

ABSTRACT
Human norovirus (NoV) accounts for 95% of nonbacterial gastroenteritis worldwide. Currently, there is no vaccine available to combat human NoV as it is not cultivable and lacks a small-animal model. Recently, we demonstrated that recombinant vesicular stomatitis virus (rVSV) expressing human NoV capsid protein (rVSV-VP1) induced strong immunities in mice (Y. Ma and J. Li, J. Virol. 85:2942–2952, 2011). To further improve the safety and efficacy of the vaccine candidate, heat shock protein 70 (HSP70) was inserted into the rVSV-VP1 backbone vector. A second construct was generated in which the firefly luciferase (Luc) gene was inserted in place of HSP70 as a control for the double insertion. The resultant recombinant viruses (rVSV-HSP70-VP1 and rVSV-Luc-VP1) were significantly more attenuated in cell culture and viral spread in mice than rVSV-VP1. At the inoculation dose of 1.0 × 106 PFU, rVSV-HSP70-VP1 triggered significantly higher vaginal IgA than rVSV-VP1 and significantly higher fecal and vaginal IgA responses than rVSV-Luc-VP1, although serum IgG and T cell responses were similar. At the inoculation dose of 5.0 × 106 PFU, rVSV-HSP70-VP1 stimulated significantly higher T cell, fecal, and vaginal IgA responses than rVSV-VP1. Fecal and vaginal IgA responses were also significantly increased when combined vaccination of rVSV-VP1 and rVSV-HSP70 was used. Collectively, these data indicate that (i) insertion of an additional gene (HSP70 or Luc) into the rVSV-VP1 backbone further attenuates the VSV-based vaccine in vitro and in vivo, thus improving the safety of the vaccine candidate, and (ii) HSP70 enhances the human NoV-specific mucosal and T cell immunities triggered by a VSV-based human NoV vaccine.
IMPORTANCE Human norovirus (NoV) is responsible for more than 95% of acute nonbacterial gastroenteritis worldwide. Currently, there is no vaccine for this virus. Development of a live attenuated vaccine for human NoV has not been possible because it is uncultivable. Thus, a live vector-based vaccine may provide an alternative vaccine strategy. In this study, we developed a vesicular stomatitis virus (VSV)-based human NoV vaccine candidate. We constructed rVSV-HSP70-VP1, coexpressing heat shock protein (HSP70) and capsid (VP1) genes of human NoV, and rVSV-Luc-VP1, coexpressing firefly luciferase (Luc) and VP1 genes. We found that VSVs with a double gene insertion were significantly more attenuated than VSV with a single VP1 insertion (rVSV-VP1). Furthermore, we found that coexpression or coadministration of HSP70 from VSV vector significantly enhanced human NoV-specific mucosal immunity. Collectively, we developed an improved live vectored vaccine candidate for human NoV which will be useful for future clinical studies.
INTRODUCTION
The Caliciviridae family includes a number of significant enteric viruses that cause gastroenteritis in humans and animals. Examples of these viruses include human norovirus (NoV), bovine NoV, porcine NoV, human sapovirus, and a newly discovered monkey calicivirus (Tulane virus [TV]) (1). It has been a challenge to work on these viruses because most of them cannot be grown in cell culture (2). Currently, human NoV and other caliciviruses are classified as category B biodefense agents by the National Institute of Allergy and Infectious Diseases (NIAID) because they are highly contagious, extremely stable, and resistant to common disinfectants, require a low infectious dose, and are associated with debilitating illness (3–5). Despite the fact that human NoV causes significant health, emotional, social, and economic burdens worldwide, there are no vaccines or antiviral drugs available to combat this infectious agent.
Development of an efficacious vaccine for human NoV has been hampered as it is not cultivable in vitro and lacks a small-animal model (2, 6, 7). The generation of human NoV virus-like particles (VLPs) has opened an alternative strategy to develop a vaccine for this virus. It has been reported that the expression of capsid gene (VP1) alone in cell culture resulted in self-assembled VLPs that are structurally and antigenically similar to native virions (8, 9). In fact, our understanding of human NoV-host interaction has been largely shaped by using VLPs to define functional receptors of human NoV, the histo-blood group antigens (10). To date, the baculovirus-insect cell expression system has been widely used for production of human NoV VLPs. Immunization of mice with VLPs orally or intranasally induced variable humoral, mucosal, and cellular immunities (7, 11–15). In fact, a baculovirus-derived VLP vaccine candidate is currently in human clinical trials (16). Volunteers that received the dry powder VLP vaccine reduced their risk of illness by 47% after exposure to human NoV. There were significant reductions in clinical norovirus illness, infection, and severity of illness in individuals who received vaccine compared with those who received the placebo. Although these studies are promising, the efficacy of this vaccine needs to be further improved. It is not known whether a VLP-based vaccine can completely protect humans from reinfection. In addition, the duration of the protection is a concern because VLPs are nonreplicating immunogens. Therefore, there is a critical need to explore other vaccine candidates such as a live vectored vaccine candidate for human NoV.
Previously, a recombinant vaccinia stomatitis virus (VSV) (rVSV-VP1) that expresses the major capsid protein of human NoV was generated (17). The yield of VLPs by the VSV expression system is approximately 10 times higher than that of the baculovirus expression system. Recombinant rVSV-VP1 was attenuated in cell culture as well as in mice compared to parental VSV. Mice inoculated with a single dose of rVSV-VP1 through intranasal and oral routes exhibited a significantly stronger humoral and cellular immune response than baculovirus-expressed VLP vaccination. In addition, mice inoculated with rVSV-VP1 had comparable levels of fecal and vaginal IgA antibodies. These findings demonstrated that the VSV expression system is not only a highly productive system to generate VLPs in vitro but also a promising vectored vaccine candidate for human NoV. However, recombinant rVSV-VP1 still causes significant body weight loss in mice even though it is attenuated compared to rVSV. In addition, whether the efficacy of this vaccine candidate can be enhanced by insertion of an adjuvant gene is not known.
To further improve the VSV-based human NoV vaccine, an adjuvant gene and human NoV VP1 were coexpressed in the VSV vector. Heat shock protein 70 (HSP70) was chosen as a vaccine adjuvant because it has been shown to modulate both innate and adaptive immune responses (18–21). Heat shock proteins (HSPs) are a family of intracellular molecular chaperones with essential functions in folding and intracellular translocation of client proteins. HSPs are classified according to mass and occur in all cell compartments, with the major inducible 70-kDa HSP (HSP70) being a predominant cytosolic member. In addition to serving as a molecular chaperone, it has been demonstrated that HSP70 supports presentation (and cross-presentation) of antigen peptides. HSP70 can enhance T and B cell-mediated adaptive immunity by stimulating maturation and cytokine secretion by antigen-presenting cells (APCs) and upregulates molecules involved in antigen presentation (major histocompatibility complex [MHC] class I and II and costimulatory markers) and adhesion (22, 23). HSP70 may also enhance safety of the vaccine vector by virtue of HSP70's ability to directly induce type I interferon (IFN) expression. The latter hypothesis is based on the observation that HSP70-mediated induction of type I IFN is correlated to reduced neurovirulence for both measles virus (MeV) (24) and VSV (25) in the mouse.
The present work tested the hypothesis that HSP70 would enhance the immune response to VP1 such that both HSP70 and VP1 are expressed from the VSV genome. Coexpression was performed by use of a double-genome insertion to express both HSP70 and VP1, and the contribution of HSP70 was confirmed by coadministration of constructs containing single-genome insertions of either HSP70 or VP1. In order to control for the effects of the double-genome insertion when measuring HSP70-mediated effects on the immune response and vaccine safety, a construct was generated in which the luciferase gene was expressed in place of the HSP70 gene. Constructs used to test the hypothesis included recombinant VSVs expressing HSP70 (rVSV-HSP70) or VP1 (rVSV-VP1), VSV coexpressing HSP70 and VP1 (rVSV-HSP70-VP1), and VSV coexpressing luciferase (Luc) and VP1 (rVSV-Luc-VP1). Subsequently, the levels of safety and efficacy of recombinants rVSV-VP1, rVSV-Luc-VP1, and rVSV-HSP70-VP1 or of coadministration of rVSV-VP1 and rVSV-HSP70 were compared in a mouse model. It was found that insertion of either HSP70 or Luc into rVSV-VP1 resulted in both in vitro and in vivo attenuation. However, HSP70, but not Luc, significantly enhanced the human NoV-specific mucosal and T cell immunities triggered by the VSV-based human NoV vaccine.
MATERIALS AND METHODS
Recombinant viruses.
Wild-type rVSV and the recombinant viruses rVSV-VP1 and rVSV-Luc were described previously (17). Recombinant rVSV-VP1 harbors the VP1 gene of human NoV at the VSV gene junction between the G and L gene sequences, whereas rVSV-Luc contains the firefly luciferase (Luc) gene at the VSV gene junction between the leader sequence and the N gene.
Plasmid construction.
Plasmids encoding VSV N (pN), P (pP), and L (pL) genes, and an infectious cDNA clone of the viral genome, pVSV1(+), were generous gifts from Gail Wertz (26). Plasmids pVSV1(+) GxxL and pVSV1(+) LexxN were kindly provided by Sean Whelan. Plasmid pVSV1(+) GxxL contains SmaI and XhoI at the G and L gene junction. Plasmid pVSV1(+) LexxN contains SmaI and XhoI at the leader sequence and N gene junction. The capsid VP1 gene of human NoV genogroup II.4 strain HS66 was kindly provided by Linda Saif. The VP1 and HSP70 genes were amplified by high-fidelity PCR and cloned into pVSV(+)GxxL at the SmaI and XhoI sites, which resulted in the construction of plasmids pVSV1(+)-VP1 and pVSV1(+)-HSP70, respectively. A third plasmid [pVSV1(+) Le-HSP70-N] was constructed by insertion of the HSP70 gene into pVSV1(+) LexxN at the leader and N gene junction. To construct a plasmid carrying both HSP70 and VP1 genes, a DNA fragment containing the VP1 gene was recovered by digestion of pVSV1(+)-VP1 with HpaI and KpnI enzymes and cloned into the same sites in pVSV1(+) Le-HSP70-N. The HSP70 gene was inserted into the leader and N gene junction, and the human NoV VP1 gene was inserted into the G and L gene junction; the resulting plasmid was designated pVSV1(+)-HSP70-VP1. Finally, the HSP70 gene was replaced by the firefly luciferase (Luc) gene in plasmid pVSV1(+)-HSP70-VP1, which resulted in the construction of pVSV1(+)-Luc-VP1. All inserted genes contained the VSV gene start and gene end sequences. All plasmid constructs were confirmed by sequencing.
Recovery and purification of recombinant VSV.
The recovery of recombinant VSV from the infectious clone was carried out as described previously (17). Briefly, BSRT7 cells were infected with a recombinant vaccinia virus (vTF7-3) expressing T7 RNA polymerase, followed by cotransfection with a plasmid carrying the VSV genome [pVSV1(+)-HSP70, pVSV1(+)-HSP70-VP1, or pVSV1(+)-Luc-VP1] and plasmids pN, pP, and pL. At 96 h posttransfection, cell culture fluids were collected and filtered through a 0.2-μm-pore-size filter, and the recombinant VSV was further amplified in BSRT7 cells. Subsequently, the recovered viruses were plaque purified as described previously. Individual plaques were isolated, and seed stocks were amplified in BSRT7 cells. The viral titer was determined by a plaque assay performed in Vero cells.
