

Background: Epac1 is a guanine nucleotide exchange factor for Rap1 activated by cAMP.
Results: We use a BRET-based method to identify modulators of Epac1 activity.
Conclusion: Molecules that displace cAMP but do not stabilize the lid of Epac are novel inhibitors.
Significance: Understanding how cAMP analogs influence movement of Epac can aid in designing inhibitors for Epac-mediated cAMP signaling.
Keywords: Bioluminescence Resonance Energy Transfer (BRET), Cyclic AMP (cAMP), Enzyme Inhibitors, Guanine Nucleotide Exchange Factor (GEF), Molecular Docking, Molecular Pharmacology, Epac1
Abstract
The signaling molecule cAMP primarily mediates its effects by activating PKA and/or exchange protein activated by cAMP (Epac). Epac has been implicated in many responses in cells, but its precise roles have been difficult to define in the absence of Epac inhibitors. Epac, a guanine nucleotide exchange factor for the low molecular weight G protein Rap, is directly activated by cAMP. Using a bioluminescence resonance energy transfer-based assay (CAMYEL) to examine modulators of Epac activity, we took advantage of its intramolecular movement that occurs upon cAMP binding to assess Epac activation. We found that the use of CAMYEL can detect the binding of cAMP analogs to Epac and their modulation of its activity and can distinguish between agonists (cAMP), partial agonists (8-chlorophenylthio-cAMP), and super agonists (8-chlorophenylthio-2′-O-Me-cAMP). The CAMYEL assay can also identify competitive and uncompetitive Epac inhibitors, e.g. (Rp)-cAMPS and CE3F4, respectively. To confirm the results with the CAMYEL assay, we used Swiss 3T3 cells and assessed the ability of cyclic nucleotide analogs to modulate the activity of Epac or PKA, determined by Rap1 activity or VASP phosphorylation, respectively. We used computational molecular modeling to analyze the interaction of analogs with Epac1. The results reveal a rapid means to identify modulators (potentially including allosteric inhibitors) of Epac activity that also provides insight into the mechanisms of Epac activation and inhibition.
Introduction
The second messenger cyclic AMP (cAMP) is an important signaling molecule involved in a wide variety of cellular processes, including proliferation, differentiation, secretion, migration, and apoptosis (1–4). Increases in cAMP occur following activation of adenylyl cyclases, which catalyze the conversion of ATP to cAMP. The effects of cAMP in mammalian cells are mediated by three effectors as follows: protein kinase A (PKA), the exchange protein activated by cAMP (Epac),2 and cyclic nucleotide-gated channels. Epac is a guanine nucleotide exchange factor (GEF) that enhances activity of Rap1 and certain other low molecular weight G proteins and has been implicated in the regulation of numerous cAMP-mediated events (5, 6). Cyclic nucleotide phosphodiesterases (PDEs) terminate cAMP signaling by catalyzing the conversion of cAMP to 5′-AMP. Regulation of the cAMP pathway is maintained by the expression and localization of adenylyl cyclases, PDEs, PKA, and Epac in distinct subcellular signaling complexes.
Because of the important roles of the cAMP signaling pathway in normal cells and in disease settings, considerable effort has been dedicated to developing tools to manipulate and study this pathway. Although much work has been done on understanding how cAMP and various cAMP analogs bind to and regulate PKA activity, the work on Epac is more limited (7).
There are two isoforms of Epac, Epac1 and Epac2, which have similar domain structures and mechanisms of activation. Epac possesses a regulatory region that sterically blocks Rap1 from interacting with the catalytic region. The regulatory region is composed of a Dishevelled, Egl-10, pleckstrin domain, which is responsible for the subcellular localization of Epac through its binding to phosphatidic acid and a cyclic nucleotide binding domain (CNBD) (8, 9). Epac2 has a second lower affinity CNBD that is involved in the subcellular localization but not in Epac2 activation. The catalytic region is composed of a CDC25 homology domain (CDC25HD), which interacts with Rap1 and facilitates the exchange of GTP for GDP, a Ras exchange motif, and a Ras association motif (10). In its unbound state, Epac is auto-inhibited, such that the CNBD interacts directly with the CDC25HD by an ionic “latch” and indirectly by a central “switchboard,” thereby sterically preventing interaction with Rap. Upon binding of cAMP, Epac undergoes a conformational change in which the regulatory domain moves away from the CDC25HD toward the Ras exchange motif, thus allowing the CDC25HD to interact with Rap and facilitate the exchange of GDP for GTP (11).
In this study, we set out to develop a rapid and inexpensive method to identify Epac1 inhibitors and examine their interaction with Epac. We took advantage of the conformational changes in Epac that are induced upon binding of cAMP as a means to assess Epac activation. Although first generation Epac-based probes of cAMP utilized FRET, constructs using BRET produce a higher signal-to-noise ratio and are better suited for the adaptation to high throughput screening (12). One such BRET-based sensor, named CAMYEL, was designed to monitor cAMP levels in living cells (13). In this sensor, human Epac1 (amino acids 149–881) is sandwiched between Renilla luciferase and citrine, a modified variant of YFP. The auto-inhibited state yields a strong BRET signal that results from the transfer of energy from luciferase to citrine. Conformational changes that occur upon activation result in a decrease in the BRET signal as the luciferase moves away from citrine. This construct lacks the Dishevelled, Egl-10, pleckstrin domain of Epac and has two mutations (T781A and F782A) that render it catalytically inactive. As a result, the construct has a better signal-to-noise ratio than do similar constructs of full-length wild-type Epac (14).
To verify that this assay measures Epac activation, we compared our findings with previous measurements of ligand binding to Epac and the activation or inhibition of Epac. Here, we show the ability of CAMYEL to identify Epac inhibitors and to predict agonism, partial agonism, and super agonism of cAMP analogs. The results provide new information regarding the binding of such analogs and the conformational changes that occur upon their interaction with Epac1.
EXPERIMENTAL PROCEDURES
Cell Lines and Transfections
Swiss 3T3 and HEK293 cells were grown in 10-cm culture dishes at 37 °C, 5% CO2 in DMEM supplemented with 10% FBS, 1% penicillin, and 1% streptomycin. Transient transfection of pcDNA3 CAMYEL into HEK293 cells was carried out using TransIT LT-1 (Mirus) according to the manufacturer's instructions. Lysis and BRET measurements were performed 48–72 h after transfection. For Rap1 pulldown assays, Swiss 3T3 cells were split in 10-cm culture dishes, allowed to adhere overnight, and then serum-starved in DMEM for 24 h before subsequent assays.
BRET/FRET Assay
HEK293 cells expressing CAMYEL were harvested and lysed in CAMYEL Assay Buffer (40 mm Hepes, pH 7.2, 140 mm KCl, 10 mm NaCl, 1.5 mm MgCl2) with 0.5% Triton X-100 and 1% Complete protease inhibitor mixture (Roche Applied Science) as described (15). After centrifugation at 20,000 × g for 10 min, the supernatant was removed and diluted to the desired volume. 100 μl was added to 96-well white plates and treated for 5 min at room temperature with the indicated treatments. Inhibitors were added 5 min before the indicated treatments. Coelenterazine was added to a final concentration of 2 μm immediately before measuring BRET. Emission from RLuc and citrine was measured simultaneously at 465 and 535 nm in a DTX-800 plate reader (Beckman Coulter). Apparent activation and inhibition constants were determined by fitting the data to a sigmoidal dose-response curve. The apparent Ki value for CE3F4 was determined by fitting the data to that for an uncompetitive inhibitor (GraphPad Prism 6). HEK293 cells expressing Epac2-cAMPS were harvested and lysed in CAMYEL Assay Buffer. After centrifugation at 20,000 × g for 10 min, the supernatant was removed and diluted to the desired volume with CAMYEL Assay Buffer. 100 μl was added to 96-well black plates and treated for 5 min at room temperature with the indicated treatments. Emission from YFP and CFP was measured simultaneously at 480 nm and 535 nm after being excited at 430 nm in an Infinite M200 plate reader (Tecan).
