide hydrolysis through the formation of hydrogen bonds to the oxygens of the catalytic Asp25 and Asp25′ residues at the active site of the HIV protease.24 Desoxy-ritonavir (2) was obtained from RTV by formation of a thiolester and subsequent free radical-mediated deoxygenation (Scheme 1).25 As expected, compound 2 was less potent than RTV in cell-based antiviral assay but retained full inhibitory activity against CYP3A. However, compound 2 still retained detective anti-HIV activity and other unfavorable properties, so after extensive structure−activity relationship studies with compound 2, we report herein the identification of cobicistat (GS-9350, 3; Scheme 2), a potent, selective, and orally bioavailable inhibitor of CYP3A that is currently undergoing clinical investigation as a pharmacoenhancer.
Scheme 2. Synthesis of Compound 3.

Conditions: (i) SO3-pyridine, Et3N, DMSO; VCl3-(THF) 3, Zn, THF. (ii) 1,1′-Thiocarbonyldiimidazole (TCDI), THF, 70 °C; P(OEt)3, 160 °C; H2, 10% Pd/C, MeOH. (iii) 1,1′-Carbonyldiimidazole, diisopropylethylamine, CH2Cl2. (iv) NaOH, MeOH; BnBr. (v) SO3-pyridine, Et3N, DMSO; morpholine, NaBH(OAc) 3, CH3CN. (vi) Diisopropylethylamine, CH3CN. (vii) NaOH, EtOH; 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, 1-hydroxy-benzotrizole, diisopropylethylamine.
Compound 3 (GS-9350, cobicistat) was prepared via the route outlined in Scheme 2 by coupling of intermediate 13 and the acid generated from 11. The synthesis of 13 starts with symmetric 1,4-diamine 6, which was prepared in five steps from commercially available Cbz-l-phenylalaninol as previously described.26 The installation of monothiazole methyl carbamate was achieved through treatment of diamine 6 with carbonate 12 using 1 equiv of base and separation from the minor biscarbamate side product to provide monocarbamate 13. The synthesis of 11 starts with carbonyl diimidazole-mediated coupling of thiazolyl amine 7 with lactone 8 to afford urea 9. Hydrolysis of lactone 9 followed by in situ protection of acid to its benzyl ester yielded urea ester 10. Oxidation of alcohol 10 to the corresponding aldehyde, followed by the reductive amination of morpholine with the resulting aldehyde led to the formation of amine 11. Finally, hydrolysis of ester 11 to the corresponding acid and subsequent coupling with the amine 13 gave compound 3.
Compound 3 was tested in both HIV-1 protease enzymatic assay and antiviral cellular assays. It is inactive against HIV-1 protease (IC50 > 30 μM) and has no inhibitory effect against HIV replication in a multicycle 5-day MT-2 HIV infection assay in the absence or presence of human serum (EC50 > 30 μM, Table 1). In addition, compound 3 showed minimal cytotoxicity, with a CC50 value above 80 μM in assays using MT-2 cells.
Table 1. Inhibition of HIV Protease and HIV Infectiona.
| compound | IC50 (μM) HIV-1 protease | EC50 (μM) MT-2 cells |
|---|---|---|
| 3 | >30 | >30 |
| 1 (RTV) | 0.0006 ± 0.0001 | 0.0133 ± 0.0047 |
| 2 | 0.0045 ± 0.001 | 0.29 ± 0.15 |
Data shown represent the means and SDs from two independent experiments.
The mode of inhibition of human CYP3A by compound 3 was extensively compared with that of RTV. Similar to RTV, 3 inhibitory effects on CYP3A may involve direct interaction and mechanism-based inhibition. Besides interacting directly at the heme group of the CYP3A enzyme, 3 is also an effective mechanism-based inhibitor of CYP3A with an inhibitory potency dependent upon its metabolism. A protocol similar to that described by Ernest et al.6 was used to measure the parameters of inactivation kinetics, kinact and KI, using midazolam as the CYP3A substrate.27 Importantly, compound 3 and RTV inactivate CYP3A similarly at both low and high concentrations and in a time- and concentration-dependent manner. The corresponding estimates of kinact and KI are presented in Table 2. These results suggest that RTV and 3 share the same mechanism of action for the inhibition of CYP3A.
