

Abstract
Spinal muscular atrophy (SMA) is a devastating neuromuscular disorder that stems from low levels of survival of motor neuron (SMN) protein. The processes that cause motor neurons and muscle cells to become dysfunctional are incompletely understood. We are interested in neuromuscular homeostasis and the stresses put upon that system by loss of SMN. We recently reported that α-COP, a member of the coatomer complex of coat protein I (COPI) vesicles, is an SMN-binding partner, implicating this protein complex in normal SMN function. To investigate the functional significance of the interaction between α-COP and SMN, we constructed an inducible NSC-34 cell culture system to model the consequences of SMN depletion and find that depletion of SMN protein results in shortened neurites. Heterologous expression of human SMN, and interestingly over-expression of α-COP, restores normal neurite length and morphology. Mutagenesis of the canonical COPI dilysine motifs in exon 2b results in failure to bind to α-COP and abrogates the ability of human SMN to restore neurite outgrowth in SMN-depleted motor neuron-like NSC-34 cells. We conclude that the interaction between SMN and α-COP serves an important function in the growth and maintenance of motor neuron processes and may play a significant role in the pathogenesis of SMA.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal-recessive neurodegenerative disease characterized by loss of alpha motor neurons and muscle wasting (1). With an incidence of approximately 1 in 6000 live births, it is the most common genetic cause of infant mortality. SMA is caused by deletion or loss-of-function mutations in the Survival of Motor Neuron (SMN1) gene (2).
The SMN protein is ubiquitously expressed and has been detected in both the nucleus and the cytoplasm. It is involved in the assembly of small nuclear ribonucleoprotein complexes (snRNPs), which regulate predominantly U11/U12 splicing events (3). Although a great deal of understanding has been gained since the identification of SMN mutations as the cause of SMA, there is still very little known about its role in motor neurons and why loss of SMN function is so detrimental to this cell type. SMN levels are high during nervous system development and decrease postnatally, indicating that SMN is crucial during this developmental window. SMN proteins have been found throughout neuronal processes in a number of cell types in culture where they interact with Gemins and RNA-binding proteins (4). Cultured primary motor neurons lacking SMN show reduced axonal outgrowth and defects in both the transport and the local translation of β-actin mRNA (5,6). Zebrafish models of SMA show dramatic alterations in motor neuron growth and branching, indicating that SMN is important in the proper formation and maintenance of motor neuronal processes and defects in neuronal pathfinding in the mouse retina show that SMN has important roles in axonogenesis in the mouse model as well (7,8).
One area that has been lacking in SMA research is a robust and reliable assay of SMN function in neuronal cell types. We have used the hybrid motor neuron-like NSC34 cell line to create a tractable model of SMA by knocking down murine SMN using a doxycycline-induced short-hairpin RNA. Previous work in our laboratory characterized the interaction between SMN protein and α-COP, a member of the COPI vesicle complex involved in trafficking of proteins between the Golgi and the endoplasmic reticulum (ER) and to and from the cell periphery (9). α-COP co-localized with SMN in axons of primary cultured neurons and knockdown of α-COP resulted in decreased levels of SMN in the growth cone of developing SH-SY5Y cells (10). In motor neuron-like NSC-34 cells, knockdown of α-COP resulted in accumulation of SMN proteins in the trans-Golgi network (11). COPI vesicle function appears to be important for motor neuron health as deletion of the COPI accessory gene Scyl1 results in a progressive motor neuron disease (12). Mice with mutations in the delta subunit of COPI, also known as archain1, develop an adult-onset neurodegenerative phenotype (13), and complete knockout of the γ-COP subunit is embryonically lethal (EUCOMM, 36616). α-COP is widely expressed throughout the brain and spinal cord, and α-COP co-immunoprecipitates with SMN in lysates from neonatal mouse brain and spinal cord (data not shown), indicating that this interaction has biological relevance in the developing nervous system. In differentiated NSC-34 cells, α-COP binds a number of mRNAs that may be present in the α-COP granules found in neurites in culture, as they have known neurite-targeting motifs (14). Taken together, these data point to α-COP as having important roles beyond Golgi–ER transport in neuronal cells. We set out to examine the role of SMN proteins in growing neuronal processes and to explore the significance of the SMN–α-COP interaction in this environment. We show here that loss of SMN in these cells reduces neurite length. This deficit can be rescued with transfection of human SMN1 or with α-COP. α-COP often interacts with its targets via dibasic motifs, general dilysines in the form of KKXX or KXK. These motifs are generally in the extreme carboxyl terminus of the target protein, but can also be found internally (15). We identified two such dilysine motifs in exon 2b of SMN that bind to α-COP, and SMN with mutations of these lysine residues fails to interact with α-COP and is unable to restore the normal neurite architecture in NSC-34 cells.
RESULTS
NSC-34 cells as a model of spinal muscular atrophy
To model SMA in vitro, we chose the NSC-34 cell line. These immortalized cells were derived from a hybrid of neuroblastoma and motor neuron-enriched embryonic mouse spinal cord cells and display many characteristics of motor neurons (16). We used the pSuperior system to create stably transfected NSC34 cells expressing the TetR protein. The highest expressing clonal line was selected and transfected with pSuperior.puro containing a short-hairpin RNA (shRNA) directed against murine SMN. Selection followed by clonal isolation resulted in NSC-34 clone #56, which produces robust knockdown of SMN protein to ∼10–20% of normal levels within 48 h (Fig. 1A). These cells maintain viability under SMN knockdown for up to 14 days, but demonstrate a marked reduction in their proliferation. The impact of SMN depletion on the ability of these cells to form neuronal processes in culture was then assessed. Cultures were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS for 48 h in the presence or absence of doxycycline to induce SMN knockdown. Following depletion of SMN protein, the cultures were plated on poly-D-lysine coated glass coverslips in DMEM/F12 with 1% FBS to induce neuronal differentiation. After 3 days of culture in neuronal differentiation media, the cells were fixed and stained with α-tubulin antibodies to visualize processes. Neurite length and number were measured for control and SMN-depleted cultures. While control cells produce lengthy neurites and mostly bipolar or pyramidal cellular morphologies, SMN-depleted cultures produced significantly shorter neurites (P < 0.01) similar to the recent reports of poor axonal outgrowth in SMN-depleted motor neuron derived from human embryonic stem cells (17) and adopted a more stellate morphology resulting in a statistically significant shift in the number of neurites per cell (P < 0.05) (Fig. 1B–E). These changes persisted as far out as 120 h in culture. Interestingly, SMN-depleted cells also display what appear to be prominent tubulin-immunopositive microtubule organizing centers, which is consistent with previous reports of altered tubulin dynamics in SMN-depleted cells (18).
