

ABSTRACT
Although the pattern recognition receptor Toll-like receptor 2 (TLR2) is typically thought to recognize bacterial components, it has been described to alter the induction of both innate and adaptive immunity to a number of viruses, including vaccinia virus (VACV). However, many pathogens that reportedly encode TLR2 agonists may actually be artifactually contaminated during preparation, possibly with cellular debris or merely with molecules that sensitize cells to be activated by authentic TLR2 agonists. In both humans and mice, the most relevant natural route of infection with VACV is through intradermal infection of the skin. Therefore, we examined the requirement for TLR2 and its signaling adaptor MyD88 in protective immunity to VACV after intradermal infection. We find that although TLR2 may recognize virus preparations in vitro and have a minor role in preventing dissemination of VACV following systemic infection with large doses of virus, it is wholly disposable in both control of virus replication and induction of adaptive immunity following intradermal infection. In contrast, MyD88 is required for efficient induction of CD4 T cell and B cell responses and for local control of virus replication following intradermal infection. However, even MyD88 is not required to induce local inflammation, inflammatory cytokine production, or recruitment of cells that restrict virus from spreading systemically after peripheral infection. Thus, an effective antiviral response does require MyD88, but TLR2 is not required for control of a peripheral VACV infection. These findings emphasize the importance of studying relevant routes of infection when examining innate sensing mechanisms.
IMPORTANCE Vaccinia virus (VACV) provides the backbone for some of the most widely used and successful viral vaccine vectors and is also related to the human pathogens Cantagalo virus and molluscum contagiosum virus that infect the skin of patients. Therefore, it is vital to understand the mechanisms that induce a strong innate immune response to the virus following dermal infection. Here, we compare the ability of the innate sensing molecule Toll-like receptor 2 (TLR2) and the signaling molecule MyD88 to influence the innate and adaptive immune response to VACV following systemic or dermal infection.
INTRODUCTION
Pattern recognition receptors (PRRs) are crucial for innate immunity, through recognition of common molecular patterns distinctive of pathogens. Activation through PRR leads to the induction of type I interferons and inflammatory cytokines. Toll-like receptors (TLRs) make up a family of PRRs that have an N-terminal extracellular domain made up of leucine-rich repeats (LRRs), a single transmembrane domain, and a cytoplasmic TIR domain shared by both the TLR and interleukin-1 receptor (IL-1R) families. The LRR domain is the main source of variability among TLRs and of genetic diversity within a single TLR (1).
Toll-like receptor 2 (TLR2) is a cell surface TLR that uniquely heterodimerizes with either TLR1 or TLR6 and directly binds adaptor protein MyD88, signaling to upregulate cytokines and chemokines that foster inflammation (2). The first TLR2 agonists identified were bacterial lipoproteins (3). In practice, TLR2 has been reported to recognize a wider range of pathogens than any other TLR, including fungi (4), protozoans (5), worms (6), Mycoplasma (7), Gram-positive and -negative bacteria (8, 9), DNA viruses (10), and RNA viruses (11), as well as host molecules such as HMGB1 (12). However, concern is growing that many reported TLR2 agonists are artifacts of possible contamination, cellular debris, or merely molecules that sensitize cells to be activated by authentic TLR2 agonists (13, 14).
TLR2 also offers diversity in its downstream signaling effects (15). In addition to inducing proinflammatory cytokines in its classical role as a MyD88-dependent cell surface receptor, it also activates type I interferon expression with both viral and bacterial ligands (16, 17), a pathway that requires internalization and may even involve the Trif adaptor molecule, rather than MyD88 (18). One virus that has been suggested to encode a TLR2 agonist(s) is vaccinia virus (VACV), a double-stranded DNA (dsDNA) orthopoxvirus that infects a variety of animals, including mice, humans, and cattle. VACV has long been a promising vector for immunization and gene therapy and is important for human health as the vaccine given to protect people from smallpox, an often-fatal disease caused by the closely related variola virus (19, 20). Although many animal studies examine immunity to VACV induced through systemic intraperitoneal (i.p.) or intravenous (i.v.) routes, the route of infection that most closely resembles that used during human immunization, infection of humans with the related poxvirus molluscum contagiosum virus (21), and the route that mimics natural infection of mice with the highly homologous orthopoxvirus ectromelia virus (ECTV), is the intradermal (i.d.) route (22). Indeed, pathogenesis experiments reveal a role for highly conserved immunomodulatory molecules following intradermal, but not other, routes of infection, indicating that this is a natural route of infection (23). In vitro myeloid cells can use TLR2 to detect an unknown component within preparations of VACV (17) and the highly attenuated vaccinia virus strain MVA (24), but the specific VACV TLR2 ligand (if any) present within these preparations remains uncharacterized.
In vivo evidence for TLR2 as a VACV sensor is very inconsistent. One study found that TLR2 and MyD88 knockout mice suffered increased viral titers despite continued high levels of serum beta interferon (IFN-β) (25), while another found no impact on viral titers but a significant decrease in serum IFN-β (26). VACV may also directly activate CD8+ T cells through TLR2 (27), suggesting that it is important for developing effective memory as well as an innate response. However, it is also reported that, although VACV stimulates CD8+ T cell activation and differentiation in a MyD88-dependent manner, the MyD88 knockout phenotype was not shared by TLR2, TLR4, TLR9, or IL-1R knockouts (28). Furthermore, all in vivo experiments on deficiencies in TLR2/MyD88 signaling thus far have used intraperitoneal or intravenous infection with large doses of VACV, which work well to induce systemic inflammation, viremia, and infection of multiple organ systems but are quite distinct from the intradermal and intranasal routes of infection that most poxviruses have evolved to utilize. Infection via the intraperitoneal or intravenous routes may allow access to innate immune cells that are not normally exposed to virus. Indeed, evidence from other pathogens (29–31) suggests that although TLR2-deficient animals are highly susceptible to systemic infection, they may respond similarly to wild-type (WT) animals when infected intradermally, even though many cell types in the skin express TLR2 (32).
In the present study, we investigated TLR2-MyD88 signaling in the murine immune system following intradermal poxvirus infection. We observed that MyD88−/− mice had enhanced viral replication and more rapid tissue destruction and were impaired in their development of CD4+ T cell and antibody responses. However, TLR2−/− mice were capable of adequately controlling virus spread, clearing virus from the skin, and establishing CD4+ T cell and antibody responses to VACV infection. Though TLR2−/− cells were impaired in responding to VACV with proinflammatory cytokines in vitro, and it is reported that TLR2−/− mice have a similar impairment early in intravenous infection (33), we found no difference between TLR2−/− and TLR2+/+ mice in recruitment of myeloid cells to the ear or in production of cytokines or interferon-stimulated genes following infection. These results suggest that any role for TLR2 in host response to vaccinia virus is superfluous when the virus enters the body through a natural route.
MATERIALS AND METHODS
Animals.
C57BL/6 mice were purchased from Charles River Laboratories. TLR2−/− (catalog no. 004650), MyD88−/− (catalog no. 009088), IL-6−/− (catalog no. 002650), LysM:cre (catalog no. 004781), and MyD88:flox (catalog no. 008888) mice were purchased from Jackson Laboratory and subsequently bred at the Hershey Medical Center. LysM:wt/cre MyD88:flox/flox mice were generated by first breeding LysM:cre and MyD88:flox mice to create LysM:cre MyD88:flox double transgenics and then breeding LysM:cre MyD88:flox mice to MyD88:flox mice. All knockout mouse strains were on the C57BL/6 background after a minimum of 12 backcrosses to this strain. All animals were maintained in the specific-pathogen-free facility of the Hershey Medical Center and treated in accordance with the National Institutes of Health and AAALAC International regulations. Food and water were provided ad libitum, with MyD88−/− mice receiving Baytril in chow and acidified water (pH 2.75) as antibacterial measures. All animal experiments and procedures were approved by the Penn State Hershey IACUC.
Viruses and infections.