Quantitative Real Time-PCR (QT-PCR)
Total RNA was isolated by TRIzol extraction (Invitrogen), and cDNA was generated using the High Capacity mRNA-cDNA system (Applied Biosystems) according to the manufacturer's instructions. QT-PCR analysis was performed on a DNA Engine Opticon 2 (Bio-Rad) using the QT-PCR Mastermix Plus for SYBR Green I kit (Eurogentec, Fremont, CA). Primers for PCR amplification were designed based on the nucleotide sequences of the respective gene target using Primer3Plus software (General Public License). The primer sequences are as follows: 18 S forward (GTAACCCGTTGAACCCCATT), and 18 S reverse (CCATCCAATCGGTAGTAGCG); Epac1 forward (CTGGACACCACTTACCAACA), and Epac1 reverse (ATTTTTGTGTCTCGGATGAGG); Epac2 forward (GGCAGGGTCTTTGGATGTTA), and Epac2 reverse (GTGCCTTGAAGTCCTTCTGC). When possible, each forward and reverse primer set was designed between multiple exons. Amplification efficiency of each primer pair was tested before analysis. Relative gene expression levels were determined using the ΔΔCT method with 18 S as the reference gene (16).
Immunoblot Analysis
Whole-cell lysates were prepared in 150 mm Na2CO3 buffer, pH 11, and homogenized by sonication. Equal amounts of protein (assayed using a dye-binding reagent; Bio-Rad) were separated by SDS-PAGE using 10% polyacrylamide precast gels (Invitrogen) and transferred to a polyvinylidene difluoride membrane with the X-Cell II blot module (Invitrogen). Membranes were blocked in PBS/Tween (1%) containing 5% nonfat dry milk and incubated with primary antibody 18 h at 4 °C. Bound antibodies were visualized using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and ECL reagent (Amersham Biosciences). Bands were compared with molecular weight standards to confirm migration of proteins at the appropriate size. Quantification of protein expression densitometry was performed using ImageJ software (National Institutes of Health, Bethesda). Epac1 (5D3), pVASP S157, and Rap1A/Rap1B antibodies were purchased from Cell Signaling (Danvers, MA).
Rap1 Activation Assay
The Rap1-GTP-binding domain (RBD) of mammalian RalGDS was expressed in Escherichia coli as a GST fusion protein as described previously (17). The purified GST-RalGDS-RBD was used for the detection of activated Rap1. 10-cm plates of Swiss 3T3 cells were washed twice with ice-cold PBS and then lysed with 1 ml of Tris/Lysis Buffer (100 mm Tris, pH 7.5, 300 mm NaCl, 50 mm MgCl2, 20% glycerol, 1% Nonidet P-40, 2 mm DTT, 2 mm vanadate, aprotinin, and 5 μg/ml leupeptin) at the indicated times. The lysates were centrifuged for 10 min, and the supernatants were incubated with 10 μg of GST-RBD on glutathione-Sepharose beads at 4 °C for 60 min. The beads were washed three times and subjected to SDS-PAGE and Western blot analysis with either an anti-pVASP mAb or an anti-Rap1 polyclonal antibody. For control of the input amount of Rap1 protein, we analyzed 20 μl of the cell lysates.
Reagents
cAMP and cAMP analogs were purchased from Biolog (Bremen, Germany).
Chembridge compounds were purchased from Chembridge (San Diego). Coelenterazine was purchased from Nanolight (Pinetop, AZ). Forskolin was purchased from Abcam Biochemicals (Cambridge, MA). Complete protease inhibitors were purchased from Roche Applied Science. Phosphatase inhibitor Mixture 3 was purchased from Sigma. Mouse reference cDNA was purchased from Zyagen (San Diego). pcDNA3 CAMYEL was a kind gift from Dr. Paul Sternweis (University of Texas Southwestern). CE3F4 was kindly provided by Frank Lezoualc'h (INSERM UMR-1048, Toulouse, France).
Molecular Modeling and Virtual Docking
The Schrödinger LigPrep Program was used to generate three-dimensional molecular models of the cAMP analog 8-CPT-N6-phenyl-cAMP (CPT-N6) (18). Using I-TASSER (19), a three-dimensional model of the Epac1 structure was produced, based on the 3CF6 PDB of Epac2 in complex with cAMP and Rap (19, 20). The Schrödinger Protein Preparation Wizard was then used for the model to yield a more accurate structure. The three-dimensional models of CPT-N6 were then screened virtually with the model of Epac1 via Induced Fit Docking (21) centered in the CNBD.
Epac1-CPT-N6 System Preparation
The top Induced Fit Docking pose with the phosphate-sugar moiety interacting with the phosphate binding cassette was saved as a pdb, and the protein-ligand system was prepared for molecular dynamics (MD). First, the topology and coordinate files were created using Amber Antechamber tools (22) and Xleap (23, 24). Next, the system was solvated in a water box of TIP3P, with the minimum distance of 10 Å between any atom of the protein-ligand system and the edge of the box. The solvated system was then neutralized before the parameter and coordinate files were saved and prepared for AMBER MD simulations.
Amber Molecular Dynamics
The solvated system was minimized via a two-step process with the Sander program. First, the protein was kept fixed, and the water positions were minimized. Second, the energy of the whole system was minimized together. Next, it was allowed to heat from 0 to 300 K during 50 ps of MD at a constant volume using the Langevin thermostat. The system was then equilibrated during 500 ps of MD at a constant pressure before production MD. This equilibrated system was then used to produce two independent MD trajectories of about 30 ns each. All MD simulations were performed with isotropic position scaling under constant pressure periodic boundary conditions, the reference pressure of 1 atm, and a pressure relaxation time of 2 ps. The system was maintained at a reference temperature of 300 K using the Langevin thermostat with a collision frequency of 2 ps−1 (25). A simple Leapfrog integrator was used to solve Newton's equations of motion and to propagate the simulation with a time step of 0.002 ps. Utilizing the Sander Amber module, the long range electrostatics interactions were handled with the particle-mesh Ewald procedure; the long range van der Waals interactions were estimated by a continuum model. See Ref. 23 for further computational details.
Root Mean Square Deviation (r.m.s.d.) Trajectory Clustering
The independent MD trajectories were further analyzed to determine the stability of the docking pose. An r.m.s.d.-based gromos clustering algorithm was used to group or “cluster” the different conformations sampled by the trajectories depending on structural similarity within the CNBD pocket (26, 27). Frames were taken from the simulation every 2 ps and aligned to the starting structure. The trajectories were then clustered based on the residues within 10 Å of the CPT-N6 ligand using an r.m.s.d. cutoff of 0.14 Å. The largest cluster was considered most representative of the trajectory.
Virtual Screening for Allosteric Epac Modulators
I-TASSER (19) was used to generate a three-dimensional model of the Epac1 structure in its inactive unbound state. This model and a library of compounds from Chembridge were prepared with the Schrödinger Protein Preparation Wizard and the LigPrep Program, respectively. A preliminary virtual screen was performed, centered on the Epac hinge, and docking poses were generated using Schrödinger Induced Fit Docking.
Statistical Analysis
Calculations and statistics were performed using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA). Numerical values are presented as means ± S.E. All experiments were performed in triplicate or quadruplicate. Analysis of data from experiments with multiple comparisons was done using analysis of variance with Bonferroni's correction. Values of p < 0.05 were considered significant.
RESULTS
Use of CAMYEL to Measure Epac Activity in Cell Lysates
CAMYEL is a BRET-based cAMP construct composed of Epac1 sandwiched between Renilla luciferase and citrine. Upon binding of cAMP, Epac1 undergoes conformational changes that result in a decrease in BRET due to luciferase moving away from citrine (Fig. 1A). We initially assessed whether CAMYEL could be used to detect Epac modulators. CAMYEL was originally designed for detection of cAMP in cells, but we tested it with cell lysates to eliminate the contribution of cell permeability. We prepared cell lysates from HEK293 cells transiently transfected with CAMYEL and then assessed BRET activity in white 96-well plates in a DTX-800 plate reader. We found that addition of cAMP decreased BRET, measured as the 465:535 nm ratio (Fig. 1B). The time course for CAMYEL activation indicated that cAMP activates the construct in <1 min and that the activated state is sustained for >60 min at room temperature. Incubation with cAMP at 37 °C for >30 min resulted in a decreased change in BRET due to the decreased concentration of cAMP, a result of its hydrolysis by PDE that could be blocked with the PDE4 inhibitor, rolipram (data not shown). Because the hydrolysis of cAMP by PDEs is temperature-dependent, we conducted all subsequent assays at room temperature and measured the activation of CAMYEL 5 min after treatments.
FIGURE 1.
