

Background: IL-6 is a profibrotic molecule, but the mechanism is unclear.
Results: IL-6 mediates fibrosis via a STAT3- and Smad3-dependent pathway mediated via a novel cytokine, Gremlin.
Conclusion: A novel pathway of IL-6 mediated fibrogenesis has been defined.
Significance: Targeting Gremlin is a new therapeutic target in fibrosis downstream of STAT3.
Keywords: Cell Biology, Collagen, Fibrogenesis, Interleukin, Signal Transduction, Interleukin-6, Extracellular
Abstract
Fibrosis is a common and intractable condition associated with various pathologies. It is characterized by accumulation of an excessive amount of extracellular matrix molecules that primarily include collagen type I. IL-6 is a profibrotic cytokine that is elevated in the prototypic fibrotic autoimmune condition systemic sclerosis and is known to induce collagen I expression, but the mechanism(s) behind this induction are currently unknown. Using healthy dermal fibroblasts in vitro, we analyzed the signaling pathways that underscore the IL-6-mediated induction of collagen. We show that IL-6 trans signaling is important and that the effect is dependent on STAT3; however, the effect is indirect and mediated through enhanced TGF-β signaling and the classic downstream cellular mediator Smad3. This is due to induction of the bone morphogenetic protein (BMP) antagonist Gremlin-1, and we show that Gremlin-1 is profibrotic and is mediated through canonical TGF-β signaling.
Introduction
Fibrosis is characterized by excessive accumulation of extracellular matrix components that include primarily collagen type I. Tissue fibrosis results from excessive scarring that ultimately leads to loss of tissue function through destruction of tissue architecture. Fibrosis is a critical component of many diseases including liver fibrosis, idiopathic pulmonary fibrosis, myelofibrosis, rheumatoid arthritis, and systemic sclerosis. Fibrosis may be the final common outcome of many autoimmune diseases, and fibrotic disease is estimated to contribute up to 45% of all deaths in the Western world; there is therefore a need for therapeutics.
Systemic sclerosis is a prototypical fibrotic disease that is characterized by activation of the immune system, autoimmunity, vasculopathy, chronic inflammation, and ultimately fibrosis (1). The cause of the disease is unknown, and there is currently no accepted treatment. However, there is now a consensus that there is an interplay between the innate and adaptive immune system that leads to differentiation of resident fibroblasts to “myofibroblasts” and the excessive deposition of extracellular matrix, primarily collagen I. Although the effector cell is the activated myofibroblast, the interplay between the immune system (innate and adaptive) and secreted profibrotic factors appears important in the pathogenesis of the disease (2). The observation of perivascular infiltrates of leukocytes at sites of fibrosis early in the disease course points to an important role of leukocytes in disease pathogenesis (3).
Interleukin-6 (IL-6) is a classic proinflammatory cytokine but is also pleiotropic. IL-6 is produced by a wide variety of cells including monocytes/macrophages, T cells, endothelial cells, fibroblasts, and hepatocytes. This cytokine is critical in many infectious disease situations and regulates a variety of cell functions such as cell proliferation, activation, and differentiation. IL-6 levels are elevated in SSc2 serum (4–6), and isolated lymphocytes spontaneously express elevated levels of IL-6. Blockade of IL-6 signaling via an anti-IL-6R antibody in sclerodermatous chronic graft versus host disease, which mimics SSc, attenuated the disease and subsequent fibrosis and increased beneficial T regulatory cells (7). The cytokine binds to its receptor IL-6R and also utilizes a common receptor, glycoprotein 130, to initiate intracellular signaling. The common cytokine receptor gp130 is shared with IL-6, Oncostatin M (OSM), IL-11, leukemia inhibitory factor (LIF), cardiotrophin-1 (CT-1), cardiotrophin-like cytokine, and ciliary neutrophilic factor (CNTF), which are a family of pleiotropic cytokines (8, 9). Although the common co-receptor gp130 is ubiquitously expressed, IL-6R is highly restricted in its expression pattern (10). IL-6R is mainly expressed by hepatocytes and a subset of T cells. This limits the repertoire of cells that are able to respond to IL-6 signaling. However, trans signaling increases the number of cells that can respond to IL-6 by the binding of soluble IL-6R, shed from cells via a sheddase, and IL-6 to gp130 to initiate signaling (10, 11). Thus cells that do not express the membrane-bound IL-6R can now respond in association with soluble IL-6R and IL-6, forming a complex. Once signaling is initiated, receptor-associated Janus kinases (JAKs) are activated, and signal transducers and activators of transcription (STATs) transcription factors are phosphorylated and translocate to the cell nucleus to coordinate gene expression by binding to STAT-responsive gene elements (12, 13). These JAKs do not possess tyrosine kinase activity themselves. The JAKs consist of JAK1, JAK2, JAK3, and TyK2. ERK can also be activated in response to IL-6 (12). Multiple JAK inhibitors are now in clinical trials to test their effects in rheumatoid arthritis. Indeed gain of function mutations in JAK2 underlie myelofibrosis and give a rationale for targeting JAK therapeutically. Although