

ABSTRACT
Lipids play a crucial role in multiple aspects of hepatitis C virus (HCV) life cycle. HCV modulates host lipid metabolism to enrich the intracellular milieu with lipids to facilitate its proliferation. However, very little is known about the influence of HCV on lipid uptake from bloodstream. Low-density lipoprotein receptor (LDLR) is involved in uptake of cholesterol rich low-density lipoprotein (LDL) particles from the bloodstream. The association of HCV particles with lipoproteins implicates their role in HCV entry; however, the precise role of LDLR in HCV entry still remains controversial. Here, we investigate the effect of HCV infection on LDLR expression and the underlying mechanism(s) involved. We demonstrate that HCV stimulates LDLR expression in both HCV-infected Huh7 cells and in liver tissue from chronic hepatitis C patients. Fluorescence activated cell sorting and immunofluorescence analysis revealed enhanced cell surface and total expression of LDLR in HCV-infected cells. Increased LDLR expression resulted in the enhanced uptake of lipoprotein particles by HCV-infected cells. Analysis of LDLR gene promoter identified a pivotal role of sterol-regulatory element binding proteins (SREBPs), in the HCV-mediated stimulation of LDLR transcription. In addition, HCV negatively modulated the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein that facilitates LDLR degradation. Ectopic expression of wild-type PCSK9 or gain-of-function PCSK9 mutant negatively affected HCV replication. Overall, our results demonstrate that HCV regulates LDLR expression at transcriptional and posttranslational level via SREBPs and PCSK9 to promote lipid uptake and facilitate viral proliferation.
IMPORTANCE HCV modulates host lipid metabolism to promote enrichment of lipids in intracellular environment, which are essential in multiple aspects of HCV life cycle. However, very little is known about the influence of HCV on lipid uptake from the bloodstream. LDLR is involved in uptake of cholesterol rich lipid particles from bloodstream. In this study, we investigated the effect of HCV on LDLR expression and the underlying mechanism triggered by the virus to modulate LDLR expression. Our observations suggest that HCV upregulates LDLR expression at both the protein and the transcript levels and that this upregulation likely contributes toward the uptake of serum lipids by infected hepatocytes. Abrogation of HCV-mediated upregulation of LDLR inhibits serum lipid uptake and thereby perturbs HCV replication. Overall, our findings highlight the importance of serum lipid uptake by infected hepatocytes in HCV life cycle.
INTRODUCTION
Hepatitis C virus (HCV) of the genus Hepacivirus and the family Flaviviridae is a single-stranded plus-sense RNA virus. Affecting 2 to 3% of the global population, HCV infection has emerged as a major health crisis. The infection is initially asymptomatic; however, chronic infection usually promotes severe liver diseases such as fibrosis, cirrhosis, and hepatocellular carcinoma (1, 2). The 9.6-kb HCV genome encodes a single polyprotein of ∼3,000 amino acids which is subsequently processed by host and viral proteases into three structural (core, E1, and E2) and seven nonstructural (P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (3). Lipids and cellular lipid storage organelles (lipid droplets) play significant role in HCV RNA replication and viral particle assembly (4–6). HCV modulates lipid metabolism in infected hepatocytes to attain intracellular enrichment of lipids necessary for viral propagation (7, 8). HCV enriches cellular lipid reserves by triggering de novo lipid biosynthesis and by reducing the catabolic breakdown and export of lipids (4, 7, 9). HCV virus is associated with lipoproteins, and lipoviral particles are highly infectious compared to the lipoprotein-free particles (6, 10).
Several studies support the involvement of low-density lipoprotein receptors (LDLR) in HCV entry; however, some discrepancies remain with regard to its precise role as receptor in the HCV entry or as facilitator of initial attachment to hepatocyte surface (10). Studies involving infection of human hepatocytes with serum-derived HCV particles strongly suggest that LDLR may mediate early steps in virus entry (11). Recently, specific role of apolipoprotein E (apoE) in virus entry has been reported via its interaction with cell surface heparin sulfate proteoglycan receptors (12). A recent study suggests that although LDLR is not essential for HCV entry, the physiological function of LDLR is crucial for the postentry events such as HCV replication (13). LDLR is a transmembrane glycoprotein that serves as a receptor for the uptake of cholesterol-containing serum lipoproteins (14). Nascent VLDL particles released by liver encounter lipoprotein lipases in the bloodstream, which hydrolyze the VLDL into intermediate-density lipoproteins (IDL). The hepatocytes then take up the IDL via interaction with LDLR. Alternatively, the hepatic lipases in the bloodstream further hydrolyze the IDL to generate LDL. The LDL contain relatively high cholesterol content and are taken up by hepatocytes via LDLR (14, 15). Some previous reports indicate the prevalence of hypo-β-lipoproteinemia and hypocholesterolemia in HCV-infected patients, suggesting an enhanced uptake of serum lipoproteins by hepatocytes in HCV-infected patients (16–18).
It is well established that sterols regulate LDLR transcription via sterol-regulatory element binding proteins (SREBPs) (14, 19). Under high-sterol conditions the transcription is repressed, whereas in low-sterol conditions the SREBPs are activated and promote transcription by binding to the sterol-regulatory element 1 (SRE-1) in the LDLR promoter (14, 19). Other cellular signaling molecules, such as growth factors, hormones, and cytokines, have also been shown to modulate LDLR transcription independent of intracellular sterol levels (20). PCSK9 and inducible degrader of LDLR (IDOL) also modulate LDLR expression by promoting the degradation of LDLR (21–23). The complete mechanism by which PCSK9 promotes LDLR degradation is currently unknown. Recent studies suggest that the LDLR-PCSK9 complex is internalized via clathrin-mediated endocytosis and then routed to lysosomes for degradation, independent of the ubiquitination and proteasomal degradation pathways (21, 22). IDOL is an E3 ubiquitin ligase and triggers the ubiquitination of conserved residues in the cytoplasmic tail of LDLR, leading to its degradation (23). Here, we investigated the effect of HCV infection on LDLR expression and the underlying mechanism(s) involved in HCV-induced stimulation of LDLR expression. We observed that HCV induced LDLR transcription via SREBPs and also downregulated PCSK9 to prevent LDLR degradation. The upregulation of LDLR expression correlated with enhanced lipid uptake in HCV-infected cells. Downregulation of LDLR by ectopic expression of PCSK9 negatively affected HCV replication.