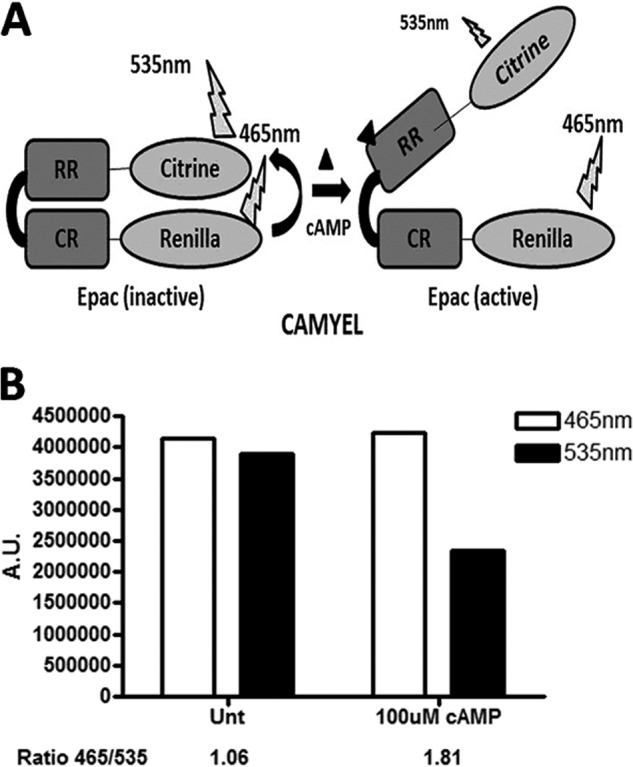
CAMYEL assay. A, schematic diagram of the activation of the CAMYEL construct: the catalytic region (CR) and regulatory region (RR) (Epac1 amino acids 149–880) with Renilla luciferase attached to the catalytic region and citrine attached to the regulatory region. B, cAMP produces a decrease in the BRET signal upon binding to CAMYEL, as measured by the decrease of 465/535 nm. A.U., arbitrary units.
To verify that CAMYEL can be used to assess modulators of Epac activity, we first determined the response to cAMP analogs with known activity toward Epac (Fig. 2B). cAMP produced a concentration-dependent reduction in BRET of CAMYEL and yielded an EC50 of 15 μm (Fig. 2A), a value similar to the 30 μm EC50 obtained for the in vitro activation of Rap1 by Epac1 (28). We normalized the ability of cAMP analogs to activate Epac, as assessed by a reduction in BRET of CAMYEL, to the reduction of BRET produced by cAMP. The Epac-specific agonist 8-CPT-2′-O-Me-cAMP (8-Me) activated CAMYEL with an EC50 of 0.5 μm and produced a larger decrease in BRET than cAMP, suggesting that 8-Me produces a more activated state of Epac than does cAMP. The larger decrease in BRET produced by 8-Me compared with cAMP is consistent with its ability to act as an agonist of Epac with greater efficacy than cAMP (29). N6-Phenyl-cAMP (N6-Phe), a PKA-specific analog, bound with an EC50 of 15 μm and produced a decrease in BRET that was only 10% of that promoted by cAMP. (Rp)-cAMPS and (Sp)-cAMPS are hydrolysis-resistant analogs of cAMP with a sulfur-substituted for the oxygen in the axial or equatorial position and act as an antagonist or agonist, respectively. (Rp)-cAMPS bound to CAMYEL with an EC50 of 68 μm but similar to N6-Phe did not stabilize the open conformation and thus produced minimal activation relative to that of cAMP (Fig. 2A). (Sp)-cAMPS activated CAMYEL to a similar maximal extent but with a slightly decreased affinity compared with cAMP.
FIGURE 2.
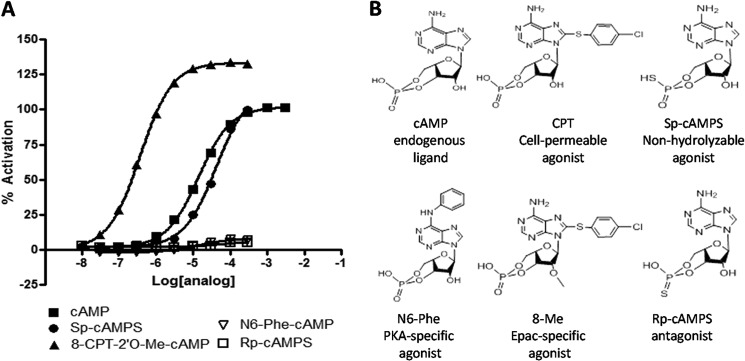
CAMYEL activation is induced by Epac agonists in a concentration-dependent manner. cAMP and cAMP analogs were added to a 96-well white plate with 100 μl of CAMYEL in HEK293 lysate and incubated for 5 min before measuring activation. A, cAMP analogs activate the CAMYEL construct in a concentration-dependent manner. B, structures of cAMP analogs used in the assay of CAMYEL activation. n = 3.
CAMYEL Can Distinguish between Partial Agonists and Agonists of Epac
To determine whether the increased binding affinity and activation by 8-Me derive from the 8-CPT or 2′-O-Me modification, we assessed the ability of CPT, 8-Me, or 2′-O-Me-cAMP (2′-O-Me) to activate CAMYEL. Relative to cAMP, 2′-O-Me had an increased ability to activate CAMYEL similar to that of 8-Me but with decreased binding affinity (Fig. 3A). The binding affinity of CPT for CAMYEL was increased relative to that of cAMP, but CPT caused a smaller decrease in BRET. This smaller decrease in BRET is consistent with evidence that the analog acts as a partial agonist and implies that the 2′-O-Me group is responsible for the increased activation although the 8-CPT produces increased binding affinity. By monitoring the total change in BRET (Fig. 3C) at saturating conditions, we were thus able to use the CAMYEL assay to detect whether cAMP analogs are agonists, “super agonists,” or partial agonists.
FIGURE 3.

CAMYEL can assess if cAMP analogs are agonists, super agonists, or partial agonists. cAMP and cAMP analogs were added and assayed as in Fig. 2. A, activation of CAMYEL by cAMP analogs. B, activation of CAMYEL is decreased by cAMP analogs modified with 8-R substitutions. C, maximum decrease in BRET (measured by 465/535 nm) in response to 300 μm cAMP, 8-Me, or CPT. D, activation assays of the cAMP analogs and their EC50 values (E) and maximum activation (F) compared with that by cAMP. n = 3, **, p < 0.01; ***, p < 0.001.
Modifications at the 8-Position of the Adenine Ring Modulate Binding Affinity and Agonism
Because analogs modified at the 8-position of the adenine ring on cAMP have altered binding affinity for Epac, we sought to determine the optimal modification to enhance binding by testing the ability of 8-CPT-cAMP, 8-Br-cAMP, and 8-piperidine-cAMP to bind and activate CAMYEL (Fig. 3B). Relative to cAMP, 8-CPT and 8-Br-cAMP had increased affinity in binding CAMYEL, and both acted as partial agonists. 8-Piperidine-cAMP had a decreased binding affinity to CAMYEL and, although a partial agonist (up to 300 μm), decreased BRET to an extent similar to that produced by cAMP. Because the highest binding affinity occurred with the 8-CPT modification, we tested if the functional group on the para-position of the phenol affects binding by assessing the ability of three cAMP analogs (8-CPT-2′-O-Me-cAMP, 8-hydroxylphenylthio-2′-O-Me-cAMP, and 8-methoxyphenylthio-2′-O-Me-cAMP) to bind and activate Epac. These analogs also have the 2′-O-Me modification that provides super agonism and were used because the corresponding analogs without 2′-O-Me were not available to us. These analogs have, respectively, a chlorine, hydroxyl, or a methoxy group in the para-position of the phenyl group. All had increased binding to CAMYEL, both in terms of affinity and maximal activity, as compared with cAMP (Fig. 3D). 8-Hydroxylphenylthio-2′-O-Me-cAMP, which bound with an EC50 of 240 nm, had the highest affinity of any cAMP analog tested (Fig. 3E). All three analogs had a similar extent of super agonism (Fig. 3F). These results indicate that substitutions at the 8-position of cAMP modulate binding affinity and can also result in partial agonism.