IL-6 trans signaling is known to cause fibrosis, the underlying molecular mechanism is unknown. In a mouse model of fibrosis, it was shown that hyperactivation of STAT3 enhanced fibrosis (14), and excessive activation of STAT3 was found in the lung tissue of patients with idiopathic lung fibrosis. Consistent with a role of STAT3 in mediating fibrosis, keloid fibroblasts have excessive IL-6 secretion and respond to IL-6 stimulation with up-regulation of collagen transcription (15). Furthermore genetic deletion of IL-6 results in reduced fibrosis in animal models of lung fibrosis (16). Indeed SSc dermal fibroblasts cultured from lesional skin of patients have elevated phosphorylated STAT3, which stays elevated in culture (17), and blockade of JAK2, which lies upstream of STAT3, reduced collagen levels in these cells and also in the bleomycin model of fibrosis (17), suggesting that JAKs play a critical role in fibrosis. Further evidence comes from the finding that hypertrophic scars from burn patients have elevated phosphorylated STAT3 levels in in situ tissue sections and also in isolated cultured hypertrophic skin fibroblasts and that a STAT3 inhibitor attenuates both collagen I expression and proliferation genes such as c-Myc (18). Thus STAT3 is important in regulating fibrosis-related genes; however, the precise molecular mechanism(s) remain to be determined. It is likely that molecular feedback loops are at play in driving the collagen deposition. To gain an understanding of the underlying molecular mechanism of IL-6 trans signaling in fibrosis, we used dermal fibroblasts to examine the role of the downstream signaling pathways utilized that lead to fibrosis.
EXPERIMENTAL PROCEDURES
Cell Culture
Dermal fibroblasts were cultured from punch biopsies taken from lesions of SSc patients (n = 3) or healthy controls undergoing breast reduction surgery. The dermal fibroblasts were isolated and cultured as described previously (19). Local ethical approval was granted for this study. Cells were maintained in RPMI medium (Sigma) supplemented with 10% (v/v) heat-inactivated serum, l-glutamine, and penicillin and streptomycin in 75-cm3 tissue culture flasks until seeding.
Chemicals
JAK kinase inhibitor Ruxolitinib was purchased from Calbiochem, and STAT1 inhibitor Fludarabine was purchased from Selleckchem. The MAPK inhibitors U0126 and SB202190 were both purchased from Cell Signaling Technology. The TGF-β receptor (TGF-βR) inhibitor SB431542 was purchased from Tocris and reconstituted in dimethyl sulfoxide (DMSO). All recombinant proteins were purchased from R&D Systems (IL-6, sIL-6R, IL-10, and Gremlin-1). The endotoxin levels were determined to be <0.01 ng/μl. Recombinant proteins were also boiled and incubated to check for contamination.
Quantitative RT-PCR
After the appropriate treatments, RNA was isolated using TRIzol according to the manufacturer's instructions. 1 μg of RNA was DNase-treated and reverse-transcribed using reverse transcriptase (Invitrogen). cDNA was then analyzed using specific primers (Collagen1A1 forward 5′-CAA GAG GAA GGC CAA GTC GAG G-3′, reverse 5′-CGT TGT CGC AGA CGC AGA T-3′; Gremlin-1 gene forward 5′-TAT GAG CCG CAC AGC CTA CA-3′, reverse 5′-GCA CCT TGG GAC CCT TTC TT-3′; and ribosomal 18 S forward 5′-CGA ATG GCT CAT TAA ATC AGT TAT GG-3′, reverse 5′-TAT TAG CTC TAG AAT TAC CAC AGT TAT CC-3′) using SYBR Green technology. Relative levels were normalized to 18 S, and -fold change was calculated using the ΔCt method.
Immunoblotting
After the appropriate treatment, cells were washed twice with cold PBS and then lysed in Laemmli buffer and separated on a 10% SDS polyacrylamide gel electrophoresis. The proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane for 1 h at 120 V, after which the membrane was blocked in 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween 20 for 1 h and the primary antibody was incubated overnight at 4 °C. Blots were then washed in TBS/Tween and incubated with the appropriate secondary antibody labeled with HRP, and then the bands visualized with chemiluminescent substrate (ECL Plus, GE Healthcare) and exposed on film (Kodak). Antibodies used were: 1:1200 phospho-Smad3 (Abcam), 1:1000 Smad3 (Abcam) phospho-p38 (Cell Signaling), 1:800 TGF-β receptor (Cell Signaling), and 1:100,000 glyceraldehyde-3 phosphate dehydrogenase (GAPDH) (Abcam). GAPDH was used to confirm equal loading.
siRNA
For the siRNA silencing experiments cells were seeded into 24-well plates, and at 50–60% confluence 100 nm Smad3 siRNA SMARTpool (Dharmacon) 100 nm or nontargeting control siRNA was transfected using DharmaFECT 1 in antibiotic and serum-free medium. Following 24 h of transfection, medium was replaced with IL-6 + sIL-6R, and after a further 24 h, the cells were harvested. For Gremlin-1 siRNA silencing, the cells were seeded, and 100 nm specific Gremlin-1 siRNA SMARTpool or 100 nm nontargeting siRNA was transfected using Dhamafect1. After 24 h, medium was replaced with nothing or IL-6 + sIL-6R; following 24 h of incubation, cells were harvested. In diseased SSc fibroblasts, cells were seeded from three donors and transfected with Gremlin-1 or nontargeting control; after 24 h, cells were harvested.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software, and one-way analysis of variance or Student's t test was applied. Data presented are the means ± S.E. For all analyses a p value of less than 0.05 was considered significant.