MATERIALS AND METHODS
Cell culture and HCV infection.
Human hepatoma cell lines Huh 7 and Huh7.5.1 were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, 100 U of penicillin/ml, and 100 μg of streptomycin/ml at 37°C in 5% CO2. The Huh7.5.1 cell line was a gift from F. Chisari (Scripps Institute, La Jolla, CA). Cell culture-derived (HCVcc) HCV JC1 strain of genotype 2a used in the present study was propagated and prepared, as described previously (24). For all immunofluorescence experiments, Huh7 cells infected with HCVcc at a multiplicity of infection (MOI) of 0.1 were used at 3 days postinfection to identify both uninfected and HCV-infected cells in the same cultures. For all other experiments, Huh7 cells infected at a high MOI of 1 were used 3 days postinfection. Huh7 cells subjected to mock infection were grown in parallel to infected cells and used as uninfected control cells.
Immunofluorescence.
Cells fixed in 4% paraformaldehyde were washed and permeabilized with 50 μM digitonin for 10 min, followed by blocking for 1 h in 1% bovine serum albumin (BSA)-PBST and then probed with primary antibody in blocking buffer overnight at 4C. After three washes in phosphate-buffered saline (PBS), the cells were stained with respective Alexa Fluor-labeled secondary antibodies (Invitrogen) for 1 h at room temperature. Antibody dilutions were as described by the manufacturer. Lipid droplets were stained with Bodipy505 (Invitrogen) in conjunction with secondary antibody staining. The nuclei were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen) and coverslips mounted on ProLong Gold Antifade (Invitrogen). Images were visualized using Olympus FluoView 1000 confocal microscope. Quantification of images was conducted with ImageJ.
Plasmids and reagents.
The luciferase reporter plasmids harboring wild-type (pLDLR-234) and mutational (pLDLR-R3, pLDLR-234-R2, pLDLR-234-R3D, and CREMU1) LDLR promoters were previously characterized (20, 25, 26). The luciferase reporter plasmids harboring PCSK9 deletion promoter D4, SRE-mut and HNF-mut, were described previously (27). The following antibodies were used: mouse monoclonal anti-core (Affinity Bioreagents), mouse monoclonal anti-HCV NS5A (a gift from Charles Rice, Rockefeller University, New York, NY), anti-HCV E2 (28), rabbit polyclonal anti-LDLR and PCSK9 antibodies (Cayman Chemical), rabbit monoclonal and polyclonal anti-LDLR (Abcam, Inc.), goat polyclonal anti-LDLR (R&D Systems) rabbit anti-βactin and rabbit anti-HA (Cell Signaling Technology). DyNAmo SYBR green PCR kit (New England BioLabs) was used for quantitative real-time PCR (qRT-PCR) analysis using the following sets of primers: GAPDH (glyceraldehyde-3-phosphate dehydrogenase) sense (5-GCCATCAATGACCCCTTCATT-3) and antisense (5-TTGACGGTGCCATGGAATTT-3), LDLR sense (5-ACTGGTGTGGAGAGGACCACC-3) and antisense (5-CAAAGGAAGACGAGGAGCAC-3), and PCSK9 sense (5-AGACCCACCTCTCGCAGTC-3) and antisense (5-GGAGTCCTCCTCGATGTAGTC-3).
Luciferase assay.
The firefly and Renilla luciferase activities in cells transfected with respective luciferase reporter constructs were determined using the dual-luciferase reporter system (Promega) according to the manufacturer's instructions. The Renilla luciferase reporter, pRL-TK, and β-galactosidase expression vector, pRSV-β-galactosidase, were used as control vectors to normalize transfection efficiency. The β-galactosidase activity was determined by using a β-galactosidase assay kit (Promega). The firefly luciferase activity was normalized against the Renilla luciferase or β-galactosidase activity.
Dil-LDL uptake.
Huh7 cells infected with HCV at an MOI of 0.1 were plated on glass coverslips at 3 days postinfection. After adherence, the cells were cultured in medium supplemented with 10% lipoprotein-deficient serum containing 10 μg of Dil (1,1′-dioctadecyl-3,3,3′3′-tetramethyl-indocarbocyanine perchlorate)-labeled LDL (Biomedical Technologies, Inc.)/ml for 5 h. The cells fixed in 4% paraformaldehyde were immunostained for HCV core and imaged using an Olympus FluoView 1000 confocal microscope.
FACS analysis.
Mock- or HCV-infected Huh7 cells were washed twice in azide buffer (PBS plus 5% BSA and 0.1% NaN3) at 4°C. Portions (100 μl) of 106 cells were aliquoted into tubes and then incubated with 20 μl of LDLR rabbit monoclonal antibody for 30 min on ice, washed twice with cold azide buffer, incubated again in the dark with 100 μl of Alexa Fluor 488-conjugated goat anti-rabbit antibody for 20 min on ice, washed again, and fixed with 2% paraformaldehyde. Antibody dilutions used were as per the manufacturer's instruction. Fluorescence-activated cell sorting (FACS) analysis was performed in a Becton Dickinson FACSCalibur dual-laser flow cytometer.
RESULTS
HCV stimulates LDLR expression.
HCV completely relies on host lipid metabolism for its proliferation (4–10). The role of LDLR in facilitating the uptake of serum lipoproteins and in promoting HCV infection is well documented (10, 14). Hence, we wanted to evaluate the effect of HCV infection on LDLR expression. We initially determined the relative levels of LDLR gene expression in mock- and HCVcc (henceforth referred to as HCV)-infected Huh7 cells by determining the relative levels of LDLR mRNA by qRT-PCR (Fig. 1A) to investigate whether HCV upregulated LDLR transcription. We observed that LDLR mRNA levels were ∼2-fold higher in HCV-infected cells compared to mock-infected cells (Fig. 1A). We then determined the protein levels of LDLR by Western blotting. In correlation to the LDLR mRNA, the protein levels were also higher in HCV-infected Huh7 cells compared to mock-infected cells (Fig. 1B). Quantification of the LDLR band intensities revealed a∼2.5-fold increase in LDLR expression in HCV-infected Huh7 cells (Fig. 1C). To investigate whether HCV induces LDLR expression in vivo, we analyzed LDLR expression in liver tissue biopsy specimens from chronic hepatitis C patients and non-HCV donors. Western blot analysis of LDLR revealed that four of five liver tissue samples from patients with chronic hepatitis C revealed significant increases in LDLR expression compared to the two liver tissues samples obtained from non-HCV donors (Fig. 1D).