Table 2. Kinetics for the Inactivation of Human Hepatic Microsomal CYP3A-Dependent Midazolam 1′-Hydroxylase Activity.
| parameter | compound 1 (RTV) | compound 3 |
|---|---|---|
| kinact (min−1) | 0.23 ± 0.06 | 0.44 ± 0.09 |
| KI (nM) | 256 ± 90 | 939 ± 353 |
| kinact/KI (min−1 μM−1) | 0.90 | 0.57 |
The mechanism-based inactivation of human CYP3A by compound 3 implies that, in the clinic, sustained CYP3A inhibition will likely be achieved even after 3 is cleared from the body and before new CYP3A enzymes are synthesized by the hepatocytes, as the half-life of CYP3A turnover has been estimated to be around 12−48 h.28In vitro data also suggest that compound 3 will inhibit CYP3A with a similar potency as RTV in human. This has recently been demonstrated in clinical studies, where 3 showed similar potency to RTV in affecting the PKs of the model CYP3A substrate, midazolam. Compound 3 also showed dose nonlinear and time-dependent PKs, consistent with it being a mechanism-based inhibitor.29
CYP3A enzymes are known to display substrate dependence in their susceptibility to inhibition. To confirm that compound 3 also retains broad substrate specificity similar to that of RTV in inhibiting CYP3A, we compared the abilities of RTV and 3 to inhibit metabolism of a diverse set of substrates using assay protocols based on current industry and regulatory guidelines.27,30 The potency of compound 3 as an inhibitor of human hepatic microsomal CYP3A was compared to that of RTV using substrates that include terfenadine, midazolam, testosterone, atazanavir (ATV), telaprevir, and elvitegravir. The inhibitory potency was either measured with marker activities for the enzymes or determined by monitoring substrate depletion. As shown in Table 3, the broad spectrum of inhibitory activity of compound 3 against human CYP3A was confirmed.
Table 3. Inhibitory Potencies against Activities Catalyzed by CYP3A and Other Major Human Hepatic Microsomal Cytochromes P450.
| calculated IC50 (μM) |
|||
|---|---|---|---|
| enzyme | activity | RTV | compound 3 |
| CYP3A | midazolam 1′-hydroxylase | 0.107 | 0.154 |
| testosterone 6β-hydroxylase | 0.116 | 0.151 | |
| terfenadine oxidase30 | 0.275 | 0.285 | |
| elvitegravir oxidase | 0.026 | 0.033 | |
| ATV oxidation | 0.040 | 0.044 | |
| telaprevir oxidation | 0.018 | 0.030 | |
| CYP1A2 | phenacetin O-deethylase | >25 | >25 |
| CYP2B6 | bupropion 4-hydroxylase | 2.9 | 2.8 |
| CYP2C8 | paclitaxel-6α-hydroxylase | 2.8 | >25 |
| CYP2C9 | tolbutamide 4-hydroxylase | 4.4 | >25 |
| CYP2C19 | S-mephenytoin 4′-hydroxylase | >25 | >25 |
| CYP2D6 | dextromethorphan O-demethylase | 2.8 | 9.2 |
The inhibitory kinetic parameters (Table 2) and broad spectrum of inhibitory activity (Table 3) of compound 3 are similar to those of RTV. It retains the characteristic of mechanism-based inhibition of CYP3A and is equipotent to RTV for the substrates tested. Inhibition studies with the most important human CYP enzymes showed no significant inhibition at concentrations likely to be achieved clinically (Table 3). In addition, compound 3 is more selective than RTV with much reduced inhibitory activity toward CYP2D6, CYP2C8, and CYP2C9.
