

Background: Engagement of CD80/CD86 on dendritic cells by CD28 on T cells induces dendritic cell production of IL-6 and IDO.
Results: The NOTCH pathway modulates activation of the PI3K pathway downstream of CD80/CD86 ligation and regulates IL-6 and IDO production.
Conclusion: Cross-talk between NOTCH and PI3K pathways modulates dendritic cell production of IL-6 and IDO.
Significance: Elucidating the molecular mechanism of NOTCH-PI3K cross-talk will have broad implications in human disease.
Keywords: Dendritic Cells; Interleukin; NOTCH Pathway; PI 3-Kinase (PI3K); Signal Transduction; Indoleamine 2,3-Dioxygenase
Abstract
Dendritic cells (DC) play a critical role in modulating antigen-specific immune responses elicited by T cells via engagement of the prototypic T cell costimulatory receptor CD28 by the cognate ligands CD80/CD86, expressed on DC. Although CD28 signaling in T cell activation has been well characterized, it has only recently been shown that CD80/CD86, which have no demonstrated binding domains for signaling proteins in their cytoplasmic tails, nonetheless also transduce signals to the DC. Functionally, CD80/CD86 engagement results in DC production of the pro-inflammatory cytokine IL-6, which is necessary for full T cell activation. However, ligation of CD80/CD86 by CTLA4 also induces DC production of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO), which depletes local pools of the essential amino acid tryptophan, resulting in blockade of T cell activation. Despite the significant role of CD80/CD86 in immunological processes and the seemingly opposing roles they play by producing IL-6 and IDO upon their activation, how CD80/CD86 signal remains poorly understood. We have now found that cross-linking CD80/CD86 in human DC activates the PI3K/AKT pathway. This results in phosphorylation/inactivation of its downstream target, FOXO3A, and alleviates FOXO3A-mediated suppression of IL-6 expression. A second event downstream of AKT phosphorylation is activation of the canonical NF-κB pathway, which induces IL-6 expression. In addition to these downstream pathways, we unexpectedly found that CD80/CD86-induced PI3K signaling is regulated by previously unrecognized cross-talk with NOTCH1 signaling. This cross-talk is facilitated by NOTCH-mediated up-regulation of the expression of prolyl isomerase PIN1, which in turn increases enzyme activity of casein kinase II. Subsequently, phosphatase and tensin homolog (which suppresses PI3K activity) is inactivated via phosphorylation by casein kinase II. This results in full activation of PI3K signaling upon cross-linking CD80/CD86. Similar to IL-6, we have found that CD80/CD86-induced IDO production by DC at late time points is also dependent upon the PI3K → AKT → NF-κB pathway and requires cross-talk with NOTCH signaling. These data further suggest that the same signaling pathways downstream of DC CD80/CD86 cross-linking induce early IL-6 production to enhance T cell activation, followed by later IDO production to self-limit this activation. In addition to characterizing the pathways downstream of CD80/CD86 in IL-6 and IDO production, identification of a novel cross-talk between NOTCH1 and PI3K signaling may provide new insights in other biological processes where PI3K signaling plays a major role.
Introduction
Antigen-specific activation of T cells is critically dependent upon costimulatory signals provided by their interaction with antigen-presenting cells, in particular dendritic cells (DC).3 CD28 engagement on T cells by its natural ligands CD80 and CD86 expressed on DC plays a central role in T cell activation by delivery of costimulation (1). The biological consequences of CD28 ligation in T cells are well described and result in proliferation, augmented metabolic efficiency, and effector function (2–4). However, it has only recently been appreciated that CD80/CD86 are not merely ligands but are also capable of signaling. In this context, it has been shown that cross-linking CD80/CD86 by CD28 induced DC production of IL-6 (5), which is required for full T cell activation (6, 7). Conversely, it has also been shown that ligation of CD80/CD86 by CTLA4 (a receptor belonging to the CD28 superfamily) results in the production of indoleamine 2,3-dioxygenase (IDO), which catabolizes the essential amino acid tryptophan to l-kynurenine, resulting in inhibition of T cell activation (8–10).