FIG 1.

HCV stimulates LDLR protein expression. (A) Quantitative real-time PCR (qRT-PCR) analysis of LDLR mRNA in mock- and HCV-infected Huh7 cells. The data presented are means ± the standard errors of the mean (SEM; n = 3). Statistical significance was determined by calculating P value using the Student unpaired two-tailed t test (*, P < 0.005). (B) Western blot analysis of LDLR in mock- and HCV-infected Huh7 cells. HCV NS5A and β-actin were used as the infection marker and the protein loading control, respectively. (C) Densitometric analysis of LDLR band shown in panel B, normalized against β-actin. (D) Western blot analysis of LDLR in liver biopsy samples from chronic hepatitis C patients (samples 3 to 7) and non-HCV donors (samples 1 and 2).
We then determined the cell surface expression of LDLR in mock- versus HCV-infected Huh7 cells. The cells were subjected to FACS analysis to probe for the cell surface expression of LDLR (Fig. 2A). We observed a dramatic shift toward right in the peak representing LDLR fluorescence in HCV-infected Huh7 cells compared to mock-infected cells (Fig. 2A, red versus blue peak), suggesting that HCV-infected cells displayed higher cell surface expression of LDLR (Fig. 2A). To further substantiate this observation, we performed immunofluorescence imaging of total cellular LDLR in HCV-infected versus uninfected cells (Fig. 2B). The HCV-infected cells displayed higher levels of LDLR expression in contrast to uninfected cells (Fig. 2B, cells marked with yellow “+” symbols versus yellow “−” symbols). The relative LDLR fluorescence intensity quantified by ImageJ analysis of five different image panels for about 20 infected versus 20 uninfected cells is shown in Fig. 2C.
FIG 2.

HCV stimulates both the surface and total expression of LDLR. (A) Mock- and HCV-infected Huh7 cells stained for cell surface LDLR were subjected to FACS. Blue and red curves represent mock- and HCV-infected cells, respectively. (B) Confocal image of HCV-infected Huh7 cells immunostained for total cellular LDLR (green) and HCV NS5A (red). Nuclei are stained with DAPI (blue). Infected and uninfected cells are demarcated by “+” and “−” signs, respectively. (C) Quantification of LDLR fluorescence intensity in uninfected versus HCV-infected cells. LDLR fluorescence intensity in ∼20 infected and ∼20 uninfected cells from independent images was quantified by ImageJ. The data represent relative fluorescence intensities expressed in arbitrary units as means ± the SEM. P values were calculated using the Student unpaired two-tailed t test (*, P < 0.05).
HCV infection promotes the uptake of Dil-LDL.
LDLR mediates the uptake of IDL and LDL from bloodstream (14, 15). Hence, an increase in LDLR expression will functionally translate into an augmented uptake of serum lipoprotein particles by the infected cells. To evaluate this, we tested whether HCV infection promotes enhanced uptake of LDL particles. HCV-infected Huh7 cells were incubated with the fluorescent probe Dil-labeled LDL (Dil-LDL) for 5 h at 37°C, as described in Materials and Methods. The immunofluorescence analysis revealed that the HCV-positive Huh7 cells (Fig. 3A; see cells labeled with yellow X in HCV core and Dil-LDL panels) displayed significantly higher levels of Dil-LDL uptake in contrast to uninfected cells. Relative quantitative analysis of Dil-LDL uptake in about 100 HCV-infected versus 100 uninfected cells from different independent images is shown Fig. 3B. The Dil-LDL uptake assay suggested that enhanced expression of LDLR in HCV-infected cells led to enhanced uptake of LDL particles by HCV-infected cells.
FIG 3.

HCV infection promotes enhanced uptake of Dil-LDL. (A) HCV-infected Huh7 cells were cultured in lipoprotein-deficient serum supplemented with 10 μg of Dil-LDL/ml for 5 h at 37°C, fixed in 4% paraformaldehyde, and immunostained for HCV core, followed by imaging using an FV1000 confocal microscope. HCV core is shown in green and Dil-LDL in red (B). Quantitative analysis of Dil-LDL fluorescence intensity in infected versus uninfected cells. Totals of 100 uninfected and 100 HCV-infected cells from independent images were analyzed using ImageJ. The data represent the relative fluorescence intensities expressed in arbitrary units as means ± the SEM. P values were calculated using the Student unpaired two-tailed t test (*, P < 0.005).
HCV-induced transcriptional upregulation of LDLR is mediated by SREBPs.