In addition to exhibiting inhibitory effects with enzymes other than CYP3A, RTV activates the pregnane X receptor (PXR), the predominant regulator of CYP3A expression. Although the net effect of RTV on human CYP3A clinically is inhibitory, this inhibitory potency is reduced upon chronic dosing, as the rate of CYP3A resynthesis is increased via induction. Other proteins induced by RTV in vivo include CYP2B6, CYP2C9, CYP2C19, UGT1A4 (UGT 1 family, polypeptide A4), and P-gp, further complicating potential drug−drug interactions.32,33 It is thus desirable to eliminate or reduce this drug interaction potential for new PK enhancers. Studies to assess the induction liability of compound 3 were performed. These studies used human hepatoma cells constitutively expressing human PXR or the aryl hydrocarbon receptor (AhR) proteins. The cells were also stably transfected with firefly luciferase as a reporter under the control of the CYP3A4 promoter (for PXR cells) or the human CYP1A2 promoter (for AhR cells). Cells were maintained in medium containing 10% (v/v) fetal bovine serum and were exposed to test compound for 24 h prior to assay of luciferase activity. Positive control inducers with a range of potencies were tested in parallel, and all were compared to the response seen with the vehicle control (0.1% v/v DMSO). The response seen with 10 μM rifampicin was considered to be 100% of the Emax.
Figure 1.
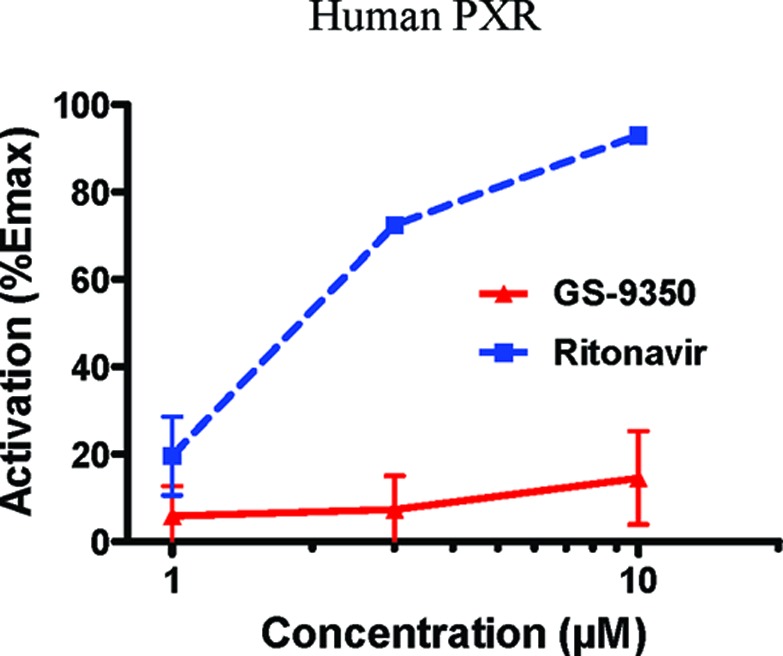
Comparison of RTV and GS-9350 (3) to activate human PXR. Values represent the means of duplicate determinations.
Neither 3 nor RTV showed significant stimulatory activity in the AhR-responsive assay. However, at 10 μM in the PXR assay, RTV exhibited 93% of Emax, and the corresponding EC50 was 1.9 μM. RTV is thus confirmed as a potent PXR agonist with the potential to achieve significant activation in human where the Cmax is around 2 μM. This is consistent with the significant induction by RTV of proteins regulated by PXR seen in the clinic. Compound 3 was considerably weaker than RTV in activating PXR with 15% of Emax detected at a 10 μM concentration. The curve shapes also suggest that 100% response with 3 would be difficult to achieve. Compound 3 is thus much weaker than RTV as an activator of PXR and is potentially less likely to cause clinical drug−drug interactions through induction.
Chronic treatment of HIV-infected patients with RTV is known to induce changes in body fat distribution (lipodystrophy), elevated cholesterol and triglycerides (hyperlipidemia), and insulin resistance, known as metabolic syndrome.34 It is believed that some of these effects are at least in part due to the direct effects of RTV on adipocytes. In vitro, RTV has been shown to affect adipocyte functions such as lipid accumulation during differentiation and insulin-stimulated glucose uptake. Therefore, compound 3 was evaluated for its effects on adipocytes in comparison with RTV. ATV, the PI used in the clinic with the least pronounced metabolic syndrome,37 was also evaluated as a comparator.