In comparison with T cell CD28 signaling, substantially less is known about CD80/CD86 signaling, especially in human DC. Constraining the understanding of CD80/CD86 signaling is that these membrane proteins have cytoplasmic tails that do not have demonstrated binding domains for other signaling molecules (with the exception of the binding of the prohibitin adaptor proteins to the CD86 tail in B cells (11)). Previous studies have demonstrated that p38 MAPK and NF-κB are activated upon CD80/CD86 ligation in murine splenic DC IL-6 production (5) and in another study that the transcription factor FOXO3A regulated IL-6 production (12). However, whether p38 MAPK, NF-κB, and FOXO3A regulate or coordinate with each other in an integrated signaling pathway in CD80/CD86-induced DC IL-6 production is unknown.
In addition to playing a central role in T cell activation, IL-6 is also a major growth/survival factor for both normal and malignant plasma cells (e.g. multiple myeloma (MM)) (13, 14). We and others have previously found that normal plasma cells (PC) and myeloma cells express CD28 and that activation of PC/MM CD28 by CD80/CD86+ DC transduced a major pro-survival signal to the PC/MM (15, 16). Furthermore, we found that PC/MM CD28-mediated CD80/CD86 cross-linking also induced DC IL-6 production (15, 16), similar to what has been reported for T cells. Paralleling these observations, it has been reported that the NOTCH-JAGGED receptor-ligand pair is also involved in myeloma-induced stromal IL-6 production (17). Thus, the importance of DC IL-6 production for both T cell activation and PC/MM survival led us to characterize how CD80 and CD86 were inducing IL-6 production, whether NOTCH1 signaling was involved, and whether IDO production was regulated through the same pathways.
EXPERIMENTAL PROCEDURES
Mice, Cell Cultures, and Flow Cytometric Analysis
Female C57BL/6J (WT) mice were purchased from The Jackson Laboratory at 5–6 weeks of age. Upon receipt, animals were housed at the Division of Laboratory Animal Resources (Roswell Park Cancer Institute) in a pathogen-free facility. All animal experiments were approved by the Roswell Park Cancer Institute Institutional Animal Care and Use Committee. Murine bone marrow mononuclear cells were differentiated as described previously (15) to obtain BMDC and were analyzed by flow cytometry for CD40, CD80, CD86, CD11b, CD11c, MHC I, and MHC II (all antibodies were conjugated to phycoerythrin and purchased from BioLegend) expression using FACSCalibur II (15). Data were analyzed using the FCS Xpress software.
Antibodies and Reagents
Antibodies for detecting p85, NOTCH1 intracellular fragment (NICD), JAGGED2, phosphorylated AKT (Thr-308), phosphorylated and total amounts of FOXO3A and PTEN, and PIN1 were purchased from Cell Signaling Technology. Pan-AKT antibody was purchased from R&D Systems, and the IDO antibody was purchased from Millipore. The anti-NRR1 antibody that blocks NOTCH1 signaling was obtained from Genentech under a material transfer agreement. The γ-secretase inhibitor DAPT, PI3K inhibitor LY-294002, and NF-κB inhibitor Bay-11-7082 were purchased from Calbiochem and used at 50 μm. The AKT inhibitor II used at 2.5 μm and the casein kinase II inhibitor IV used at 50 μg/ml were both purchased from Calbiochem. All inhibitors were added to DC cultures for 2 h before the addition of CD28-Ig. CD28-Ig was purified from spent medium of COS-7 cells transfected with plasmids expressing CD28-Ig (gift from Peter S. Linsley, AVI Biopharma, Inc.) and was used at 10 μg/ml.
Culture and Flow Cytometry of Human Mo-DC
Monocytes were purified from normal human blood obtained under protocols approved by the Institutional Review Board of Roswell Park Cancer Institute, as described previously (16). They were differentiated to human DC in RPMI 1640 media with GM-CSF (10 ng/ml, Sigma) and IL-4 (1000 units/ml, R&D Systems) for 7 days and were analyzed for expression of CD14, CD11b, CD11c, CD80, CD83, CD86, CD1a, MHC I, and MHC II (all conjugated to PE, Beckman Coulter). Cells were stained with anti-NOTCH1 antibody (clone A6, Thermo Scientific) or anti-JAGGED2 (N-19, Santa Cruz Biotechnology). Goat anti-mouse secondary antibody-PE (Jackson ImmunoResearch) was used to detect NOTCH1, and goat anti-rabbit FITC was used to detect JAGGED2.