VACV (strain WR) stocks were produced in 143B.TK− cell monolayers and ultracentrifuged through a 45% sucrose cushion to purify viral particles from other cellular debris. For intradermal infection, mice were sedated using ketamine-xylazine and injected in each ear pinna with 104 PFU of VACV in a volume of 10 μl. For i.v. and i.p. infection, mice were injected with 106 or 107 PFU of VACV in a volume of 500 μl. For assaying neutralizing antibodies in serum, VACV-NP-SIINFEKL-GFP (34) was used to infect B6/WT-3 cells. Wild-type VACV (Western Reserve strain) was used in all other experiments.
To assess dermal pathogenesis, ear thickness was measured using a 0.0001-in. dial micrometer (Mitutoyo), and lesion progression was measured using a ruler. To analyze the presence of replicating virus, organs were harvested, subjected to three freeze-thaw cycles in Hanks balanced salt solution (HBSS), ground in a 7-ml Dounce homogenizer, and sonicated. Lysate was placed on a monolayer of 143B.TK− cells, and plaques were counted 2 days later. 143B.TK− and B6/WT-3 cells were maintained in Dulbecco modified Eagle medium (DMEM) with 5% fetal bovine serum (FBS) (HyClone).
Flow cytometry of cells at the site of infection.
Ear pinnae were cut into strips and digested in 1 mg/ml collagenase XI (Sigma) for 60 min at 37°C. Live cells were blocked and stained on ice in 2.4G2 cell supernatant containing 10% normal mouse serum (Sigma). Antibodies included CD11b (M1/70), CD11c (N418), CD8 (53-6.7), B220 (RA3-6B2), and Ly6G (1A8) from eBioscience and Ly6C (clone AL-21), CD45.2 (104), CD19 (1D3), CD90.2 (53-2.1), NK1.1 (PK136), IL12p40/p70 (C15.6), and IL-6 (MP5-20F3) from BD Pharmingen. Phycoerythrin (PE)-Cy7-streptavidin (BD) was used to label biotin-conjugated antibodies. Dendritic cell (DC) subsets were identified as follows: B220+ “plasmacytoid” DCs (CD19− CD90− NK1.1− CD11c+ CD11b− B220+), CD11b+ “conventional” DCs (CD19− CD90− NK1.1− CD11c+ CD11b+ B220− CD8−), and CD8α+ “lymphoid resident” DCs (CD19− CD90− NK1.1− CD11c+ CD11b− B220− CD8+). Sample acquisition was performed with an LSRII flow cytometer (BD), and data were analyzed with FlowJo software (TreeStar).
Analysis of neutralizing antibodies in serum.
Mice were deeply sedated with ketamine-xylazine, and a 25-gauge 5/8-in. needle was inserted below the xiphoid process to draw 400 to 800 μl blood from the heart. Serum was obtained by repeated centrifugation to remove all cells, followed by at least one freeze-thaw cycle. Serum was serially diluted in normal mouse serum and incubated at 37°C with VACV-NP-SIINFEKL-GFP. After 1 h, virus-serum mixtures were added to WT-3 cells at a multiplicity of infection (MOI) of 10:1 and incubated on a rotating rack at 37°C. Infectivity was measured as the percentage of green fluorescent protein-positive (GFP+) cells after 6 h of incubation with virus. Sample acquisition was performed with a FACSCanto flow cytometer (BD), and data were analyzed with FlowJo.
Intracellular cytokine staining.
Spleens were digested using 1 mg/ml collagenase D (Roche) for 30 min at 37°C and a 10-min ammonium-chloride-potassium (ACK) lysis to lyse red blood cells. The resulting splenocytes were stimulated with infectious VACV at an MOI of 10 in DMEM with 5% FBS for a total of 6 h. During the last 4 h of incubation, 10 μg/ml brefeldin A (Sigma) was added to the medium. To detect intracellular cytokines, cells were fixed in 1% formaldehyde (Acros Organics) and stained and washed in 2.4G2 cell supernatant containing 10% normal mouse serum and 0.5% saponin (Sigma). Sample acquisition was performed with an LSRII flow cytometer, and data were analyzed using FlowJo.
ELISpots.
Previously defined VACV major histocompatibility complex (MHC) class I and II epitopes (35) were screened for reactivity against splenocytes from WT C57BL6, TLR2−/−, and MyD88−/− mice at 10 days post-VACV ear infection. Splenocytes from naive mice were used as antigen-presenting cells (APCs). Naive splenocytes were incubated with the indicated peptide (final concentration, 2 μg/ml) in 37°C, 5% CO2. VACV-primed and peptide-pulsed splenocytes were then coincubated overnight, and IFN-γ-positive T cell responses were assayed by IFN-γ enzyme-linked immunosorbent spot assay (ELISpot) (BD). Spots were counted using ImmunoSpot software (CTL). Peptides were synthesized by New England Peptide (Gardner, MA).
Real-time PCR.
The extraction of total RNA from tissue and reverse transcription of RNA were carried out as described previously (36), and the PCR was done as before (37). The following dye combinations were used for detection and data normalization: 6-carboxyfluorescein (FAM) (reporter for IFNs, MX-1, and IL-6) and hexachlorofluorescein (HEX) (reporter for glyceraldehyde-3-phosphate dehydrogenase [GAPDH]).
RESULTS
TLR2 is superfluous in controlling intradermal VACV infection.
To examine the role of TLR2 in poxvirus recognition, we infected mice via the natural intradermal (i.d.) route in the ear pinnae with 104 PFU of VACV and monitored pathology and viral replication. TLR2−/− mice were indistinguishable from wild-type mice in the degree and rate of ear swelling (Fig. 1A). Following the initial period of skin infection in which uncontrolled viral replication spurs recruitment of inflammatory monocytes, a lesion develops and is observed on both the inoculated convex surface and the smoother concave surface of the ear. Viral replication decreases past day 5 of infection and is no longer detectable once the lesion is resolved by loss of necrotic tissue (22). Tissue damage develops more aggressively in many immunodeficient mouse strains (38–40), but in TLR2−/− mice, we observed that the pathology developed similarly to that in wild-type (WT) mice (Fig. 1B). At no stage of intradermal infection was there significantly more VACV replication in TLR2−/− mice than in WT mice (Fig. 1C), although 7 of 12 TLR2−/− mice, compared to 4 of 12 WT mice, had some detectable virus remaining on day 16. VACV normally remains restricted to the ear pinnae following i.d. infection, but components of the immune system are required to prevent systemic spread (38), and these components may be distinct from those that control virus replication locally. Therefore, we also looked for evidence that TLR2 restricted the spread of virus from the ear pinnae both by quantification of virus titers in the primary site of replication following systemic infection, the ovaries, and by evaluation of morbidity following VACV infection. In multiple experiments, TLR2−/− mice had no detectable VACV in ovaries above that detected in WT mice (Fig. 1D), did not lose weight (Fig. 1E), and had no visible signs of systemic infection such as tail lesions. These data indicate that a successful innate response to a natural route of VACV infection did not depend on TLR2.
FIG 1.