Basal and cholesterol-mediated human LDLR gene expression is transcriptionally controlled by a 177-bp (−142 to +35) LDLR promoter 5′ of the transcription start site (20). The regulatory elements identified in this promoter region include the three GC-rich imperfect 16-bp repeats and two TA-rich TATA-like sequences and a sterol-independent regulatory element (SIRE), downstream (20). To determine the primary transcription factors involved in HCV-mediated transcriptional upregulation of LDLR gene expression, we used the previously characterized wild-type and mutational LDLR promoter luciferase reporters (Fig. 4A) (20, 25, 26). Mock- and HCV-infected Huh7 cells were transfected with the respective LDLR promoter luciferase reporters, together with the β-galactosidase expression vector. The luciferase activity was normalized against β-galactosidase activity to correct for variations in transfection efficiency. The results show that HCV was able to trigger the luciferase reporter activity in the wild-type LDLR promoter (LDLR234) and other mutated promoters except in the LDLR234-R2 and LDLR-R3 promoters (Fig. 4B), suggesting that these two mutant promoters lack the binding site for the transcription factor targeted by HCV. LDLR234-R2 contains mutation in the repeat 2 region that harbors the SRE-1 element, the binding site for SREBPs (20), suggesting that SREBPs are required for HCV-mediated upregulation of LDLR gene transcription. LDLR-R3 is a deletion mutant promoter spanning nucleotides −52 to +13 of the LDLR promoter, which harbors only the repeat 3 (Sp1 binding region) and TATA-like sequences and the SIRE element (20). HCV was not able to upregulate reporter activity in LDLR-R3 (Fig. 4A and B), suggesting that the transcription factor Sp1 (required for basal transcription), TATA-like sequences and SIRE region are not involved in HCV-mediated upregulation of LDLR transcription. In correlation to this observation, the luciferase activity of LDLR234-R3D mutant promoter (mutated in repeat 3, the binding site for Sp1) and the CREMU1 mutant (which contains mutation in cyclic AMP responsive element in the SIRE region) was upregulated by HCV (Fig. 4B), suggesting that transcription factors other than SREBPs do not play a role in HCV-mediated LDLR gene transcription. Altogether, these observations also rule out the role of sterol-independent mechanisms in HCV-mediated upregulation of LDLR gene transcription.
FIG 4.

HCV stimulates LDLR gene transcription via SREBPs. (A) As schematic representation of the wild-type and mutational constructs of LDLR promoter luciferase reporters. The schematic representation has been adapted and modified from previous publication (26). (B) Mock- and HCV-infected Huh7 cells were transiently transfected with the respective wild-type and mutational luciferase reporter constructs of LDLR promoter, together with pRSV-β-galactosidase. After 24 h, the luciferase and β-galactosidase activities were determined. The firefly luciferase activity was normalized against β-galactosidase activity to correct for variation in transfection efficiency. The data shown are means ± the SEM (n = 3). P values were calculated using the Student unpaired two-tailed t test, * P < 0.05, ** P < 0.0005.
HCV downregulates PCSK9 expression at protein level.
Previous studies have shown that LDLR expression at posttranslational level is also regulated by PCSK9. PCSK9 mediates the degradation of LDLR by nonproteasomal mechanism (21–23). In addition to the transcriptional induction of LDLR gene expression whether HCV could influence LDLR protein levels by downregulating PCSK9, was investigated. To evaluate this, we initially determined the relative levels of PCSK9 mRNA in mock- and HCV-infected Huh7 cells (Fig. 5A). We observed that HCV infection did not significantly alter PCSK9 mRNA levels, which were only marginally above the levels found in mock-infected cells (Fig. 5A). However, Western blot analysis revealed that PCSK9 protein expression was reduced in HCV-infected cells in comparison to mock-infected cells (Fig. 5B). PCSK9 is a direct transcriptional target of SREBPs (29), and it is well known that HCV stimulates SREBPs activation (7, 30). Intriguingly, we did not observe any significant upregulation of PCSK9 mRNA levels in HCV-infected cells (Fig. 5A). PCSK9 gene expression is also transcriptionally induced by hepatocyte nuclear factor 1α (HNF1α), and both SREBPs and HNF1α have been shown to cooperatively modulate PCSK9 gene transcription (27). HCV has been shown to downregulate HNF1α activation (31); hence, we wanted to determine whether HCV prevents the SREBPs-mediated transcriptional stimulation of PCSK9 via HNF1α. To evaluate this hypothesis, we analyzed the PCSK9 promoter activity in HCV-infected and uninfected Huh7 cells using the previously characterized deletion and mutational PCSK9 promoter luciferase reporters (Fig. 5C) (27). In correlation to the marginal increase in PCSK9 mRNA levels, we also observed a marginal stimulation of luciferase activity of the PCSK9 D4 deletion promoter construct (equivalent to wild-type promoter D1) in HCV-infected cells (Fig. 5D). The luciferase reporter activity of the SRE-mutant and HNF-mutant constructs of PCSK9 promoter in HCV-infected cells was marginally reduced and increased, respectively (Fig. 5D). Overall, our results suggest that in HCV infection, SREBPs only marginally stimulate PCSK9 promoter activity and that HNF1α does not influence the marginal stimulation of PCSK9 promoter.
FIG 5.

HCV downregulates PCSK9 at protein level. (A) qRT-PCR analysis of relative levels of PCSK9 mRNA normalized against GAPDH mRNA in mock- and HCV-infected Huh7 cells. The data shown are means ± the SEM of three independent experiments. (B) Western blot analysis of PCSK9 in mock- and HCV-infected Huh7 cells. (C) Schematic representation of deletion and mutational constructs of PCSK9 promoter luciferase reporters. The schematic representation has been adapted and modified from previous publication (27). (D) Mock- and HCV-infected and Huh7 cells were transiently transfected with respective wild-type and mutational luciferase reporter constructs of PCSK9 promoter together with pRSV-β-galactosidase. After 24 h, the luciferase and β-galactosidase activities were determined. The firefly luciferase activity was normalized against the β-galactosidase activity to correct for variations in transfection efficiency. The data shown are means ± the SEM (n = 3). P values were calculated using Student unpaired two-tailed t test (*, P < 0.005; **, P < 0.0005). (E). Analysis of PCSK9 ubiquitination. Mock- and HCV-infected cells were transiently transfected with wild-type PCSK9-Flag and HA-ubiquitin expression vectors. At 36 h posttransfection, the cells were either left untreated or treated with 20 μM MG132 for 5 h. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody, followed by Western blotting with anti-HA antibody. A total of 15% of the total immunoprecipitation input was used for Western blot analysis with anti-HA antibody. (F) Mock- and HCV-infected cells were either left untreated or received 20 μM MG132 treatment for 5 h, and the PCSK9 levels were determined by Western blotting. (G) Western blot analysis of cIAP1 in mock- and HCV-infected Huh7 cells. HCV Core/NS5A was used as an infection marker, and β-actin was used as an internal loading control.