T Cell Proliferation Assay
T cell proliferation assay was performed as described previously (18). Briefly, normal human T cells (2 × 105 cells/well) were cultured with human DC (7 × 105 cells/well) that were immature or matured with LPS (10 ng/ml) for 72 h. This was followed by addition of [3H]thymidine (purchased from PerkinElmer Life Sciences) for 18 h to assess T cell proliferation.
DC-Jurkat Cocultures
DC (7 × 105 cells/well) were cultured in medium alone, with 50 μm DAPT, or were transfected with scramble siRNA/NOTCH1 siRNA. After 24 h, DC were further cultured alone or with Jurkat cells (2 × 105 cells/well) that were untransfected or transfected with scramble/JAGGED2 siRNA.
ELISA for IL-6, IL-23, and IFN-γ
Cell culture supernatants from DC cultured in medium, CD28-Ig, or the indicated inhibitors were collected after 4 h and were assayed by ELISA to detect IL-6 as described previously (16). IL-23 and IFN-γ was detected using supernatants of cells cultured for 48 h. Briefly, ELISA plates coated with the IL-6 capture antibody were blocked with 1% BSA and incubated with culture supernatants. This was followed by incubation with the IL-6 detection antibody, and streptavidin-HRP was added. IL-6 was measured based on a colorimetric reaction at 450 nm. ELISA reagents for IL-6 were purchased from R&D Systems, and reagents for IL-23 and IFN-γ were purchased from eBioscience.
Quantitative-Real Time PCR
Total RNA that was isolated using TRIzol LS reagent (Invitrogen), as per the manufacturer's instructions, was used to generate cDNA (iScript cDNA synthesis, Bio-Rad), as per the manufacturer's protocol. Gene expression was determined using CK II-specific primers (forward, 5′-AGCGATGGGAACGCTTTGTCC-3′, and reverse, 5′-CATCCCAAGGGGGTTGGCAGC-3′) using quantitative-PCR (Bio-Rad). Data are represented as fold change relative to untreated DC, normalized to actin.
Reverse Transcriptase PCR (RT-PCR)
Extraction of RNA and synthesis of cDNA were performed as described above. Gene expression was determined using gene-specific primers as follows: HES-1: forward, 5′-AAACCCTCAGCACTTGCTC-3′, and reverse, 5′-TCACCTCGTTCATGCACTC-3′; CK-II: forward, 5′-AGCGATGGGAACGCTTTGTCC-3′, and reverse, 5′-CATCCCAAGGGGGTTGGCAGC-3′); FOXO3A: forward, 5′-CGGCGGCGGGCTGTCTC, and reverse, 5′-AGTGGGCGATGGCTGGGATGG; and PIN1: forward, 5′-CTGATCAACGGCTACATCCA-3′, and reverse, 5′-TCAAATGGCTTCTGCATCTG-3′ using RT-PCR (Eppendorf AG). Data are represented as relative change in mRNA expression to actin.
Western Blot
Western blot analysis was performed as described previously (19). Briefly, cells were lysed in RIPA buffer, and the amount of protein was quantified using Bradford reagent (Bio-Rad). SDS-PAGE was performed, and membranes were probed with the indicated antibodies. Secondary antibodies were obtained from Promega.
siRNA Knockdown
DC were transfected with siRNA for p85, AKT1, NOTCH1, JAGGED-2, or PIN1 (ON-TARGET SMARTpool, Thermoscientific) using the human dendritic cell transfection kit from Lonza as per the manufacturer's instructions. Scramble siRNA was used as control. After 24 h, DC were cultured +/− CD28-Ig (10 μg/ml) for an additional 24 h. Cell supernatants were collected to perform ELISA for IL-6, and cell lysates were used to perform Western blots to detect the percentage of knockdown of the protein of interest. Jurkat cells were transfected with siRNA for JAGGED2 or scramble siRNA using the Lipofectamine kit (Invitrogen) as per the manufacturer's instructions. After 24 h, a second transfection was performed to sustain the knockdown of JAGGED2, and the cells were further cultured for 24 h. The transfected cells were then cocultured with DC. Densitometry was performed to quantify the percentage of knockdown by using the ImageJ software.