Use of CAMYEL to Identify Inhibitors of Epac
We next set out to determine whether the CAMYEL assay can be used to identify Epac inhibitors. (Rp)-cAMPS is an antagonist of CNBDs in proteins. We tested its ability to act as a competitive inhibitor on Epac by assessing a range of concentrations for inhibition of CAMYEL preactivated with 100 μm cAMP for 5 min and found that (Rp)-cAMPS acts as a weak competitive inhibitor (i.e. in the high micromolar range (Fig. 4A)). (Rp)-8-CPT-cAMPS ((Rp)-CPT), an (Rp)-cAMPS analog with a chlorophenylthio substitution at the 8 position of the adenine ring of cAMP, inhibited CAMYEL with an IC50 of 39 μm. Increasing concentrations of (Rp)-CPT shifted the concentration curve for activation of CAMYEL by cAMP to the right without changing the maximal activation, results indicative of a competitive inhibitor (Fig. 4B). To determine the disassociation constant for 8-CPT-(Rp)-cAMPS, we conducted Schild analysis (Fig. 4C). The slope of the Schild plot was 1.0, confirming that (Rp)-cAMPS is a competitive inhibitor. The intercept on the abscissa was −5.5, corresponding to a Ki of 3.2 μm. Thus, the use of CAMYEL can detect competitive inhibitors of Epac and their disassociation constant can be determined by Schild analysis.
FIGURE 4.
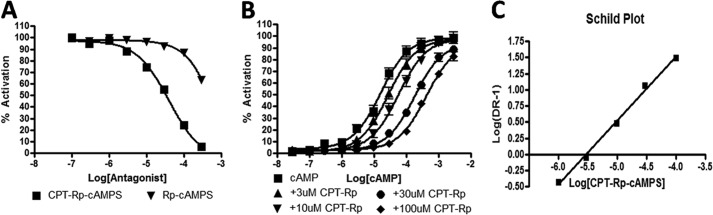
(Rp)-substituted cAMP analogs are competitive inhibitors, and addition of 8-CPT enhances binding affinity. cAMP and cAMP analogs were added and assayed as in Fig. 2. A, (Rp)-cAMPS and (Rp)-CPT inhibit CAMYEL preactivated by 100 μm cAMP in a concentration-dependent manner. B, (Rp)-CPT was preincubated with CAMYEL for 5 min before adding cAMP. The concentration-dependent activation of CAMYEL by cAMP was determined 5 min after activation by cAMP. C, Schild plot obtained from B. n = 3–6.
We used the recently identified uncompetitive inhibitor CE3F4 to test the ability of CAMYEL to identify allosteric inhibitors of Epac (30). Preincubation with 3, 10, or 20 μm CE3F4 reduced the maximal decrease in BRET produced by cAMP in a concentration-dependent manner and yielded a Ki of 5.2 μm (Fig. 5A). CE3F4 was also able to inhibit the activation of CAMYEL by 8-Me or CPT in a concentration-dependent manner with Ki values of 16.1 μm and 2.3 μm, respectively (Fig. 5, B and C). In combination with the data obtained for (Rp)-CPT, these results demonstrate that the use of CAMYEL can identify competitive and uncompetitive (allosteric) inhibitors of Epac.
FIGURE 5.
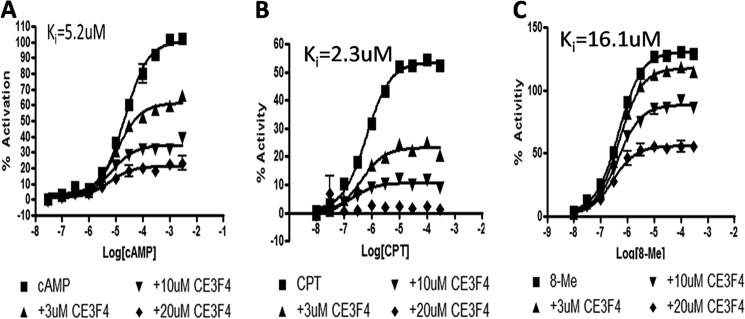
CE3F4, an Epac-specific inhibitor, inhibits CAMYEL activation. CAMYEL was preincubated with CE3F4 (3, 10, and 20 μm) for 5 min before assessing the ability of cAMP (A), 8-Me (B), or CPT (C) to activate CAMYEL. CE3F4 inhibited the ability of the Epac agonists to activate CAMYEL in a concentration-dependent manner. n = 3.
N6-Modified Analogs Are Competitive Inhibitors of Epac
The amino group of the adenine ring of cAMP is important for interaction with the “lid” of Epac protein and for stabilizing the active conformation (20). Because N6-Phe-cAMP did not fully activate CAMYEL but bound to CAMYEL with an affinity similar to that of cAMP, we tested if a similar interaction occurred for other N6-modified cAMP analogs (Fig. 6A). Several such analogs (up to 300 μm) did not show significant activation (Fig. 6B). However, the compounds bound to some extent, competing with 100 μm cAMP with affinities in the following rank-order: Phe > monobutyryl > benzene = benzoyl > aminoethyl (Fig. 6C). Although both N6-monobutyryl-cAMP and N6-Phe-cAMP bound and inhibited Epac, their binding affinities were similar to that of cAMP. Thus, these would not be suitable as competitive inhibitors for Epac in vivo. To determine whether modifications at the 8 position would increase their binding affinity, we tested the ability of 8-CPT-N6-Phe-cAMP (CPT-N6) to inhibit CAMYEL. CPT-N6 inhibited CAMYEL with an IC50 of 2.2 μm whereas N6-Phe had an IC50 of ∼100 μm (Fig. 6D). Activation of CAMYEL by cAMP in the presence of increasing amounts of CPT-N6 shifted the concentration-response curve to the right without changing the maximal activation, consistent with competitive inhibition (Fig. 6E). Schild analysis confirmed that CPT-N6 acts as a competitive inhibitor (Ki = 0.5 μm (Fig. 6F)). Thus N6-modified analogs can inhibit Epac activity, especially if such analogs contain modifications at the 8 position that enhance binding.
FIGURE 6.

N6-Modified cAMP analogs are competitive inhibitors of Epac1, as assessed in the CAMYEL assay. A, structures of the adenine ring of cAMP showing various N6 substitutions. N6-Modified cAMP analogs were tested for their ability to activate CAMYEL (B) or inhibit 100 μm cAMP-activated CAMYEL (C). D, CAMYEL activated by 100 μm cAMP is inhibited by N6-Phe or CPT-N6 in a concentration-dependent manner. E, CPT-N6 inhibits CAMYEL activation by cAMP in a concentration-dependent manner. F, Schild plot obtained from data shown in E. n = 3–6. AE, aminoethyl; Bn, benzene; Bnz, benzoyl; Br, bromine; MB, monobutyryl.
cGMP has a carbonyl group in place of the N6 amino group on cAMP and is a competitive inhibitor for Epac at high concentrations. To determine whether cGMP derivatives might be leads for Epac inhibitors, we tested cGMP, CPT-cGMP, and CPT-2′-O-Me-cGMP for their ability to activate (Fig. 7A) or inhibit (Fig. 7B) Epac. cGMP and CPT-cGMP were unable to activate Epac at any concentration tested. CPT-2′-O-Me-cGMP may be a weak partial agonist at >300 μm. CPT-cGMP and CPT-2′-O-Me-cGMP were able to inhibit Epac (IC50 = 75 μm and 360 μm, respectively) but at concentrations significantly higher than that of CPT-N6. For this reason we did not pursue cGMP analogs as Epac inhibitors.
FIGURE 7.
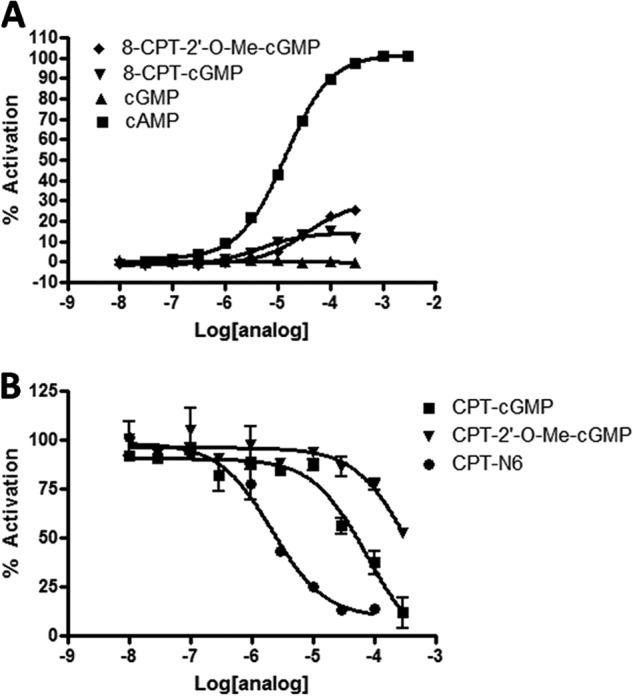
cGMP analogs as inhibitors of Epac1, as assessed in the CAMYEL assay. A, concentration-dependent activation of CAMYEL by cGMP and cGMP analogs. B, concentration-dependent inhibition by cGMP and cGMP analogs of CAMYEL preactivated with 100 μm cAMP. n = 3.