The lipid accumulation assay monitored normal lipid accumulation in cultured human adipocytes following induction of differentiation in the presence of tested PIs for 9 days. RTV showed a clear effect with an EC50 of 16 μM (Table 4). In contrast, both 3 and ATV exhibited no effect at a concentration up to 30 μM. The glucose uptake assay monitored insulin-stimulated glucose uptake in mouse adipocytes in the presence of 10 μM RTV, ATV, or 3. RTV showed a pronounced effect at this concentration. In contrast, the effects on glucose uptake by 3 and ATV were significantly less. The minimal adverse effects of 3 in these assays suggest a lower potential for toxicity related to altered lipid metabolism as compared to RTV.
Table 4. Inhibition of Cell Functions in Adipocytesa.
| compound | lipid accumulation EC50 (μM) | glucose uptake (% inhibition at 10 μM) |
|---|---|---|
| 3 | >30 | 9.5 ± 6.4 |
| 1 (RTV) | 16 ± 8 | 55 ± 10 |
| ATV | >30 | 0.4 ± 0.9 |
Data shown represent the mean and s.d. from at least 3 independent experiments.
Compound 3 has a greatly improved aqueous solubility as compared with RTV at both neutral (pH 7.4: 75 vs ∼2.0 μg/mL) and acidic (pH 2.2: >6500 vs 3.1 μg/mL) conditions. Compound 3 has been formulated as a tablet, as well as coformulated with other agents.
Both compound 3 and RTV exhibit poor metabolic stability with preclinical species in vitro. Their metabolic stability is concentration-dependent, and both can inhibit their own metabolism at high concentrations. The PK of compound 3 in preclinical animals is consistent with this, as high clearance is observed at low doses. Volumes of distribution are moderate. Because of mechanism-based inhibition of human CYP3A and consequent self-inhibition of their clearance, the absorption potential in human is the more relevant PK parameter for 3 and RTV. The absorption potential was evaluated in portal vein cannulated dogs. Results indicated that the absorption of 3 is above 50% in this species, comparable to those of RTV. Compound 3 was therefore expected to have a high absorption potential in humans.
In summary, compound 3 (cobicistat, GS-9350) is a potent and selective human CYP3A inhibitor that lacks significant anti-HIV activity. In vitro studies also suggest that 3 may have a lower potential for causing undesired drug−drug interactions and lipid disorders than RTV. On the basis of these results, compound 3 was selected as a clinical candidate for further development. It has been coformulated as a tablet with elvitegravir/emtricitabine/tenofovir DF, a single tablet fixed-dose regimen (FDR) known as Quad, which is currently being tested in the clinic. The size of Quad is smaller than Atripla. In phase I studies, at 100 and 200 mg once daily dosing, 3 reduced the clearance of midazolam by 90 and 95%, respectively.16,29 When used as a component of the integrase FDR Quad, 3 (150 mg once daily) enhanced the PK of elvitegravir to similar levels (Ctrough) as boosted with 100 mg of once daily RTV.16,29 Additionally, 3 boosted plasma concentrations of ATV to levels that were bioequivalent to those obtained when coadministered with 100 mg of once daily RTV.39 Compound 3 is currently being evaluated in phase II studies in HIV-1-infected subjects, both as a component of Quad and as a pharmacoenhancer for ATV.
Acknowledgments
We thank William Lee, Randy Vivian, You-Chul Choi, Jennifer Chau, Quynh Iwata, Chris Yang, Eugene Eisenberg, Leah Tong, Swami Swaminathan, Chin Tay, Tomas Cihlar, Roy Bannister, Carina Cannizzaro, Joanna Koziara, and Kathy Brendza for their contributions and Weidong Zhong for his valuable suggestions on the manuscript.