Single-cycle growth curves.
Confluent BSRT7 cells were infected with individual viruses at a multiplicity of infection (MOI) of 10. After 1 h of absorption, the inoculum was removed, the cells were washed twice with Dulbecco's modified Eagle's medium (DMEM), fresh DMEM (supplemented with 2% fetal bovine serum) was added, and the infected cells were incubated at 37°C. Aliquots of the cell culture fluid were removed at specific time points, and the virus titer at each time point was determined by plaque assay in Vero cells.
Analysis of protein synthesis.
Confluent BSRT7 cells were infected with rVSV, rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, or rVSV1-Luc-VP1 at an MOI of 10 as described above. At 3 h postinfection, cells were washed with methionine- and cysteine-free (M−C−) medium and incubated with fresh M−C− medium supplemented with actinomycin D (15 μg/ml). After 1 h of incubation, the medium was replaced with M−C− medium supplemented with EasyTag 35S-Express (Perkin-Elmer, Wellesley, MA) (4 μCi/ml). After 4 h of incubation, cytoplasmic extracts were prepared and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously. Labeled proteins were detected either by autoradiography or by using a phosphorimager.
RT-PCR.
Viral RNA was extracted from rVSV, rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, or rVSV1-Luc-VP1 using an RNeasy minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Two primers (5′-CGAGTTGGTATTTATCTTTGC-3′ and 5′-GTACGTCATGCGCTCATCG-3′) were designed to target the VSV G gene at position 4524 and the L gene at position 4831 (numbering refers to the complete VSV Indiana genome sequence), respectively. These two primers were used to detect the insertion of VP1 or HSP70 genes at the G and L gene junction. Another two primers [VSV(+)1-17, 5′-ACGAAGACAAACAAACC-3′, and VSV(-), 5′-CCTCATTTGCAGGAAG-3′] were designed to target the VSV leader sequence at position 1 and the N gene at position 115. These two primers were used to detect the HSP70 or Luc gene which was inserted between the VSV leader and N gene junction. Reverse transcription-PCR (RT-PCR) was performed using a OneStep RT-PCR kit (Qiagen). The amplified products were analyzed by 1% agarose gel electrophoresis.
Western blotting.
BSRT7 cells were infected with rVSV, VSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, or rVSV-Luc-VP1 at an MOI of 10 as described above. At specific time points postinfection, cells were lysed in buffer containing 5% β-mercaptoethanol, 0.01% NP-40, and 2% SDS. Proteins were separated by 12% SDS-PAGE and transferred to a Hybond enhanced chemiluminescence nitrocellulose membrane (GE Healthcare, Piscataway, NJ) in a Mini Trans-Blot electrophoretic transfer cell (Bio-Rad, Hercules, CA). The blot was probed with guinea pig anti-human NoV VP1 antiserum (a generous gift from Xi Jiang) at a dilution of 1:5,000, followed by horseradish peroxidase-conjugated goat anti-guinea pig IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:20,000. The HSP70 protein was detected with mouse anti-HSP70 monoclonal antibody (Enzo Life Sciences, Farmingdale, NY) at 1:2,000, followed by horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (Thermo Scientific). The blot was developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and exposed to Kodak BioMax MR film.
Production and purification of VLPs by a baculovirus expression system.
Purification of human NoV VLPs from insect cells using a baculovirus expression system was performed as described previously (17). Briefly, Spodoptera frugiperda (Sf9) cells were infected with baculovirus expressing the human NoV VP1 protein at an MOI of 10, and the infected Sf9 cells and cell culture supernatant were harvested at 6 days postinoculation. The VLPs were purified from cell culture supernatant and cell lysate by ultracentrifugation through a 40% (wt/vol) sucrose cushion, followed by CsCl isopycnic gradient (0.39 g/cm3) ultracentrifugation. Purified VLPs were analyzed by SDS-PAGE, Western blotting, and electron microscopy (EM). The protein concentrations of the VLPs were measured using Bradford reagent (Sigma Chemical Co., St. Louis, MO).
Animal experiment 1.
Thirty-five 4-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories, Wilmington, MA) were randomly divided into seven groups (five mice per group). Mice in groups 1, 2, 3, 4, and 5 were inoculated with 106 PFU of rVSV, rVSV-Luc, rVSV-VP1, rVSV-HSP70, and rVSV-HSP70-VP1, respectively. Mice in group 6 were inoculated with 100 μg of VLPs (purified from the baculovirus expression system). Mice in group 7 were inoculated with 50 μl of DMEM and served as unimmunized controls. All mice were inoculated through the combination of intranasal and oral routes. Half of the inoculum was administered intranasally, and the other half was administered orally. After inoculation, the animals were evaluated on a daily basis for mortality, weight loss, and the presence of any symptoms of VSV infection, including ruffled fur, hyperexcitability, tremors, circling, and paralysis. Blood samples were collected from each mouse weekly by facial vein bleed, and serum was isolated for IgG antibody detection. Fecal and vaginal swab homogenate samples were isolated weekly for the detection of norovirus-specific IgA. At 5 weeks postinoculation, all mice were sacrificed. Spleens were isolated from each mouse, and mononuclear cell (MNC) suspensions were prepared for T cell proliferation analyses. (All animal studies in this study were approved by the Institutional Animal Care and Use Committee [protocol no. 2009A0160] at The Ohio State University.)
Animal experiment 2.
Twenty-five 4-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories) were randomly divided into five groups (five mice per group). Mice in groups 1 and 2 were inoculated with 106 PFU of rVSV-VP1 or rVSV-HSP70-VP1. Mice in groups 3 and 4 were inoculated with 5 × 106 PFU of rVSV-VP1 or rVSV-HSP70-VP1. Mice in group 5 were inoculated with 50 μl of DMEM and served as unimmunized controls. The remaining procedure was identical to that used for animal experiment 1.
Animal experiment 3.
Fifteen 4-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories) were randomly divided into three groups (five mice per group). Mice in group 1 were inoculated with 106 PFU of rVSV-VP1. Mice in group 2 were inoculated with 106 PFU of rVSV-VP1 and 106 PFU of rVSV-HSP70. Mice in group 3 were inoculated with 50 μl of DMEM and served as unimmunized controls. The remaining procedure was identical to that used for animal experiment 1.
Animal experiment 4.
Thirty-two 4-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories) were randomly divided into 4 groups (8 mice per group). Mice in groups 1, 2, and 3 were inoculated with rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1, respectively. Mice in group 4 were inoculated with DMEM and served as uninfected controls. Each mouse was inoculated intranasally at a dose of 106 PFU in a volume of 50 μl. After inoculation, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection. The severity of clinical signs associated with VSV infection was scored based on the following criteria: grade 3 (severe) was characterized by ruffled fur, hyperexcitability, tremors, circling, and paralysis; grade 2 (moderate) was characterized by ruffled fur with neurological symptoms such as circling; grade 1 (mild) was characterized by ruffled fur, with no neurological symptoms; and grade 0 mice exhibited no symptoms. The body weight for each mouse was monitored on a daily basis. At days 3 and 5 postinoculation, 4 mice from each group were euthanized. The brain from each mouse was collected for virus titration by plaque assay.
Animal experiment 5.
Twenty 6-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories) were randomly divided into four groups (five mice per group). Mice in groups 1, 2, and 3 were inoculated intranasally with 106 PFU of rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1. Mice in groups 4 were inoculated with 50 μl of DMEM and served as unimmunized controls. The remaining procedure was identical to that used for animal experiment 1.
T cell proliferation assay.
Ninety-six-well plates were coated with 50 μl of highly purified human NoV VLPs (10 μg/ml) in 200 mM NaCO3 buffer (pH 9.6) at 4°C overnight. After homogenization, spleen cells were washed twice with phosphate-buffered saline (PBS) and plated in triplicate at 5 × 105 cells/well in a 96-well plate in RPMI 1640 medium with 2% naive mouse serum. After 48 h of incubation at 37°C, 0.5 μCi of [3H]thymidine was added to each well and incubated for 16 h. Following incubation, cells were transferred onto glass filters and counted with a Betaplate counter (Wallac, Turku, Finland). The stimulation index (SI) was calculated as the mean of the following ratio: incorporation of [3H]thymidine in human NoV VLP-stimulated cells/incorporation of [3H]thymidine in nonstimulated cells (in cpm).
Serum IgG ELISA.
Ninety-six-well plates were coated with 50 μl of highly purified human NoV VLPs (7.5 μg/ml) in 50 mM NaCO3 buffer (pH 9.6) at 4°C overnight. Individual serum samples were tested for human NoV-specific IgG on VLP-coated plates. Briefly, serum samples were 2-fold serially diluted and added to VLP-coated wells. After incubation at room temperature for 1 h, the plates were washed five times with PBS-Tween (0.05%), followed by incubation with 50 μl of goat anti-mouse IgG horseradish peroxidase-conjugated secondary antibody (Sigma) at a dilution of 1:80,000 for 1 h. Plates were washed and developed with 75 μl of TMB (3,3′,5,5′-tetramethylbenzidine), and the optical density (OD) at 450 nm was determined using an enzyme-linked immunosorbent assay (ELISA) plate reader. Endpoint titer values were determined as the reciprocal of the highest dilution that had an absorbance value greater than the background level (DMEM control).
HBGA blocking assay.
A histo-blood group antigen (HBGA) blocking assay was performed to measure the ability of serum antibodies to inhibit human NoV VLP binding to H type 1 synthetic carbohydrates using a method described previously (27). Briefly, human NoV VLPs (0.32 μg/ml) were incubated with an equal volume of serial 2-fold diluted serum from mice. Neutravidin-coated, 96-well microtiter plates were coated with 2.5 μg/ml of synthetic polyvalent Lewis d (H type 1)-polyacrylamide (PAA)-biotin (Glycotech) for 1 h at 22°C. Plates were washed 3 times with 0.1 mol/liter sodium phosphate buffer (pH 6.4). The serum-VLP solutions were added to the plates and incubated at 4°C for 2 h. Plates were washed, and then human NoV-specific rabbit polyclonal serum (provided by Xi Jiang) (dilution, 1:5,000) was incubated for 1 h at 4°C. The plates were washed again, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (Sigma) (dilution, 1:5,000) was added and the mixture was incubated for 1 h at 4°C. The signal was then developed by adding tetramethylbenzidine peroxidase liquid substrate (Kirkegaard and Perry Laboratory) and stopped after 10 min of incubation at 22°C by adding 1 mol/liter phosphoric acid. Optical density (OD) was measured at 450 nm with the use of a SpectraMax M5 plate reader (Molecular Devices). Blank wells were incubated with buffer instead of serum-VLP and served as a negative control, whereas VLP binding to carbohydrates in the absence of a serum sample was used as a positive control. The 50% blocking titer (BT50), defined as the titer at which the OD value (after subtraction of the blank) was 50% of the OD value of the positive control, was determined for each sample.