Comparison of Inhibitors
(Rp)-cAMPS can bind CNBDs but does not activate them, due to the inability of the analog to properly interact with the phosphate binding cassette. N6-Phe-cAMP can bind to CNBDs and activate them but cannot stabilize the active conformation of Epac as a consequence of decreased interaction with the lid. We sought to verify that (Rp)-cAMPS is an antagonist that does not activate the CNBD although N6-Phe would be able to interact with the CNBD of Epac and activate the isolated domain. To test this, we used Epac2-cAMPS, a FRET-based sensor that has the CNBD-B of Epac2 sandwiched between CFP and YFP. Activation of the CNBD is detected by a decrease in FRET between CFP and YFP. cAMP induced a concentration-dependent decrease in FRET (EC50 = 1.9 μm) (Fig. 8A). N6-Phe-cAMP activated the CNBD to the same extent as cAMP but with higher apparent affinity (EC50 = 0.2 μm). (Rp)-cAMPS did not activate the CNBD but instead increased the FRET signal (EC50 = 176 μm), implying that it stabilizes the closed, inhibited state of the CNBD. Pretreatment of Epac2-cAMPS with (Rp)-CPT (100 μm) increased the apparent affinity of cAMP, indicating that the (Rp)-cAMPS analog can be competitive inhibitors for Epac2 as well as Epac1. Comparison of cAMP activation of CAMYEL in the presence of 10 μm CPT-N6 or 10 μm (Rp)-CPT revealed that CPT-N6 has a greater ability than (Rp)-CPT to shift the curve to the right and increase the apparent affinity of cAMP (Fig. 8B). To determine the affinity of the different inhibitors, we compared their ability to inhibit CAMYEL activated by 100 μm cAMP. CPT-N6 and CE3F4 had similar affinities with IC50 values of 2.2 μm and 7.3 μm, respectively (Fig. 8C), although (Rp)-CPT had a lower affinity (IC50 = 39 μm).
FIGURE 8.

Comparison of inhibitors of Epac, as assessed using Epac2-cAMPS or the CAMYEL assay. A, cAMP, (Rp)-cAMPS, or N6-cAMP-Phe were added to 100 μl of Epac2-cAMPS lysate in a black 96-well plate. After 5 min, their ability to activate Epac2-CNBD was measured (results normalized to cAMP). B, activation of CAMYEL by cAMP preincubated for 5 min with 10 μm CPT-N6 or 10 μm (Rp)-CPT. C, CAMYEL was activated with 100 μm cAMP before assessment of the ability of CE3F4, CPT-N6, and (Rp)-CPT to inhibit CAMYEL. n = 3.
Verification of Inhibition of Epac by cAMP Analogs in Cells
(Rp)-CPT and CPT-N6 were able to inhibit Epac in vitro, but we thought it was important to test their ability to inhibit endogenous cAMP signaling in cells. To test this, we employed Swiss 3T3 cells, which express mRNA (assessed by QT-PCR) of Epac1 and Epac2 (Fig. 9A). Epac1 was the main isoform expressed (ΔCt = 23 versus Epac2 with ΔCt = 35), corresponding to ∼4000-fold more Epac1 mRNA than Epac2 mRNA. Analysis of protein expression showed abundant Epac1 protein but no detectable Epac2 protein (data not shown). Swiss 3T3 cells express Rap1, which can be activated by stimulants that increase cAMP (31). Incubation of Swiss 3T3 cells with the adenylyl cyclase activator, forskolin, increased Rap1 activation and the phosphorylation of VASP at serine 157 (Fig. 9B). Treatment of cells with 50 μm N6-Phe (a PKA-specific analog) increased VASP phosphorylation but did not change Rap1 activation, indicating that PKA mediates VASP phosphorylation (32). Treatment of the cells with 50 μm 8-Me (an Epac-specific analog), increased Rap1 activation but did not promote VASP phosphorylation, confirming that Epac mediates Rap1 activation. Treatment of the cells with 50 μm CE3F4 for 30 min prevented the activation of Rap1 by forskolin without changing VASP phosphorylation, further indicating that Epac activates Rap and PKA phosphorylates VASP (Fig. 9E). These results led us to test the activation or inhibition of Epac1 and PKA activity by simultaneously monitoring Rap1 activation or VASP phosphorylation, respectively, via activation of the cAMP pathway by forskolin alone or in the presence of putative inhibitors. We tested the ability of (Rp)-CPT or CPT-N6 to inhibit Epac1 activation. Treatment of the cells with either 50 μm CPT-N6 (Fig. 10A) or 100 μm (Rp)-CPT (Fig. 10B) prevented the activation of Rap1 by forskolin. Thus, the findings obtained from the CAMYEL assay in vitro predict Epac activation and inhibition in cells.
FIGURE 9.

Epac1 activates Rap1 and PKA activation mediates VASP phosphorylation in Swiss 3T3 cells. A, QT-PCR analysis of Epac1 and Epac2 mRNA expression in Swiss 3T3 cells and mouse reference cDNA. B, Rap1 activation and VASP phosphorylation were assessed by Western blotting of Swiss 3T3 cells that were untreated (Unt) or incubated with 8-Me, N6, or forskolin (Fsk) for 5 min. C, pooled results (n = 3) for Rap1 activation. D, VASP phosphorylation by 8-Me, N6, or forskolin. E, inhibition of forskolin-promoted Rap1 activation but not VASP phosphorylation by 20 μm CE3F4. F and G, pooled results for effect of 20 μm CE3F4 on forskolin-promoted Rap1 activation and VASP phosphorylation, respectively n = 4, **, p < 0.01.
FIGURE 10.

CPT-N6 and (Rp)-CPT inhibit forskolin-promoted Epac activation in Swiss 3T3 cells. Swiss 3T3 cells were incubated with 50 μm CPT-N6 (A and B) or 100 μm (Rp)-CPT (C and D) for 30 min and were then untreated (Unt) or incubated with forskolin (Fsk) (10 μm). Cells were harvested 5 min later, and the levels of activated Rap1 were quantified. B and D, show pooled results for n = 5, *, p < 0.01.
Molecular Model of CPT-N6 Docking Pose
To obtain further insight regarding the binding of the CPT-N6 analog to Epac1 and because a structure of Epac1 has not been solved, we undertook a computational molecular modeling approach based on a solved structure for activated Epac2. The three-dimensional model of active Epac1 was generated with I-TASSER and had an estimated accuracy of 0.52 ± 0.15 (TM-score, where a TM-score <0.17 suggests a random similarity and a TM-score >0.5 typically corresponds to the same fold or domain in SCOP/CATH) (33). The Protein Prep Wizard and LigPrep Program of the Maestro 9.2 (Schrödinger, LLC) module were used to prepare the Epac1 model and CPT-N6, respectively. The Schrödinger Induced Fit Docking procedure was used to generate predicted binding poses and scores (34). Among the results where the phosphate-sugar moiety interacted with the phosphate binding cassette (PBC), the highest Glide XP score for CPT-N6 docked into the CNBD of the Epac1 model was −10.1 kcal/mol (35). Taking this top pose, the protein-ligand system was prepared for molecular dynamics (MD). The two independent 30-ns trajectories were then clustered by r.m.s.d. near the CNBD to identify the most representative structures of the simulation. These structures are also the most stable conformations during these MD runs, suggesting that they may be possible binding poses for the protein-ligand complex. These two poses were also very similar, even though the trajectories were run independently (Fig. 11B). As shown in Fig. 11, C and D, residues around the PBC that appear to form hydrogen bonds with the CPT-N6 analog include Gly-311, Gln-312, Leu-313, Ala-314, Arg-321; there is a possibility for interaction between the CPT moiety and Leu-356. The structure most representative of trajectory 1 (Fig. 11C) includes two waters able to hydrogen bond with the ligand phosphate group, where that of trajectory 2 (Fig. 11D) shows only one water molecule hydrogen bonded to it. Both appear to be very stably bound to the PBC but lack any bonds to the “lid” similar to what is seen between (Sp)-cAMPS and Epac2 (Fig. 11A).