Figure 1.

Knockdown of SMN in NSC-34 cells reduces neurite outgrowth. Western blot of lysates from NSC-34 clone #56 shows reduction of SMN proteins following treatment with 2 µg/ml doxycycline compared with β-actin, reducing levels by 81% after 48 h as shown by quantification values below the blot (A). SMN knockdown significantly reduces average neurite length (P < 0.01 by Student's t-test) following 3 days in neurite outgrowth medium (B). The distribution of the number of neurites per cell is significantly shifted (P < 0.05 by Chi-squared test) in doxycyline-treated culture following 3 days in neurite outgrowth medium compared with controls (C). Immunofluorescence with antibodies against α-tubulin demonstrates the consistent phenotype of short, spiky SMN-depleted cells compared with the long, mostly bipolar control cultures.
To determine whether the neurite outgrowth phenotype in NSC-34 clone #56 was SMN-dependent, two experiments were carried out. First, cells from the parent NSC-34 clone expressing only the TetR protein were cultured in the presence and absence of doxycycline. No knockdown of SMN protein was detectable following doxycycline treatment. When cells were grown in neuronal differentiation media, no alteration in neurite length or number was detectable in doxycycline-treated cultures (data not shown). Moreover, we were able to rescue the defects in both neurite outgrowth and number by transiently transfecting the cultures with eGFP-tagged human SMN (a generous gift from Philip J Young) on the day of neuronal induction. The eGFP-SMN is mainly cytoplasmic and punctate, and fluorescence extends throughout the length of the cell body and neurites. Transfection with human SMN fully rescues neurite length and significantly shifted the distribution of neurites per cell toward that of control cultures (Fig. 2). These experiments combine to show that a decreased neurite length and altered morphology in NSC-34 clone #56 upon doxycycline treatment is a result of SMN depletion. Interestingly, eGFP-SMN bearing the E134 K mutation, which is associated with severe disease presentation (3), did not rescue neurite outgrowth in these cells, while there was modest rescue seen with a construct containing the A2G mutation (Fig. 3), which is associated with a milder SMA phenotype (19). Similar rescue of neurite number was seen with the A2G mutant and the wild-type SMN, but no significant rescue of neurite number was seen in cells transfected with the E139 K mutant as determined by Chi-squared analysis. Western blot analysis from cultures transfected with wild-type, A2G or E139 K eGFP-SMN shows that expression levels were comparable and the eGFP-SMN constructs are not affected by the doxycycline-induced knockdown of murine SMN (Supplementary Material, Fig. S2).
Figure 2.
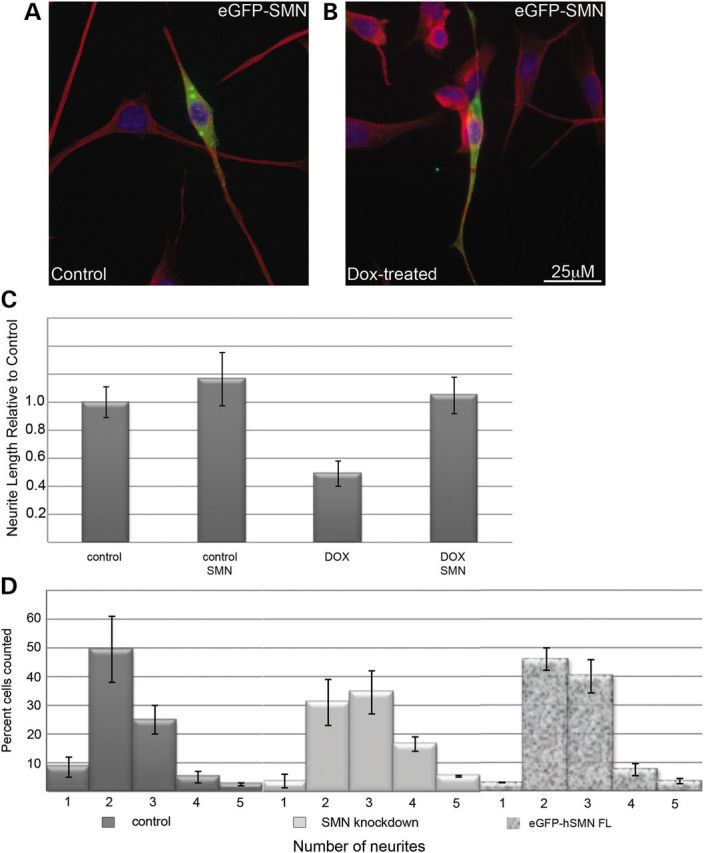
Human SMN restored normal growth in NSC-34 #56 cultures. Cell processes were visualized with immunofluorescent detection of α-tubulin (red) and nuclei visualized in blue DAPI stain. Transfection with eGFP-hSMN followed by 72 h in neurite outgrowth medium demonstrates increased neurite outgrowth in transfected cells compared with un-transfected neighbors (A and B). Measurement of the average neurite length shows that eGFP-hSMN significantly rescues (P < 0.05 compared with non-transfected neighbors) neurite length in doxycycline-treated cultures (C). Comparison of eGFP-positive cells to non-transfected neighbors reveals a statistically significant (P < 0.05 by Chi-squared analysis) shift in the distribution of neurites per cell in doxycycline-treated cultures compared with untransfected neighbors (D).
Figure 3.