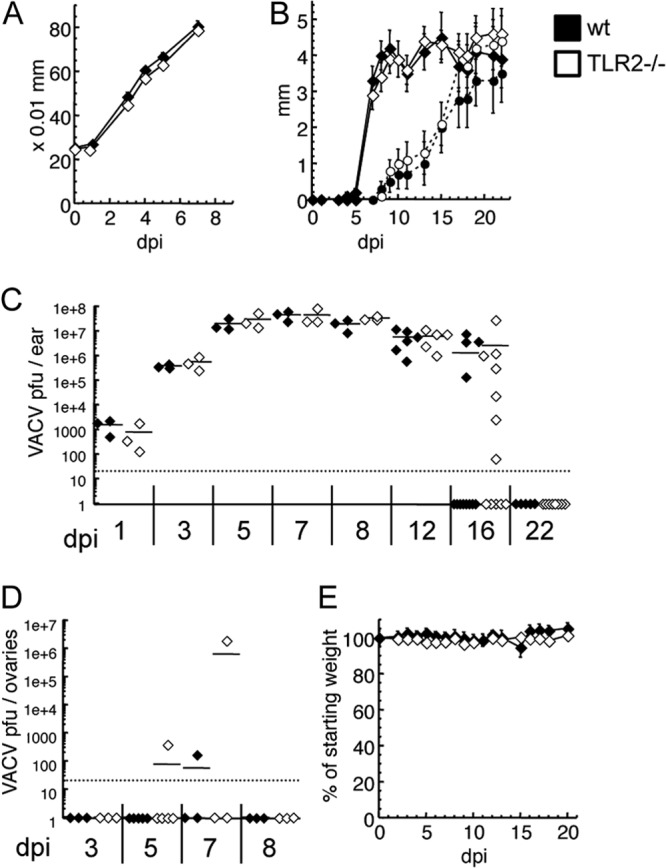
TLR2 does not contribute significantly to the innate response to intradermal VACV infection. C57BL/6 (black) or TLR2−/− (white) mice were infected intradermally with 104 PFU VACV in each ear pinna. (A) Ear swelling was monitored with a thickness gauge until tissue loss began (n = 10 ears). (B) The size of lesions (solid lines) and subsequent loss of tissue (dotted lines) were monitored for 3 weeks (n = 10 ears). Results are representative of two independent experiments. (C and D) On the specified day postinfection, mice were sacrificed; ears (C) and ovaries (D) were freeze-thawed 3 times, homogenized, and sonicated to release virions; and serial dilutions were applied to cell monolayers for VACV titer determination. (E) Weight loss was monitored for 3 weeks (n = 5). Dotted lines indicate the limit of detection for virus plaque titer assays. dpi, day postinfection.
TLR2 contributes to activation of dendritic cells by VACV in vitro but not recruitment of myeloid cells in vivo.
The results above led us to inquire as to whether our stock of VACV was capable of activating dendritic cells through TLR2 in vitro. We harvested splenocytes from naive TLR2−/− or WT mice and stimulated them with VACV before adding brefeldin A to block cytokine secretion. Cells were stained with antibodies for CD11c and other markers of dendritic cell subsets to divide DCs (CD19− CD90− NK1.1− CD11c+) into “plasmacytoid” DCs (CD11b− B220+), “conventional” DCs (CD11b+ B220− CD8−), and CD8α+ “lymphoid resident” DCs (CD11b− B220− CD8+). The cells were also stained to reveal production of the proinflammatory cytokines IL-6 and IL-12. Though VACV is not as potent a stimulus as ECTV (not shown), more IL-6 and IL-12 were detected in VACV-stimulated cells than in unstimulated cells. TLR2−/− DCs expressed lower levels of IL-6 (Fig. 2A) and IL-12 (Fig. 2B) than did WT DCs. Although it has been suggested that many reported TLR2 ligands are artifacts of contamination (13), we observed that the cytokine production triggered by sucrose cushion-purified VACV was at least as TLR2 dependent as that produced following exposure of splenocytes to crude lysate from VACV-infected cells (not shown).
FIG 2.
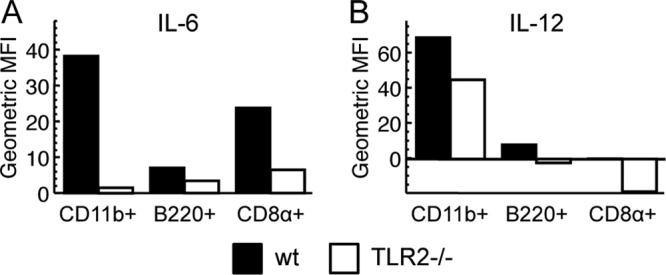
TLR2 contributes to in vitro activation of dendritic cells by VACV. Splenocytes pooled from three naive C57BL/6 (black) or TLR2−/− (white) mice were treated with brefeldin A to block secretion, stimulated for 6 h with VACV at an MOI of 10, and stained to measure intracellular IL-6 (A) or intracellular IL-12 (B) production by dendritic cell (CD19− NK1.1− CD90− CD11c+) subsets. The three subsets are “conventional” CD11b+ DCs (CD11c+ CD11b+ B220− CD8α−), “plasmacytoid” B220+ DCs (CD11c+ CD11b− B220+), and “lymphoid-resident” CD8α+ DCs (CD11c+ CD11b− B220− CD8α+). Values shown are geometric mean fluorescent intensity (MFI), normalized to geometric MFI of unstimulated cells. Cells stimulated with lysate from uninfected cells gave results similar to those for unstimulated cells. These results are representative of three independent experiments.
Our in vitro results concur with other investigators' findings that TLR2 is important early in infection for production of proinflammatory cytokines, particularly IL-6 (25, 33). We investigated whether this deficit had practical importance in the recruitment of cells to the site of acute infection. The normal course of cell recruitment in the intradermal VACV model involves minimal swelling until day 3; an influx of classical inflammatory monocytes which peaks at day 5; then development of lesions and entry of tissue-protective CD11b+ Ly6G+ cells, which are attracted by unknown mechanisms and outnumber inflammatory monocytes by day 7; and a large number of lymphocytes (especially CD8+ T cells), along with continued influx of Ly6G+ cells, on days 9 to 11 (38). We used flow cytometry to characterize cells in the infected ears as infiltrating myeloid cells (CD45+ FSCHI CD11b+ CD19− CD90− NK1.1−) and then as dendritic cells (CD11c+), inflammatory monocytes (CD11c− Ly6CHI Ly6G−), or tissue-protective Ly6G+ cells (CD11c− Ly6CMED Ly6G+) (Fig. 3A). For each of these subsets, WT and TLR2−/− mice showed similar adherence to the predicted kinetics of cell recruitment (Fig. 3B to D). These data indicate that any role for TLR2 in stimulating the secretion of chemoattractants is not essential for attracting the cells most important for controlling VACV infection.
FIG 3.

Lack of TLR2 does not significantly affect recruitment of innate immune cells to the site of intradermal vaccinia virus infection. C57BL/6 (black) or TLR2−/− (white) mice were infected intradermally with VACV in each ear pinna. On days 2, 5, 7, and 9 postinfection, mice were sacrificed and cells from ear pinnae were stained for flow cytometry. Myeloid cell subsets were identified by CD45 positivity, morphology, and the gating strategy depicted in panel A. For each mouse, the number of CD11c+ cells (B), CD11b+ Ly6C+G− cells (C), and CD11b+ Ly6C+G+ cells (D) was calculated as a percentage of all CD45+ FSCHI cells. These results are representative of two independent experiments with 2 to 3 samples in each group.
One of the most obvious reported deficiencies in TLR2−/− mice challenged i.v. with VACV is their impaired secretion of IL-6 (33) or production of type I interferons (17). Therefore, to expand our investigations, we measured induction of transcripts of type I interferons, the interferon-inducible gene MX-1, or IL-6, on day 1 or day 7 after infection with VACV in the ear of TLR2−/− mice versus wild-type mice by quantitative PCR. We found that induction of IL-6 and MX-1 transcripts was not reduced in TLR2−/− mice versus those in WT mice on either day 1 or day 7 postinfection. Similarly, induction of type I interferons in WT or TLR2−/− mice was not reduced (and may have been slightly enhanced) on day 1 postinfection and was in a similar range on day 7 postinfection. To expand our results, we examined the induction of transcripts in mice lacking MyD88 or in LysM:wt/cre MyD88:flox/flox mice, which have the MyD88 gene deleted specifically in myeloid cells, including those that infiltrate the site of infection. Surprisingly, we found little difference in any of the transcripts examined in either knockout strain (Fig. 4), indicating a MyD88-independent redundant pathway in the induction of these genes following i.d. VACV infection. Furthermore, IL-6 was not important for control of VACV following i.d. infection as virus titers and recruitment of myeloid cells were similar in IL-6 knockout and wild-type mice (not shown).