RESULTS
JAKs and STATs Drive IL-6-induced Collagen Expression
We and others had previously demonstrated that incubation with IL-6 and sIL-6R leads to Collagen1A1 expression in healthy dermal fibroblasts in vitro (19, 20). We tested the hypothesis that chemical blockade of JAK 1 using the clinical JAK inhibitor Ruxolitinib would reduce IL-6 trans signaling-induced collagen induction downstream of receptor engagement. Indeed incubation with IL-6 or sIL-6R alone did not affect collagen I expression in dermal fibroblasts; however, incubation with both IL-6 and sIL-6R led to an increased expression of collagen I 6.83-fold, and preincubation with Ruxolitinib attenuated this induction of collagen down to 2.98-fold as compared with control (p = 0.001). Ruxolitinib alone was changed by 1.23-fold as compared with control (Fig. 1A).
FIGURE 1.

IL-6 trans signaling drives fibrogenesis. A, dermal fibroblasts were seeded and treated with IL-6 (20 ng/ml) or sIL-6R (25 ng/ml) alone or in combination or pretreated with JAK inhibitor Ruxolitinib (Rux) for 24 h, after which cells were then analyzed for Collagen1A1 mRNA. B, cells were treated with IL-6 (20 ng/ml) or sIL-6R (25 ng/ml) alone or in combination or pretreated with the STAT1-specific inhibitor Fludarabine (Fluda, 50 ng/ml) for 24 h, after which cells were harvested and analyzed for Collagen1A1 mRNA. C, cells were treated with IL-6 (20 ng/ml) or sIL-6R (25 ng/ml) alone or in combination or pretreated with the MAPK inhibitor U0126 (20 μm) for 24 h. Cells were then harvested, and Collagen1A1 levels were analyzed. Data are expressed as mean ± S.E. (n = 4) ***, p = 0.001.
Because IL-6 can also activate STAT1 to a lesser degree than STAT3, we examined the role of STAT1 in collagen induction by pharmacological blockade of STAT1 with Fludarabine. Fludarabine is a potent and specific STAT1 inhibitor (21) and is used as an antileukemia drug. Fig. 1B shows that IL-6 and sIL-6R induce collagen I expression but that pretreatment with Fludarabine does not reduce this increased expression. Thus IL-6-mediated collagen expression is independent of STAT1 activation.
ERK has been reported to be affected by IL-6 incubation, and we therefore wanted to examine the role of ERK in IL-6 trans signaling induction of collagen I. Using the ERK inhibitor U0126 that targets the ERK pathway, we found that this did not attenuate the IL-6-mediated increase in Collagen1A1 induction (Fig. 1C).
Collagen Time Course Demonstrates Secondary Effects
Because the collagen induction via IL-6 trans signaling appeared elevated after 24 h of stimulation, we performed a time course analysis to see whether this effect was mediated earlier. Time course analysis of collagen mRNA expression revealed that 24 h was the peak stimulation after IL-6 + sIL-6R and that after this time collagen decreased (Fig. 2A). Promoter analysis suggested that STAT3 does not have a binding site in the collagen promoter. Based on this we felt the effect was indirect.
FIGURE 2.
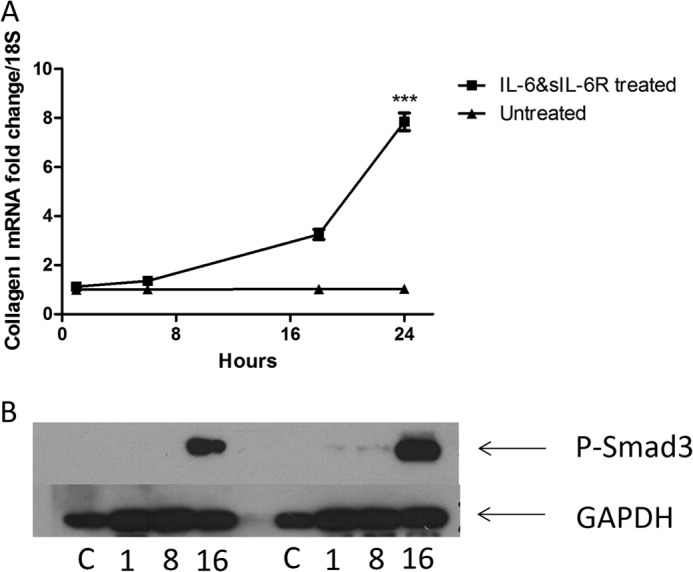
Delayed kinetics in IL-6 signaling. A, dermal fibroblasts were exposed to IL-6 (20 ng/ml) and sIL-6R (25 ng/ml) or nothing, and after 6, 18, and 24 h, RNA was harvested and collagen 1A1 levels were quantified normalized to 18 S. Data are expressed as the mean ± S.E. B, representative Western blot of phospho-Smad3 from two independent experiments, GAPDH was used a loading control. C = control. The numbers indicate the times. n = 3. ***, p = 0.001 (t test).