An SMN mutant associated with severe SMA does not restore neurite outgrowth. Compared with transfection with wild-type eGFP-hSMN (A) transfection with eGFP-SMN carrying the pathogenic E134 K mutation failed to restore neurite outgrowth in SMN-depleted cultures compared with untransfected neighbors. eGFP-hSMN bearing the A2G mutation (C) was indistinguishable from wild-type. Neurites were visualized with immunofluorescence against α-tubulin (red) and nuclei were visualized with DAPI (blue). Quantification of the neurite length from three separate experiments shows no significant rescue from E134 K mutant SMN compared with non-transfected neighbors in doxycyline-treated cultures (D). Chi-squared analysis shows that similar to eGFP-SMN wild-type, SMN A2G shifts the number of neuritis per cell to a more normal distribution (P < 0.05 compared with non-transfected knockdown cultures) but SMN E139 K cells continue to display an increase in the number of neurite per cell relative to control cultures (E).
The SMN protein contains a number of domains that are known to mediate its subcellular localization and its ability to interact with various binding partners (20,21). To determine which regions of SMN are important for its ability to rescue neurite outgrowth in our NSC34 model, we used a series of eGFP-tagged human SMN constructs lacking exons 1 through 6. SMN lacking exon 7 was very unstable as has been previously reported (22), and could not be expressed in these cells at detectable levels for the required 3 days in cultures and thus was removed from the analysis. As seen in Figure 4A and quantified in Figure 4G, SMN lacking exon 1 (SMN Δ1) adopted a punctate cytoplasmic distribution, extending throughout the cell body and neurites. This construct was able to rescue neurite outgrowth and was indistinguishable from full-length eGFP-tagged SMN. Similar results were seen with constructs lacking exons 4 or 5. Although SMN lacking exon 5 adopted a much more diffuse subcellular distribution, both SMN Δ4 and SMN Δ5 rescued neurite outgrowth in doxycycline-treated cultures. In contrast, SMN lacking exons 2b, 3 or 6 displayed diffuse nuclear and cytoplasmic distribution and failed to restore neurite outgrowth or normal morphology in doxycycline-treated cultures (Fig. 4B, D, F and G).
Figure 4.
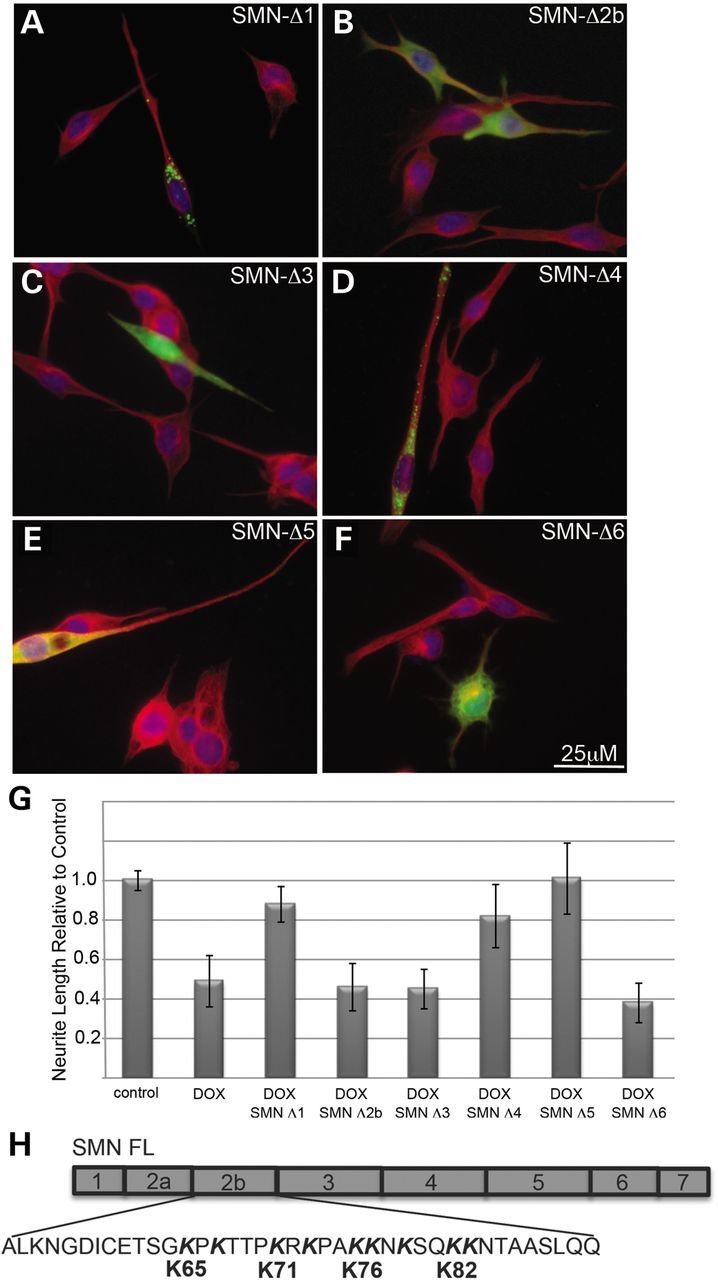
SMN lacking exons 2b, 3 or 6 fails to rescue neurite outgrowth. Transfection of SMN-depleted NSC-34 #56 cultures with a series eGFP-hSMN deletion constructs shows that deletion of exons 1, 4 or 5 has no impact on the distribution of the proteins within the cell or its ability to restore neurite outgrowth to doxycyline-treated cultures compared with non-transfected neighbors (A, D and E). However, SMNΔ5 produced a more diffuse pattern compared with the normal punctate fluorescence seen with wild-type SMN. Neurites were visualized with immunofluorescence using antibodies to α-tubulin (red) and nuclei were visualized with DAPI (blue). Measurement of the neurite length from three separate experiments shows significant rescue of neurite length (P < 0.05 by Student's t-test) in cells transfected with SMNΔ1, SMNΔ4 or SMNΔ5 compared with non-transfected neighbors in doxycycline-treated cultures (G). Potential α-COP-binding motifs in exon 2b (H)
We were particularly interested in the failure of SMN Δ2b to rescue neurite outgrowth in SMN-depleted NSC34 clone #56 cultures. Our previous in vitro binding experiments demonstrated that SMN exon 2b was sufficient for binding of α-COP (10). In support of the role of a-COP in neurite outgrowth, over-expression human α-COP co-transfected with eGFP in either control or SMN-depleted NSC34 clone #56 cultures resulted in a significant rescue of neurite length, similar to the rescue achieved with transfection of human SMN (Fig. 5).