FIG 4.
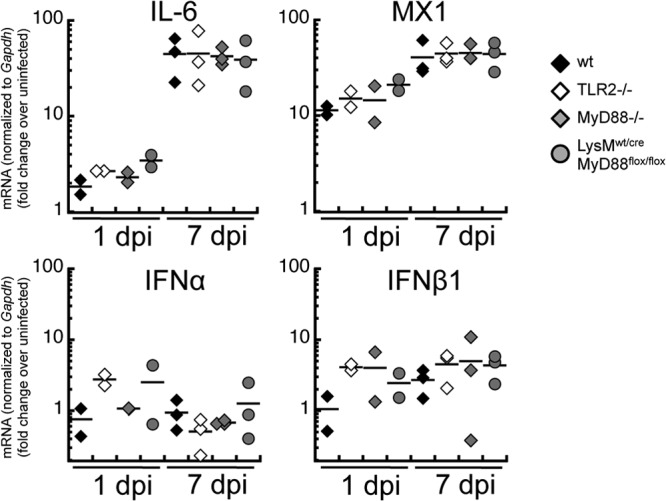
MyD88-independent induction of inflammatory genes at the site of infection. Female mice of WT, TLR2−/−, MyD88−/−, and LysMwt/cre MyD88flox/flox strains were infected in each ear pinna with 104 PFU of band-purified VACV strain WR. At 24 h or 7 days postinfection, the right ear was cut into strips and frozen at −80°C in RNAlater. Total RNA was extracted and reverse transcribed into cDNA which was used as the template for quantitative real-time PCR, with primers specific for IL-6 (Il6), MX-1 (Mx1), IFN-α (all IFN-α transcripts except Ifna4), IFN-β1 (Ifnb1), or the housekeeping gene Gapdh. Sample sizes were n = 4 for naive ears, n = 2 for day 1, and n = 3 for day 7. Results shown are typical of two independent experiments.
TLR2 may play a limited role in control of virus dissemination following systemic VACV infection.
To attempt to reconcile our above findings with the published work that describes a role for TLR2 in control of VACV infection, we systemically infected TLR2−/− mice and WT mice with much larger inocula (up to 3 logs higher) than those used intradermally and monitored mice for 32 days for morbidity or mortality. During systemic VACV infection (i.p. or i.v.), the initial stage of immune mobilization is spurred by viremia and viral replication in internal organs and may be followed by skin lesions visible on the tails of mice (34, 41, 42)—akin to the progression of ECTV infection or smallpox but markedly less virulent (43, 44). In our system, i.v. infection with 107 PFU is followed by weight loss that reaches a peak on day 5 postinfection. By day 5, the tails of infected mice become swollen and appear limp, and individual pocks are not evident until day 6, when they become distinguishable as discolored or ulcerated hairless spots. By day 9, the swelling recedes, and pocks begin to heal and appear as pale patches.
Using 12 mice in each group, we saw that VACV-infected TLR2−/− and WT mice displayed similar kinetics of weight loss and recovery (Fig. 5A). All mice survived infection, but TLR2 knockouts had a statistically greater number of pocks on the tails (Fig. 5B) and were more prone to losing skin altogether near the tail tip (Fig. 5C). MyD88−/− mice were also more susceptible to i.v. infection, suffering severe weight loss and mortality (Fig. 5D), which was notably increased in comparison to both WT and TLR2−/− mice.
FIG 5.

TLR2 contributes to defense against disseminated vaccinia virus infection. (A to C) C57BL/6 (black) or TLR2−/− (white) mice were infected with 107 PFU of VACV i.v. and monitored for weight loss (A) and pathogenesis (n = 12). Pocks were observed only on the tail and were counted daily (B), along with the observation that some mice had significant loss of necrotic tissue at the tail tip (C). (D) C57BL/6 (black) or MyD88−/− (gray) mice were infected with 107 PFU VACV i.v. and monitored for weight loss and pathogenesis (n = 4). Daggers indicate MyD88−/− mouse mortality. The one MyD88−/− mouse alive at 10 days p.i. continued to survive until sacrificed (21 days p.i.). (E) On the specified days after infection with 107 PFU of VACV i.v., mice were sacrificed, ovaries were homogenized, and serial dilutions were applied to cell monolayers to determine VACV titer. (F) Mice were infected with the specified inoculum of VACV through the specified route. At 3 days postinfection, mice were sacrificed, ovaries and livers were homogenized, and the VACV titer was measured as stated above. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
VACV is normally cleared from the liver and other internal organs after about a week (45, 46) but is particularly tropic for ovarian follicles and can be detected there after it has been cleared from all other organs (47). We saw that TLR2−/− and WT mice infected with VACV i.v. had similar titers during the peak of viral replication, and there was no evidence that TLR2−/− mice were slower to clear the infection (Fig. 5E). In addition to i.v. infected mice, we looked at VACV replication at an early/peak time point (72 h) in mice infected with 107 PFU or 106 PFU i.p. In none of these infection models did TLR2−/− mice have more viral replication in either the liver or the ovaries (Fig. 5F). Other studies have examined VACV levels in the liver following infection (17, 33). The liver is one of the major sites of replication for ECTV (48), but following infection with VACV, titers in the liver typically do not approach those observed in the ovaries. As expected, we found that titers in the liver were 3 to 5 logs lower than those found in the ovaries, and there was no discernible difference between titers found in WT and TLR2−/− mice. Our data suggest that TLR2−/− mice are capable of controlling VACV infection in the internal organs, but the observation of increased lesions on TLR2−/− infected mice supports a model in which TLR2 signaling, probably in phagocytes, limits the dissemination of the virus.
TLR2-independent MyD88 signaling is important for adaptive anti-VACV immunity.
Although it appears that TLR2 is superfluous for viral control and an innate immune response, we examined whether it may play a role in establishing the adaptive immunity that makes VACV such a valuable tool for immunization. CD8 T cell responses have been described to be dependent upon cell-intrinsic MyD88 following intranasal infection (28), a model in which the T cell response is required for protection. Following i.d. infection, T cells alone are not required for protection or to restrict VACV spread from the ear (38, 49), but we examined the role of TLR2 and MyD88 in regulation of CD4 responses (which do have a role following i.v. infection [42]) following i.d. infection. We used a library of peptides containing class II-restricted epitopes that are targeted by a significant number of T cells after VACV infection of mice (35). The number of functional splenic T cells targeting each peptide, 10 days postinfection, was assayed using gamma interferon ELISpot. Similar numbers of T cells targeting each of 17 class II-restricted epitopes detected at levels above background (Fig. 6) were found in in TLR2−/− mice and WT mice. However, in MyD88−/− mice, all 17 epitopes stimulated fewer T cells than in WT mice, with a significant difference for 7 of the 17.
FIG 6.
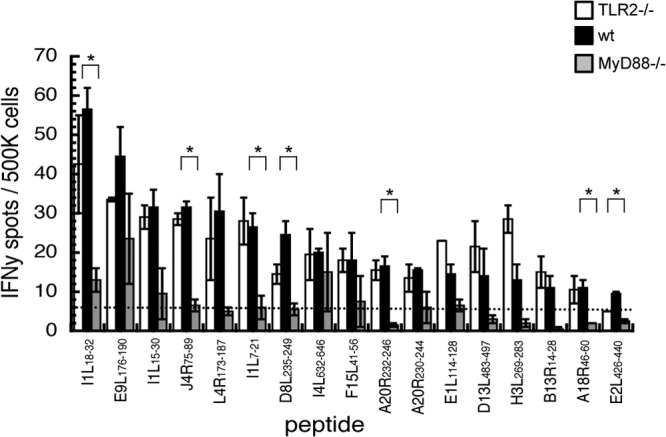
TLR2-independent MyD88 signaling is required for an optimal anti-VACV CD4 T cell response. C57BL/6 (black), TLR2−/− (white), or MyD88−/− (gray) mice were infected intradermally with VACV in each ear pinna. On day 10 postinfection, mice were sacrificed and splenocytes were stimulated by naive splenocytes that had been pulsed with a library of MHC class II-restricted poxviral peptides. Each bar represents pooled T cells from three spleens, with error bars showing the standard error of the mean for two experimental replicates. *, P < 0.05 in unpaired t test of C57BL/6 and MyD88−/− samples.