Fecal IgA ELISA.
For each stool sample, human NoV-specific and total fecal IgA levels were determined as described previously (17). Fecal pellets were diluted 1:2 (wt/vol) in PBS containing 0.1% Tween and a Complete EDTA-free proteinase inhibitor cocktail tablet (Roche, Mannheim, Germany). Samples were mixed twice for 30 s using a vortex device and clarified twice by centrifugation at 10,000 × g for 10 min. Ninety-six-well plates were coated with 50 μl of highly purified human NoV VLPs (1 μg/ml) in 50 mM NaCO3 buffer (pH 9.6) at 4°C overnight for detection of human NoV-specific IgA, while total fecal IgA was determined by capturing all fecal extract IgA molecules with 1 μg/ml sheep anti-mouse IgA (Sigma). To block nonspecific protein binding, the plates were incubated for 4 h at 4°C with 10% (wt/vol) dry milk–PBS (10% Blotto). The level of IgA was calculated from a standard curve that was determined by the absorbance values of the mouse IgA standard (Sigma). The human NoV-specific IgA level was expressed in nanograms per milliliter, and each corresponding total IgA level was expressed in micrograms per milliliter.
Vaginal IgA ELISA.
Ninety-six-well plates were coated with human NoV VLPs in selected columns as described above. After an overnight blocking at 4°C with 5% Blotto, 75 μl of an undiluted vaginal sample per well or a 1:5 dilution of the sample was added, and the sample was serially diluted 2-fold on the plate and incubated for 2 h at 37°C. The remaining protocol was identical to that described above for the human NoV-specific fecal IgA ELISA or the serum IgG ELISA.
Quantitative and statistical analyses.
Quantitative analysis was performed either by densitometric scanning of autoradiographs or by using a Typhoon PhosphorImager and ImageQuant TL software (GE Healthcare, Piscataway, NJ). Each experiment was performed three to six times. Statistical analysis was performed by one-way multiple comparisons using Minitab 16 statistical analysis software (Minitab Inc., State College, PA). A P value of <0.05 was considered statistically significant.
RESULTS
Recovery of recombinants rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1.
Previously, we recovered recombinant rVSV-VP1 and rVSV-Luc, in which human NoV VP1 or firefly luciferase (Luc) genes were inserted at the G-L gene junction and the leader-N gene junction, respectively (17). Using a similar approach, we successfully recovered three additional recombinant viruses, rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1 (Fig. 1A). Recombinant rVSV-HSP70 harbors the HSP70 gene at the G-L gene junction. Recombinant rVSV-HSP70-VP1 contains double insertions with HSP70 at the leader-N gene junction and VP1 at the G-L gene junction. Recombinant rVSV-Luc-VP1 contains double insertions with Luc at the leader-N gene junction and VP1 at the G-L gene junction. As shown in Fig. 1B, the plaque sizes of these recombinant viruses in Vero cells differed, depending on the site of the insertion. Recombinants rVSV-VP1, rVSV-Luc, and rVSV-HSP70 formed plaques of similar sizes, but these plaques were smaller than those of wild-type rVSV (P < 0.05). Interestingly, the doubly inserted recombinant viruses, rVSV-HSP70-VP1 and rVSV-Luc-VP1, formed much smaller plaques than rVSV-VP1 (P < 0.05) (Fig. 1B). After 24 h of incubation, rVSV formed plaques that were 4.1 ± 0.4 mm (mean ± standard deviation) in diameter. The average plaque sizes for rVSV-VP1, rVSV-Luc, and rVSV-HSP70 were 1.7 ± 0.6 mm, 1.2 ± 0.4 mm, and 1.6 ± 0.3 mm, respectively, after 48 h of incubation. However, the plaque sizes for rVSV-HSP70-VP1 and rVSV-Luc-VP1 were 0.8 ± 0.4 mm and 1.1 ± 0.5 mm, respectively, after 48 h of incubation; the difference was not statistically significant (P > 0.05). This result suggests that growth of virus containing double genome insertions (i.e., rVSV-HSP70-VP1 and rVSV-Luc-VP1) is impaired relative to that of rVSV-VP1. To confirm that the recovered virus indeed contained the target genes, viral genomic RNA was extracted from each recombinant virus followed by RT-PCR using primers annealing either to the VSV leader sequence and N gene or to the G and L genes. As shown in Fig. 1C, a 2.1-kb cDNA band containing the VP1 gene and a 2.3-kb band containing the HSP70 gene were amplified from genomic RNA extracted from rVSV-HSP70-VP1 and rVSV-HSP70, respectively, indicating that VP1 or HSP70 was inserted at the G and L gene junction in these two recombinant viruses. In addition, a 2.1-kb cDNA band containing the HSP70 gene was amplified from rVSV-HSP70-VP1 using primers annealing to the VSV leader and the N gene, indicating that HSP70 was inserted at the gene junction between the leader and the N gene. Similarly, a 1.8-kb band (Luc) and a 1.9-kb band (VP1) were amplified from rVSV-Luc-VP1. Subsequently, the amplified cDNA was purified and sequenced, confirming that VP1, HSP70, or Luc was indeed inserted into the targeted position in VSV genome. Finally, the entire genome of each recombinant was sequenced to confirm that no additional mutations had been introduced.
FIG 1.

Recovery of rVSV-HSP70-VP1, rVSV-HSP70, and rVSV-Luc-VP1. (A) Construction of plasmids. For rVSV-VP1 and rVSV-HSP70, VP1 and HSP70 were inserted at the G-L gene junction in their respective VSV genomes. For rVSV-HSP70-VP1, the HSP70 and VP1 genes were inserted at gene junctions between leader and N and between G and L, respectively. For rVSV-Luc-VP1, the luciferase and VP1 genes were inserted at gene junctions between leader and N and between G and L, respectively. le, VSV leader sequence; N, nucleocapsid gene; P, phosphoprotein gene; M, matrix protein gene; G, glycoprotein gene; L, large polymerase gene; tr, VSV trailer sequence. (B) Plaque diameter of recombinant viruses. Plaques of rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1 were developed after 48 h of incubation compared to rVSV, which was developed after 24 h of incubation. An average size of 20 plaques for each recombinant virus is shown. (C) Amplification of the inserted genes from recombinant viruses by RT-PCR. Genomic RNA was extracted from each virus. The VP1 gene was amplified by RT-PCR using two primers annealing to the G and L genes. The HSP70 gene was amplified from rVSV-HSP70 using the two primers annealing to the G and L genes. The HSP70 or Luc gene was amplified from rVSV-HSP70-VP1 or rVSV-Luc-VP1 using two primers annealing to the leader and N sequences.
Single-step growth curves of recombinant VSVs in cell culture.
The kinetics of release of infectious virus for each recombinant VSV were compared by using a single-step growth assay in BSRT7 cells. As shown in Fig. 2A, recombinants rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1 had a delay in the exponential rise in viral progeny release and the time at which peak viral progeny was observed relative to those of rVSV. Titers of these recombinants were decreased by 1 to 2 logs relative to rVSV titers during the first 4 to 18 h postinfection. Wild-type rVSV reached a peak titer (4.6 × 109 PFU/ml) at 12 h postinfection. Recombinant rVSV-VP1 reached a peak titer of 4.0 × 109 PFU/ml at approximately 24 h postinfection. However, recombinant rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1 reached peak titers of 2.5 × 109, 7.2 × 108, and 1.2 × 109 PFU/ml, respectively, at approximately 30 to 36 h postinfection. Overall, there is no significant difference in the growth curves for rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1 (P > 0.05).
FIG 2.

Growth and protein synthesis of recombinant VSVs in BSRT7 cells. (A) Single-step growth curve of recombinant VSVs. Confluent BSRT7 cells were infected with individual virus at an MOI of 10. Samples of supernatant were harvested at the indicated intervals over a 48-h time period, and the virus titer was determined by plaque assay. Titers represent the averages of the results of three independent single-step growth experiments. (B) Expression of VP1 and HSP70 by rVSV-VP1, rVSV-HSP70, and rVSV-HSP70-VP1. BSRT-7 cells were infected with each recombinant virus at an MOI of 10. Proteins were metabolically labeled by incorporation of [35S]methionine-cysteine in the presence of actinomycin D. Cytoplasmic extracts were harvested at 5 h postinfection, and proteins were analyzed by SDS-PAGE and detected using a phosphorimager. Arrows denote VP1 protein, and stars denote HSP70 or Luc protein. (C) The level of VSV structural protein expression for recombinant viruses relative to parental rVSV. Data were generated in three independent experiments. For each protein, the mean ± the standard deviation is shown as a percentage of that observed for rVSV. (D) The level of VP1 or HSP70 protein synthesized by recombinant VSVs. The VP1 synthesized by rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1 and the HSP70 synthesized by rVSV-HSP70 and rVSV-HSP70-VP1 were quantified. Data were generated in three independent experiments.
Characterization of the expression of VP1 and HSP70 by the VSV vector.
The expression of VP1 and HSP70 by the VSV vector was determined by [35S] metabolic labeling in virus-infected cells. As shown in Fig. 2B, rVSV synthesized five viral proteins, L, G, P, N, and M. In rVSV-VP1-infected cells, an additional protein band with a molecular mass of approximately 58 kDa was detected (Fig. 2B, lane 2), which correlates to the size of the VP1 protein of human NoV. In rVSV-HSP70-infected cells (Fig. 2B, lane 4), an additional 70-kDa protein band was detected, and this is the correct size for HSP70. In rVSV-HSP70-VP1-infected cells, two additional protein bands with molecular masses of 70 kDa and 58 kDa were detected (Fig. 2B, lane 6), which are masses consistent with the sizes of HSP70 and VP1, respectively. In rVSV-Luc-VP1-infected cells, two additional protein bands with molecular masses of 65 kDa and 58 kDa were detected (Fig. 2B, lane 8), which are masses consistent with the sizes of Luc and VP1, respectively. As shown in Fig. 2C, all recombinants synthesized significantly lower levels of native VSV proteins (particularly L, P, and G proteins) than rVSV. In addition, rVSV-HSP70-VP1 and rVSV-Luc-VP1 expressed much lower levels of VSV proteins than rVSV-VP1 and rVSV-HSP70, suggesting again that they were more attenuated in cell culture. Quantitative analysis showed approximately 30% to 68%, 35% to 65%, 10% to 55%, and 12% to 50% expression of VSV proteins by rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc-VP1, respectively, relative to expression of the wild-type rVSV (Fig. 2C). We also quantified the VP1 synthesized by rVSV-VP1 and rVSV-HSP70-VP1 and the HSP70 synthesized by rVSV-HSP70 and rVSV-HSP70-VP1. Result showed that rVSV-HSP70-VP1 synthesized approximately 13 times more HSP70 protein than rVSV-HSP70 (Fig. 2D), which was due to the fact that rVSV-HSP70-VP1 harbors HSP70 gene at the leader and N gene junction whereas rVSV-HSP70 contains HSP70 at the G and L gene junction. This is also consistent with the VSV protein synthesis strategy, in which the abundance of gene expression decreases with the distance from the 3′ end to the 5′ end of the VSV genome. Similarly, the level of VP1 synthesized by rVSV-VP1 was approximately 5 times more than that synthesized by rVSV-HSP70-VP1 and rVSV-Luc-VP1 (Fig. 2D). This is because the additional insertion of the HSP70 or Luc gene in the rVSV-VP1 backbone resulted in further transcription attenuation of the downstream genes.