Figure 5.

Over-expression of α-COP increases neurite length in SMN-depleted cells. Transfection of NSC-34 56 cultures with human α-COP increased neurite outgrowth compared with non-transfected neighbors (A). α-COP was co-transfected with eGFP to identify transfected cells compared with control neighbors. In SMN-depleted cells, co-transfection of eGFP and α-COP restored neurite outgrowth compared with un-transfected neighbors (B). Neurites were visualized with immunofluorescence to α-tubulin (red) and nuclei were visualized with DAPI (blue). Measurement of the neurite length from three separate experiments shows significant rescue (P < 0.05 by Student's t-test) of neurite length in cells transfected with eGFP and α-COP in SMN-depleted cultures (C).
As shown in Figure 4, SMN lacking exon 2b fails to rescue neurite outgrowth. Several proteins that bind to α-COP contain characteristic lysine-rich motifs (K–K or K×K) (23) and exon 2b contains these lysine motifs at amino acids 65–x–67, 71–x–73, 76–77-x-79 and 82–83 (Fig. 5H). To determine whether these putative a-COP binding residues affect the ability of SMN to rescue neurite outgrowth, we used two incomplete deletions of exon 2b. The first, SMN Δ2b-1, lacks only the first eight amino acids of exon 2b (ALKNGDIC) and retains all four potential α-COP binding lysine motifs. As shown in Figure 6A, eGFP-hSMN Δ2b-1 adopts a cytoplasmic punctate pattern similar to full-length SMN and is able to rescue neurite outgrowth in SMN-depleted cultures (Fig. 6C). The second construct, SMN Δ2b-2 contained a larger deletion within exon 2b that removes lysines 65–76 but retains lysines 82–83. This construct demonstrated a diffuse nuclear and cytoplasmic staining pattern and failed to neurite outgrowth (Fig. 6B and C). This indicates that the region of exon 2b containing the putative α-COP binding residues is critical for the ability to rescue neurite outgrowth.
Figure 6.
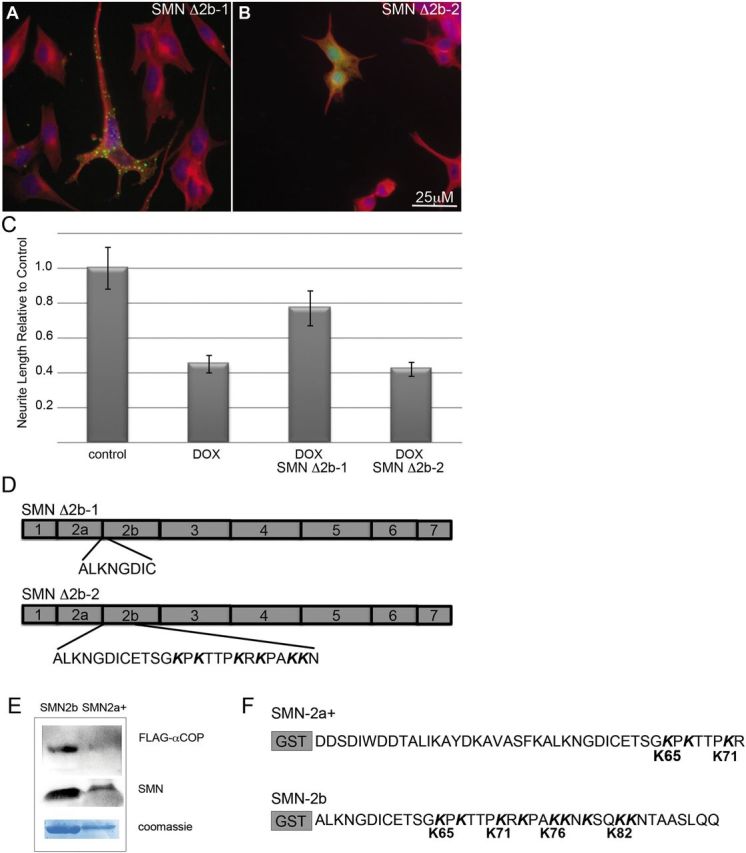
Dilysine motifs in SMN exon 2b mediate neurite rescue and α-COP binding. eGFP-hSMN constructs with deletions within exon 2b that either retained (Δ2b-1) or lacked the dilysine motifs (Δ2b-2) as diagramed in (D) were transfected into doxycycline-treated NSC-34 #56 cultures (A and B). Only SMNΔ2b-1, which includes all the dilysine motifs, demonstrated the normal punctate, cytoplasmic distribution and rescued neurite outgrowth in SMN-depleted cultures compared with non-transfected neighbors. Neurites were visualized with immunofluorescence to α-tubulin (red) and nuclei were visualized with DAPI (blue). Measurement of neurite length from three separate experiments shows significant rescue (P < 0.05 by Student's t-test) of neurite length in cells transfected with SMND2b-1 in SMN-depleted cultures (C). GST fusions of SMN exon 2a along with the first 20 amino acids of exon 2b (GST-2a+), which contains only lysines 65 and 71 greatly reduced binding to FLAG- α-COP compared with GST-SMN exon 2b which retains all four dilysine motifs (E and F).
To examine the ability of these lysine residues to bind a-COP protein in vitro, we used GST-tagged fragments of SMN exon 2. The first was a fusion of exon 2a with the first 20 amino acids of exon 2b (SMN 2a+). This contained lysine 65, 67 and 71 but lacked the lysines from 76–82. We compared the behavior of this 2a+ fragment with GST-tagged exon 2b containing all four lysines. Flag-tagged human α-COP was transcribed and translated in vitro, and the resulting reticulocyte lysate was incubated with GST-tagged SMN 2b or SMN 2a+ immobilized on glutathione beads. Blotting bound proteins with an anti-FLAG antibody showed that exon 2b successfully bound α-COP but exon 2a+ that lacks lysines 76–82 did not (Fig. 6D). These experiments together demonstrate that lysines 76, 77 and 79 appear to be crucial for both binding of α-COP to SMN and for SMN-dependent neurite outgrowth.