Delayed Smad3 Phosphorylation in Treated Cultures
We examined whether phosphorylated Smad3 was phosphorylated using Western blotting. This revealed that only after 16 h of stimulation with IL-6 + sIL-6R were there significant phosphorylation levels in the dermal fibroblast (Fig. 2B).
TGF-β1 Levels Are Not Elevated
Because phosphorylated Smad 3 levels were only detectable after 16 h following IL-6 + sIL-6R stimulation, we examined the expression of TGF-β1. Quantitative RT-PCR was used to identify the mRNA for TGF-β1, and this was surprisingly found not to be significantly elevated (Fig. 3A) at the time points examined.
FIGURE 3.

TGF-β1 or receptor levels are not elevated. A, TGF-β1 levels were quantified at different time points after incubation with IL-6 (20 ng/ml) and sIL-6R (25 ng/ml). B, TGF-β1 receptor II levels were quantified by quantitative RT-PCR after stimulation with IL-6 (20 ng/ml) and sIL-6R (25 ng/ml). C, representative immunoblot of TGF-βR II after stimulation with IL-6 (20 ng/ml) and sIL-6R (25 ng/ml). C = control untreated. Time is indicated in hours. Data are expressed as the mean ± S.E. n = 4. No significant differences were found.
TGF-β Receptor II Levels
Because the TGF-β1 levels were not elevated, we also examined the levels of the TGF-β receptor II in the dermal fibroblasts. Again there were no significant differences at both the transcriptional mRNA and the protein levels (Fig. 3, B and C).
Blockade of TGF-β Signaling Reduces Collagen Elevation by IL-6 + sIL-6R
Because activation of Smad3 was occurring secondary to IL-6 trans signaling, we used SB431542 to inhibit TGF-β signaling via both TGF-β receptors. Pretreatment with SB431542 significantly reduced IL-6 + sIL-6R-induced collagen expression (p = <0.0001) (Fig. 4A). DMSO (0.01%) was used as a vehicle control; this had no effect on the cells.
FIGURE 4.

IL-6 trans signaling is mediated via Smad3. A, fibroblasts were seeded and treated with IL-6 (20 ng/ml) or sIL-6R (25 ng/ml) or in combination or pretreated with the TGF-βR II inhibitor SB431542, SB431542 alone, or DMSO vehicle control (0.01%) for 24 h, and then Collagen1A1 levels were quantified. B, cells were seeded and then transfected with siRNA against total Smad3 or nontargeting control (100 nm) with DharmaFECT 1, and 14 h later, cells were incubated with IL-6 + sIL-6R (25 ng/ml). 24 h later, the cells were harvested, and Collagen1A1 was analyzed. C, representative immunoblot for Smad3 48 h after siRNA transfection (100 nm siRNA) C = control. n = 3. Data are the mean ± S.E. ***, p < 0.0001.
siRNA Knockdown of Smad3 Attenuates IL-6-induced Collagen Synthesis
One of the downstream signaling pathways utilized by the canonical signaling pathway through which TGF-β operates is Smad3; this is an intracellular transcription factor that is phosphorylated and translocates into the nucleus to facilitate gene transcription. Using specific siRNA against total Smad3, it was shown that IL-6 + sIL-6R-induced collagen expression was significantly attenuated as compared with a nontargeting control siRNA after 24 h (Fig. 4B). Knockdown of Smad3 was highly efficient (Fig. 4C).
siRNA Targeting of Gremlin-1 Reduces the Induction of Collagen
Gremlin-1 is a BMP antagonist. Using specific siRNA directed against Gremlin-1, it was shown that the induction of collagen by IL-6 + sIL-6R was significantly diminished, but a nontargeting control siRNA did not reduce the induction of collagen (Fig. 5A).
FIGURE 5.

Gremlin is downstream of IL-6. A, fibroblasts were transfected with specific siRNA against Gremlin-1 or nontargeting control (100 nm) with DharmaFECT 1. After 24 h, medium was removed and replaced with IL-6 + sIL-6R (25 ng/ml). 24 h later, cells were harvested, and Collagen1A1 was quantified. B, Gremlin mRNA levels were quantified after 1 and 16 h of incubation with IL-6 (20 ng/ml) + sIL-6R (25 ng/ml). C, Gremlin levels in dermal fibroblasts from healthy control or SSc patients. Data are expressed as the mean ± S.E. n = 3 ***, p < 0.001; *, p < 0.05.