FIGURE 11.

Molecular model of CPT-N6 bound to Epac1. A, solved structure of Epac2 CNBD (green) with (Sp)-cAMPS (PDB code 3CF6). B, representative structures from the halo-MD of CPT-N6 docked to the Epac1 model (trajectory 1 in purple and trajectory 2 in orange) are shown for comparison. C, closer look at the CPT-N6-bound Epac1 models from trajectory 1. D, closer look at the CPT-N6-bound Epac1 models from trajectory 2, showing potential interactions. Ligands and water molecules are shown as sticks, protein residues as lines, and bonds as dotted lines. Protein secondary structure is shown as a schematic in A and B. Residues are labeled for Epac1 and Epac 2 (in parentheses) for comparison with C and D. Figures were created using PyMOL.
Virtual Screening of Compounds That Interact at the Epac Hinge Identifies a Possible Allosteric Inhibitor
We undertook a similar approach to generate a model of Epac1 in its inactive state. A library of compounds from Chembridge was then virtually docked to the hinge region of this Epac1 model so as to identify compounds that might be allosteric inhibitors. The 133 compounds that had the highest score were tested experimentally.
Compound 5225554, a barbituric acid derivative (Fig. 12A), had the best activity as an inhibitor in the CAMYEL assay. Further studies using this assay revealed that compound 5225554 bound in a noncompetitive manner (Fig. 12B). Experiments conducted with Swiss 3T3 cells indicated that 5225554 inhibits forskolin-stimulated Rap1 activation without altering VASP phosphorylation (Fig. 12, C and D). The Schrödinger Induced Fit Docking generated high scoring poses suggesting that compound 5225554 fits nicely into a pocket near the Epac hinge and residues Leu-315 and Phe-342 (Leu-408 and Phe-435 in Epac 2). Fig. 12E shows possible hydrogen bonding between the compound and residues Ser-282 and Val-337. Fig. 12F shows a slightly different possible pose where 5225554 may form bonds with residues His-248, Asp-309, and Gln-Q312. The CAMYEL assay can thus identify novel inhibitors of Epac1, whose site of action is not in the cAMP-binding site but likely in the hinge region of the protein.
FIGURE 12.

Chembridge compound 5225554 is an allosteric inhibitor of Epac. A, structure of 5225554. B, preincubation with increasing concentrations of 5225554 decreases cAMP-promoted activation of CAMYEL. C, Swiss 3T3 cells were treated with 100 μm compound 5225554 for 30 min prior to incubation with forskolin (Fsk) (10 μm) for 5 min. Activated Rap1 and VASP phosphorylation were then assessed by Western blotting. D and E, pooled results (n = 3) for effect of 5225554 on forskolin-promoted Rap1 activation and VASP phosphorylation, respectively. F and G, virtual docking of compound 5225554 near the hinge region in the inactive Epac1 model. Protein secondary structure is shown as a schematic. 5225554 is yellow, surrounding residues are shown as lines. Leu-315 and Phe-342 are shown as sticks to highlight proximity; black dotted lines depict potential hydrogen bonding. PyMOL was used to generate F and G. *, p < 0.05; **, p < 0.01; Unt, untreated.
DISCUSSION
These results show that the use of the CAMYEL assay with cell lysates provides a means to assess conformational changes that occur upon ligand binding and to identify modulators of Epac activity. Although Epac-cAMP sensors, such as CAMYEL, are used to assess Epac activation in cells, we tested whether it can provide a method to detect different modes of inhibition and of agonism. The EC50 values for cAMP analogs obtained for activation of CAMYEL are similar to those obtained for their activation of Rap1 in an in vitro Epac GEF assay (36). The absolute affinities are somewhat higher than those obtained by displacement of cAMP but the relative affinities compared with cAMP are similar (Table 1) (37, 38).
TABLE 1.
Binding affinity of cAMP analogs, as assessed by CAMYEL
The table shows the affinity of cAMP analogs compared with cAMP as measured by the CAMYEL assay (described under “Experimental Procedures”). The ability of the analogs to activate or inhibit CAMYEL compared with cAMP is also shown. The relative affinity of the analogs as measured by displacement of radioactive cAMP as reported in Refs. 37, 38 is shown for comparison with the values obtained by the CAMYEL assay. The abbreviations used are as follows: AE, aminoethyl; Bn, benzene; Bnz, benzoyl; Br, bromine; MB, monobutyryl; NA, not applicable.
| Compounds | Abbreviation | EC50 | IC50 | Maximal activation | Change in affinity compared with cAMP | Reported affinity compared with cAMPa,b |
|---|---|---|---|---|---|---|
| μm | μm | |||||
| cAMP | cAMP | 15 μm | NA | 100 | 1 | 2.9 μma |
| Compounds with eight substitutions | ||||||
| 8-Bromine-cAMP | 8-Br | 3.5 | 60 | 3.71 | 8.10a | |
| 8-Piperidino-cAMP | 8-PIP | 78 | 89 | 0.17 | 0.21a | |
| 8-Chlorophenylthio-cAMP | CPT | 0.59 | 54 | 22.03 | 65.00a | |
| 8-Chlorophenylthio-2′-O-methyl-cAMP | 8-Me | 0.43 | NA | 130 | 30.23 | 4.60a |
| 8-Hydroxyphenylthio-2′-O-methyl-cAMP | 8-HPT-Me | 0.23 | NA | 132 | 56.52 | 6.90b |
| 8-Methoxyphenylthio-2′-O-methyl-cAMP | 8-MeOPT-Me | 0.46 | NA | 126 | 28.26 | 7.10b |
| Compound N6 substitutions | ||||||
| N6-Phenol-cAMP | N6-Phe | 13 | 100 | 8 | 1.00 | 2.80a |
| N6-Benzene-cAMP | N6-Bn | 9.9 | 100 | 22 | 1.31 | |
| N6-Benzoyl-cAMP | N6-Bnz | 9.6 | 300 | NA | 1.35 | 1.30b |
| N6-Aminoethyl-cAMP | AE-cAMP | >300 | ||||
| 2-Aza-ϵ-cAMP | Aza-cAMP | 300 | ||||
| 8-Chlorophenylthio-N6-phenol-cAMP | CPT-N6 | 0.27 | 3 | 12 | 48.15 | 110.00b |
| N6-Monobutyrl-cAMP | N6-MB | 64 | >300 | 6 | ||
| Compounds with thiophosphates | ||||||
| (Rp)-cAMPS | (Rp)-cAMPS | 68 | >300 | 6 | 0.19 | |
| (Sp)-cAMPS | (Sp)-cAMPS | 41 | 105 | |||
| (Rp)-CPT-cAMPS | (Rp)-CPT | 39 | ||||
| Compounds with 2′ substitutions | ||||||
| 2′O-Me-cAMP | 2′-Me-cAMP | 50 | 125 | 0.26 | 0.12a | |
| 2′-dcAMP | 2-dcAMP | 600 | 92 | 0.02 | 0.003a | |
| 2′-F-cAMP | 2-F-cAMP | 400 | 88 | 0.03 | ||
| Compounds based on cGMP | ||||||
| cGMP | 250 | 0.05 | 0.08a | |||
| 8-CPT-cGMP | CPT-cGMP | 4.9 | 30 | 14 | 2.65 | |
| 8-CPT-2′-O-Me-cGMP | 8-Me-cGMP | 33 | 300 | 29 | 0.39 | |
Previous methods to identify Epac inhibitors have involved analysis of the displacement of fluorescent or radioactive cAMP analogs from purified Epac protein (39, 40). Although this approach can identify competitive inhibitors, it only measures binding interactions and does not provide information about the mechanism of inhibition of the competitor and thus, is likely unable to detect allosteric inhibitors that do not displace the ligand. Those prior studies involved the use of purified protein, which is expensive (in terms of time and effort) to obtain. Our method is a novel, rapid, and cost-effective way to identify modulators of Epac activity in cell lysates. Thus, the CAMYEL assay provides a means to reliably measure the binding of analogs to Epac and simultaneously measure their ability to activate it.