We used site-directed mutagenesis to mutate each of the putative a-COP binding dibasic motifs in eGFP-tagged SMN. Alanine mutations were introduced into GST-tagged exon 2b, although in this case, only lysines 76 and 82 were altered. In cultured SMN-depleted cells, eGFP-tagged SMN K65A and K71A were indistinguishable from wild-type proteins, adopting a cytoplasmic punctate appearance with fluorescence throughout the length of the cell body and neuritic processes, and significant rescue of neurite length compared with non-transfected neighbors (Fig. 7A and B). In contrast, SMN K76A and SMN K82A predominantly appear to diffuse, evenly distributed throughout the nucleus and the cytoplasm, and fail to rescue neurite outgrowth in SMN-depleted cultures (Fig. 7C and D). When these mutations were introduced into GST-tagged SMN exon 2b, they destroyed the binding of in vitro translated FLAG-a-COP (Fig. 7F). We infer that dilysine motifs 76–77 and 82–83 mediate α-COP binding to SMN exon 2b and are important for rescue of neurite outgrowth in SMN-depleted NSC34 cells. When transfected in HeLa cells, these proteins co-immunoprecipitate at equal levels with Gemin2 when compared to wild type SMN (Supplementary Material, Fig. S1a and b). When co-transfected into NSC-34 cells along with HA-tagged human SMN, all mutants equally co-immunoprecipitated with antibodies against HA (Supplementary Material, Fig. S1d), implying that despite being within the exon 2b self-association domain (21), these retain the ability to oligomerize. Finally, when expressed in HeLa cells, the eGFP-SMN mutants all co-localize with Gemin2-positive puncta (Supplementary Material, Fig. S1c). Taken together, these data demonstrate that we have disturbed the COPI binding in exon 2b without significantly altering other SMN protein–protein interactions.
Figure 7.
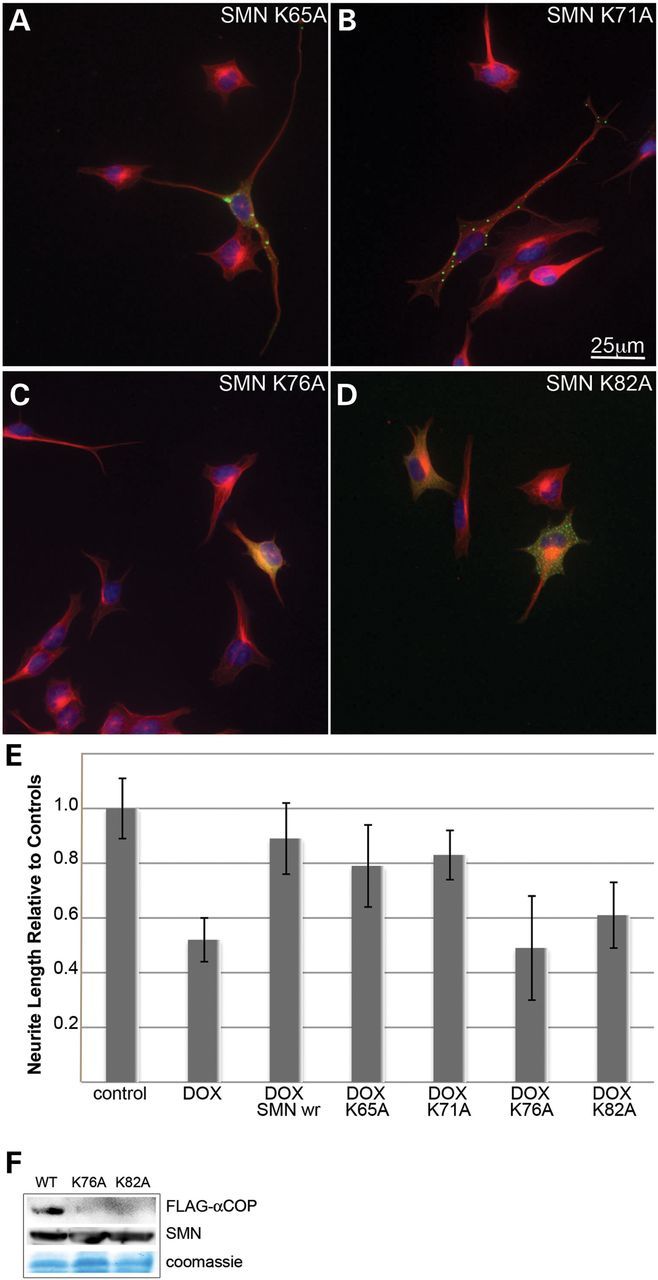
Lysine 76 and 82 in SMN exon 2b mediate neurite outgrowth and alpha-COP binding. Mutation of either K76A or K82A in eGFP-hSMN resulted in a diffuse cytoplasmic localization and reduced ability to restore neurite outgrowth compared with mutations of K65A or K71A (A–D). Measurement of neurite length from three separate experiments shows significant rescue (P < 0.05 by Student's t-test) of neurite length in cells transfected with K65A or K71A mutant SMN but no rescue from either K76A or K82A (E). GST fusions of SMN exon 2b carrying either the K76A or K82A mutation fail to bind FLAG- α-COP (F).
DISCUSSION
We have shown that NSC-34 cells provide a suitable, reproducible model for the in vitro study of SMN protein function using the formation and maintenance of neurites as the phenotypic readout. Depletion of SMN protein levels has been shown to result in decreased neurite outgrowth in several cell culture models including NSC34 cells (18,24,25), thereby providing one of the most consistent biological markers of SMN depletion. In our system, depletion of SMN in these cells to approximately 20% of normal levels leads to a pronounced defect in neurite formation under reduced serum conditions, which can be rescued by transfection with shRNA-resistant human SMN or a-COP. We, and others have shown that knockdown of α-COP redistributes SMN within the cell, leading to decreased levels of SMN protein in growth cones of SH-SY5Y cells and an accumulation of SMN protein at the trans-Golgi network in differentiated NSC-34 cells (10,11). The role of COPI trafficking in motor neuron disease has become increasingly apparent as new animal models become available. A recent knockout of the COPI co-factor Scyl1 produced adult-onset motor neuron disease with TDP-43 pathology reminiscent of ALS (12). This underscores the possibility that modification of COPI function could have therapeutic significance in motor neuron diseases beyond SMA, a prospect that takes on increasing likelihood as more proteins associated with ALS such as FUS are found to also interact with SMN (26).