To investigate how HCV reduced PCSK9 protein levels, we determined whether HCV promoted PCSK9 degradation via the proteasome pathway. Mock- and HCV-infected Huh7 cells were cotransfected with PCSK9-Flag and hemagglutinin (HA)-ubiquitin expression vectors. At 48 h posttransfection, one set of cells was treated with the proteasome inhibitor MG132 (20 μM for 5 h). The cell lysates were subjected to immunoprecipitation with anti-flag antibody and immunoblotted with anti-HA antibody (Fig. 5E). The results reveal that MG132 treatment promoted the accumulation of undegraded ubiquitinated proteins in both uninfected and HCV-infected cells. HA-ubiquitinated PCSK9-Flag was almost undetectable in both uninfected and HCV-infected untreated cells; however, MG132 treatment promoted modestly higher accumulation of HA-ubiquitinated PCSK9-Flag in HCV-infected cells, suggesting that HCV promotes the rapid degradation of PCSK9 via ubiquitination (Fig. 5E). To further substantiate this result, we investigated whether proteasome inhibition by MG132 treatment rescues PCSK9 protein levels in HCV-infected cells. We observed that 20 μM MG132 treatment for 5 h significantly rescued PCSK9 levels in HCV-infected cells (Fig. 5F). A recent report suggests that cellular inhibitor of apoptosis protein 1 (cIAP1) binds and promotes PCSK9 degradation and that c-IAP1 also acts as an E3 ubiquitin ligase of PCSK9 (32). We investigated whether HCV promotes cIAP1 expression to facilitate PCSK9 degradation. Western blot analysis of cIAP1 in cell lysates obtained from mock- and HCV-infected Huh7 cells reveal a modest increase in cIAP1 protein levels in HCV-infected cells (Fig. 5G).
HCV stimulates LDLR expression initially at protein level, followed by transcriptional stimulation of LDLR gene.
Since posttranslational regulation is a more rapid means of regulating active protein levels, we then investigated if HCV promotes LDLR expression initially at protein level subsequently, followed by the transcriptional stimulation of LDLR gene. Huh7 cells were either mock infected or infected with high titer (MOI = 5) of HCVcc. The cells were collected at 24, 36, and 48 h postinfection, and the mRNA and protein levels of LDLR were determined by qRT-PCR (Fig. 6A) and Western blot analysis (Fig. 6C). The PCSK9 levels were also determined in the same samples by Western blotting (Fig. 6C). To analyze LDLR promoter activity, Huh7 cells transfected with luciferase reporter construct of LDLR promoter were either mock infected or infected with high titer of HCVcc (MOI = 5), and LDLR promoter activity was determined in cells collected at 24, 36, and 48 h postinfection as described in Materials and Methods (Fig. 6B). The results reveal that HCV marginally stimulates LDLR protein levels at 36 h postinfection, which is paralleled by a concomitant decrease in PCSK9 levels (Fig. 6C, D, and E). The increase in LDLR mRNA levels is observed at 48 h postinfection (Fig. 6A), which is further, substantiated by a corresponding increase in LDLR promoter activity at 48 h postinfection (Fig. 6B). Overall, these results suggest that HCV stimulates LDLR expression first at the protein level, which is further complemented by subsequent transcriptional stimulation of LDLR gene
FIG 6.

HCV initiates upregulation of LDLR expression at protein level, followed by transcriptional stimulation of LDLR gene. (A) Huh7 cells were either mock infected or infected with HCVcc at an MOI of 5, cells were collected at the indicated time points postinfection, and the LDLR mRNA levels were determined by qRT-PCR of total cellular RNA. LDLR mRNA was normalized against GAPDH mRNA. (B) Huh7 cells transiently transfected with luciferase reporter construct of LDLR promoter, together with pRSV-β-galactosidase, were either mock infected or infected with HCVcc at an MOI of 5. The cells were collected at the indicated time points postinfection, and the firefly luciferase and β-galactosidase activities in the cell lysates were determined as described in ‘Materials and Methods. The luciferase activity was normalized against the β-galactosidase activity to normalize variations in transfection efficiency. The data presented are means ± the SEM (n = 3). P values were calculated by using the Student unpaired two-tailed t test (*, P < 0.05; **, P < 0.005). (C) Western blot analysis of LDLR and PCSK9 in cell lysates obtained from cells described in panel A. HCV NS5A was used as an infection marker, and β-actin was used as a protein loading control. (D and E) Densitometric analysis of the respective LDLR (D) and PCSK9 (E) Western blots shown in panel C. Band intensities were normalized against β-actin bands and are expressed as percentage of the control.
Ectopic expression of wild-type PCSK9 and gain-of-function mutant PCSK9 negatively affected HCV replication.
A recent study suggests that although LDLR is not essential for HCV entry the physiological function of LDLR is crucial for the postentry events such as HCV replication (13). Lipid plays a crucial and indispensable role in HCV replication and maturation (4–10), and an increase in LDLR expression will functionally translate into an augmented uptake of serum lipoprotein particles by the infected cells and enrichment of intracellular milieu with lipids. Hence, we wanted to test our hypothesis that HCV promotes LDLR expression to facilitate HCV replication. Wild-type PCSK9 and the gain-of-function mutant D374Y were ectopically expressed in full-length replicon cells (FL-Feo) of HCV-genotype 2a. At 48 h posttransfection, the luciferase activity in the cell lysate was determined as described in Materials and Methods. The results show that the ectopic expression of both the wild-type PCSK9 and the gain-of-function mutant D374Y reduced HCV replication by >50% (Fig. 7A). Interestingly, the gain-of-function mutant D374Y was only slightly more potent than wild-type PCSK9 in effecting HCV replication (Fig. 7A). We also determined the effect of wild-type PCSK9 and D374Y mutant on HCV replication in HCVcc-infected Huh7 cells. To circumvent the effect of wild-type PCSK9 and D374Y mutant on HCVcc entry, we initially infected the cells with HCVcc for 4 h, followed by transfection with PCSK9 expression vectors. At 3 days postinfection, HCV replication was determined by qRT-PCR of HCV RNA as previously described (33). In accordance with the results obtained in full-length replicon cells, we observed inhibition of HCV replication in cells ectopically expression wild-type PCSK9 or D374Y mutant (Fig. 7B). The levels of LDLR in mock- and HCV-infected Huh7 cells ectopically expressing wild-type PCSK9 or D374Y mutant were determined by Western blotting (Fig. 7C). As expected, the LDLR protein levels were reduced in cells expressing the wild-type PCSK9 and D374Y mutant, and the reduction was more pronounced in cells expressing the gain-of-function mutant D374Y (Fig. 7C).