CD4 T cell-dependent production of antibody is required for effective protection following i.v. challenge with VACV (42), so we examined whether the lack of MyD88 (or TLR2) that had an effect upon CD4 responses impacted the production of neutralizing antibody. We isolated serum either during acute i.d. infection (10 days postinfection) or after mice had successfully resolved the infection (32 days postinfection). To assay for the presence of neutralizing antibodies, we made serial dilutions of cell-free serum and then mixed serum with a strain of VACV engineered to express GFP upon infection of a target cell. Virus-serum mixtures were incubated with the murine WT-3 cell line, and the percentage of GFP+ cells was determined by flow cytometry. At both postinoculation time points (Fig. 7A and B), sera from WT and TLR2−/− mice were equally capable of neutralizing VACV infectivity. However, MyD88−/− sera had significantly lower levels of neutralizing antibody both during acute infection (Fig. 7C) and during the early stages of humoral memory (Fig. 7D). Therefore, our conclusions on the significance of MyD88 signaling for adaptive immunity are limited to B cells and CD4+ T cells. These data indicate that TLR2 alone does not play a role in establishing adaptive immunity to VACV, but at least one receptor upstream of MyD88 does play a major role.
FIG 7.

TLR2-independent MyD88 signaling is required for optimal humoral immunity following VACV infection. (A and B) C57BL/6 (black) or TLR2−/− (white) mice were infected intradermally with VACV in each ear pinna. Mice were sacrificed on day 10 (A) or day 32 (B) postinfection. Serial dilutions of serum were incubated with VACV-NP-S-EGFP to test their ability to neutralize viral infectivity. Serum-virus mixtures were applied to WT-3 cells for 6 h at an MOI of 10:1, and the percentage of cells that became enhanced GFP positive (EGFP+) was quantified using flow cytometry. (C and D) C57BL/6 (black) or MyD88−/− (gray) mice were infected intradermally with VACV in each ear pinna. Mice were sacrificed on day 10 (C) or day 32 (D) postinfection, and neutralizing antibodies in serum were measured as stated above. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
TLR2-independent MyD88 signaling is needed for optimal control of intradermal VACV infection.
If redundancy in the innate immune system means that TLR2 alone is not required, there may still be a requirement for TLR2 in combination with other MyD88-dependent pathways. Synergy between two or more TLRs has been found to be important in other viral infections (10, 50). Therefore, we looked for evidence that other MyD88-dependent receptors are involved in controlling VACV replication and pathogenesis in this natural route of infection.
When observing the pathogenesis of intradermal VACV, we saw that the kinetics of ear swelling were the same in MyD88−/− and WT mice (Fig. 8A), suggesting that there was no deficit in the immediate inflammatory mediators responsible for vascular permeability. Ear lesions spread at similar rates in the two mouse strains, but tissue loss proceeded at a slightly higher rate in MyD88-deficient mice, suggesting more tissue necrosis (Fig. 8B). When virus titers in the ear pinnae were measured on day 3, 5, or 7 postinfection, MyD88−/− mice showed significantly more viral replication (Fig. 8C), a particularly profound finding as virus replication occurs similarly despite large differences in the inoculating dose (22). This difference was not detected on day 10, due partially to MyD88−/− mice losing more of the tissue in which the virus replicates. The enhanced viral replication in MyD88 knockouts was not observed in LysM:wt/cre MyD88:flox/flox mice, which have the MyD88 gene deleted specifically in myeloid cells (Fig. 8D). Finally, we looked for viral replication in the ovaries on day 5 postinfection and saw no VACV in either MyD88−/− or wild-type mice (data not shown), further suggesting that there is no impairment in the function of the myeloid phagocytes that block viral spread from the site of infection. However, these data show a clear role for TLR2-independent MyD88 signaling in the timely control of VACV replication at the site of active infection, as well as in the mobilization of lymphocytes to protect against future poxvirus challenges.
FIG 8.

TLR2-independent MyD88 signaling contributes to clearance of VACV during intradermal infection. C57BL/6 (black) or MyD88−/− (gray) mice were infected intradermally with VACV in each ear pinna. (A) Ear swelling was monitored with a thickness gauge until tissue loss began (n = 10 ears). (B) The size of lesions (solid lines) and subsequent loss of tissue (dotted lines) were monitored for 3 weeks (n = 10 ears). (C) On the specified day postinfection, mice were sacrificed, ears were homogenized, and serial dilutions were applied to cell monolayers for VACV titer determination. (D) C57BL/6 (black) or LysMwt/cre MyD88flox/flox (shaded) mice were infected with VACV as stated above, and at day 7, mice were sacrificed and ears were homogenized for titer determination. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
DISCUSSION
A lack of TLR2 has been shown to confer increased susceptibility to a wide range of infections (51). However, the majority of these publications examine bacterial infections. Among the few viruses with established TLR2 ligands are measles virus (52), hepatitis C virus (HCV) (53), Epstein-Barr virus (54), and multiple herpesviruses bearing glycoproteins gB and gL/gH (55). There is some evidence that TLR2 polymorphisms influence the success of measles vaccination (56), and HCV patients with a deletion in the 5′ untranslated region (UTR) of TLR2 have more risk of hepatocellular carcinoma (57). Epstein-Barr virus (EBV), HCV, and measles virus do not infect mice, but following herpes simplex virus 1 (HSV-1) infection, TLR2−/− mice appear to have a deficit in proinflammatory cytokine secretion, making them more susceptible to periocular herpetic lesions but more resistant to corneal keratitis after eye infection (58), as well as more resistant to encephalitis after intracranial infection (59). With another herpesvirus, mouse cytomegalovirus (MCMV), one study observed enhanced susceptibility in TLR2 knockouts (60), in contrast to other findings that MyD88 knockouts are susceptible but TLR2 knockouts are not (61, 62). These are among the few in vivo studies showing viral susceptibility in TLR2 knockout mice.
Although VACV is known to be recognized by TLR2 in vitro, the evidence gleaned from in vivo experiments is inconsistent. In addition to repeated observations that proinflammatory cytokine production is blunted in TLR2 knockouts, Yang and colleagues have seen enhanced viral replication in ovaries of TLR2−/− mice at day 2 or 3 post-i.p. infection (25, 63), while Nolan and colleagues saw that viral replication was similar at early time points post-i.v. infection but persisted for longer in TLR2−/− mice (33). In this study, we indirectly confirm that TLR2−/− mice are marginally more susceptible to VACV dissemination, by observing enhanced skin pathogenesis on the tail, but we did not find any difference in viral titers at either early or late time points. Differences between our results and those of others may be the result of many variations in the experimental protocol, including the type of cell culture cells used to grow virus, the method of purifying virus, and the strain of mice used to measure virus pathogenesis and titers. Also, the Nolan laboratory found delayed VACV clearance by measuring viral gene expression with a luciferase reporter assay, while we measured infectious viral particles with a plaque assay. In addition, Barbalat et al. proposed that TLR2-mediated recognition of VACV by myeloid cells induced significant production of type I IFN and that this controlled virus replication in vivo (17). However, we found little induction of type I IFN following i.d. infection with VACV, a result that echoes a previous observation following i.v. infection (64), and any induction observed was independent of both TLR2 and MyD88.