We identified exon 2b of SMN as sufficient for binding α-COP, and noted that exon 2b contained a series basic lysine and arginine amino acids 65, 71, 76/77 and 82/83. Dilysines are the preferred motifs that mediate binding to COPI vesicles (27,28). We found that mutation of the dilysine KK motifs in exon 2b at amino acids 76/77 and 82/83, but not the dibasic or variant motifs K×K or K×R, reduced α-COP binding in vitro and concomitantly destroyed the ability of human SMN to restore neurite outgrowth in the SMN-depleted NSC-34 cells. Importantly, these mutant proteins retain the ability to bind Gemin2 and wild-type SMN. We believe that these mutations act exclusively by interfering with α-COP binding and thus transport of SMN.
Our experiments also support the importance of the Tudor domain in SMN exon 3 in supporting neurite outgrowth, as has been shown previously by expressing exon 3 mutations associated with severe SMA (29). Our NSC-34 cell assay is also sensitive to mutations associated with a more severe SMA clinical presentation as the E134 K mutation failed to restore neurite outgrowth.
We have previously shown that α-COP co-localizes with SMN in developing neurites in culture and in primary retinal neurons. Reduced levels of α-COP limited the presence of SMN at the growth cone of developing processes. In our NSC-34 cell model of SMA, a minimal amount of SMN protein remains, sufficient to prevent cell death for at least 14 days. Transfection with α-COP may promote the trafficking of the residual SMN protein to the developing neurite, allowing for support of the growing structure. SMN mutants with the putative α-COP binding lysines mutated fail to rescue neurite outgrowth and fail to bind α-COP in vitro. We believe that this is specific to the interaction between SMN and a-COP as transfection with β-COP, which does not directly bind SMN, failed to increase neurite length in this system. These results suggest that α-COP-dependent vesicles might be limiting for this transport pathway. Alternatively, over-expression of α-COP may aid in the transport of co-factors important to the function on the remaining SMN. We recently reported that α-COP is associated with a number of specific mRNAs in differentiated NSC-34 cells, and the increased trafficking of these transcripts, such as β-actin (6,30,31), may provide support for the SMN-depleted neurite even in the absence of direct dependence on SMN.
It was recently shown that developing primary neurons lacking SMN1 fail to induce local translation of β-actin in response to extracellular laminin cues (6). It had previously been suggested that reduced levels of β-actin mRNA in the processes of developing SMA neurons was a failure of mRNA trafficking, but this recent report suggests an additional failure of local cell signaling. α-COP has been shown to be critical for proper endocytosis and nuclear translocation of the endothelial growth fact receptor (EGFR) (32). Whether this α-COP function depends on SMN binding is unclear, but if response to extracellular cues is blunted in SMN-depleted cells, additional α-COP may help promote the peripheral function of what little SMN protein remains. Even in conjunction with factors that increase SMN2 levels, increased COPI function may serve to deliver more SMN to the machinery needed for the maintenance of motor neurons.
The final test of the significance of α-COP binding to SMA disease pathology must be undertaken in an animal model. We have plans to introduce wild-type and dilysine-mutant SMN into a severe mouse model of SMA to determine whether these mutations are unable to restore neuromuscular function in vivo. We are also exploring the possibility of rescuing the SMA phenotype by transgenic over-expression of α-COP.
To date, most research into pharmacological therapeutic intervention has relied on the SMN2 gene and attempts to increase its ability to produce full-length SMN. However, many of the compounds being tested lack specificity and have the potential for harmful off-target effects. The COPI vesicle complex assembly and function are regulated by G-protein coupled proteins, making this system a potentially tractable pharmacological target. If increases in COPI levels or assembly rate can promote the trafficking of SMN protein to the growing neuronal processes, this may allow even minimal amounts of SMN to sustain the proper growth and maintenance of alpha motor neurons in SMA patients.
MATERIALS AND METHODS
Generation of SMN knockdown NSC-34 cells
To establish an inducible system for knocking down the expression of mouse SMN gene, we started with generating motor neuron-like cells NCS-34 that stably expresses Tet repressor protein (tet-R). For that purpose, NSC-34 cells were transfected with pcDNA6/TR. Individual clones were isolated after 4 weeks of selection with 10 mg/ml of blasticidin S HCl (Invitrogen). To identify a clone with the highest level of tet-R we used a reporter construct, pcDNA4/TO-luciferase. In this construct Firefly luciferase is expressed from a CMV promoter that contains tet-R binding sites. More than 100 of randomly selected individual clones were screened for their ability to repress luciferase expression. Briefly, each clone was transiently transfected with pcDNA4/TO-luciferase and allowed to grow in the absence or presence of 1 mg/ml doxycycline (Sigma). Approximately 30 h after transfection cells were lysed in Luciferase lysis buffer (Promega), and assayed using a Firefly Luciferase assay kit (Promega). NSC-34 clone #4 (NSC-34–4) that showed the highest difference (∼10 times) in luciferase amount when grown with and without doxycycline was subsequently used to establish the inducible shRNA expression system. To express shRNAs of interest, we used a pSUPERIOR.puro plasmid (Oligoengine). In this plasmid, shRNA is expressed from the H1 promoter that contains the tet-R binding site. The shRNA GGAGAAUGAAAGUCAAGUU that targets the junction of exon3/exon4 was cloned into a pSUPERIOR.puro vector following manufacturer's recommendations. Also, shRNA that targets Photinus pyralis luciferase mRNA sequence CGUACGCGGAAUACUUCG was used as a negative control. The silencing efficiency of the shRNA was first confirmed using a psiCHECK-moSMN construct, in which mouse SMN cDNA was fused to the Renilla luciferase gene using XhoI and NotI restriction sites in the psiCHECK-2 plasmid (Promega). Briefly, NSC34–4 clone was co-transfected with psiCHECK-moSMN (25 ng) and the pSUPERIOR.puro-shRNA (175 ng) in a 24-well plate, and luciferase activity was measured 30 and 50 h later using a Dual-Glo Luciferase assay (Promega). The shRNA caused >90% decrease in luciferase activity when compared with the control shRNA as well as mock-transfected cells. The pSUPERIOR.puro-shRNA construct was transfected into NSC-34–4 cells and individual clones were isolated after 4 weeks of selection with 2.5 mg/ml of puromycin. To test for SMN knockdown, >30 randomly selected individual clones were tested by RT–PCR for mRNA levels after being grown for 3 days in the absence or presence of 2 mg/ml doxycycline. Clones with a significant decrease in SMN mRNA levels upon doxycycline addition in growth medium were then used in western blot analysis. Clone NSC-34–4 #56 that expresses shRNA was selected for subsequent experiments.