The kinetics of VP1 expression by rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1 were monitored by Western blotting. Briefly, BSRT7 cells were infected with rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1 at an MOI of 10, and cell lysates were harvested at various time points. The cell lysates were analyzed by SDS-PAGE, followed by Western blotting using a polyclonal antibody against the VP1 protein. As shown in Fig. 3A, high levels of VP1 protein were detected in cells infected by rVSV-VP1 at 8 h postinfection. The VP1 expression gradually increased to 30 h postinfection. In contrast, VP1 expression in rVSV-HSP70-VP1 and rVSV-Luc-VP1 was significantly delayed and the level of VP1 in rVSV-HSP70-VP1 and rVSV-Luc-VP1 was significantly lower than that in rVSV-VP1. VP1 was not detectable in rVSV-HSP70-VP1 at 8 h postinfection, and only low levels of VP1 were detected at 12 h postinfection. No VP1 protein was detected in rVSV-Luc-VP1-infected cells until 18 h postinfection. Quantitative analysis showed that the VP1 level of rVSV-HSP70-VP1 and rVSV-Luc-VP1 was approximately five times lower than that of rVSV-VP1 at 24 and 30 h postinfection (Fig. 3A, bottom panel), which is consistent with the 35S metabolic labeling experiment (Fig. 2B). No significant difference between rVSV-HSP70-VP1 and rVSV-Luc-VP1 in VP1 expression was observed from 18 h to 30 h postinfection (P > 0.05).
FIG 3.
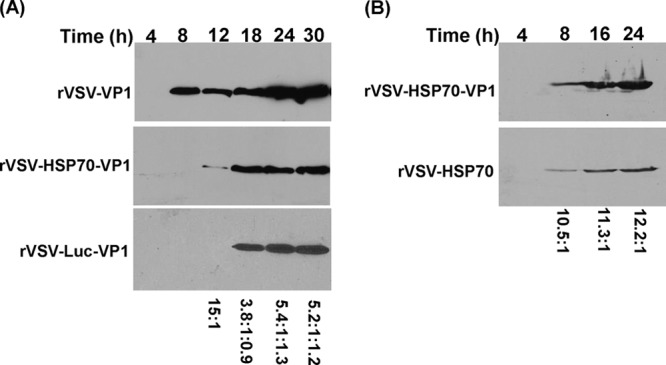
Kinetics of VP1 and HSP70 expression in virus-infected cells. (A) Kinetics of VP1 protein expression. BSRT-7 cells were infected with rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1 at an MOI of 10. Cytoplasmic extracts were harvested at indicated time points. Equal amounts of total cytoplasmic lysate were analyzed by SDS-PAGE, followed by Western blotting using guinea pig anti-human NoV VP1 antiserum. The ratio of VP1 produced by rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1 is shown in the bottom. A representative blot is shown. (B) Kinetics of HSP70 expression. BSRT-7 cells were infected with rVSV-HSP70 or rVSV-HSP70-VP1 at an MOI of 10. Cytoplasmic extracts were harvested at indicated time points. Equal amounts of total cytoplasmic lysate were analyzed by SDS-PAGE, followed by Western blotting using mouse anti-HSP70 antibody. The ratio of HSP70 produced by rVSV-HSP70-VP1 and rVSV-HSP70 is shown in the bottom. A representative blot is shown.
The kinetics of HSP70 expression by rVSV-HSP70 and rVSV-HSP70-VP1 were monitored by Western blotting. As shown in Fig. 3B, HSP70 protein was detectable in cells infected by both viruses at 8 h postinfection. The HSP70 expression gradually increased between 8 and 24 h postinfection. However, the HSP70 level in rVSV-HSP70-VP1-infected cells was approximately 10 to 12 times higher than that in the rVSV-HSP70-infected cells at the three time points (8, 16, and 24 h postinfection) (Fig. 3B, bottom panel), which is consistent with the 35S metabolic labeling experiment (Fig. 2B).
These results demonstrated that (i) both VP1 and HSP70 proteins were highly expressed by the VSV vector; (ii) the VP1 protein level expressed by rVSV-HSP70-VP1 and rVSV-Luc-VP1 was approximately 5 times lower than that expressed by rVSV-VP1; and (iii) the HSP70 protein expression level of rVSV-HSP70-VP1 was approximately 13 times higher than that of rVSV-HSP70. Collectively, the combination of decreased viral plaque size, delayed single-step viral replication, and reduced VSV protein synthesis suggests that expression of rVSV-HSP70-VP1 and rVSV-Luc-VP1 is more attenuated than that of rVSV-HSP70 and rVSV-VP1 in cell culture.
Recombinant rVSV-HSP70-VP1 expression is more attenuated than rVSV-VP1 expression in a mouse model.
Recombinants rVSV-VP1, rVSV-HSP70, rVSV-HSP70-VP1, and rVSV-Luc were inoculated into mice at a dose of 106 PFU through a combination of intranasal and oral routes. After inoculation, animals were evaluated daily for weight loss and the presence of clinical signs. As shown in Fig. 4, significant differences in mouse body weight changes were observed during days 2 to 14 postinoculation. Mice inoculated with rVSV-Luc had the greatest magnitude and duration of weight loss (P < 0.05) among those inoculated with all the recombinant viruses tested. The rVSV-Luc-inoculated mice exhibited mild clinical signs of VSV infection (i.e., ruffled hair coat) at days 4 to 10 postinoculation. Mice inoculated with rVSV-VP1 and rVSV-HSP70 also showed a moderate weight loss (P < 0.05) at up to 8 days postinoculation. The magnitude of weight loss in these two groups was similar to that seen with rVSV-Luc at 8 days postinoculation but was more transient. For these two groups, weight gain was observed after day 6 postinoculation. No significant clinical signs of VSV infection were observed in these two groups. In contrast, the rVSV-HSP70-VP1 treatment group had significantly less weight loss than the rVSV-VP1 and rVSV-HSP70 treatment groups (P < 0.05) at days 4 to 10 postinoculation, and the body weight of the mice in this group recovered quickly at 1 week postinoculation. In addition, no significant clinical signs were observed during the entire experimental period. Mice inoculated with DMEM and VLPs did not have any weight loss or clinical signs of disease. This experiment indicates that all recombinant viruses were attenuated in mice compared to wild-type rVSV, which causes severe disease and death in the mice. However, rVSV-HSP70-VP1 was more attenuated in mice than other recombinants, including rVSV-VP1, rVSV-HSP70, and rVSV-Luc.
FIG 4.

Dynamics of mouse body weight after inoculation with recombinant viruses. Five BALB/c mice in each group were inoculated with 106 PFU of rVSV-Luc, rVSV-VP1, rVSV-HSP70, or rVSV-HSP70-VP1 or with 100 μg of VLPs (purified from insect cells expressed by baculovirus) through the combination of intranasal and oral routes. Body weight for each mouse was evaluated every other day for 5 weeks. The average body weight of five mice is shown.
Recombinant rVSV-HSP70-VP1 triggered similar levels of serum IgG, T cell, and fecal IgA responses but a significantly higher vaginal IgA response than rVSV-VP1 at a dose of 106 PFU.
To compare the human NoV-specific antibodies triggered by rVSV-VP1 and rVSV-HSP70-VP1, blood samples were isolated from each mouse and the serum IgG antibody response was determined by ELISA. The geometric mean titers (GMT) were calculated for each group of mice. As shown in Fig. 5A, mice inoculated with rVSV-VP1 and rVSV-HSP70-VP1 triggered significantly higher serum IgG responses than mice inoculated with baculovirus-produced VLPs at 2 to 5 weeks postinoculation (P < 0.05). Both rVSV-VP1 and rVSV-HSP70-VP1 had a high level of antibody at 1 week postinoculation and had a continuous increase in antibody titers through week 5. Mice inoculated with 100 μg of baculovirus-expressed VLPs had similar levels of human NoV-specific IgG antibodies at week 1 postinoculation. However, the IgG antibody in the VLP group had decreased by week 2. As controls, mice inoculated with rVSV-Luc, rVSV-HSP70, and DMEM did not have human NoV-specific serum IgG antibody responses during the experimental period. Results showed that rVSV-VP1 and rVSV-HSP70-VP1 had IgG antibody responses comparable to and antibody responses significantly stronger than those of the traditional VLP-based vaccine candidate.
FIG 5.

Immune responses induced by VSV-based human NoV vaccines. (A) Serum IgG immune responses. Groups of five BALB/c mice were inoculated with either 106 PFU of rVSV-VP1 or rVSV-HSP70-VP1 or 100 μg of VLPs through the combination of intranasal and oral routes. Serum samples were collected weekly and analyzed by ELISA using human NoV-specific serum IgG antibody. Data are expressed as geometric mean titers (GMT) determined for five mice. Error bars at each time point represent the standard deviations between the results determined for the mice. (B) T cell proliferative responses. Spleen cells were harvested from all mice in each group at week 5 postinoculation and were stimulated with human NoV VLPs. T cell proliferation was measured by [3H]thymidine incorporation. The stimulation index (SI) was calculated as the mean of the following ratio: proliferation of human NoV VLP-stimulated cells/proliferation of cells in medium in cpm. Data are expressed as the means of the results from five mice ± the standard deviations. Asterisks indicate statistical significance (P < 0.05). (C) Fecal IgA responses. Fecal samples were collected from all mice at week 5 postinoculation. Samples were diluted in PBS, mixed using a vortex device, and clarified by centrifugation. Human NoV-specific antibody and total IgA antibody levels were detected by ELISA. The ratio between human NoV-specific IgA and total IgA was calculated for each mouse. Data are expressed as average titers from all mice ± standard deviations. Numbers of IgA-positive mice are indicated above the error bars. (D) Vaginal IgA responses. Vaginal samples were collected at week 5 postinoculation from each mouse, and human NoV-specific and total IgA antibody levels were determined by ELISA. The level of vaginal IgA is shown as the ratio between human NoV-specific IgA and total IgA. Data are expressed as the average titer for all mice ± standard deviations.
To compare the cellular immune responses, the spleen was isolated from each mouse at week 5 postinoculation, and the cellular immune responses were measured using a T cell proliferation assay. As shown in Fig. 5B, mice inoculated with rVSV-VP1 and rVSV-HSP70-VP1 exhibited human NoV-specific T cell proliferation levels higher than those seen with traditional VLP-based vaccines. All mice in the rVSV-VP1 and rVSV-HSP70-VP1 groups had strong human NoV-specific T cell responses, with average stimulation indices of 9.6 and 12.0, respectively. In contrast, only three of the five mice in the VLP group had a T cell immune response, with an average stimulation index of 4.8. As controls, mice inoculated with rVSV-Luc, rVSV-HSP70, and DMEM had background levels of T cell immune responses. There was no significant difference between the rVSV-VP1 and rVSV-HSP70-VP1 groups in the NoV-specific T cell proliferative response (P > 0.05). Therefore, these data demonstrate that rVSV-VP1 and rVSV-HSP70-VP1 induce similar levels of T cell immune responses that are stronger than those induced by the VLP-based vaccine candidate.