Casein Kinase II Assay
DC were lysed in RIPA buffer containing 0.5% Nonidet P-40, followed by brief sonication. The lysates were used to assay enzyme activity by measuring the CK II substrate peptide (RRRDDDSDDD, provided in the enzyme activity kit, Millipore) phosphorylation by estimating the transfer of γ-phosphate of [γ-32P]ATP by the enzyme. The activity was measured using a luminescence counter (Microbeta Trilux from PerkinElmer Life Sciences), and data were represented as units/min/mg.
EMSA for NF-κB Activity
EMSA was performed as described previously (19). Briefly, cells were lysed in RIPA buffer, and the lysates were incubated in a mixture containing Buffer D, dIdC, and BSA. This was followed by incubation with 32P-labeled primer containing consensus NF-κB-binding sites (5′-GATCCAACGGCAGGGGAATTCCCCTCTCCTTA-3′). For supershift assay, anti-p50 or anti-p65 (Santa Cruz Biotechnology) antibodies were added to the lysates. Samples were run on a 4% polyacrylamide gel and transferred to a filter paper. The dried gel was exposed to an x-ray film overnight at −80 °C.
IDO Activity Assay
DC (1 × 105 cells/well) were cultured with IFN-γ (1000 units/ml) in media containing 100 mm tryptophan for 2.5 h followed by addition of CTLA4-Ig (50 μg/ml) or CD28-Ig (50 μg/ml) and the indicated inhibitors. After 48 h, cell culture supernatants were assayed for l-kynurenine levels as a readout of IDO activity that was measured based on a colorimetric reaction at 490 nm.
Statistical Analysis
Student's t test was performed for statistical analysis using a two-tailed, equal variance test.
RESULTS
Ligation of CD80 and/or CD86 on DC Induces IL-6 Expression via Activation of PI3K/AKT Pathway
In these studies, we examined DC differentiated from primary human monocytes treated with GM-CSF + IL-4. These Mo-DC expressed characteristic DC markers and were immature as indicated by the absence of the maturation marker CD83 and also by low/medium expression of other maturation markers CD40 and MHC II (Fig. 1A). They were capable of driving T cell proliferation (the functional hallmark of DC), and this was significantly increased if they were matured by the addition of LPS (Fig. 1B).
FIGURE 1.
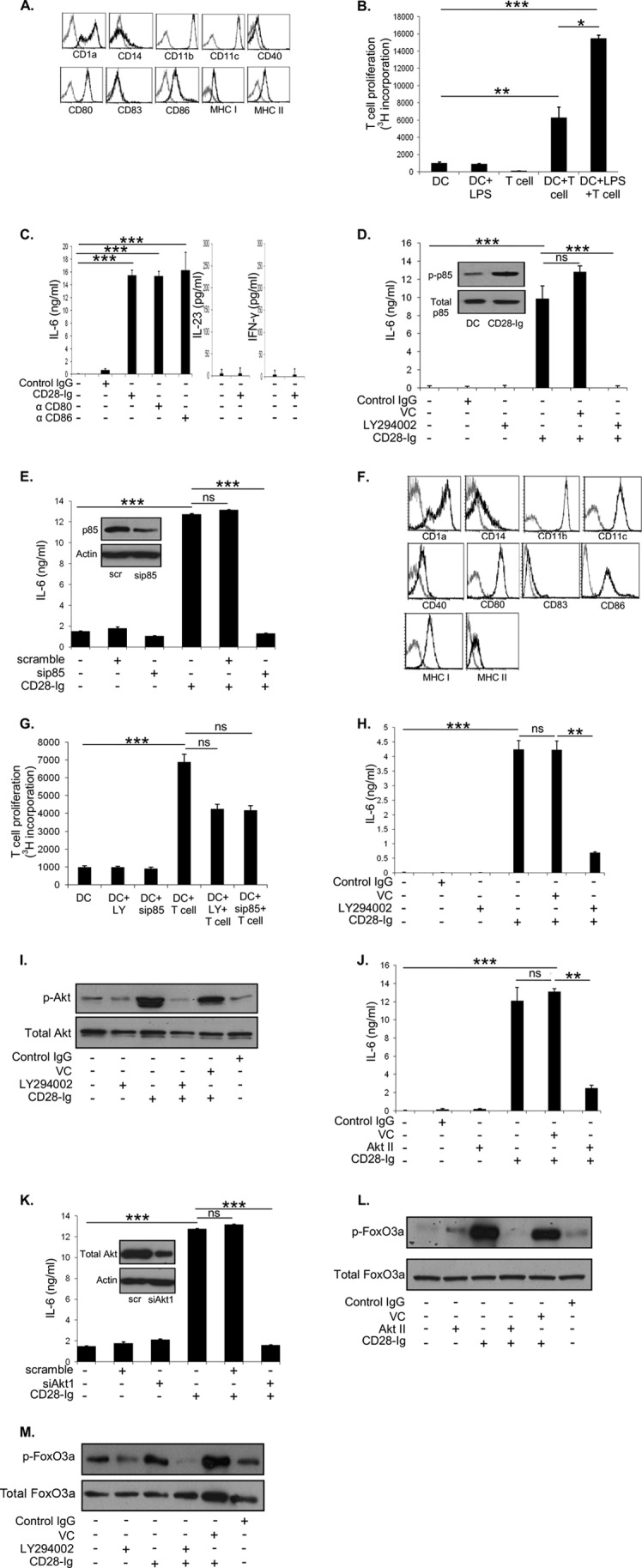
A, flow cytometric analysis of Mo-DC cultured in medium for expression of CD1a, CD11b, CD11c, CD14, CD40, CD80, CD83, CD86, MHC I, and MHC II. B, T cell proliferation assay was performed by determining the amount of thymidine incorporated by proliferating T cells when cultured with DC +/− LPS. C, ELISA for IL-6 of cell culture supernatants of Mo-DC +/− control IgG, CD28-Ig, anti-CD80 antibody, or anti-CD86 antibody. ELISA for IL-23 and IFN-γ of cell culture supernatants of Mo-DC cultured +/− CD28-Ig. D, Western blots were performed to detect phosphorylated p85 and total p85 using cell lysates from Mo-DC cultured in medium or with CD28-Ig (inset). ELISA for IL-6 of cell culture supernatants from Mo-DC cultured with vehicle control (VC, DMSO), control IgG, or the PI3K inhibitor LY294002 +/− CD28-Ig. E, Western blots were performed to detect p85 and actin using cell lysates from Mo-DC transfected with scramble siRNA or p85 siRNA (inset). ELISA for IL-6 of cell culture supernatants of Mo-DC cultured with scramble siRNA, p85 siRNA +/− CD28-Ig, is shown. F, flow cytometric analysis of Mo-DC cultured with the PI3K inhibitor LY294002 for expression of CD1a, CD1b, CD11c, CD14, CD40, CD80, CD83, CD86, MHC I, and MHCII. G, T-cell proliferation assay was performed by determining the amount of thymidine incorporated by proliferating T cells when cultured with DC cultured in medium +/− the PI3K inhibitor LY294002 or siRNA p85 and further cultured with T cells. H, ELISA for IL-6 of cell culture supernatants from murine BMDC (VC, DMSO), control IgG, or the PI3K inhibitor LY294002 +/− CD28-Ig. I, Western blots were performed to detect phosphorylated and total AKT using lysates from Mo-DC cultured with vehicle control (VC, DMSO), control IgG, or the PI3K inhibitor LY294002 +/− CD28-Ig. J, ELISA for IL-6 of cell culture supernatants of Mo-DC cultured with vehicle control (VC, DMSO), control IgG, or the AKT inhibitor AKT II +/− CD28-Ig. K, Western blots were performed to detect total AKT and actin using lysates from DC transfected with scramble siRNA or AKT1 siRNA (inset). ELISA for IL-6 of cell culture supernatants of Mo-DC cultured with scramble siRNA, AKT1, siRNA +/− CD28-Ig. L, Western blots were performed to detect phosphorylated and total FOXO3a using cell lysates from Mo-DC cultured with vehicle control (VC, DMSO), control IgG or the AKT inhibitor AKT II +/− CD28-Ig. M, Western blots were performed to detect phosphorylated and total FOXO3a using cell lysates from Mo-DC cultured with vehicle control (VC, DMSO), or the PI3K inhibitor. Results shown are representative of three experiments. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To elucidate the mechanism of CD80/CD86 signaling in DC, we used the recombinant fusion protein CD28-Ig (the extracellular domain of CD28 fused to the Fc part of IgG1), which specifically binds CD80 and CD86 and elicits IL-6 production in murine DC (20). We found that CD28-Ig significantly up-regulated IL-6 production by human Mo-DC compared with control IgG (Fig. 1C). Addition of anti-CD80 or anti-CD86 mAb similarly induced IL-6 production, demonstrating that either CD80 or CD86 activation is sufficient to elicit full IL-6 production in Mo-DC (in contrast to what we (15) and others (5) have found for murine DC) (Fig. 1C). Previously, it was reported in murine DC that ligation of CD80/CD86 by CD28-Ig slightly up-regulated IL-23 and significantly increased IFN-γ production (5). However, we found that human Mo-DC did not produce either of the cytokines (Fig. 1C). In considering what downstream pathway(s) might be involved, previous studies reported activation of PI3K upon CD86 activation in IgG1 production by murine B cells (21). Based on