Neurite outgrowth assays
Prior to induction of neurite outgrowth, cells were maintained in either vehicle or doxycycline-treated (2 mg/ml) growth medium (DMEM, 10% FBS) for 48 h to reduce the levels of SMN. Following induction of knockdown, the cells were plated on PDL-coated 18 mm glass coverslips in Neurite outgrowth media (DMEM/F12, 1% FBS) at a density of 20 000 cells/ml. eGFP-SMN and alpha-COP expression vectors (1 µg DNA per well) were introduced at the time of plating by reverse transfection with Lipofectamine 2000 for 6 h to yield a transfection efficiency of 5–10%. Following transfection, the media was replaced with either vehicle or doxycycline-treated neurite outgrowth media and replaced every 48 h. Following 72 h of neurite extension, cells were fixed in 4% paraformaldehyde. For immunohistochemistry, fixed cells were incubated in blocking buffer (PBS + 5% normal goat serum and 1% Triton-X 100). Neurites were stained with a monoclonal antibody against α-tubulin (1:10 000) followed by goat anti-mouse AlexaFluor 598 fluorescent secondary. Images were captured at ×20 magnification using the QuickPro software, and the neurite length was determined using the measure function. Neurites were measured from at least 50 cells from two separate culture dates. Statistically significant changes in neurite length were determined by Student's t-test. Significant shifts in the number of neurites per cell relative to control cultures were determined by Chi-squared analysis. All error bars represent the standard error of the mean.
Site-directed mutagenesis
Mutations were introduced into previously described eGFP-SMN and GST-SMN exon 2b constructs using the QuickChange system. The following primer pairs were generated to introduce the K65A, K71A, K76A and K82A mutations into these vectors: K65AF 5′-GTGAAACTTCGGGTGCACCAAAAACCACACC-3′, K65AR 5′-GGTGTGGTTTTTGGTGCACCCGAAGTTTCAC-3′, K71AF 5′-GTAAACCAAAAACCACACCTGCAAGAAAACCTGCTAAGAAG -3′, K71AR 5′-CTTCTTAGCAGGTTTTCTTGCAGGTGTGGTTTTTGGTTTAC-3′, K76AF 5′-CACCTAAAAGAAAACCTGCTGCCGCGAATAAAAGCCAAAAGAAG-3′, K76AR 5′-CTTCTTTTGGCTTTTATTCGCGGCAGCAGGTTTTCTTTTAGGTG-3′, K82AF 5′-CTAAGAAGAATAAAAGCCAACAGCAGAATACTGCAGCTTCCTTAC-3′, K82AR 5′-GTAAGGAAGCTGCAGTATTCTGCTGTTGGCTTTTATTCTTCTTAG-3′.
Expression of GST-fusion proteins
GST-SMN exon 2a+ and GST-SMN exon 2b fusions were cloned into pGex4T-3 as previously described. The constructs were transformed into DH5a cells, and a single colony was isolated and grown to OD600. Cells were collected and lysed following 3 h of IPTG induction, and GST fusions were bound to High Performance Glutathione Sepharose (GE Healthcare) for 1 h at 4°C. Protein expression was confirmed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) of 20 µl of beads in 2× sodium dodecyl sulphate (SDS) sample buffer on a 4–15% Tris–Glycine gel followed by visualization with Coomassie blue stain. The GST-SMN fragments run at ∼32 and 31 kD, respectively. For α-COP-binding studies, FLAG-tagged human α-COP was cloned into the pTNT vector as previously described and expressed from the T7 promoter using the TNT T7 Quick Coupled Transcription/Translation System (Promega). Immobilized GST–SMN fusions were incubated with FLAG-α-COP protein for 4 h at 4°C and washed five times. Bound proteins were removed in 2× SDS sample buffer and separated by SDS–PAGE on a 4–15% Tris–glycine gradient gel. Proteins were transferred to PVDF membrane and visualized by western blotting with an anti-FLAG antibody (M2, 1:2500) or antibodies against SMN exon 2 (MANSMA 1).
FUNDING
Funding from the Muscular Dystrophy Association and the University of Indiana School of Medicine supported this work.