Treatment of cells with IL-6 + sIL-6R resulted in a significant increase in Gremlin-1 levels at 16 h following stimulation (Fig. 5B). Analysis of unstimulated SSc fibroblasts revealed a 3-fold increase as compared with healthy control cultures (p = 0.0478, Student's t test) (Fig. 5C).
Recombinant Gremlin-1 Is Profibrotic
Because we could reduce collagen induction via Smad3 signaling inhibition and siRNA against the BMP antagonist Gremlin, we sought to determine whether direct addition of Gremlin induces collagen in dermal fibroblasts. Indeed incubation of 1 μg of recombinant Gremlin-1 led to a 5.3-fold increase of collagen as compared with untreated cultures (Fig. 6A). We also demonstrated that the addition of Gremlin induces robust phosphorylation of Smad3 after 1 h of incubation and that recombinant Gremlin also induced phospho-p38 at similar time points (Fig. 6B).
FIGURE 6.

Recombinant Gremlin (rGremlin) is profibrotic. A, dermal fibroblasts were seeded and treated with recombinant Gremlin (1 μg/ml). 24 h later, the cells were harvested, and Collagen1A1 levels were quantified. B, representative immunoblot of phospho-Smad3 (p-Smad3), phospho-p38 (p38), and GAPDH after stimulation with Gremlin-1 (Grem, 1 μg/ml). C, fibroblasts were seeded and incubated with Gremlin-1 (1 μg/ml) or pretreated with the TGF-βR inhibitor SB431542 (10 μm) prior to Gremlin-1 incubation or treated with SB431542 alone (10 μm) or with DMSO vehicle alone. 2 h later, Collagen1A1 levels were quantified. D, cells were seeded and incubated with Gremlin-1 (1 μg/ml) or pretreated with SB202 or SB202 alone or with DMSO vehicle control. After 24 h, Collagen1A1 levels were quantified. E, cells were seeded and transfected with Smad3-specific siRNA or nontargeting control (100 nm) with DharmaFECT 1. After 24 h, medium was removed and replaced with medium containing recombinant Gremlin-1 (1 μg/ml). Following 24 h of incubation, cells were harvested, and Collagen1A1 quantified. Data are expressed as the mean ± S.E. n = 4 ***, p < 0.0001.
Blockade of canonical TGF-β signaling with SB431542 significantly reduced Gremlin-induced collagen expression by IL-6 trans signaling (p = <0.001) (Fig. 6C). However, blockade with the p38 inhibitor SB202190 (10 μm) did not reduce the induction of collagen (Fig. 6D). siRNA-mediated silencing of Smad3 revealed that this is the main signaling molecule regulating Gremlin-1-mediated collagen (Fig. 6E).
IL-10 Does Not Induce Gremlin-1 Up-regulation
To confirm the specificity of IL-6 for the induction of Gremlin, we used another STAT3-inducing cytokine, IL-10, to determine the specificity. IL-10 did not induce Gremlin-1 expression at any time point analyzed and mirrored control levels (Fig. 7A).
FIGURE 7.
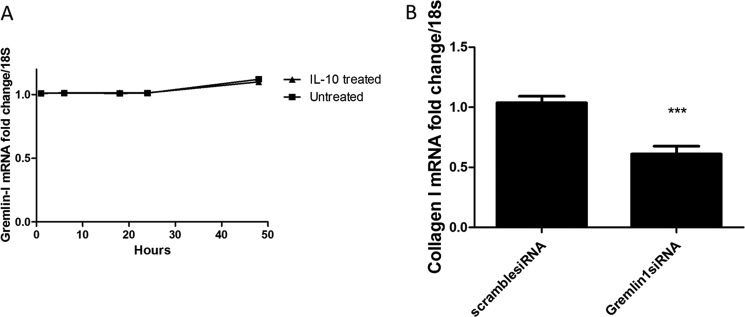
Effect is IL-6-specific, and siRNA-mediated silencing of Gremlin reduces fibrosis. A, cells were seeded and incubated with recombinant IL-10 (a STAT3 cytokine), and at set times, the cells were harvested, and Collagen1A1 levels quantitated by quantitative RT-PCR. No significant differences were found. B, diseased SSc fibroblasts were seeded and transfected with siRNA against Gremlin-1 (100 nm) or nontargeting control siRNA with DharmaFECT 1. After 24 h, Collagen1A1 was quantified, and -fold change was compared with nontargeting control siRNA. Data are expressed as the mean ± S.E. n = 3 ***, p < 0.001, Student's t test.
Silencing of Gremlin-1 Reverses the Profibrotic Phenotype in SSc Dermal Fibroblasts
Because the SSc fibroblasts are autonomous and produce excessive collagen I in vitro and because we have demonstrated elevated levels of the novel cytokine Gremlin in these cells, we used siRNA against Gremlin-1 and examined the levels of collagen following knockdown. Fig. 7B demonstrates that silencing of Gremlin reduced collagen transcripts by 50% as compared with scrambled control siRNA (p = 0.01, Student's t test).