Our results show that in addition to the ability of the CAMYEL assay to measure binding affinity, it can also be used to predict the efficacy of activators based on differences in the maximal decrease in BRET. We found that 8-CPT produces a 50% smaller decrease whereas 8-Me shows a 125% larger decrease in BRET than does cAMP. The overall changes in BRET correlate with the decreased (50%) and increased (300%) GEF activity induced by 8-CPT and 8-Me, respectively, indicating that the CAMYEL assay can predict if analogs are agonists, partial agonists, or super agonists. The differing maximal decreases in BRET may result from ability of the compounds to produce a smaller or greater open state of the protein or perhaps from a shift in the equilibrium between the activated and inactivated states by the compounds. Although this assay cannot distinguish between those two possibilities, nor can it determine the exact extent of change in enzymatic activity, CAMYEL provides a means to assess modulators of Epac activity and to infer potential effects on the conformation and activity of the protein.
We found that the CAMYEL assay is particularly well suited to identify competitive inhibitors of Epac. (Rp)-cAMPS, a phosphorothioate analog of cAMP with a sulfur in the equatorial position of the phosphate, is an antagonist of CNBDs, based on its inability to interact properly with the phosphate binding cassette (PBC) of CNBDs, which is necessary for their activation (41). The addition of 8-CPT to (Rp)-cAMPS ((Rp)-CPT) enhances its binding to Epac and its ability to act as a competitive inhibitor in vitro (Fig. 4) and in vivo (Fig. 10). (Rp)-CPT also antagonizes PKA and cyclic nucleotide-gated channels and thus is a general antagonist of CNBDs (42).
Our results show that CAMYEL can also identify allosteric modulators of Epac. The uncompetitive inhibitor CE3F4 reduced the maximal activation of CAMYEL and slightly shifted the EC50 of cAMP to the left in a concentration-dependent manner, results indicative of an uncompetitive inhibitor. We further showed that the apparent Ki of CE3F4 depends on the mode of activation of Epac. This result suggests that either the binding affinity changes between the different activated states or perhaps that 8-Me induces a more stable active open form that is more difficult to close.
We verified our in vitro results using Swiss 3T3 cells, which express Rap1 and Epac (Fig. 9). We chose these cells because we found that they have a more dynamic range of Rap1 activation upon cAMP stimulation than do several other cell types we tested (data not shown). As a result, we were able to assess activation or inhibition of PKA or Epac1 in parallel by monitoring phosphorylation of VASP or activation of Rap1, respectively, in response to forskolin. CE3F4, CPT-N6, and (Rp)-CPT all inhibited Rap1 activation induced by forskolin (Figs. 9 and 10). The concentrations necessary for inhibition by cAMP analogs are higher than in our in vitro assays, perhaps reflecting the contribution of cell permeability for the cAMP analogs.
In the crystal structure of Epac2 activated by (Sp)-cAMPS, the amino group of cAMP interacts with a conserved VLVLE motif in the CNBD (termed the “lid”) that is crucial for Epac activation. N6-Substituted cAMP analogs can activate the CNBD of Epac but, due to an inability to interact with the lid and stabilize the activated state, they cannot overcome autoinhibition constraints (43). N6-Phe activated Epac2-cAMPS, in which the CNBD-B is sandwiched between YFP and CFP, lack the residues necessary to stabilize the auto-inhibited state. N6-Phe and cAMP activated this construct similarly, although (Rp)-cAMPS could not and increased FRET, consistent with the idea that it stabilizes the inactive form of Epac. Because this increase in energy transfer did not occur in the CAMYEL assay, we infer that interactions between the regulatory and catalytic domains contribute to stabilizing the auto-inhibited state, causing full-length Epac protein to be predominantly in the closed state when not bound to cAMP. This suggests that the two analogs work via two different mechanisms to inhibit Epac as follows: (Rp)-cAMPS inhibit by displacing cAMP and not activating the CNBD, although N6-Phe activates the CNBD but cannot interact with the lid properly to overcome the auto-inhibition necessary for Epac activation.
Because CPT-N6 was a more efficient inhibitor of Epac than (Rp)-CPT, we sought to determine how it binds to Epac. Fig. 11B shows a potential binding mode for CPT-N6 to a model of activated Epac1. From the MD trajectories, the conformations of the protein-ligand system were clustered based on r.m.s.d. at the CNBD of Epac1 to identify the most stable states. The top cluster from each 30-ns trajectory shows CPT-N6 with the phosphate sugar extensively interacting with the PBC and surrounding residues. As in the PDB of Epac2 with (Sp)-cAMPS, both top clusters show CPT-N6 with possible water interactions. However, the CPT-N6 cAMP analog does not appear to interact with the Epac1 lid, which is necessary to stabilize the active conformation. A possible explanation for the activity of CPT-N6, which binds well to Epac but inhibits its activation, may be that it stably interacts with the PBC, blocking cAMP, but not with the lid and therefore cannot stabilize the active conformation. This idea is consistent with the experimental results because the 8-CPT moiety enhances binding, although N6-Phe inhibits Epac activation. The interaction of Leu-356 with the 8-CPT moiety instead of the adenine base may explain the partial agonism of 8-CPT-cAMP as this interaction is crucial for the full activation of Epac. Even if the Leu-356 residue (part of the CNBD lid) is stabilized by the CPT moiety, the Ras exchange motif side of the lid was not able to securely interact with the analog in our MD simulations. There may be an intermediate state, between the inactive and active structures, in which CPT-N6 binds, but the full active conformation is not achieved and/or stabilized.
Previous studies show that bending of the hinge in the CNBD of Epac induced by cAMP binding is important for Epac activation and is maintained by hydrophobic interactions formed among Leu-408, Phe-435, and Leu-439 (which are conserved in Epac1) (44). An F435G Epac mutant possesses GEF activity close to that of the wild-type protein activated with cAMP and can partially activate Rap1 even without cAMP, suggesting that Phe-435 is restrictive enough to prevent GEF activity without cAMP, but it permits full bending of the hinge with cAMP present. However, an F435W mutation, with the bulkier tryptophan residue, inhibits Epac bending, resulting in a decrease in catalytic activity. Hence, this hinge region appears to be an attractive region to target with Epac inhibitors.
Investigation of three-dimensional Epac models indicate that there may be a “druggable” region near these hinge residues, based on preliminary virtual screening and experimental studies (data not shown). The assay described here provides a useful means to identify such modulators and succeeded in identifying the barbituric acid derivative 5225554 as an allosteric inhibitor of Epac. 5225554 decreased maximal activation of CAMYEL without changing the EC50, results indicative of a noncompetitive inhibitor. This is in agreement with the predicted binding at the hinge region instead of the cAMP binding pocket. Use of these complementary approaches suggests that targeting this area with small molecules can identify allosteric inhibitors of Epac.
The two cAMP effectors, PKA and Epac, are important intracellular signal transducers involved in signal transduction from Gs-coupled G protein-coupled receptors. Although multiple PKA inhibitors are available, first generation Epac inhibitors are just now being discovered. Compared with receptors and kinases, GEFs and their respective GTPases are not generally considered to be classical “druggable” targets due to the lack of suitable binding pockets (45). The CBND of Epac presents a target for small molecules that will act as competitive inhibitors. The discovery of CE3F4 and 5225554 indicates that Epac activity can also be modulated by allosteric effectors. The CAMYEL assay provides a rapid and cost-effective assay to identify both types of these inhibitors and also contributes information regarding the mechanism for Epac activation. Thus, this assay may prove to be useful for identifying novel inhibitors of Epac.
Acknowledgments
We thank Dr. Paul Sternweis for the pcDNA3 CAMYEL plasmid. We thank Dr. Frank Lezoualc'h for the CE3F4 compound.
This work was supported, in whole or in part, by National Institutes of Health Training Grant 5T32DK007541-27 and Grant GM31749 (to J. A. M.). This work was also supported by National Science Foundation Grant MCB1020765 (to J. A. M.), the Howard Hughes Medical Institute, the National Biomedical Computation Resource, the Center for Theoretical Biological Physics, and National Science Foundation Supercomputer Centers.