FIG 7.
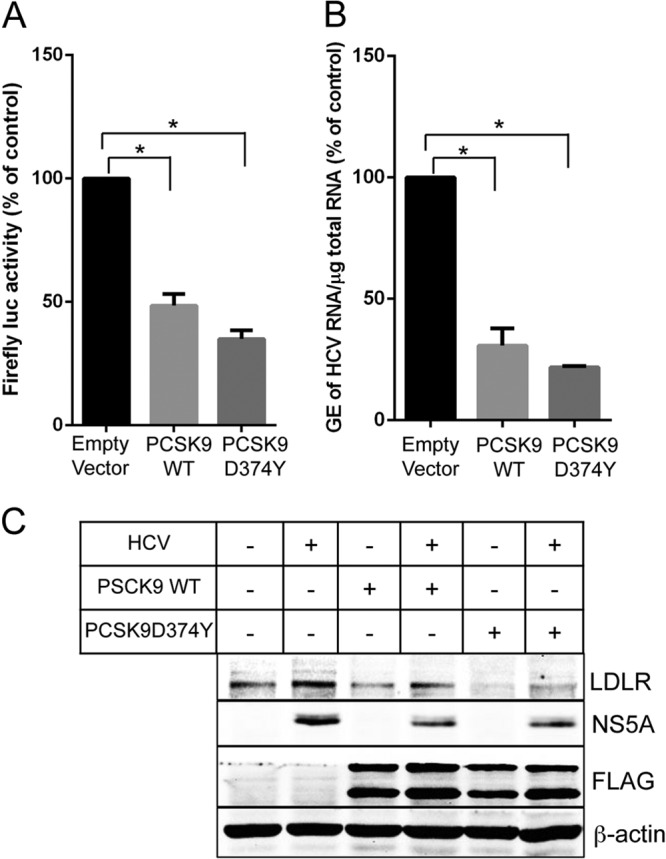
Ectopic expression of wild-type PCSK9 or D374Y mutant negatively affects HCV replication. (A) Full-length replicon cells (FL-Feo cells) of HCV genotype 2a were transiently transfected with wild-type or D374Y mutant PCSK9-Flag expression vector, and at 48 h posttransfection the firefly luciferase activity in cell lysates was determined. (B) At 4 h postinfection, HCVcc-infected Huh7 cells were transfected with wild-type or D374Y mutant PCSK9-Flag expression vector, and at 72 h postinfection the HCV replication was determined by qRT-PCR analysis of HCV RNA. The data presented are means ± the SEM (n = 3). P values were calculated by using a Student unpaired two-tailed t test (*, P < 0.0005). (C) Western blot analysis of LDLR and FLAG-tagged wild-type and mutant PCSK9 in mock- and HCV-infected cells untransfected or transiently transfected with wild-type PCSK9-Flag or D374Y PCSK9-Flag expression vector. HCV NS5A was used as an infection marker, and β-actin was used as protein loading control.
DISCUSSION
HCV modulates host lipid metabolism to promote the intracellular repertoire of lipids essential in multiple aspects of HCV life cycle, which include entry, replication, maturation, and egress (4–10). HCV is known to promote de novo lipid biosynthesis via SREBPs (30) and halt the catabolic breakdown of lipids (34, 35). However, very little is known about the influence of HCV on lipid uptake from bloodstream. LDLR is a protein primarily responsible for uptake of cholesterol-rich lipoproteins from bloodstream (14, 15). Several reports indicate that HCV particles are associated with lipoproteins, and the association with lipoproteins determines their specific infectivity (6, 10). These observations emphasize the plausible role of LDLR in HCV entry. However, conflicting reports exist on the role of LDLR in HCV entry (10).
Data from a clinical study suggests that LDLR expression correlates with viral load in HCV-infected patients (36). The high prevalence of hypocholesterolemia and hypo-β-lipoproteinemia in HCV-infected patients suggests a higher hepatic uptake of serum lipoproteins in HCV patients (16–18). These studies signify the intricate relation between HCV infection and LDLR expression. However, many viruses, including HCV, follow superinfection exclusion, which is the ability of an established viral infection to interfere infection with another virus (37). This notion suggests that the HCV-mediated stimulation of LDLR expression may not essentially promote reinfection of an infected hepatocyte but may be involved in viral processes downstream of entry. A recent study conducted in the HCV cell culture model corroborates this hypothesis and suggests that although LDLR is not involved in HCV entry, the physiological function of LDLR as a lipid-providing receptor is necessary for the establishment of productive infection (13). Neutralization of LDLR function in HCV-infected cell with specific antibody resulted in lowering of cellular cholesterol levels and an increase in the ratio of phosphatidylethanolamine to phosphatidylcholine with an associated decline in HCV replication (13). Cholesterol and phospholipids are essential components of new membranes and may be required for the formation of replication complexes, and the inhibition of lipid and cholesterol biosynthetic pathways have been shown to perturb HCV replication (4–7).
Upregulation of LDLR expression by HCV will result in higher uptake of cholesterol-rich lipoproteins from bloodstream and will positively influence the cholesterol content and other lipids in infected hepatocytes. Augmenting the expression of LDLR is one of the major mechanisms through which cells promote LDL uptake in response to cholesterol-depleted conditions (14, 19). We observed that HCV stimulated both the total and cell surface expression of LDLR (Fig. 1 and 2). The increase in LDLR expression in HCV-infected cells reflected in an increased uptake of Dil-LDL particles (Fig. 3), suggesting that the HCV-mediated induction of LDLR expression functional contributes to an increased uptake of lipoproteins. However, the increase in Dil-LDL uptake did not linearly correlate with the 2.5-fold increase in LDLR protein level, suggesting that other factors also influence lipoprotein uptake. It is also very likely that rapid saturation of Dil fluorescence signal makes it difficult to derive an exact linear correlation between LDLR protein levels and Dil-LDL uptake.