To compare the mucosal immune responses, human NoV-specific and total IgA levels in fecal and vaginal extracts were determined by ELISA as described in Materials and Methods. The level of IgA response was expressed as the ratio between human NoV-specific IgA and total IgA. Prior to antigen inoculation, there was no human NoV-specific IgA in either fecal or vaginal samples from any mice. Figure 5C shows the fecal IgA antibody response at week 5 postinoculation. Only two of five mice had an IgA response in the VLP vaccination groups. All mice in the rVSV-VP1 group developed human NoV-specific IgA, while three of the five mice in the rVSV-HSP70-VP1 group exhibited an IgA response. Figure 5D shows the vaginal IgA antibody response at week 5 postinoculation. All mice in the VLP, rVSV-VP1, and rVSV-HSP70-VP1 groups had a high level of human NoV-specific vaginal IgA antibody. Responses to rVSV-HSP70-VP1 were significantly higher than those observed with VLP and rVSV-VP1 (P < 0.05). None of the mice in the rVSV-Luc, rVSV-HSP70, and DMEM groups showed human NoV-specific fecal and vaginal IgA antibody over the entire experimental period. Therefore, HSP70 enhances vaginal mucosal immunity against human NoV at a dose of 106 PFU/mouse.
Recombinant rVSV-HSP70-VP1 stimulated higher T cell, fecal, and vaginal IgA responses than rVSV-VP1 at a dose of 5 × 106 PFU.
The experiments described above clearly demonstrated that rVSV-HSP70-VP1 and rVSV-VP1 stimulated similar levels of humoral, T cell, and fecal mucosal immunity in mice despite the fact that the level of VP1 protein produced by rVSV-HSP70-VP1 was approximately 5 times less than that of rVSV-VP1, suggesting that HSP70 may play a role in enhancing the immune response triggered by VP1 protein. To further evaluate the vaccine dose response for the two most promising candidates (rVSV-VP1 and rVSV-HSP70-VP1), we increased the vaccination dosage to 5 × 106 PFU per mouse and examined the safety, serum IgG levels, T cell proliferative responses, and mucosal IgA responses as described above. As shown in Fig. 6, a high dose (5 × 106 PFU) of rVSV-VP1 caused significantly more weight loss than a low dose (106 PFU) of rVSV-VP1 (P < 0.05) during days 6 to 18 postinoculation. However, no significant weight differences were seen with the two doses of rVSV-HSP70-VP1 (P > 0.05). Overall, mice receiving 5 × 106 PFU of rVSV-VP1 had weight losses that were more severe than those seen with 5 × 106 PFU of rVSV-HSP70-VP1 (P < 0.05), suggesting that rVSV-HSP70-VP1 was more attenuated than rVSV-VP1. Furthermore, mice inoculated with 5 × 106 PFU of rVSV-HSP70-VP1 did not exhibit any clinical signs of VSV infection whereas mice inoculated with 5 × 106 PFU of rVSV-VP1 exhibited mild clinical signs (i.e., ruffled hair coat).
FIG 6.

Dynamics of mouse body weight after inoculation with recombinant viruses. Five BALB/c mice in each group were inoculated with 106 PFU of rVSV-VP1, 106 PFU of rVSV-HSP70-VP1, 5 × 106 PFU of rVSV-VP1, or 5 × 106 PFU of rVSV-HSP70-VP1. Body weight for each mouse was measured every other day for 5 weeks. The average body weight of five mice per group is shown.
As shown in Fig. 7A, mice inoculated with 5 × 106 and 106 PFU of rVSV-HSP70-VP1 and rVSV-VP1 stimulated similar levels of serum IgG antibody (P > 0.05), suggesting that an increased vaccination dose did not significantly enhance serum antibody responses. Since human NoV cannot be grown in cell culture, there is no standard virus neutralization assay available. Thus, we used a HBGA blocking assay to determine whether serum antibody had potential antiviral capacity. As shown in Fig. 7B, serum samples from vaccinated groups, but not those from the DMEM control group, blocked the binding of human NoV VLPs to HBGA receptors. There was no significant difference in GMTs of BT50 (50% blocking titer) during the 5-week period (P > 0.05), which was consistent with the levels of serum IgG antibodies determined by ELISA.
FIG 7.

The effects of immunization doses on serum IgG immune response triggered by VSV-based human NoV vaccine. (A) Serum IgG antibody responses. Groups of five BALB/c mice were inoculated with 106 PFU of rVSV-VP1, 106 PFU of rVSV-HSP70-VP1, 5 × 106 PFU of rVSV-VP1, or 5 × 106 PFU of rVSV-HSP70-VP1. Serum samples were collected weekly and analyzed by ELISA using human NoV-specific serum IgG antibody. Data are expressed as geometric mean titers (GMT) determined for five mice. Error bars at each time point represent the standard deviations of the results of the comparisons between mice. (B) Serum HBGA blocking antibody. The ability of serum antibodies to inhibit human NoV VLP binding to HBGAs, the functional receptor of human NoV, was measured by an ELISA as described in Materials and Methods. The 50% blocking titer (BT50), defined as the titer at which the OD value (after subtraction of the blank) was 50% of the OD of the positive control, was determined for each sample. Error bars at each time point represent the standard deviations of the results of the comparisons between mice.
Fecal and vaginal IgA antibody responses were also determined. As shown in Fig. 8A, mice inoculated with 5 × 106 PFU of rVSV-HSP70-VP1 stimulated fecal IgA antibody responses that were significantly higher than those of any of the other vaccination groups (P < 0.05). The level of vaginal IgA in 5 × 106 PFU of rVSV-VP1, 5 × 106 PFU of rVSV-VP1-HSP70, and 106 PFU of rVSV-HSP70-VP1 was significantly higher than that in 106 PFU of rVSV-VP1 (P < 0.05) (Fig. 8B). In addition, the level of vaginal IgA in 5 × 106 PFU of rVSV-VP1-HSP70 was significantly higher than that in 106 PFU of rVSV-HSP70-VP1 (P < 0.05) (Fig. 8B). Interestingly, mice inoculated with 5 × 106 PFU of rVSV-HSP70-VP1 had significantly (P < 0.05) enhanced T cell proliferative responses to norovirus antigen compared to all other vaccination groups, including 5 × 106 PFU of rVSV-VP1, 106 PFU of rVSV-VP1, and 106 PFU of rVSV-HSP70-VP1 (Fig. 8C). However, there was no significant difference (P > 0.05) in T cell immune responses between mice vaccinated with 5 × 106 PFU of rVSV-VP1, 106 PFU of rVSV-VP1, or 106 PFU of rVSV-HSP70-VP1. As controls, mice inoculated with DMEM and rVSV-HSP70 defined background levels for NoV-specific serum IgG antibody, HBGA blocking antibody, T cell proliferation, or fecal and vaginal IgA antibody responses. Thus, these data demonstrated that HSP70 enhanced T cell and mucosal immunities but not humoral antibody responses to NoV at an inoculation dose of 5 × 106 PFU per mouse.
FIG 8.

The effects of immunization doses on mucosal and T cell immune responses triggered by VSV-based human NoV vaccine. (A) Fecal IgA responses. Fecal samples were collected from all mice at week 5 postinoculation. Human NoV-specific and total IgA antibody levels were determined by ELISA. The ratio between human NoV-specific IgA and total IgA was calculated for each mouse. Data are expressed as average titers of IgA-positive mice ± standard deviations. (B) Vaginal IgA responses. Vaginal samples were collected at week 5 postinoculation from each mouse, and human NoV-specific and total IgA antibody levels were determined by ELISA. The level of vaginal IgA is shown as log 10 (ratio between human NoV-specific IgA and total IgA). Data are expressed as average titers of IgA-positive mice ± standard deviations. (C) T cell proliferative responses. Spleen cells were harvested from all mice in each group at week 5 postinoculation and stimulated with human NoV VLPs. T cell proliferation was measured by [3H]thymidine incorporation. The stimulation index (SI) was calculated as the mean of the following ratio: proliferation of human NoV VLP-stimulated cells/proliferation of cells in medium in cpm. Data are expressed as the means of the results determined for five mice ± the standard deviations. Asterisks indicate statistical significance (P < 0.05).
Enhanced mucosal immune responses by a combination of rVSV-VP1 and rVSV-HSP70 vaccination.
If HSP70 indeed enhances the immune response triggered by the VSV-based human NoV vaccine, we may expect similar enhancement when administering a combined vaccination of rVSV-HSP70 and rVSV-VP1. Mice receiving a combination of 106 PFU of rVSV-HSP70 and 106 PFU of rVSV-VP1 had significantly less weight loss than mice that received 106 PFU of rVSV-VP1 alone (P < 0.05) (Fig. 9). As shown in Fig. 10A, mice receiving the combination vaccination showed stimulated levels of serum IgG antibody similar to those seen with rVSV-VP1 vaccination alone (P > 0.05). Similarly, there was no significant difference between combination vaccination and rVSV-VP1 vaccination alone in serum HBGA blocking titers (P > 0.05) (Fig. 10B). However, the combination vaccination triggered significantly higher fecal IgA (Fig. 11A) and vaginal IgA (Fig. 11B) responses than rVSV-VP1 vaccination alone (P < 0.05). In addition, there was no significant difference in the T cell immune responses triggered by these two vaccination strategies (P > 0.05) (Fig. 11C). These data suggest that combined vaccination of rVSV-HSP70 and rVSV-VP1 (at ratio of 1:1) enhanced human NoV-specific mucosal immunity.
FIG 9.

Dynamics of mouse body weight after inoculation with recombinant viruses for animal study 3. Five BALB/c mice in each group were inoculated with either 106 PFU of rVSV-VP1 or the combination of 106 PFU of rVSV-VP1 and 106 PFU of rVSV-HSP70. Body weight for each mouse was evaluated every other day for 5 weeks. The average body weight of five mice per group is shown.
FIG 10.

The effects of coimmunization on serum IgG response triggered by VSV-based human NoV vaccines. (A) Serum IgG antibody response. Groups of five BALB/c mice were inoculated with either 106 PFU of rVSV-VP1 or the combination of 106 PFU of rVSV-VP1 and 106 PFU of rVSV-HSP70. Serum samples were collected weekly and analyzed by ELISA using human NoV-specific serum IgG antibody. Data are expressed as geometric mean titers (GMT) of five mice per group. Error bars at each time point represent the standard deviations of the means. (B) Serum HBGA blocking antibody. The ability of serum antibodies to inhibit human NoV VLP binding to HBGAs was measured by an ELISA as described in Materials and Methods. The 50% blocking titer (BT50) was determined for each sample. Data are expressed as means ± standard deviations for each treatment group at the indicated time point.
FIG 11.