DISCUSSION
IL-6 is described as a classic proinflammatory cytokine; however, it is also profibrotic, but the exact underlying mechanism is still undefined. Fibrotic diseases such as SSc are characterized by uncontrolled activation of fibroblasts that secrete huge quantities of extracellular matrix following inflammation. It is known that levels of IL-6 are elevated in SSc and that this correlates with skin score (20). In this study we demonstrated that IL-6 trans signaling leads to collagen increases through an indirect mechanism that is mediated through the BMP antagonist Gremlin-1, which reveals a novel pathway. Blockade of STAT3 reduced IL-6-induced collagen induction both in this study and in keloid fibroblasts in a previous study (22). However, it is independent of STAT1. Moreover blockade of MAPK signaling with U0126 did not reduce collagen levels. This is not in accordance with Khan et al. (20). We and others have reported that phospho-STAT3 is elevated in SSc dermal fibroblasts in culture and that they maintain this elevated constitutive STAT3 activation in serial passages (17, 19). Hyperactivation of STAT3 in genetically modified mice in which they have “switched on” hyperactive STAT3 constantly shows that if you treat them with bleomycin, their lung fibrosis is exacerbated, thereby underscoring the role of IL-6 signaling in fibrosis (14). The molecular mechanism for this is unknown. Furthermore, genetic deletion of SOCS3, the endogenous negative regulator of STAT3, exacerbates graft versus host disease, a model of SSc (23).
Because it had been reported that IL-6 can induce expression of TGF-β1 in liver cells (24, 25), at least at the mRNA level, we examined TGF-β1 expression after IL-6 stimulation. However, it appeared that TGF-β1 mRNA was not increased after IL-6 trans signaling. Therefore another mechanism other than IL-6-mediated TGF-β1 must be increasing the collagen expression. The time course of the induction of collagen expression was delayed, indicating that this was an indirect effect rather than a direct transcriptional activation of collagen. Furthermore there was a delayed phosphorylation of Smad3, a critical downstream mediator of fibrosis. Once Smad3 is phosphorylated, it dimerizes with Smad4 and then shuttles to the nucleus to regulate gene expression.
Gremlin-1 is a BMP antagonist and contains a highly conserved cysteine-rich domain and is a member of the TGF-β superfamily of cystine knot cytokines. Gremlin-1 has been found to predict adverse outcome in heart failure, is highly correlated to myocardial fibrosis grade (26), and was initially identified as being induced under rather high glucose conditions (27). Interestingly Gremlin has been found to be overexpressed in idiopathic pulmonary fibrosis (28), a devastating disease. Gremlin-1 is an antagonist of BMPs including BMP4 and BMP7, and BMP7 is the antagonist of TGF-β1 signaling and has been found to inhibit TGF-β1-induced renal fibrosis in the kidney (29) and also the eye (30). Gremlin has also been suggested to be a specific marker of activated stellate cells by transcriptome analysis (31). Also overexpression of Gremlin has been found to inhibit BMP4, thus leading to enhanced TGF-β signaling and extracellular matrix deposition in primary open angle glaucoma (32). However, a non-BMP-dependent role has recently come to light. A recent study found that Gremlin can bind the VEGF receptor2 to induce angiogenesis (33); it likely that Gremlin utilizes other pathways. We have found a non-BMP-dependent pathway of Gremlin in dermal fibroblasts. We demonstrated that siRNA-mediated knockdown of Gremlin-1 resulted in reductions of IL-6 trans signaling-induced collagen expression. We further show that IL-6 induces Gremlin and that SSc dermal fibroblasts have elevated constitutive levels of Gremlin. Gremlin utilizes the TGF-β signaling pathway and is dependent on Smad3 for its action but not p38. Although phosphorylation of p38 occurred, this pathway appears to play no role in the induction of collagen. Therefore Gremlin utilizes only the canonical signaling pathway of TGF-β. Reactivation of developmental genes may occur in fibrotic diseases similar to cancer. Only one study on the bleomycin model of fibrosis (a mimic of SSc) has shown altered levels of Gremlin in a gene arrays analysis, so this represents an important finding. Importantly in SSc fibroblasts siRNA-mediated reduction of Gremlin reduced collagen by over 50%.
Also of note is the fact that Gremlin is additionally associated with the development of pulmonary hypertension and haplodeficiency in a mouse model that augments hypertension and vascular remodeling (34). This is important as many SSc patients also develop pulmonary hypertension, which carries a high mortality rate. Thus targeting Gremlin may be beneficial on two fronts: first, the modulation of fibrogenesis, and second, the augmentation of pulmonary hypertension. Moreover haplodeficiency of Gremlin also reduces diabetic nephropathy and associated damage markers and the fibrotic protein connective tissue growth factor (35). To confirm that the modulation of Gremlin levels by STAT3 was IL-6-specific, we used another STAT3 utilizing cytokine IL-10. We found over the time course that IL-10 could not induce Gremlin levels (or collagen). Therefore the profibrotic nature of STAT3 activation is cytokine-specific, although both cytokines use STAT3 downstream of receptor engagement. Thus although different cytokines may utilize the same STAT3 transcription factor, the ultimate response is different.