We observed that HCV upregulated LDLR gene transcription through SREBPs (Fig. 4A). It is well known that SREBPs are activated in low-sterol conditions, leading to the upregulation of lipid biosynthetic pathways and LDLR expression to promote internalization of cholesterol-rich lipoproteins from bloodstream, suggesting that the expression of LDLR on the cell surface of hepatocytes is inversely related to the cellular cholesterol concentration (14, 19, 20). This is the major mechanism through which statins work in reducing LDL-cholesterol levels in plasma by upregulating LDLR expression via inhibition of cholesterol synthesis (38). Interestingly, HCV activates SREBPs in normal physiologic conditions (30, 39) through a mechanism not completely elucidated, leading to the intracellular accumulation of lipids via enhanced synthesis and probably enhanced uptake by LDLR. Our observation that HCV upregulates LDLR gene transcription via SREBPs (Fig. 4) highly correlates with this notion. Intracellular accumulation of lipids promotes lipid droplet formation, the cellular lipid storage organelles that play a pivotal role in HCV virus particle assembly and secretion processes (5–7). Many reports show the accumulation of lipid droplets in HCV-infected cells (34, 40).
LDLR is also regulated at the protein level by PCSK9 and IDOL (21–23). PCSK9 and IDOL are transcriptional targets of SREBPs and LXR, respectively (23, 29). Although SREBPs are activated during HCV infection (30), we did not observe any significant increase in PCSK9 mRNA levels in HCV-infected cells (Fig. 5A), suggesting a complex modulation of PCSK9 gene transcription by HCV. More detailed studies are required to completely understand the effect of HCV on the transcriptional modulation of PCSK9. Although there was no significant alteration in PCSK9 mRNA levels, the protein expression was downregulated in HCV-infected cells (Fig. 5A and B), suggesting that HCV targets PCSK9 at the protein level. Our observations suggest that HCV targets PCSK9 for proteasomal degradation, probably by upregulating the levels of the E3-ubiquitin ligase of PCSK9, cIAP1 (Fig. 5G). However, at this juncture we cannot rule out any other additional mechanism(s) that play a role in HCV-mediated downregulation of PCSK9. Using a high MOI of infection, we observed upregulation of LDLR protein levels as early as 36 h postinfection, followed by an additional increase at 48 h (Fig. 6C). Although the LDLR protein levels increased at 36 h postinfection, the LDLR mRNA and promoter activity were upregulated at 48 h postinfection (Fig. 6A and B), suggesting that the early increase in LDLR protein levels is mediated by the downregulation of PCSK9, and the subsequent transcriptional upregulation of LDLR gene further complements this increase. In correlation to this hypothesis, the increase in LDLR protein levels at 36 h postinfection was associated with a concomitant decline in PCSK9 levels (Fig. 6C)
HCV promotes LDLR expression probably to promote lipid uptake from bloodstream to enrich the hepatocyte intracellular milieu with lipids to facilitate HCV replication. In agreement with this hypothesis, we observed that perturbation of LDLR expression by ectopic expression of wild-type PCSK9 or the gain-of-function D347Y mutant inhibited HCV replication in both full-length replicon cells and HCVcc-infected Huh7 cells (Fig. 7A and B). Overall, our study suggests that serum-lipid uptake is one of the primary means of intracellular lipid enrichment in HCV infection. Interestingly, PCSK9 overexpression has been used as a strategy to inhibit HCV infection by preventing entry (41). In addition, the effect of PCSK9 overexpression on events downstream of entry may also contribute to the anti-HCV potential of PCSK9 making it an attractive target for the therapeutic intervention of HCV infection.
In conclusion, our observations highlight the stimulation of LDLR expression by HCV and signify the potential involvement of the LDLR in HCV infection with future implications for LDLR in anti-HCV therapy. The assumption that the alpha interferon currently used in anti-HCV therapy partly functions through the downregulation of LDLR (42) further supports the need to develop novel, less-toxic anti-HCV therapeutic strategies targeting LDLR.
ACKNOWLEDGMENTS
This study is supported by National Institutes of Health grants AI085087, DK077704, and DK08379 to A.S.
We gratefully acknowledge Jay Horton, University of Texas Southwestern Medical Center, Dallas, Texas, for kindly providing Flag-tagged expression vectors of wild-type PCSK9 and gain-of-function mutant D374Y.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567. 10.1016/S1473-3099(05)70216-4 [DOI] [PubMed] [Google Scholar]
- 2.Negro F, Alberti A. 2011. The global health burden of hepatitis C virus infection. Liver Int. 31(Suppl 2):1–3. 10.1111/j.1478-3231.2011.02537.x. [DOI] [PubMed] [Google Scholar]
- 3.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463. 10.1038/nrmicro1645 [DOI] [PubMed] [Google Scholar]
- 4.Ye J. 2007. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 3:e108. 10.1371/journal.ppat.0030108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLauchlan J. 2009. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta 1791:552–559. 10.1016/j.bbalip.2008.12.012 [DOI] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 19:95–103. 10.1016/j.tim.2010.11.005 [DOI] [PubMed] [Google Scholar]
- 7.Syed GH, Amako Y, Siddiqui A. 2010. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol. Metab. 21:33–40. 10.1016/j.tem.2009.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Pene V, Krishnamurthy S, Cha H, Liang TJ. 2013. Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nat. Med. 19:722–729. 10.1038/nm.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:15669–15674. 10.1073/pnas.202608199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burlone ME, Budkowska A. 2009. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J. Gen. Virol. 90:1055–1070. 10.1099/vir.0.008300-0 [DOI] [PubMed] [Google Scholar]
- 11.Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, Roitelman J, Barbaras R, Graber P, Ghersa P, Smolarsky M, Funaro A, Malavasi F, Larrey D, Coste J, Fabre JM, Sa-Cunha A, Maurel P. 2007. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 46:411–419. 10.1016/j.jhep.2006.09.024 [DOI] [PubMed] [Google Scholar]