The effects of coimmunization on mucosal and T cell responses triggered by VSV-based human NoV vaccines. (A) Fecal IgA responses. Fecal samples were collected from all mice at week 5 postinoculation. Human NoV-specific and total IgA antibody levels were detected by ELISA. The ratio between human NoV-specific IgA and total IgA was calculated for each mouse. Data are expressed as average titers of IgA-positive mice ± standard deviations. (B) Vaginal IgA responses. Vaginal samples were collected at week 5 postinoculation from each mouse, and human NoV-specific and total IgA antibody levels were determined by ELISA. The level of vaginal IgA is shown as the ratio between human NoV-specific IgA and total IgA. Data are expressed as average titers of IgA-positive mice ± standard deviations. (C) T cell proliferative responses. Spleen cells were harvested from all mice in each group at week 5 postinoculation and stimulated with human NoV VLPs. T cell proliferation was measured by [3H]thymidine incorporation. The stimulation index (SI) was calculated as the mean of the following ratio: proliferation of human NoV VLP-stimulated cells/proliferation of cells in medium in cpm. Data are expressed as the means of the results determined for five mice ± the standard deviations. Asterisks indicate statistical significance (P < 0.05).
Recombinant rVSV-HSP70-VP1 stimulated significantly higher mucosal immunity than rVSV-Luc-VP1.
Finally, we directly compared the levels of safety and immunogenicity of rVSV-HSP70-VP1 and rVSV-Luc-VP1 as a control for the effects of a double-genome insertion, where VP1 is expressed between the VSV G and L gene and a second gene is expressed from between the leader and N gene sequence. Briefly, rVSV-HSP70-VP1 and rVSV-Luc-VP1 were inoculated into mice at dose of 106 PFU and mouse body weight changes were monitored over the course of 5 weeks. At week 5 postvaccination, serum IgG, fecal and vaginal IgA, and T cell immune responses were determined. As shown in Fig. 12, there was no significant difference between rVSV-HSP70-VP1 and rVSV-Luc-VP1 in mouse body weight changes (P > 0.05), suggesting that these two recombinant viruses had similar degrees of attenuation in vivo. Recombinant rVSV-VP1 caused significantly more body weight losses at days 10 and 12 postinoculation than rVSV-HSP70-VP1 and rVSV-Luc-VP1 (P < 0.05). Consistent with the results of the combination (rVSV-HSP70 and rVSV-VP1) vaccination strategy, the fecal and vaginal IgA responses seen with rVSV-HSP70-VP1 were significantly higher than those seen with rVSV-Luc-VP1 (P < 0.05) (Fig. 13). However, there was no significant difference in the levels of serum IgG response and T cell proliferation among rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1 (data not shown). These results demonstrated that the enhanced mucosal immunity that is induced by rVSV-HSP70-VP1 is due to the HSP70 and is not a nonspecific effect of a second genome insertion between the leader and N gene of VSV-VP1.
FIG 12.

Dynamics of mouse body weight after inoculation with recombinant viruses containing single versus double insertions. Five BALB/c mice in each group were inoculated with 106 PFU of rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1. Body weight for each mouse was evaluated every other day for 5 weeks. The average body weight of five mice per group is shown.
FIG 13.

Recombinant rVSV-HSP70-VP1 triggered significantly higher fecal and vaginal IgA levels than rVSV-Luc-VP1. (A) Fecal IgA responses. Fecal samples were collected from all mice at week 5 postinoculation. Human NoV-specific and total IgA antibody levels were detected by ELISA. Data are expressed as the means of the results determined for five mice ± the standard deviations. (B) Vaginal IgA responses. Vaginal samples were collected at week 5 postinoculation from each mouse, and human NoV-specific and total IgA antibody levels were determined by ELISA. Data are expressed as the means of the results determined for five mice ± the standard deviations. Asterisks indicate statistical significance (P < 0.05).
Spread of rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1 in mice.
Wild-type VSV causes systemic infection and fatal encephalitis in mice. After intranasal inoculation, VSV infects olfactory neurons in the nasal mucosa and subsequently enters the central nervous system (CNS) through the olfactory nerves. The virus may then be disseminated to other areas in the brain, with brain viral burden being correlated with the severity of VSV-associated clinical symptoms. To further evaluate the safety of rVSV-VP1, rVSV-HSP70-VP1, and rVSV-Luc-VP1, we compared their abilities to establish an acute brain infection. Mice were inoculated intranasally with these recombinant VSVs, and, at days 3 and 5 postinoculation, mice were euthanized and brain viral titers were quantitated by plaque analysis. As shown in Fig. 14, all four mice in the rVSV-VP1 group had infectious virus in brain at days 3 and 5 postinfection, with average titers of 3.1 log10 PFU/ml and 3.7 log10 PFU/ml, respectively. In contrast, only one of four rVSV-HSP70-VP1-infected mice had infectious virus in the brain at days 3 and 5 postinfection, with average titers of 1.3 log10 PFU/ml and 0.7 log10 PFU/ml, respectively. Similarly, one of four rVSV-Luc-VP1-infected mice had infectious virus in the brain at days 3 and 5 postinfection, with average titers of 0.7 log10 PFU/ml and 0.8 log10 PFU/ml, respectively. There was no significant difference between the rVSV-HSP70-VP1 and rVSV-Luc-VP1 brain viral burdens (P > 0.05). This result demonstrated that rVSV-HSP70-VP1 and rVSV-Luc-VP1 were significantly more attenuated in mice than rVSV-VP1.
FIG 14.

Spread of recombinant virus in brains of mice. Mice were inoculated intranasally with 106 PFU of rVSV-VP1, rVSV-HSP70-VP1, or rVSV-Luc-VP1. At days 3 and 5 postinoculation, 4 mice from each group were euthanized. The brain from each mouse was collected for virus titration by plaque assay. Data are expressed as the mean viral titers of four mice ± the standard deviations. Numbers of virus-positive mice for each group are indicated.
DISCUSSION
HSP70 enhances the safety of VSV-based human NoV vaccine.
One of the major concerns of VSV-based vaccines is safety, particularly since VSV is neurotropic. VSV infects a wide range of wild and domestic animals such as cattle, horses, deer, and pigs and produces vesicular lesions in the mouth, teats, and feet. Although VSV does not induce vesicular lesions in mice, it does cause a multisystemic infection that may include fatal encephalitis. After intranasal inoculation, VSV infects olfactory neurons in the nasal mucosa and subsequently enters the central nervous system (CNS) through the olfactory nerves (28). The virus is then disseminated to other areas in the brain through retrograde and possibly anterograde transneuronal transport, ultimately causing an acute brain infection (29). Mortality, pathology, and viral burden in lung and brain tissues are dependent on the age, species, and gender of the mice.
A variety of approaches have been employed to attenuate VSV, including gene rearrangement (30), M gene mutation (31), G protein cytoplasmic tail truncations (32), G protein deletions (33), and combinations thereof. These approaches decreased viral replication in cell culture as well as virulence in mice. In this study, it was found that double insertion of foreign genes (insertion of HSP70 and VP1 or of Luc and VP1) into the VSV vector resulted in further attenuation of VSV in cell culture as well as in mice. Recombinants rVSV-HSP70-VP1 and rVSV-Luc-VP1 formed much smaller plaques, replicated less efficiently, and synthesized fewer VSV proteins in cell culture than rVSV-VP1. Mice infected with rVSV-HSP70-VP1 and rVSV-Luc-VP1 had less body weight loss, exhibited no clinical signs of VSV infection, and showed significantly diminished viral titers in brains. There was no significant difference between rVSV-HSP70-VP1 and rVSV-Luc-VP1 in attenuation in vitro and in vivo. Thus, insertion of multiple genes in the VSV genome backbone can serve as an additional approach to further attenuate VSV vector.
One basis for the attenuation observed in rVSV-HSP70-VP1 and rVSV-Luc-VP1 may be the transcriptional attenuation that is associated with insertion of a gene at the 3′ end of the viral genome, in this case, HSP70 or Luc between the leader and N gene sequence. Other studies have shown that insertion of the Luc gene at the 3′ end of VSV genome decreases transcription of downstream genes, which in turn impairs viral replication in vitro and in vivo (34). Given the fact that rVSV-HSP70-VP1 and rVSV-Luc-VP1 had similar levels of attenuation, transcriptional attenuation would appear to play a dominant role in the attenuation of rVSV-HSP70-VP1 that overshadows any effects of HSP70 on enhancing viral clearance. For both measles virus (MeV) and VSV, expression of HSP70 diminishes the neurovirulence in mice that is associated with increased production of type I interferon and increased viral clearance from brain (24, 25, 35, 36). A model for this protective mechanism was established in which HSP70 in the virus-infected cell is released to engage Toll-like receptors on uninfected brain macrophages. The result is the induction of antigen presentation functions and production of type I interferon, which combine to enhance viral clearance (25, 36). The ability of HSP70 to promote clearance of VSV would account for the protection observed following coadministration of 106 PFU of rVSV-VP1 and rVSV-HSP70 to mice; there was significantly less weight loss than was seen with mice that received 106 PFU of rVSV-VP1 alone (P < 0.05). It will be interesting to determine whether HSP70 can enhance the safety of VSV-vectored vaccines through a type I IFN-mediated antiviral mechanism.
HSP70 enhances the immunogenicity of the viral vaccine.
HSP70 has been shown to be a strong adjuvant for a number of viral vaccines, including porcine reproductive and respiratory syndrome virus (PRRSV) (37), Japanese encephalitis (JE) virus (38, 39), Hantaan virus (40), respiratory syncytial virus (RSV) (41), and measles virus (24). PRRSV causes immunosuppression in pigs. It was found that recombinant adenoviruses expressing GP3/GP5 of highly pathogenic PRRSV and HSP70 stimulated stronger humoral immunity and cytokine responses (gamma IFN [IFN-γ] and interleukin-4 [IL-4]) and provided protection against virulent PRRSV challenge in pigs. In contrast, adenoviruses expressing GP3/GP5 alone are insufficient to induce protective immune responses in pigs. Codelivery of HSP70 with an RSV subunit vaccine (G1F/M2) induced significant higher levels of neutralizing antibodies and cytotoxic T lymphocyte (CTL) activity than unadjuvanted G1F/M2. Fusion of HSP70 to the nucleocapsid protein (NP) of Hantaan virus (HSP70-NP) or codelivery of HSP70 and NP proteins (HSP70+NP) elicited significantly higher NP-specific antibody titers and higher frequencies of IFN-γ-producing cells and CTL activities in vivo than conventional NP vaccination alone (40). HSP70 also enhanced T lymphocyte proliferation and IFN-γ and IL-4 secretion and produced virus-specific memory B cells and long-lasting antibodies when fused with the envelope (E) protein of JE virus (38).