We have also conducted a novel investigation of the significance of TLR2 and MyD88 via the more physiologically relevant intradermal route of infection with VACV. In this system, unlike artificially induced systemic infection, TLR2 proved to be completely redundant for restricting the pathogenesis caused by VACV. Typically, we used a much higher dose (106 PFU) when infecting i.v. than when infecting i.d. (104 PFU). However, even when we infected i.d. with 106 PFU, a dose that is known to allow systemic spread of VACV (22), we did not observe a difference in pathogenesis between WT and TLR2−/− mice (not shown). Therefore, it is likely that the route of infection, rather than the size of the viral inoculum, accounts for the observed differences. When investigated further for deficiencies of the innate or adaptive immune system, TLR2 knockouts were also indistinguishable from WT mice in recruitment of myeloid cells to the site of infection, proliferation of VACV-specific T cells, and production of VACV-neutralizing serum antibodies. Finally, with respect to control of the virus, at no point from day 1 to day 22 postinfection was there a significant difference between WT and TLR2−/− mice in their viral titers in the ear, and there was no sign that TLR2 signaling was needed to block systemic spread of the virus. Therefore, we believe that many of the in vitro observations of a role for TLR2 in recognition of VACV may stem from contamination in virus preparations. However, this does not rule out a minor redundant role for TLR2 in recognition of viral components or danger signals induced by virus-infected cells during an in vivo infection, a process that can also be accomplished by other TLR or inflammasome components.
MyD88−/− mice also do not permit systemic spread of the virus in our infectious model. However, they have a marked impairment in the production of CD4+ T cells and antibodies, and they are less able to control viral replication in infected tissue. The 1.5-log increase in virus found in the ear on day 7 in mice lacking MyD88 is important, as even inoculation with doses that differ by 3 to 4 logs produces similar levels of virus in the ear at this time point (22). Our discovery that TLR2-independent MyD88 signaling is important for anti-VACV immunity is compatible with an earlier study using a variety of CD8 T cell assays, which found that T cell expansion is impaired in MyD88−/− mice post-i.p. infection, but we did not see this phenotype in TLR2 or IL-1R knockouts (28). Here, we found an effect on MHC class II-restricted responses, indicating a MyD88-dependent process involved in priming CD4 T cells following intradermal infection. Therefore, our results do have a common theme in that both the CD4 and CD8 T cell responses are MyD88 dependent and TLR2 and IL-1R independent. This may indicate that the crucial MyD88 signaling is downstream of one or more members of the IL-1R family (IL-1R, IL-18R, and ST2). Exogenous IL-18 helps mice resolve VACV infection (65, 66), although the importance of IL-18 and IL-1β in this infection model is blunted by the virus's repertoire of inflammasome inhibitors and soluble IL-18 and IL-1β binding proteins (67). However, since the B16R protein is specific for IL-1β (68), IL-1R may be more important in its role as receptor for the alarmin IL-1α. Indeed, overexpression of IL-1α helps to resolve intradermal VACV infection (69). Recent findings also suggest a role for ST2, which recognizes the alarmin IL-33 rather than an inflammasome product (70). Alternately, there may be redundancy in MyD88 signaling, with some cell types responding to viral ligands or alarmins via TLR2, and some cell types responding to inflammasome products or alarmins via IL-1R family members. We have demonstrated that production of MyD88 by cells expressing LysM, which include inflammatory monocytes and “tissue-protective” myeloid cells recruited to the site of infection, is not important for control of virus replication, prevention of systemic spread of VACV, induction of the innate response, or a tissue protective response. Indeed, MyD88 deficiency did not allow systemic spread of the virus following i.d. infection, indicating that the recruitment and activation of the mononuclear phagocytes that we have previously shown to restrict VACV spread from the skin (38) are MyD88 independent. The identity of the innate sensor that recruits and activates these cells remains unknown.
Our data indicate that an increase in VACV virulence requires multiple host MyD88-dependent pathways to be abrogated, an observation that is common in the immune response to many viruses. For the dsDNA herpesviruses HSV-1, MCMV, and murid herpesvirus 68 (MHV-68), more than one TLR contributes to control of the infection (10, 50, 71). For example, during i.p. MCMV infection, no effect of TLR7 single knockout is detectable, but TLR7/9 double knockouts are more susceptible than TLR9 single knockouts (72). Similar results were seen with intranasal HSV-1, with TLR2 knockouts impaired in cytokine production but not impaired in survival or viral clearance, while TLR2/9 double knockouts had worse outcomes than did single knockouts (10). In influenza virus infection, TLR7 alone seems to be required for successful clearance of a primary infection (73), but inflammasome signaling through IL-1R is important for protective immunity (74). MyD88−/− mice are far more susceptible to lymphocytic choriomeningitis virus (LCMV) infection, while TLR7/9 double knockouts and IL-1R knockouts show only partial susceptibility (75, 76). The oncolytic effects of vesicular stomatitis virus (VSV), abrogated similarly by both IFN-α/βR and MyD88 knockout, are not abrogated by removing either IL-1R or any of the individual TLRs likely to recognize VSV (77). And, even in a case where the protective effect of MyD88 is independent of IFN-α/β (rabies virus), MyD88 signaling requires a combination of TLR7 and at least one other receptor (78).
Our results are compatible with an immune system in which, for each pathogen, one or more pattern recognition receptors respond with an immediate warning signal, which primes the immune system for a robust response once more pronounced indicators of infection develop, such as viral replication and necrosis. TLR2 likely assists in early immune mobilization by upregulating proinflammatory cytokines and IFN-α/β, as well as pro-IL-1β and pro-IL-18 that can then be cleaved and secreted by inflammasomes. However, during a physiological VACV challenge there would be sufficient levels of these factors, even in the absence of TLR2, to combat the infection equally well as long as inflammasome activation still occurs. In this scenario, TLR2 is necessary only when the body is challenged by an artificially high inoculum and may be more useful as part of a redundant suite of PRRs that are continually activated by the various constituents of the microbiome to maintain a baseline level of innate immune readiness. ECTV, another orthopoxvirus, is one example of a virus for which the loss of a single TLR (TLR9) has a major effect on susceptibility even with a small inoculum (26, 79), but there are few other viruses in that category and VACV does not appear to be one of them.
ACKNOWLEDGMENTS
We thank Irene Reider and Jennifer Mellinger for expert technical support; Nate Sheaffer in the Hershey Medical Center flow cytometry core facility; Lauren Weiler for maintaining the MyD88 knockout mouse colony; and Karen Briar, Robin Goshorn, Jeanette Mohl, Tim Cooper, and Tiffany Whitcomb for essential animal handling and veterinary assistance.
This work was supported by NIH grants U19 AI083008 to L.J.S., AI 056094 and AI070537 to C.C.N., and training grant 5 T32 CA60395-15 (principal investigator Harriet Isom).