Supplementary Material
ACKNOWLEDGEMENTS
All eGFP-SMN constructs were a generous gift from Dr Robert Morse in the laboratory of Dr Philip Young at Peninsula Medical School, Exeter, UK.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Coovert D.D., Le T.T., McAndrew P.E., Strasswimmer J., Crawford T.O., Mendell J.R., Coulson S.E., Androphy E.J., Prior T.W., Burghes A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 3.Buhler D., Raker V., Luhrmann R., Fischer U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum. Mol. Genet. 1999;8:2351–2357. doi: 10.1093/hmg/8.13.2351. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H.L., Pan F., Hong D., Shenoy S.M., Singer R.H., Bassell G.J. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Todd A.G., Morse R., Shaw D.J., McGinley S., Stebbings H., Young P.J. SMN, Gemin2 and Gemin3 associate with beta-actin mRNA in the cytoplasm of neuronal cells in vitro. J. Mol. Biol. 2010;401:681–689. doi: 10.1016/j.jmb.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 6.Rathod R., Havlicek S., Frank N., Blum R., Sendtner M. Laminin induced local axonal translation of beta-actin mRNA is impaired in SMN-deficient motoneurons. Histochem. Cell Biol. 2012;138:737–748. doi: 10.1007/s00418-012-0989-1. [DOI] [PubMed] [Google Scholar]
- 7.Liu H., Beauvais A., Baker A.N., Tsilfidis C., Kothary R. Smn deficiency causes neuritogenesis and neurogenesis defects in the retinal neurons of a mouse model of spinal muscular atrophy. Dev. Neurobiol. 2011;71:153–169. doi: 10.1002/dneu.20840. [DOI] [PubMed] [Google Scholar]
- 8.McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck R., Rawet M., Wieland F.T., Cassel D. The COPI system: molecular mechanisms and function. FEBS Lett. 2009;583:2701–2709. doi: 10.1016/j.febslet.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 10.Peter C.J., Evans M., Thayanithy V., Taniguchi-Ishigaki N., Bach I., Kolpak A., Bassell G.J., Rossoll W., Lorson C.L., Bao Z.Z., et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum. Mol. Genet. 2011;20:1701–1711. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ting C.H., Wen H.L., Liu H.C., Hsieh-Li H.M., Li H., Lin-Chao S. The spinal muscular atrophy disease protein SMN is linked to the Golgi network. PLoS One. 2012;7:e51826. doi: 10.1371/journal.pone.0051826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelletier S., Gingras S., Howell S., Vogel P., Ihle J.N. An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J. Neurosci. 2012;32:16560–16573. doi: 10.1523/JNEUROSCI.1787-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X., Kedlaya R., Higuchi H., Ikeda S., Justice M.J., Setaluri V., Ikeda A. Mutation in archain 1, a subunit of COPI coatomer complex, causes diluted coat color and Purkinje cell degeneration. PLoS Genet. 2010;6:e1000956. doi: 10.1371/journal.pgen.1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Todd A.G., Lin H., Ebert A.D., Liu Y., Androphy E.J. COPI transport complexes bind to specific RNAs in neuronal cells. Hum. Mol. Genet. 2013;22:729–736. doi: 10.1093/hmg/dds480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trujillo C., Taylor-Parker J., Harrison R., Murphy J.R. Essential lysine residues within transmembrane helix 1 of diphtheria toxin facilitate COPI binding and catalytic domain entry. Mol. Microbiol. 2010;76:1010–1019. doi: 10.1111/j.1365-2958.2010.07159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cashman N.R., Durham H.D., Blusztajn J.K., Oda K., Tabira T., Shaw I.T., Dahrouge S., Antel J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z.B., Zhang X., Li X.J. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013;23:378–393. doi: 10.1038/cr.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen H.L., Lin Y.T., Ting C.H., Lin-Chao S., Li H., Hsieh-Li H.M. Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum. Mol. Genet. 2010;19:1766–1778. doi: 10.1093/hmg/ddq058. [DOI] [PubMed] [Google Scholar]
- 19.Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M., et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell. Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hua Y., Zhou J. Modulation of SMN nuclear foci and cytoplasmic localization by its C-terminus. Cell Mol. Life Sci. 2004;61:2658–2663. doi: 10.1007/s00018-004-4300-z. [DOI] [PubMed] [Google Scholar]
- 21.Morse R., Shaw D.J., Todd A.G., Young P.J. Targeting of SMN to Cajal bodies is mediated by self-association. Hum. Mol. Genet. 2007;16:2349–2358. doi: 10.1093/hmg/ddm192. [DOI] [PubMed] [Google Scholar]
- 22.Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 23.Eugster A., Frigerio G., Dale M., Duden R. The alpha- and beta’-COP WD40 domains mediate cargo-selective interactions with distinct di-lysine motifs. Mol. Biol. Cell. 2004;15:1011–1023. doi: 10.1091/mbc.E03-10-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briese M., Esmaeili B., Sattelle D.B. Is spinal muscular atrophy the result of defects in motor neuron processes. Bioessays. 2005;27:946–957. doi: 10.1002/bies.20283. [DOI] [PubMed] [Google Scholar]
- 25.Bosio Y., Berto G., Camera P., Bianchi F., Ambrogio C., Claus P., Di Cunto F. PPP4R2 regulates neuronal cell differentiation and survival, functionally cooperating with SMN. Eur. J. Cell Biol. 2012;91:662–674. doi: 10.1016/j.ejcb.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Yamazaki T., Chen S., Yu Y., Yan B., Haertlein T.C., Carrasco M.A., Tapia J.C., Zhai B., Das R., Lalancette-Hebert M., et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez M., Scales S.J., Kreis T.E., Perez F. Membrane recruitment of coatomer and binding to dilysine signals are separate events. J. Biol. Chem. 2000;275:29162–29169. doi: 10.1074/jbc.M003630200. [DOI] [PubMed] [Google Scholar]
- 28.Letourneur F., Gaynor E.C., Hennecke S., Demolliere C., Duden R., Emr S.D., Riezman H., Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 29.Kotani T., Sutomo R., Sasongko T.H., Sadewa A.H., Gunadi Minato T., Fujii E., Endo S., Lee M.J., Ayaki H., et al. A novel mutation at the N-terminal of SMN Tudor domain inhibits its interaction with target proteins. J. Neurol. 2007;254:624–630. doi: 10.1007/s00415-006-0410-x. [DOI] [PubMed] [Google Scholar]
- 30.Fallini C., Bassell G.J., Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 2012;1462:81–92. doi: 10.1016/j.brainres.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Todd A.G., Lin H., Ebert A.D., Liu Y., Androphy E.J. COPI transport complexes bind to specific RNAs in neuronal cells. Hum. Mol. Genet. 2013;22:729–736. doi: 10.1093/hmg/dds480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y.N., Wang H., Yamaguchi H., Lee H.J., Lee H.H., Hung M.C. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem. Biophys. Res. Commun. 2010;399:498–504. doi: 10.1016/j.bbrc.2010.07.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.