In this study, we found that HSP70 enhanced the mucosal IgA responses of human NoV vaccine when it was coexpressed from a VSV vector. We demonstrated adjuvant effects of HSP70 on mucosal immunity using four independent experiments. First, at the dose of 1.0 × 106 PFU, rVSV-HSP70-VP1 produced significantly higher vaginal IgA levels than rVSV-VP1. Second, at the dose of 5.0 × 106 PFU, rVSV-HSP70-VP1 produced significantly higher fecal IgA levels than rVSV-VP1. Third, coadministration of rVSV-HSP70 and rVSV-VP1 triggered significantly stronger fecal and vaginal mucosal immunity than rVSV-VP1 alone. Finally, rVSV-HSP70-VP1 triggered significantly higher fecal and vaginal IgA levels than rVSV-Luc-VP1. We realized that inclusion of HSP70 in the VSV vector appears to increase the vaginal IgA responses preferentially over the fecal IgA responses (Fig. 5 and 8). It is possible that the ELISA is more sensitive for detecting IgA in the vaginal fluids than in the fecal samples, which are more complicated matrices. It is also possible that different organs may respond to the antigen stimulation differently. In addition, significant differences in the variability of the ratio between NoV-specific IgA and total IgA were observed among different experiments (see, for example, Fig. 11 and 13). This was probably due to the ages of the mice used in each experiment. Importantly, inclusion of HSP70 did significantly (P < 0.05) enhance the IgA responses in both experiments (Fig. 11 and 13). Besides the adjuvant effect, HSP70 may promote the viral gene expression which leads to synthesizing of more VP1 protein and thus triggering of a stronger IgA immune response. In fact, HSP70 was shown to interact with the N protein of measles virus, a nonsegmented negative-sense (NNS) RNA virus, which resulted in enhanced viral gene expression (42, 43). It is possible that HSP70 can interact with the N protein of VSV, which also is an NNS RNA virus. We observed that expression of VP1 in virus-infected cells occurred earlier with rVSV-HSP70-VP1 than with rVSV-Luc-HSP70 (Fig. 3A), although there was no significant difference between these two viruses in VP1 expression levels at later time points (P > 0.05).
IgA is produced in large quantities at mucosal surfaces, including the gastrointestinal tract, genitourinary tract, prostate, and respiratory epithelium, and is found in tears, saliva, and colostrum. It is generally believed that a robust mucosal immune response is required for the eradiation and resolution of mucosal pathogens. For example, poliovirus induces a secretory IgA response that can neutralize the virus and is associated with decreases in virus shedding in stool (44, 45). Mucosal IgA correlates with protection against polio infection (46). Studies in rotavirus, another major cause of gastroenteritis, found that the presence of mucosal rotavirus-specific IgA strongly correlates with less-severe disease and prevention of rotavirus infection in humans (47, 48). However, the importance of IgA in protection against human NoV infections has been difficult to prove since it is not cultivable and lacks a suitable animal model. Epidemiological and human volunteer studies suggest a link between mucosal IgA induced either by infection or by vaccine candidates and protective immunity from human NoV infection (49–51). Many studies have shown that human NoV-specific antibody can be detected in fecal and vaginal extracts in mice and human volunteers following vaccination of VLP-based vaccine candidates, and that response has been used as an indicator to evaluate the efficacy of a vaccine candidate (13, 51). In human NoV-infected gnotobiotic pigs, anti-NoV IgA was detected as early as 6 days following virus challenge and the severity of diarrhea was correlated with intestinal IgA antibody titers (52). In gnotobiotic calves, it was found that both NoV-specific IgA and IgA-secreting cells were present 28 days following NoV challenge (53). In a recent human clinical trial of a VLP-based vaccine candidate, it was found that the fecal IgA titer correlated with the efficacy of protection following a human NoV challenge (16). These data suggest that human NoV-specific IgA levels at the intestinal surface are correlated with either the cessation of virus excretion or protection against infection and disease. In addition, it will be interesting to determine whether vaginal IgA is correlated with the immunoprotection of norovirus in the future.
We also found that HSP70 can potentially enhance human NoV-specific T cell immunity. At the same inoculation dose (1 × 106 PFU), rVSV-HSP70-VP1 and rVSV-VP1 triggered similar levels of specific cellular immunity in spite of the fact that VP1 expression by rVSV-HSP70-VP1 was approximately 5-fold less than that of rVSV-VP1. Coadministration of rVSV-HSP70 and rVSV-VP1 at the dose of 1 × 106 PFU did not significantly enhance T cell immune response compared to rVSV-VP1 vaccination alone. Similarly, there was no significant difference in T cell immunity between rVSV-HSP70-VP1 and rVSV-Luc-VP1 at the dose of 1 × 106 PFU. However, we found that mice immunized with 5 × 106 PFU of rVSV-HSP70-VP1 generated significantly higher T cell immunity than those immunized with rVSV-VP1 (P < 0.05). Previously, it has been shown that HSP70 can enhance the serum IgG antibody response to a number of viruses, including RSV (41), Hantaan virus (40), and JE virus (38, 39). Unexpectedly, we did not observe a significant increase in NoV-specific serum IgG antibody or HBGA blocking antibody levels in any of the experiments, suggesting that HSP70 plays a minor role in the NoV-specific IgG response. Since our experiments were conducted at a dose of 1 × 106 or 5 × 106 PFU per mouse, it will be of great interest to investigate the effects of lower vaccination doses on T cell and serum IgG antibody responses.
VSV as a vector to delivery viral vaccines.
In the last decade, VSV has been proven to be an excellent vector for delivery of foreign antigens as live vaccines, oncolytic therapy, and gene delivery (54–56). VSV is suitable for delivery of vaccines against the following three categories of viral pathogen: viruses that cause persistent infection such as HIV (55, 57) and HCV (58); viruses that are highly lethal such as that causing severe acute respiratory syndrome (SARS) (59, 60) and Ebola and Marburg viruses (61); and viruses that cannot be grown in cell culture such as human NoV (17). The drawback for the VSV vector is that there is little experience with VSV administration in humans. However, at least three independent phase I human clinical trials are currently being carried out to test the safety, immune response, and effectiveness of the VSV-based vaccine and oncolytic therapy in human. Two VSV-based HIV vaccine strategies (using VSV-Indiana HIV gag vaccine and HIV DNA vaccine followed by a VSV-gag vaccine boost) are currently being tested in healthy, HIV-uninfected adults for the phase I clinical trials. Recombinant VSV expressing beta interferon has been approved for phase I clinical trials to treat patients with liver cancer. It seems clear that detailed information on safety, replication, pathogenesis, and immunogenicity of VSV-based vaccines in humans will be forthcoming.
In summary, it was found that (i) insertion of HSP70 into VSV vector further attenuates the VSV-based vaccine in cell culture in the mouse model and (ii) HSP70 enhances the human NoV-specific mucosal immunity triggered by VSV-based human NoV vaccine.
ACKNOWLEDGMENTS
This study was supported by a grant from OSU Public Health Preparedness for Infectious Diseases (PHPID) to J.L. and M.O.
We thank Gail Wertz for her generous gift of VSV infectious clone and Sean Whelan for providing pVSV(+)GxxL plasmid. We thank Linda Saif for the VP1 gene of human norovirus GII.4 strain HS66, Xi Jiang for human norovirus VP1 antibody, and Bernard Moss for the vTF7-3 virus.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Farkas T, Sestak K, Wei C, Jiang X. 2008. Characterization of a rhesus monkey calicivirus representing a new genus of Caliciviridae. J. Virol. 82:5408–5416. 10.1128/JVI.00070-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MPG, Estes MK. 2004. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85:79–87. 10.1099/vir.0.19478-0 [DOI] [PubMed] [Google Scholar]
- 3.Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2008. Norovirus pathogenesis: mechanisms of persistence and immune evasion in human populations. Immunol. Rev. 225:190–211. 10.1111/j.1600-065X.2008.00680.x [DOI] [PubMed] [Google Scholar]
- 4.Estes MK, Prasad BV, Atmar RL. 2006. Noroviruses everywhere: has something changed? Curr. Opin. Infect. Dis. 19:467–474. 10.1097/01.qco.0000244053.69253.3d [DOI] [PubMed] [Google Scholar]
- 5.Koopmans M. 2008. Progress in understanding norovirus epidemiology. Curr. Opin. Infect. Dis. 21:544–552. 10.1097/QCO.0b013e3283108965 [DOI] [PubMed] [Google Scholar]
- 6.Li J, Predmore A, Divers E, Lou F. 2012. New interventions against human norovirus: progress, opportunities, and challenges. Annu. Rev. Food Sci. Technol. 3:331–352. 10.1146/annurev-food-022811-101234 [DOI] [PubMed] [Google Scholar]
- 7.Estes MK, Ball JM, Guerrero RA, Opekun AR, Gilger MA, Pacheco SS, Graham DY. 2000. Norwalk virus vaccines: challenges and progress. J. Infect. Dis. 181(Suppl 2):S367–S373. 10.1086/315579 [DOI] [PubMed] [Google Scholar]
- 8.Jiang X, Wang M, Graham DY, Estes MK. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. 10.1126/science.286.5438.287 [DOI] [PubMed] [Google Scholar]
- 10.Tan M, Jiang X. 2010. Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLoS Pathog. 6:e1000983. 10.1371/journal.ppat.1000983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia M, Farkas T, Jiang X. 2007. Norovirus capsid protein expressed in yeast forms virus-like particles and stimulates systemic and mucosal immunity in mice following an oral administration of raw yeast extracts. J. Med. Virol. 79:74–83. 10.1002/jmv.20762 [DOI] [PubMed] [Google Scholar]
- 12.Ball JM, Hardy ME, Atmar RL, Conner ME, Estes MK. 1998. Oral immunization with recombinant Norwalk virus-like particles induces a systemic and mucosal immune response in mice. J. Virol. 72:1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerrero RA, Ball JM, Krater SS, Pacheco SE, Clements JD, Estes MK. 2001. Recombinant Norwalk virus-like particles administered intranasally to mice induce systemic and mucosal (fecal and vaginal) immune responses. J. Virol. 75:9713–9722. 10.1128/JVI.75.20.9713-9722.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamoto N, Tanaka T, Natori K, Takeda N, Nakata S, Jiang X, Estes MK. 2002. Cross-reactivity among several recombinant calicivirus virus-like particles (VLPs) with monoclonal antibodies obtained from mice immunized orally with one type of VLP. J. Clin. Microbiol. 40:2459–2465. 10.1128/JCM.40.7.2459-2465.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang XR, Buehner NA, Hutson AM, Estes MK, Mason HS. 2006. Tomato is a highly effective vehicle for expression and oral immunization with Norwalk virus capsid protein. Plant Biotechnol. J. 4:419–432. 10.1111/j.1467-7652.2006.00191.x [DOI] [PubMed] [Google Scholar]