- 12.Jiang J, Wu X, Tang H, Luo G. 2013. Apolipoprotein e mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS One 8:e67982. 10.1371/journal.pone.0067982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J. 2012. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55:998–1007. 10.1002/hep.25501 [DOI] [PubMed] [Google Scholar]
- 14.Goldstein JL, Brown MS. 2009. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29:431–438. 10.1161/ATVBAHA.108.179564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chappell DA, Medh JD. 1998. Receptor-mediated mechanisms of lipoprotein remnant catabolism. Prog. Lipid Res. 37:393–422. 10.1016/S0163-7827(98)00017-4 [DOI] [PubMed] [Google Scholar]
- 16.Serfaty L, Andreani T, Giral P, Carbonell N, Chazouilleres O, Poupon R. 2001. Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 34:428–434. 10.1016/S0168-8278(00)00036-2 [DOI] [PubMed] [Google Scholar]
- 17.Clark PJ, Thompson AJ, Vock DM, Kratz LE, Tolun AA, Muir AJ, McHutchison JG, Subramanian M, Millington DM, Kelley RI, Patel K. 2012. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology 56:49–56. 10.1002/hep.25631 [DOI] [PubMed] [Google Scholar]
- 18.Petit JM, Benichou M, Duvillard L, Jooste V, Bour JB, Minello A, Verges B, Brun JM, Gambert P, Hillon P. 2003. Hepatitis C virus-associated hypobetalipoproteinemia is correlated with plasma viral load, steatosis, and liver fibrosis. Am. J. Gastroenterol. 98:1150–1154. 10.1016/S0002-9270(03)00106-0 [DOI] [PubMed] [Google Scholar]
- 19.Desvergne B, Michalik L, Wahli W. 2006. Transcriptional regulation of metabolism. Physiol. Rev. 86:465–514. 10.1152/physrev.00025.2005 [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Briggs MR, Kraemer FB. 2006. Elucidation of an SRE-1/SREBP-independent cellular pathway for LDL-receptor regulation: from the cell surface to the nucleus. Future Cardiol. 2:605–612. 10.2217/14796678.2.5.605 [DOI] [PubMed] [Google Scholar]
- 21.Maxwell KN, Fisher EA, Breslow JL. 2005. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc. Natl. Acad. Sci. U. S. A. 102:2069–2074. 10.1073/pnas.0409736102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. 2007. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282:18602–18612. 10.1074/jbc.M702027200 [DOI] [PubMed] [Google Scholar]
- 23.Zelcer N, Hong C, Boyadjian R, Tontonoz P. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325:100–104. 10.1126/science.1168974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413. 10.1073/pnas.0504877103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Ahlborn TE, Briggs MR, Kraemer FB. 2000. Identification of a novel sterol-independent regulatory element in the human low density lipoprotein receptor promoter. J. Biol. Chem. 275:5214–5221. 10.1074/jbc.275.7.5214 [DOI] [PubMed] [Google Scholar]
- 26.Li H, Liu J. 2009. Identification of heterogeneous nuclear ribonucleoprotein K as a transactivator for human low density lipoprotein receptor gene transcription. J. Biol. Chem. 285:17789–17797. 10.1074/jbc.M109.082057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. 2009. Hepatocyte nuclear factor 1α plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 284:28885–28895. 10.1074/jbc.M109.052407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- 29.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. 2003. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U. S. A. 100:12027–12032. 10.1073/pnas.1534923100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waris G, Felmlee DJ, Negro F, Siddiqui A. 2007. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81:8122–8130. 10.1128/JVI.00125-07 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Matsui C, Shoji I, Kaneda S, Sianipar IR, Deng L, Hotta H. 2012. Hepatitis C virus infection suppresses GLUT2 gene expression via downregulation of hepatocyte nuclear factor 1α. J. Virol. 86:12903–12911. 10.1128/JVI.01418-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Liu L, Hornby D. 2012. c-IAP1 binds and processes PCSK9 protein: linking the c-IAP1 in a TNF-alpha pathway to PCSK9-mediated LDLR degradation pathway. Molecules 17:12086–12101. 10.3390/molecules171012086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amako Y, Syed GH, Siddiqui A. 2011. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J. Biol. Chem. 286:11265–11274. 10.1074/jbc.M110.182097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Syed GH, Siddiqui A. 2011. Effects of hypolipidemic agent nordihydroguaiaretic acid on lipid droplets and hepatitis C virus. Hepatology 54:1936–1946. 10.1002/hep.24619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia-Brandt L, Juge-Aubry CE, Meier CA, Hadengue A, Negro F. 2006. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment Pharmacol. Ther. 23:107–114. 10.1111/j.1365-2036.2006.02729.x [DOI] [PubMed] [Google Scholar]
- 36.Petit JM, Minello A, Duvillard L, Jooste V, Monier S, Texier V, Bour JB, Poussier A, Gambert P, Verges B, Hillon P. 2007. Cell surface expression of LDL receptor in chronic hepatitis C: correlation with viral load. Am. J. Physiol. Endocrinol. Metab. 293:E416–E420. 10.1152/ajpendo.00091.2007 [DOI] [PubMed] [Google Scholar]
- 37.Tscherne DM, Evans MJ, von Hahn T, Jones CT, Stamataki Z, McKeating JA, Lindenbach BD, Rice CM. 2007. Superinfection exclusion in cells infected with hepatitis C virus. J. Virol. 81:3693–3703. 10.1128/JVI.01748-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scharnagl H, Marz W. 2005. New lipid-lowering agents acting on LDL receptors. Curr. Top. Med. Chem. 5:233–242. 10.2174/1568026053544524 [DOI] [PubMed] [Google Scholar]
- 39.Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. 2009. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 284:9237–9246. 10.1074/jbc.M808773200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulant S, Douglas MW, Moody L, Budkowska A, Targett-Adams P, McLauchlan J. 2008. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 9:1268–1282. 10.1111/j.1600-0854.2008.00767.x [DOI] [PubMed] [Google Scholar]
- 41.Labonte P, Begley S, Guevin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. 2009. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 50:17–24. 10.1002/hep.22911 [DOI] [PubMed] [Google Scholar]
- 42.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. U. S. A. 96:12766–12771. 10.1073/pnas.96.22.12766 [DOI] [PMC free article] [PubMed] [Google Scholar]