Footnotes
Published ahead of print 8 January 2014
REFERENCES
- 1.Kang JY, Lee JO. 2011. Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 80:917–941. 10.1146/annurev-biochem-052909-141507 [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 3.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285:736–739. 10.1126/science.285.5428.736 [DOI] [PubMed] [Google Scholar]
- 4.Calich VL, Pina A, Felonato M, Bernardino S, Costa TA, Loures FV. 2008. Toll-like receptors and fungal infections: the role of TLR2, TLR4 and MyD88 in paracoccidioidomycosis. FEMS Immunol. Med. Microbiol. 53:1–7. 10.1111/j.1574-695X.2008.00378.x [DOI] [PubMed] [Google Scholar]
- 5.Carrera-Silva EA, Guinazu N, Pellegrini A, Cano RC, Arocena A, Aoki MP, Gea S. 2010. Importance of TLR2 on hepatic immune and non-immune cells to attenuate the strong inflammatory liver response during Trypanosoma cruzi acute infection. PLoS Negl. Trop. Dis. 4:e863. 10.1371/journal.pntd.0000863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M, Gao Y, Du X, Zhang D, Ji M, Wu G. 2011. Toll-like receptor (TLR) 2 and TLR4 deficiencies exert differential in vivo effects against Schistosoma japonicum. Parasite Immunol. 33:199–209. 10.1111/j.1365-3024.2010.01265.x [DOI] [PubMed] [Google Scholar]
- 7.Love W, Dobbs N, Tabor L, Simecka JW. 2010. Toll-like receptor 2 (TLR2) plays a major role in innate resistance in the lung against murine Mycoplasma. PLoS One 5:e10739. 10.1371/journal.pone.0010739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mele T, Madrenas J. 2010. TLR2 signalling: at the crossroads of commensalism, invasive infections and toxic shock syndrome by Staphylococcus aureus. Int. J. Biochem. Cell Biol. 42:1066–1071. 10.1016/j.biocel.2010.03.021 [DOI] [PubMed] [Google Scholar]
- 9.Zamboni DS, Campos MA, Torrecilhas AC, Kiss K, Samuel JE, Golenbock DT, Lauw FN, Roy CR, Almeida IC, Gazzinelli RT. 2004. Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J. Biol. Chem. 279:54405–54415. 10.1074/jbc.M410340200 [DOI] [PubMed] [Google Scholar]
- 10.Lima GK, Zolini GP, Mansur DS, Freire Lima BH, Wischhoff U, Astigarraga RG, Dias MF, das Gracas Almeida Silva M, Bela SR, do Valle Antonelli LR, Arantes RM, Gazzinelli RT, Bafica A, Kroon EG, Campos MA. 2010. Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am. J. Pathol. 177:2433–2445. 10.2353/ajpath.2010.100121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuevas CD, Lavanya M, Wang E, Ross SR. 2011. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 85:11058–11068. 10.1128/JVI.05304-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piccinini AM, Midwood KS. 2010. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010:672395. 10.1155/2010/672395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erridge C. 2010. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 87:989–999. 10.1189/jlb.1209775 [DOI] [PubMed] [Google Scholar]
- 14.Zähringer U, Lindner B, Inamura S, Heine H, Alexander C. 2008. TLR2—promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213:205–224. 10.1016/j.imbio.2008.02.005 [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–295. 10.1126/science.1183021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietrich N, Lienenklaus S, Weiss S, Gekara NO. 2010. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One 5:e10250. 10.1371/journal.pone.0010250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbalat R, Lau L, Locksley RM, Barton GM. 2009. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10:1200–1207. 10.1038/ni.1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aubry C, Corr SC, Wienerroither S, Goulard C, Jones R, Jamieson AM, Decker T, O'Neill LA, Dussurget O, Cossart P. 2012. Both TLR2 and TRIF contribute to interferon-beta production during Listeria infection. PLoS One 7:e33299. 10.1371/journal.pone.0033299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy RB, Ovsyannikova IG, Jacobson RM, Poland GA. 2009. The immunology of smallpox vaccines. Curr. Opin. Immunol. 21:314–320. 10.1016/j.coi.2009.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lousberg EL, Diener KR, Brown MP, Hayball JD. 2011. Innate immune recognition of poxviral vaccine vectors. Expert Rev. Vaccines 10:1435–1449. 10.1586/erv.11.121 [DOI] [PubMed] [Google Scholar]
- 21.Senkevich TG, Bugert JJ, Sisler JR, Koonin EV, Moss B. 1996. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813–816. 10.1126/science.273.5276.813 [DOI] [PubMed] [Google Scholar]
- 22.Tscharke DC, Smith GL. 1999. A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J. Gen. Virol. 80:2751–2755 [DOI] [PubMed] [Google Scholar]
- 23.Tscharke DC, Reading PC, Smith GL. 2002. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J. Gen. Virol. 83:1977–1986 [DOI] [PubMed] [Google Scholar]
- 24.Delaloye J, Roger T, Steiner-Tardivel QG, Le Roy D, Knaup Reymond M, Akira S, Petrilli V, Gomez CE, Perdiguero B, Tschopp J, Pantaleo G, Esteban M, Calandra T. 2009. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 5:e1000480. 10.1371/journal.ppat.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Zhu J, Martinez J, Huang X, Yang Y. 2007. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 109:619–625. 10.1182/blood-2006-06-027136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutherland DB, Ranasinghe C, Regner M, Phipps S, Matthaei KI, Day SL, Ramshaw IA. 2011. Evaluating vaccinia virus cytokine co-expression in TLR GKO mice. Immunol. Cell Biol. 89:706–715. 10.1038/icb.2010.157 [DOI] [PubMed] [Google Scholar]
- 27.Quigley M, Martinez J, Huang X, Yang Y. 2009. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood 113:2256–2264. 10.1182/blood-2008-03-148809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, De Trez C, Flynn R, Ware CF, Croft M, Salek-Ardakani S. 2009. The adaptor molecule MyD88 directly promotes CD8 T cell responses to vaccinia virus. J. Immunol. 182:6278–6286. 10.4049/jimmunol.0803682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collazo CM, Sher A, Meierovics AI, Elkins KL. 2006. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8:779–790. 10.1016/j.micinf.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 30.Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. 2006. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74:3657–3662. 10.1128/IAI.02030-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller LS, O'Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. 2006. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24:79–91. 10.1016/j.immuni.2005.11.011 [DOI] [PubMed] [Google Scholar]
- 32.Miller LS. 2008. Toll-like receptors in skin. Adv. Dermatol. 24:71–87. 10.1016/j.yadr.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Gorman WE, Sampath P, Simonds EF, Sikorski R, O'Malley M, Krutzik PO, Chen H, Panchanathan V, Chaudhri G, Karupiah G, Lewis DB, Thorne SH, Nolan GP. 2010. Alternate mechanisms of initial pattern recognition drive differential immune responses to related poxviruses. Cell Host Microbe 8:174–185. 10.1016/j.chom.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antón LC, Schubert U, Bacik I, Princiotta MF, Wearsch PA, Gibbs J, Day PM, Realini C, Rechsteiner MC, Bennink JR, Yewdell JW. 1999. Intracellular localization of proteasomal degradation of a viral antigen. J. Cell Biol. 146:113–124. 10.1083/jcb.146.1.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moutaftsi M, Bui HH, Peters B, Sidney J, Salek-Ardakani S, Oseroff C, Pasquetto V, Crotty S, Croft M, Lefkowitz EJ, Grey H, Sette A. 2007. Vaccinia virus-specific CD4+ T cell responses target a set of antigens largely distinct from those targeted by CD8+ T cell responses. J. Immunol. 178:6814–6820 [DOI] [PubMed] [Google Scholar]
- 36.Xu RH, Rubio D, Roscoe F, Krouse TE, Truckenmiller ME, Norbury CC, Hudson PN, Damon IK, Alcami A, Sigal LJ. 2012. Antibody inhibition of a viral type 1 interferon decoy receptor cures a viral disease by restoring interferon signaling in the liver. PLoS Pathog. 8:e1002475. 10.1371/journal.ppat.1002475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubio D, Xu RH, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, Balachandran S, Alcami A, Norbury CC, Sigal LJ. 2013. Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell Host Microbe 13:701–710. 10.1016/j.chom.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer MA, Davies ML, Reider IE, Heipertz EL, Epler MR, Sei JJ, Ingersoll MA, Rooijen NV, Randolph GJ, Norbury CC. 2011. CD11b(+), Ly6G(+) cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog. 7:e1002374. 10.1371/journal.ppat.1002374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iborra S, Izquierdo HM, Martinez-Lopez M, Blanco-Menendez N, Reis e Sousa C, Sancho D. 2012. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J. Clin. Invest. 122:1628–1643. 10.1172/JCI60660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian T, Dubin K, Jin Q, Qureshi A, King SL, Liu L, Jiang X, Murphy GF, Kupper TS, Fuhlbrigge RC. 2012. Disruption of TNF-alpha/TNFR1 function in resident skin cells impairs host immune response against cutaneous vaccinia virus infection. J. Invest. Dermatol. 132:1425–1434. 10.1038/jid.2011.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutt LM. 1975. The immune response to infection with vaccinia virus in mice. I. Infection and the production of antibody neutralizing cell-associated and cell-free virus. J. Hyg. (Lond.) 74:301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu R, Johnson AJ, Liggitt D, Bevan MJ. 2004. Cellular and humoral immunity against vaccinia virus infection of mice. J. Immunol. 172:6265–6271 [DOI] [PubMed] [Google Scholar]