Supporting Information Available
Biological assays, cytochrome P450 inhibition determination, inactivation of human CYP3A activity and induction assessment, preclinical PKs, experimental procedures, and analytical data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Hammer S. M.; Eron J. J.; Reiss P.; Schooley R. T.; Thompson M. A.; Walmsley S.; Cahn P.; Fischl M. A.; Gatell J. M.; Hirsh M. S.; Jacobsen D. M.; Montaner J. S. G.; Richman D. D.; Yeni P. G.; Volberding P. A.. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the international AIDS society USA panel. J. Am. Med. Assoc. 2008, 300, 555−570 and references therein. [DOI] [PubMed] [Google Scholar]
- Panel on Antiretroviral Guidelines for Adults and Adolescent, Department of Health and Human Services (DHHS). Guidelines for the Use of Antiretroviral Agents in HIV-1 Infected Adults and Adolescents; December 1, 2009; pp 1−168 and references therein. Available at ContentFiles/AdultandAdolescentGL.pdf. [Google Scholar]
- Willig J. H.; Abroms S.; Westfall A. O.; Routman J.; Adusumilli S.; Varshney M.; Allison J.; Chatham A.; Raper J. L.; Kaslow R. A.; Saag M. S.; Mugavero M. J.. Increased regimen durability in the era of once-daily fixed-dose combination antiretroviral therapy. AIDS 2008, 22, 1951−1960 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parienti J.-J.; Bangsberg D. R.; Verdon R.; Gardner E. M.. Better adherence with once-daily antiretroviral regimens: A meta-analysis. Clin. Infect. Dis. 2009, 48, 484−488 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber J. G.Using pharmacokinetics to optimize antiretroviral drug-drug interactions in the treatment of human immunodeficiency virus infection. Clin. Infect. Dis. 2000, 30 (Suppl. 2), S123−S129 and references therein. [DOI] [PubMed] [Google Scholar]
- Cooper C. L.; van Heeswijk R. P. G.; Gallicano K.; Cameron D. W. A review of low-dose ritonavir in protease inhibitor combination therapy. Clin. Infect. Dis. 2003, 36, 1585–1592. [DOI] [PubMed] [Google Scholar]
- Youle M.Overview of boosted protease inhibitors in treatment-experienced HIV-infected patients. J. Antimicrob. Chemother. 2007, 60, 1195−1205 and references therein. [DOI] [PubMed] [Google Scholar]
- Busse K. H.; Penzak S. R.. Pharmacological enhancement of protease inhibitor with ritonavir: An update. Expert Rev. Clin. Pharmacol. 2008, 1, 533−545 and references therein. [DOI] [PubMed] [Google Scholar]
- Kempf D. J.; Marsh K. C.; Kumar G.; Rodrigues A. D.; Denissen J. F.; McDonald E.; Kukulka M. J.; Hsu A.; Granneman G. R.; Baroldi P. A.; Sun E.; Pizzuti D.; Plattner J. J.; Norbeck D. W.; Leonard J. M. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke L. L.; Durol A. L. B.; Duan S. X.; Greenblatt D. J. Potent mechanism based inhibition of human CYP3A in vitro by amprenavir and ritonavir. Eur. J. Clin. Pharmacol. 2000, 56, 259–261. [DOI] [PubMed] [Google Scholar]
- Ernest C. S. 2nd; Hall S. D.; Jones D. R. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J. Pharmacol. Exp. Ther. 2005, 312, 583–591. [DOI] [PubMed] [Google Scholar]
- Obach R. S.; Walsky R. L.; Venkatakrishnan K. Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos. 2007, 35, 246–255. [DOI] [PubMed] [Google Scholar]
- Mathias A. A.; West S.; Hui J.; Kearney B. P. Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin. Pharmacol. Ther. 2009, 85, 64–70. [DOI] [PubMed] [Google Scholar]
- Xu L.; Desai M. C. Pharmacokinetic enhancers for HIV drugs. Curr. Opin. Invest. Drugs 2009, 10, 775–786. [PubMed] [Google Scholar]
- Murray B. P. Mechanism-based inhibition of CYP3A4 and other cytochromes P450. Annu. Rep. Med. Chem. 2009, 44, 535–551. [Google Scholar]