- 16.Atmar RL, Bernstein DI, Harro CD, Al-Ibrahim MS, Chen WH, Ferreira J, Estes MK, Graham DY, Opekun AR, Richardson C, Mendelman PM. 2011. Norovirus vaccine against experimental human Norwalk virus illness. N. Engl. J. Med. 365:2178–2187. 10.1056/NEJMoa1101245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y, Li J. 2011. Vesicular stomatitis virus as a vector to deliver virus-like particles of human norovirus: a new vaccine candidate against an important noncultivable virus. J. Virol. 85:2942–2952. 10.1128/JVI.02332-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Lin Y, Liao C, Hsieh S. 2000. Modulatory effects of the human heat shock protein 70 on DNA vaccination. J. Biomed. Sci. 7:412–419. 10.1007/BF02255816 [DOI] [PubMed] [Google Scholar]
- 19.Todryk SM, Gough MJ, Pockley AG. 2003. Facets of heat shock protein 70 show immunotherapeutic potential. Immunology 110:1–9. 10.1046/j.1365-2567.2003.01725.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciupitu AM, Petersson M, O'Donnell CL, Williams K, Jindal S, Kiessling R, Welsh RM. 1998. Immunization with a lymphocytic choriomeningitis virus peptide mixed with heat shock protein 70 results in protective antiviral immunity and specific cytotoxic T lymphocytes. J. Exp. Med. 187:685–691. 10.1084/jem.187.5.685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumaraguru U, Gierynska M, Norman S, Bruce BD, Rouse BT. 2002. Immunization with chaperone-peptide complex induces low-avidity cytotoxic T lymphocytes providing transient protection against herpes simplex virus infection. J. Virol. 76:136–141. 10.1128/JVI.76.1.136-141.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milani V, Noessner E, Ghose S, Kuppner M, Ahrens B, Scharner A, Gastpar R, Issels RD. 2002. Heat shock protein 70: role in antigen presentation and immune stimulation. Int. J. Hyperthermia 18:563–575. 10.1080/02656730210166140 [DOI] [PubMed] [Google Scholar]
- 23.Millar DG, Garza KM, Odermatt B, Elford AR, Ono N, Li Z, Ohashi PS. 2003. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat. Med. 9:1469–1476. 10.1038/nm962 [DOI] [PubMed] [Google Scholar]
- 24.Carsillo T, Carsillo M, Niewiesk S, Vasconcelos D, Oglesbee M. 2004. Hyperthermic pre-conditioning promotes measles virus clearance from brain in a mouse model of persistent infection. Brain Res. 1004:73–82. 10.1016/j.brainres.2003.12.041 [DOI] [PubMed] [Google Scholar]
- 25.Kim MY, Ma Y, Zhang Y, Li J, Shu Y, Oglesbee M. 2013. hsp70-dependent antiviral immunity against cytopathic neuronal infection by vesicular stomatitis virus. J. Virol. 87:10668–10678. 10.1128/JVI.00872-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. U. S. A. 92:8388–8392. 10.1073/pnas.92.18.8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeck A, Kavanagh O, Estes MK, Opekun AR, Gilger MA, Graham DY, Atmar RL. 2010. Serological correlate of protection against norovirus-induced gastroenteritis. J. Infect. Dis. 202:1212–1218. 10.1086/656364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiss CS, Plakhov IV, Komatsu T. 1998. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann. N. Y. Acad. Sci. 855:751–761. 10.1111/j.1749-6632.1998.tb10655.x [DOI] [PubMed] [Google Scholar]
- 29.Cornish TE, Stallknecht DE, Brown CC, Seal BS, Howerth EW. 2001. Pathogenesis of experimental vesicular stomatitis virus (New Jersey serotype) infection in the deer mouse (Peromyscus maniculatus). Vet. Pathol. 38:396–406. 10.1354/vp.38-4-396 [DOI] [PubMed] [Google Scholar]
- 30.Wertz GW, Perepelitsa VP, Ball LA. 1998. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. U. S. A. 95:3501–3506. 10.1073/pnas.95.7.3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. 2008. Immune response in the absence of neurovirulence in mice infected with m protein mutant vesicular stomatitis virus. J. Virol. 82:9273–9277. 10.1128/JVI.00915-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang X, Zhang S, Sun X, Li J, Sun T. 2012. Evaluation of attenuated VSVs with mutated M or/and G proteins as vaccine vectors. Vaccine 30:1313–1321. 10.1016/j.vaccine.2011.12.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnell MJ, Johnson JE, Buonocore L, Rose JK. 1997. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell 90:849–857. 10.1016/S0092-8674(00)80350-5 [DOI] [PubMed] [Google Scholar]
- 34.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:61–119 [DOI] [PubMed] [Google Scholar]
- 35.Carsillo T, Carsillo M, Traylor Z, Rajala-Schultz P, Popovich P, Niewiesk S, Oglesbee M. 2009. Major histocompatibility complex haplotype determines hsp70-dependent protection against measles virus neurovirulence. J. Virol. 83:5544–5555. 10.1128/JVI.02673-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim MY, Shu Y, Carsillo T, Zhang J, Yu L, Peterson C, Longhi S, Girod S, Niewiesk S, Oglesbee M. 2013. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J. Virol. 87:998–1009. 10.1128/JVI.02710-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Jiang P, Li Y, Wang X, Cao J, Zeshan B. 2009. HSP70 fused with GP3 and GP5 of porcine reproductive and respiratory syndrome virus enhanced the immune responses and protective efficacy against virulent PRRSV challenge in pigs. Vaccine 27:825–832. 10.1016/j.vaccine.2008.11.088 [DOI] [PubMed] [Google Scholar]
- 38.Fei-fei G, Jian W, Feng X, Li-ping S, Quan-yun S, Jin-ping Z, Pu-yan C, Pei-hong L. 2008. Japanese encephalitis protein vaccine candidates expressing neutralizing epitope and M.T hsp70 induce virus-specific memory B cells and long-lasting antibodies in swine. Vaccine 26:5590–5594. 10.1016/j.vaccine.2008.07.104 [DOI] [PubMed] [Google Scholar]
- 39.Ge FF, Qiu YF, Yang YW, Chen PY. 2007. An hsp70 fusion protein vaccine potentiates the immune response against Japanese encephalitis virus. Arch. Virol. 152:125–135. 10.1007/s00705-006-0822-z [DOI] [PubMed] [Google Scholar]
- 40.Li J, Li KN, Gao J, Cui JH, Liu YF, Yang SJ. 2008. Heat shock protein 70 fused to or complexed with hantavirus nucleocapsid protein significantly enhances specific humoral and cellular immune responses in C57BL/6 mice. Vaccine 26:3175–3187. 10.1016/j.vaccine.2008.02.066 [DOI] [PubMed] [Google Scholar]
- 41.Zeng R, Zhang Z, Mei X, Gong W, Wei L. 2008. Protective effect of a RSV subunit vaccine candidate G1F/M2 was enhanced by a HSP70-like protein in mice. Biochem. Biophys. Res. Commun. 377:495–499. 10.1016/j.bbrc.2008.10.002 [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Glendening C, Linke H, Parks CL, Brooks C, Udem SA, Oglesbee M. 2002. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 76:8737–8746. 10.1128/JVI.76.17.8737-8746.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Bourhis JM, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. 2005. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 337:162–174. 10.1016/j.virol.2005.03.035 [DOI] [PubMed] [Google Scholar]
- 44.Keller R, Dwyer JE. 1968. Neutralization of poliovirus by IgA coproantibodies. J. Immunol. 101:192–202 [PubMed] [Google Scholar]
- 45.Onorato IM, Modlin JF, McBean AM, Thoms ML, Losonsky GA, Bernier RH. 1991. Mucosal immunity induced by enhance-potency inactivated and oral polio vaccines. J. Infect. Dis. 163:1–6. 10.1093/infdis/163.1.1 [DOI] [PubMed] [Google Scholar]
- 46.Buisman AM, Abbink F, Schepp RM, Sonsma JA, Herremans T, Kimman TG. 2008. Preexisting poliovirus-specific IgA in the circulation correlates with protection against virus excretion in the elderly. J. Infect. Dis. 197:698–706. 10.1086/527487 [DOI] [PubMed] [Google Scholar]
- 47.Hjelt K, Grauballe PC, Paerregaard A, Nielsen OH, Krasilnikoff PA. 1987. Protective effect of preexisting rotavirus-specific immunoglobulin A against naturally acquired rotavirus infection in children. J. Med. Virol. 21:39–47. 10.1002/jmv.1890210106 [DOI] [PubMed] [Google Scholar]
- 48.Matson DO, O'Ryan ML, Herrera I, Pickering LK, Estes MK. 1993. Fecal antibody responses to symptomatic and asymptomatic rotavirus infections. J. Infect. Dis. 167:577–583. 10.1093/infdis/167.3.577 [DOI] [PubMed] [Google Scholar]
- 49.Wyatt RG, Greenberg HB, Dalgard DW, Allen WP, Sly DL, Thornhill TS, Chanock RM, Kapikian AZ. 1978. Experimental infection of chimpanzees with the Norwalk agent of epidemic viral gastroenteritis. J. Med. Virol. 2:89–96. 10.1002/jmv.1890020203 [DOI] [PubMed] [Google Scholar]
- 50.Okhuysen PC, Jiang X, Ye L, Johnson PC, Estes MK. 1995. Viral shedding and fecal IgA response after Norwalk virus infection. J. Infect. Dis. 171:566–569. 10.1093/infdis/171.3.566 [DOI] [PubMed] [Google Scholar]
- 51.Tacket CO, Sztein MB, Losonsky GA, Wasserman SS, Estes MK. 2003. Humoral, mucosal, and cellular immune responses to oral Norwalk virus-like particles in volunteers. Clin. Immunol. 108:241–247. 10.1016/S1521-6616(03)00120-7 [DOI] [PubMed] [Google Scholar]
- 52.Souza M, Cheetham SM, Azevedo MS, Costantini V, Saif LJ. 2007. Cytokine and antibody responses in gnotobiotic pigs after infection with human norovirus genogroup II.4 (HS66 strain). J. Virol. 81:9183–9192. 10.1128/JVI.00558-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Souza M, Azevedo MS, Jung K, Cheetham S, Saif LJ. 2008. Pathogenesis and immune responses in gnotobiotic calves after infection with the genogroup II.4-HS66 strain of human norovirus. J. Virol. 82:1777–1786. 10.1128/JVI.01347-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J. Virol. 73:3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, Montefiori D, Roberts A, Buonocore L, Rose JK. 2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106:539–549. 10.1016/S0092-8674(01)00482-2 [DOI] [PubMed] [Google Scholar]
- 56.Majid AM, Ezelle H, Shah S, Barber GN. 2006. Evaluating replication-defective vesicular stomatitis virus as a vaccine vehicle. J. Virol. 80:6993–7008. 10.1128/JVI.00365-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan GS, McKenna PM, Koser ML, McLinden R, Kim JH, McGettigan JP, Schnell MJ. 2005. Strong cellular and humoral anti-HIV Env immune responses induced by a heterologous rhabdoviral prime-boost approach. Virology 331:82–93. 10.1016/j.virol.2004.10.018 [DOI] [PubMed] [Google Scholar]
- 58.Buonocore L, Blight KJ, Rice CM, Rose JK. 2002. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J. Virol. 76:6865–6872. 10.1128/JVI.76.14.6865-6872.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faber M, Lamirande EW, Roberts A, Rice AB, Koprowski H, Dietzschold B, Schnell MJ. 2005. A single immunization with a rhabdovirus-based vector expressing severe acute respiratory syndrome coronavirus (SARS-CoV) S protein results in the production of high levels of SARS-CoV-neutralizing antibodies. J. Gen. Virol. 86(Pt 5):1435–1440. 10.1099/vir.0.80844-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. 2005. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology 340:174–182. 10.1016/j.virol.2005.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geisbert TW, Daddario-DiCaprio KM, Geisbert JB, Reed DS, Feldmann F, Grolla A, Stroeher U, Fritz EA, Hensley LE, Jones SM, Feldmann H. 2008. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 26:6894–6900. 10.1016/j.vaccine.2008.09.082 [DOI] [PMC free article] [PubMed] [Google Scholar]