In conclusion IL-6 trans signaling leads to activation of STAT3 via phosphorylation translocation to the nucleus and activation of target genes, and Gremlin is induced and activates the canonical TGF-β signaling cascade via Smad3 activation that leads to collagen expression in an autocrine loop. It is of interest that a major pharmaceutical company have a therapeutic antibody directed against Gremlin-1, which may be a possible therapeutic in SSc. This may be more specific than general JAK inhibition, which has been shown to be effective in an animal model of SSc (17). Confirming target activation in the patient may be of benefit before the commencement of treatment.
Footnotes
- SSc
- systemic sclerosis
- IL-6R
- IL-6 receptor
- sIL-6R
- soluble IL-6R
- BMP
- bone morphogenetic protein
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Bhattacharyya S., Wei J., Varga J. (2012) Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol. 8, 42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Reilly S., Hügle T., van Laar J. M. (2012) T cells in systemic sclerosis: a reappraisal. Rheumatology 51, 1540–1549 [DOI] [PubMed] [Google Scholar]
- 3. Kräling B. M., Maul G. G., Jimenez S. A. (1995) Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology 63, 48–56 [DOI] [PubMed] [Google Scholar]
- 4. Needleman B. W., Wigley F. M., Stair R. W. (1992) Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor α, and interferon-γ levels in sera from patients with scleroderma. Arthritis Rheum. 35, 67–72 [DOI] [PubMed] [Google Scholar]
- 5. Ihn H., Sato S., Fujimoto M., Kikuchi K., Takehara K. (1995) Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch. Dermatol. Res. 287, 193–197 [DOI] [PubMed] [Google Scholar]
- 6. Matsushita T., Hasegawa M., Hamaguchi Y., Takehara K., Sato S. (2006) Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J. Rheumatol. 33, 275–284 [PubMed] [Google Scholar]
- 7. Le Huu D., Matsushita T., Jin G., Hamaguchi Y., Hasegawa M., Takehara K., Fujimoto M. (2012) IL-6 blockade attenuates the development of murine sclerodermatous chronic graft-versus-host disease. J. Invest. Dermatol. 132, 2752–2761 [DOI] [PubMed] [Google Scholar]
- 8. Pennica D., Shaw K. J., Swanson T. A., Moore M. W., Shelton D. L., Zioncheck K. A., Rosenthal A., Taga T., Paoni N. F., Wood W. I. (1995) Cardiotrophin-1: biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 270, 10915–10922 [DOI] [PubMed] [Google Scholar]
- 9. DeChiara T. M., Vejsada R., Poueymirou W. T., Acheson A., Suri C., Conover J. C., Friedman B., McClain J., Pan L., Stahl N., Ip N. Y., Yancopoulos G. D. (1995) Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 83, 313–322 [DOI] [PubMed] [Google Scholar]
- 10. Jones S. A., Horiuchi S., Topley N., Yamamoto N., Fuller G. M. (2001) The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 15, 43–58 [DOI] [PubMed] [Google Scholar]
- 11. O'Reilly S., Ciechomska M., Cant R., Hügle T., van Laar J. M. (2012) Interleukin-6, its role in fibrosing conditions. Cytokine Growth Factor Rev. 23, 99–107 [DOI] [PubMed] [Google Scholar]
- 12. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Reilly S., Cant R., Ciechomska M., van Laar J. M. (2013) Interleukin-6: a new therapeutic target in systemic sclerosis[quest]. Clin. Trans. Immunol. 2, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Donoghue R. J., Knight D. A., Richards C. D., Prêle C. M., Lau H. L., Jarnicki A. G., Jones J., Bozinovski S., Vlahos R., Thiem S., McKenzie B. S., Wang B., Stumbles P., Laurent G. J., McAnulty R. J., Rose-John S., Zhu H. J., Anderson G. P., Ernst M. R., Mutsaers S. E. (2012) Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 4, 939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghazizadeh M., Tosa M., Shimizu H., Hyakusoku H., Kawanami O. (2007) Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J. Invest. Dermatol. 127, 98–105 [DOI] [PubMed] [Google Scholar]
- 16. Saito F., Tasaka S., Inoue K., Miyamoto K., Nakano Y., Ogawa Y., Yamada W., Shiraishi Y., Hasegawa N., Fujishima S., Takano H., Ishizaka A. (2008) Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am. J. Respir. Cell Mol. Biol. 38, 566–571 [DOI] [PubMed] [Google Scholar]
- 17. Dees C., Tomcik M., Palumbo-Zerr K., Distler A., Beyer C., Lang V., Horn A., Zerr P., Zwerina J., Gelse K., Distler O., Schett G., Distler J. H. (2012) JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 64, 3006–3015 [DOI] [PubMed] [Google Scholar]