- 43.Buller RML, Palumbo GJ. 1991. Poxvirus pathogenesis. Microbiol. Rev. 55:80–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman JL, Nichols DK, Martinez MJ, Raymond JW. 2010. Animal models of orthopoxvirus infection. Vet. Pathol. 47:852–870. 10.1177/0300985810378649 [DOI] [PubMed] [Google Scholar]
- 45.Reading PC, Moore JB, Smith GL. 2003. Steroid hormone synthesis by vaccinia virus suppresses the inflammatory response to infection. J. Exp. Med. 197:1269–1278. 10.1084/jem.20022201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Remakus S, Sigal LJ. 2011. Gamma interferon and perforin control the strength, but not the hierarchy, of immunodominance of an antiviral CD8+ T cell response. J. Virol. 85:12578–12584. 10.1128/JVI.05334-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Adams YF, Croft M. 2011. Preferential replication of vaccinia virus in the ovaries is independent of immune regulation through IL-10 and TGF-beta. Viral Immunol. 24:387–396. 10.1089/vim.2011.0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esteban DJ, Buller RM. 2005. Ectromelia virus: the causative agent of mousepox. J. Gen. Virol. 86:2645–2659. 10.1099/vir.0.81090-0 [DOI] [PubMed] [Google Scholar]
- 49.Hickman HD, Reynoso GV, Ngudiankama BF, Rubin EJ, Magadan JG, Cush SS, Gibbs J, Molon B, Bronte V, Bennink JR, Yewdell JW. 2013. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe 13:155–168. 10.1016/j.chom.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crane MJ, Gaddi PJ, Salazar-Mather TP. 2012. UNC93B1 mediates innate inflammation and antiviral defense in the liver during acute murine cytomegalovirus infection. PLoS One 7:e39161. 10.1371/journal.pone.0039161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliveira-Nascimento L, Massari P, Wetzler LM. 2012. The role of TLR2 in infection and immunity. Front. Immunol. 3:79. 10.3389/fimmu.2012.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S. 2002. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76:8729–8736. 10.1128/JVI.76.17.8729-8736.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. 2004. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127:1513–1524. 10.1053/j.gastro.2004.08.067 [DOI] [PubMed] [Google Scholar]
- 54.Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. 2009. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182:851–859 [DOI] [PubMed] [Google Scholar]
- 55.Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G. 2012. Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-κB. J. Virol. 86:6555–6562. 10.1128/JVI.00295-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ovsyannikova IG, Haralambieva IH, Vierkant RA, Pankratz VS, Jacobson RM, Poland GA. 2011. The role of polymorphisms in Toll-like receptors and their associated intracellular signaling genes in measles vaccine immunity. Hum. Genet. 130:547–561. 10.1007/s00439-011-0977-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nischalke HD, Coenen M, Berger C, Aldenhoff K, Muller T, Berg T, Kramer B, Korner C, Odenthal M, Schulze F, Grunhage F, Nattermann J, Sauerbruch T, Spengler U. 2012. The toll-like receptor 2 (TLR2) −196 to −174 del/ins polymorphism affects viral loads and susceptibility to hepatocellular carcinoma in chronic hepatitis C. Int. J. Cancer 130:1470–1475. 10.1002/ijc.26143 [DOI] [PubMed] [Google Scholar]
- 58.Sarangi PP, Kim B, Kurt-Jones E, Rouse BT. 2007. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: role of the toll pathway in early inflammatory events in stromal keratitis. J. Virol. 81:11128–11138. 10.1128/JVI.01008-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA. 2012. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 86:2273–2281. 10.1128/JVI.06010-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. 2006. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 80:4286–4291. 10.1128/JVI.80.9.4286-4291.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 175:6723–6732 [DOI] [PubMed] [Google Scholar]
- 62.Hokeness-Antonelli KL, Crane MJ, Dragoi AM, Chu WM, Salazar-Mather TP. 2007. IFN-alphabeta-mediated inflammatory responses and antiviral defense in liver is TLR9-independent but MyD88-dependent during murine cytomegalovirus infection. J. Immunol. 179:6176–6183 [DOI] [PubMed] [Google Scholar]
- 63.Martinez J, Huang X, Yang Y. 2010. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog. 6:e1000811. 10.1371/journal.ppat.1000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waibler Z, Anzaghe M, Frenz T, Schwantes A, Pohlmann C, Ludwig H, Palomo-Otero M, Alcami A, Sutter G, Kalinke U. 2009. Vaccinia virus-mediated inhibition of type I interferon responses is a multifactorial process involving the soluble type I interferon receptor B18 and intracellular components. J. Virol. 83:1563–1571. 10.1128/JVI.01617-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gherardi MM, Ramirez JC, Esteban M. 2003. IL-12 and IL-18 act in synergy to clear vaccinia virus infection: involvement of innate and adaptive components of the immune system. J. Gen. Virol. 84:1961–1972. 10.1099/vir.0.19120-0 [DOI] [PubMed] [Google Scholar]
- 66.Tanaka-Kataoka M, Kunikata T, Takayama S, Iwaki K, Ohashi K, Ikeda M, Kurimoto M. 1999. In vivo antiviral effect of interleukin 18 in a mouse model of vaccinia virus infection. Cytokine 11:593–599. 10.1006/cyto.1998.0453 [DOI] [PubMed] [Google Scholar]
- 67.Perdiguero B, Esteban M. 2009. The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 29:581–598. 10.1089/jir.2009.0073 [DOI] [PubMed] [Google Scholar]
- 68.Alcamí A, Smith GL. 1996. A mechanism for the inhibition of fever by a virus. Proc. Natl. Acad. Sci. U. S. A. 93:11029–11034. 10.1073/pnas.93.20.11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tian T, Liu L, Freyschmidt EJ, Murphy GF, Kupper TS, Fuhlbrigge RC. 2009. Overexpression of IL-1alpha in skin differentially modulates the immune response to scarification with vaccinia virus. J. Invest. Dermatol. 129:70–78. 10.1038/jid.2008.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, Kreutzfeldt M, Hegazy AN, Schrick C, Fallon PG, Klemenz R, Nakae S, Adler H, Merkler D, Lohning M, Pinschewer DD. 2012. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 335:984–989. 10.1126/science.1215418 [DOI] [PubMed] [Google Scholar]
- 71.Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J. 2010. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS One 5:e13742. 10.1371/journal.pone.0013742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M. 2008. Cutting edge: overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J. Immunol. 180:5799–5803 [DOI] [PubMed] [Google Scholar]
- 73.Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P. 2012. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J. Gen. Virol. 93:555–559. 10.1099/vir.0.039065-0 [DOI] [PubMed] [Google Scholar]
- 74.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79–87. 10.1084/jem.20081667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, Akira S. 2008. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82:196–206. 10.1128/JVI.01640-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou S, Kurt-Jones EA, Cerny AM, Chan M, Bronson RT, Finberg RW. 2009. MyD88 intrinsically regulates CD4 T-cell responses. J. Virol. 83:1625–1634. 10.1128/JVI.01770-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, Vile R. 2011. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol. Ther. 19:150–158. 10.1038/mt.2010.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, Faber M, Dietzschold B, Hooper DC. 2011. The role of toll-like receptors in the induction of immune responses during rabies virus infection. Adv. Virus Res. 79:115–126. 10.1016/B978-0-12-387040-7.00007-X [DOI] [PubMed] [Google Scholar]
- 79.Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O'Keeffe M, Hochrein H. 2008. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J. Clin. Invest. 118:1776–1784. 10.1172/JCI33940 [DOI] [PMC free article] [PubMed] [Google Scholar]