- DeJesus E.; Berger D.; Markowitx M.; Cohen C.; Hawkins T.; Ruane P.; Elion R.; Farathing C.; Zhong L.; Cheng A. K.; McColl D.; Kearney B. P. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naïve and treatment-experienced patients. JAIDS 2006, 43, 1–5. [DOI] [PubMed] [Google Scholar]
- Ramanathan S.; Abel S.; Tweedy S.; West S.; Hui J.; Kearney B. P. Pharmacokinetic interaction of ritonavir-boosted elvitegravir and maraviroc. JAIDS 2010, 53, 209–214. [DOI] [PubMed] [Google Scholar]
- Pozniak A. L.; Boffito M.; Russell D.; Ridgway C.; Muirhead G. A novel probe drug interaction study to investigate the effect of selected antiretroviral combinations on the pharmacokinetics of a single oral dose of maraviroc in HIV-positive subjects. Br. J. Clin. Pharmacol. 2008, 65Suppl. 154–59and references therein.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierling J.; Poordad F.; Lawitz E.. Once daily narlaprevir (SCH 900518) in combination with PEGINTRON (peginterferon alfa-2b)/ribavirin for treatment-naïve subjects with genotype-1 CHC: Interim results from NEXT-1, a Phase 2a study. AASLD 2009, Abstract #LB4. [Google Scholar]
- For illustration that ritonavir binds to HIV protease, see Kempf D. J.; Marsh K. C.; Denissen J. F.; McDonald E.; Vasavanonda S.; Flentge C. A.; Green B. E.; Fino L.; Park C. H.; Kong X.-P.; Wideburg N. E.; Saldivar A.; Ruiz L.; Kati W. M.; Sham H. L.; Robins T.; Stewart K. D.; Hsu A.; Plattner J. J.; Leonard J. M.; Norbeck D. W.. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 2484−2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D. H. R.; McCombie S. W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc. Perkins I 1975, 1574. [Google Scholar]
- Xu L.; Desai M. C.; Liu H. A novel and efficient synthesis of chiral C2-symmetric 1,4-diamines. Tetrahedron Lett. 2009, 50, 552–554. [Google Scholar]
- Bjornsson T. D.; Callaghan J. T.; Einolf H. J.; Fischer V.; Gan L.; Grimm S.; Kao J.; King S. P.; Miwa G.; Ni L.; Kumar G.; McLeod J.; Obach R. S.; Roberts S.; Roe A.; Shah A.; Snikeris F.; Sullivan J. T.; Tweedie D.; Vega J. M.; Walsh J.; Wrighton S. A. The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. 2003, 31, 815−832 and references therein. [DOI] [PubMed] [Google Scholar]
- Yang J.; Liao M.; Shou M.; Jamei M.; Yeo K. R.; Tucker G. T.; Rostami-Hodjegan A.. Cytochrome P450 turnover: Regulation of synthesis and degradation, methods for determining rates, and implications for the predictions of drug interactions. Curr. Drug Metab. 2008, 9, 384−93 and references therein. [DOI] [PubMed] [Google Scholar]
- Mathias A.; German P.; Murray B. P.; Wei L.; Jain A.; West S.; Warren D; Hui J.; Kearney B. P. Pharmacokinetics and pharmacodynamics of GS-9350: A novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010, 87, 322–329. [DOI] [PubMed] [Google Scholar]
- FDA, HHS. Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling, 2006; http://www.fda.gov/cder/guidance/6695dft.pdf. [DOI] [PubMed]
- Kharasch E. D.; Mitchell D.; Coles R.; Blanco R. Rapid clinical induction of hepatic cytochrome P4502B6 activity by ritonavir. Antimicrob. Agents Chemother. 2008, 52, 1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisy M. M.; Yakiwchuk E. M.; Hughes C. A. Induction effects of ritonavir: Implications for drug interactions. Ann. Pharmacother. 2008, 42, 1048–1059. [DOI] [PubMed] [Google Scholar]
- Carr A.; Samaras K.; Burton S.; Law M.; Freund J.; Chisholm D. J.; Cooper D. A. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 1998, 12, F51–F58. [DOI] [PubMed] [Google Scholar]
- Gazzard B.; Moyle G. Dose atazanavir cause lipodystrophy?. J. HIV Ther. 2004, 9, 41–44. [PubMed] [Google Scholar]
- Ramanathan S.; Warren D.; Wei L.; Kearney B. P.. Pharmacokinetic boosting of atazanavir with the pharmacoenhancer GS-9350 versus ritonavir. ICAAC 2009, Abstract #A1-1301. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.