- Epac
- exchange protein activated by cAMP
- 8-Me
- 8-CPT-2′-O-Me-cAMP
- BRET
- bioluminescence resonance energy transfer
- CAMYEL
- cAMP sensor using YFP-Epac-RLuc
- CDC25HD
- CDC25 homology domain
- CNBD
- cyclic nucleotide binding domain
- CPT
- chlorophenylthio
- CPT-N6
- 8-CPT-N6-phenyl-cAMP
- GEF
- guanine nucleotide exchange factor
- PBC
- phosphate binding cassette
- PDE
- phosphodiesterase
- Phe
- phenyl
- QT-PCR
- quantitative real time-PCR
- r.m.s.d.
- root-mean-square deviation
- Br
- bromine
- MD
- molecular dynamics
- PDB
- Protein Data Bank
- CFP
- cyan fluorescent protein
- N6-Phe
- N6-phenyl-cAMP.
REFERENCES
- 1. Insel P. A., Zhang L., Murray F., Yokouchi H., Zambon A. C. (2012) Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. Oxf. Engl. 204, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Billington C. K., Ojo O. O., Penn R. B., Ito S. (2013) cAMP regulation of airway smooth muscle function. Pulm. Pharmacol. Ther. 26, 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beavo J. A., Brunton L. L. (2002) Cyclic nucleotide research–still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3, 710–718 [DOI] [PubMed] [Google Scholar]
- 4. Ozaki N., Shibasaki T., Kashima Y., Miki T., Takahashi K., Ueno H., Sunaga Y., Yano H., Matsuura Y., Iwanaga T., Takai Y., Seino S. (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2, 805–811 [DOI] [PubMed] [Google Scholar]
- 5. Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., Graybiel A. M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 6. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 7. Schwede F., Maronde E., Genieser H., Jastorff B. (2000) Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 87, 199–226 [DOI] [PubMed] [Google Scholar]
- 8. Qiao J., Mei F. C., Popov V. L., Vergara L. A., Cheng X. (2002) Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J. Biol. Chem. 277, 26581–26586 [DOI] [PubMed] [Google Scholar]
- 9. Consonni S. V., Gloerich M., Spanjaard E., Bos J. L. (2012) cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 109, 3814–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu C., Takahashi M., Li Y., Song S., Dillon T. J., Shinde U., Stork P. J. (2008) Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell. Biol. 28, 7109–7125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Rooij J., Rehmann H., van Triest M., Cool R. H., Wittinghofer A., Bos J. L. (2000) Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J. Biol. Chem. 275, 20829–20836 [DOI] [PubMed] [Google Scholar]
- 12. Boute N., Jockers R., Issad T. (2002) The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 23, 351–354 [DOI] [PubMed] [Google Scholar]
- 13. Jiang L. I., Collins J., Davis R., Lin K.-M., DeCamp D., Roach T., Hsueh R., Rebres R. A., Ross E. M., Taussig R., Fraser I., Sternweis P. C. (2007) Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282, 10576–10584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ponsioen B., Zhao J., Riedl J., Zwartkruis F., van der Krogt G., Zaccolo M., Moolenaar W. H., Bos J. L., Jalink K. (2004) Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 5, 1176–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matthiesen K., Nielsen J. (2011) Cyclic AMP control measured in two compartments in HEK293 cells: phosphodiesterase K(M) is more important than phosphodiesterase localization. PLoS One 6, e24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franke B., Akkerman J. W., Bos J. L. (1997) Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 16, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeLano W. L. (2013) LigPrep, Version 2.6, Schrödinger, LLC, New York [Google Scholar]
- 19. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rehmann H., Arias-Palomo E., Hadders M. A., Schwede F., Llorca O., Bos J. L. (2008) Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 455, 124–127 [DOI] [PubMed] [Google Scholar]
- 21. Henzler A. M., Rarey M. (2010) In pursuit of fully flexible protein-ligand docking: modeling the bilateral mechanism of binding. Mol. Inform. 29, 164–173 [DOI] [PubMed] [Google Scholar]
- 22. DeLano W. L. (2013) Protein Preparation Wizard, Epik Version 2.4, Impact Version 5.9, and Prime Version 3.2, Schrödinger, LLC, New York [Google Scholar]
- 23. Case D. A., Darden T. A., Cheatham T. E., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 24. Wang J., Wang W., Kollman P. A., Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 25. Pastor R. W., Brooks B. R., Szabo A. (1988) An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Physiol. 65, 1409–1419 [Google Scholar]
- 26. Christen M., Hünenberger P. H., Bakowies D., Baron R., Bürgi R., Geerke D. P., Heinz T. N., Kastenholz M. A., Kräutler V., Oostenbrink C., Peter C., Trzesniak D., van Gunsteren W. F. (2005) The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 26, 1719–1751 [DOI] [PubMed] [Google Scholar]
- 27. Daura X., Gademann K., Jaun B., Seebach D., van Gunsteren W. F., Mark A. E. (1999) Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. Engl. 38, 236–240 [Google Scholar]
- 28. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., Bos J. L. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906 [DOI] [PubMed] [Google Scholar]
- 29. Courilleau D., Bouyssou P., Fischmeister R., Lezoualc'h F., Blondeau J.-P. (2013) The (R)-enantiomer of CE3F4 is a preferential inhibitor of human exchange protein directly activated by cyclic AMP isoform 1 (Epac1). Biochem. Biophys. Res. Commun. 440, 443–448 [DOI] [PubMed] [Google Scholar]
- 30. Courilleau D., Bisserier M., Jullian J.-C., Lucas A., Bouyssou P., Fischmeister R., Blondeau J.-P., Lezoualc'h F. (2012) Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 287, 44192–44202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Altschuler D. L., Ribeiro-Neto F. (1998) Mitogenic and oncogenic properties of the small G protein Rap1b. Proc. Natl. Acad. Sci. U.S.A. 95, 7475–7479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sechi A. S., Wehland J. (2004) ENA/VASP proteins: multifunctional regulators of actin cytoskeleton dynamics. Front. Biosci. 9, 1294–1310 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y., Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 34. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R. (2006) Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 49, 534–553 [DOI] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2013) Small-Molecule Drug Discover Suite 2013-3, Glide, Version 6.1, Schrödinger, LLC, New York [Google Scholar]
- 36. Rehmann H., Schwede F., Døskeland S. O., Wittinghofer A., Bos J. L. (2003) Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J. Biol. Chem. 278, 38548–38556 [DOI] [PubMed] [Google Scholar]
- 37. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H.-G., Døskeland S. O. (2003) cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402 [DOI] [PubMed] [Google Scholar]
- 38. Dao K. K., Teigen K., Kopperud R., Hodneland E., Schwede F., Christensen A. E., Martinez A., Døskeland S. O. (2006) Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 281, 21500–21511 [DOI] [PubMed] [Google Scholar]
- 39. Tsalkova T., Mei F. C., Cheng X. (2012) A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS One 7, e30441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McPhee I., Gibson L. C., Kewney J., Darroch C., Stevens P. A., Spinks D., Cooreman A., MacKenzie S. J. (2005) Cyclic nucleotide signalling: a molecular approach to drug discovery for Alzheimer's disease. Biochem. Soc. Trans. 33, 1330–1332 [DOI] [PubMed] [Google Scholar]
- 41. Wu J., Jones J. M., Nguyen-Huu X., Ten Eyck L. F., Taylor S. S. (2004) Crystal structures of RIα subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry 43, 6620–6629 [DOI] [PubMed] [Google Scholar]
- 42. Gjertsen B. T., Mellgren G., Otten A., Maronde E., Genieser H. G., Jastorff B., Vintermyr O. K., McKnight G. S., Døskeland S. O. (1995) Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1β action. J. Biol. Chem. 270, 20599–20607 [DOI] [PubMed] [Google Scholar]
- 43. Harper S. M., Wienk H., Wechselberger R. W., Bos J. L., Boelens R., Rehmann H. (2008) Structural dynamics in the activation of Epac. J. Biol. Chem. 283, 6501–6508 [DOI] [PubMed] [Google Scholar]
- 44. Tsalkova T., Blumenthal D. K., Mei F. C., White M. A., Cheng X. (2009) Mechanism of Epac activation: structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J. Biol. Chem. 284, 23644–23651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vigil D., Cherfils J., Rossman K. L., Der C. J. (2010) Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer 10, 842–857 [DOI] [PMC free article] [PubMed] [Google Scholar]