- 18. Ray S., Ju X., Sun H., Finnerty C. C., Herndon D. N., Brasier A. R. (2013) The IL-6 trans-Signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J. Invest. Dermatol. 133, 1212–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hügle T., O'Reilly S., Simpson R., Kraaij M. D., Bigley V., Collin M., Krippner-Heidenreich A., van Laar J. M. (2013) Tumor necrosis factor-costimulated T lymphocytes from patients with systemic sclerosis trigger collagen production in fibroblasts. Arthritis Rheum. 65, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khan K., Xu S., Nihtyanova S., Derrett-Smith E., Abraham D., Denton C. P., Ong V. H. (2012) Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann. Rheum. Dis. 71, 1235–1242 [DOI] [PubMed] [Google Scholar]
- 21. Frank D. A., Mahajan S., Ritz J. (1999) Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med. 5, 444–447 [DOI] [PubMed] [Google Scholar]
- 22. Lim C. P., Phan T. T., Lim I. J., Cao X. (2006) Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene 25, 5416–5425 [DOI] [PubMed] [Google Scholar]
- 23. Hill G. R., Kuns R. D., Raffelt N. C., Don A. L., Olver S. D., Markey K. A., Wilson Y. A., Tocker J., Alexander W. S., Clouston A. D., Roberts A. W., MacDonald K. P. (2010) SOCS3 regulates graft-versus-host disease. Blood 116, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ogata H., Chinen T., Yoshida T., Kinjyo I., Takaesu G., Shiraishi H., Iida M., Kobayashi T., Yoshimura A. (2006) Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-β1 production. Oncogene 25, 2520–2530 [DOI] [PubMed] [Google Scholar]
- 25. Aoki H., Ohnishi H., Hama K., Shinozaki S., Kita H., Yamamoto H., Osawa H., Sato K., Tamada K., Sugano K. (2006) Existence of autocrine loop between interleukin-6 and transforming growth factor-β1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 99, 221–228 [DOI] [PubMed] [Google Scholar]
- 26. Mueller K. A., Tavlaki E., Schneider M., Jorbenadze R., Geisler T., Kandolf R., Gawaz M., Mueller I. I., Zuern C. S. (2013) Gremlin-1 identifies fibrosis and predicts adverse outcome in patients with heart failure undergoing endomyocardial biopsy. J. Card. Fail. 19, 678–684 [DOI] [PubMed] [Google Scholar]
- 27. McMahon R., Murphy M., Clarkson M., Taal M., Mackenzie H. S., Godson C., Martin F., Brady H. R. (2000) IHG-2, a mesangial cell gene induced by high glucose, is human Gremlin: regulation by extracellular glucose concentration, cyclic mechanical strain, and transforming growth factor-β1. J. Biol. Chem. 275, 9901–9904 [DOI] [PubMed] [Google Scholar]
- 28. Koli K., Myllärniemi M., Vuorinen K., Salmenkivi K., Ryynänen M. J., Kinnula V. L., Keski-Oja J. (2006) Bone morphogenetic protein-4 inhibitor Gremlin is overexpressed in idiopathic pulmonary fibrosis. Am. J. Pathol. 169, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeisberg M., Hanai J., Sugimoto H., Mammoto T., Charytan D., Strutz F., Kalluri R. (2003) BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9, 964–968 [DOI] [PubMed] [Google Scholar]
- 30. Fuchshofer R., Yu A. H., Welge-Lüssen U., Tamm E. R. (2007) Bone morphogenetic protein-7 is an antagonist of transforming growth factor-β2 in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 48, 715–726 [DOI] [PubMed] [Google Scholar]
- 31. Boers W., Aarrass S., Linthorst C., Pinzani M., Elferink R. O., Bosma P. (2006) Transcriptional profiling reveals novel markers of liver fibrogenesis: Gremlin and insulin-like growth factor-binding proteins. J. Biol. Chem. 281, 16289–16295 [DOI] [PubMed] [Google Scholar]
- 32. Wordinger R. J., Fleenor D. L., Hellberg P. E., Pang I. H., Tovar T. O., Zode G. S., Fuller J. A., Clark A. F. (2007) Effects of TGF-β2, BMP-4, and Gremlin in the trabecular meshwork: implications for glaucoma. Invest. Ophthalmol. Vis. Sci. 48, 1191–1200 [DOI] [PubMed] [Google Scholar]
- 33. Mitola S., Ravelli C., Moroni E., Salvi V., Leali D., Ballmer-Hofer K., Zammataro L., Presta M. (2010) Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 116, 3677–3680 [DOI] [PubMed] [Google Scholar]
- 34. Cahill E., Costello C. M., Rowan S. C., Harkin S., Howell K., Leonard M. O., Southwood M., Cummins E. P., Fitzpatrick S. F., Taylor C. T., Morrell N. W., Martin F., McLoughlin P. (2012) Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation 125, 920–930 [DOI] [PubMed] [Google Scholar]
- 35. Roxburgh S. A., Kattla J. J., Curran S. P., O'Meara Y. M., Pollock C. A., Goldschmeding R., Godson C., Martin F., Brazil D. P. (2009) Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes 58, 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]