this, we investigated the role of PI3K in CD80/CD86-induced DC IL-6 production by inhibiting PI3K activity with the small molecule inhibitor LY294002. We found that CD28-Ig markedly induced phosphorylation of p85, the regulatory subunit of PI3K which indicates PI3K activity, when compared with Mo-DC cultured in medium (Fig. 1D, inset). Whereas CD28-Ig significantly up-regulated DC IL-6 production compared with vehicle control or control IgG, inhibiting PI3K activity significantly inhibited this (Fig. 1D). Similarly, siRNA-mediated silencing of the p85 regulatory subunit of PI3K (90% knockdown by densitometry compared with scrambled siRNA, Fig. 1E, inset), significantly decreased DC IL-6 production (Fig. 1E). Of note, inhibiting PI3K activity did not affect the phenotype of Mo-DC (Fig. 1F) when compared with Mo-DC cultured in medium (Fig. 1A). Mo-DC-induced T cell proliferation was reduced upon culturing Mo-DC with the PI3K inhibitor LY294002 or siRNA p85 (Fig. 1G). Although the decrease we observed was not significant, it showed a trend reflecting reduced T cell proliferation. To confirm that the effect we saw on IL-6 production was not limited to human Mo-DC, we cultured murine BMDC with LY294002 and/or murine CD28-Ig, and found the effect of blocking PI3K activity on IL-6 production in human DC was recapitulated (Fig. 1H).
Downstream of PI3K activation, phosphatidylinositol 1,4,5-phosphate (PIP3) produced by PI3K recruits AKT to the plasma membrane, where it is phosphorylated/activated by PDK1 (22). As predicted, we found that blocking PI3K activity decreased AKT phosphorylation irrespective of the presence of CD28-Ig (Fig. 1I). As seen for LY294002, inhibiting AKT activation by the small molecule inhibitor AKT II caused significant down-regulation of CD28-Ig-induced DC IL-6 production (Fig. 1J). siRNA-mediated silencing of AKT1 (95% knockdown, Fig. 1K, inset) also significantly reduced CD28-Ig-mediated induction of IL-6 in DC (Fig. 1K). Downstream of AKT activation, it has been previously reported that the transcriptional factor FOXO3A, which has been reported to regulate DC IL-6 production (12), is directly phosphorylated by AKT (23). Phosphorylated FOXO3A is shuttled out of the nucleus and retained in the cytoplasm, preventing it from repressing its transcriptional targets, including IL-6 (23). We found that whereas CD28-Ig increased phosphorylation of FOXO3A compared with control, blocking AKT activity completely inhibited this effect (Fig. 1L), and blocking PI3K activity also reversed CD28-Ig-mediated phosphorylation of FOXO3A (Fig. 1M).
Involvement of FOXO3A and NF-κB Downstream of PI3K/AKT in IL-6 Production
It has been previously shown that CTLA4-Ig (a soluble fusion protein consisting of an IgG tail fused to the extracellular domain of CTLA4, a member of the CD28 superfamily that also binds CD80 and CD86) down-regulated TLR7-induced IL-6 production via up-regulation of nuclear FOXO3A, which in turn suppressed IL-6 transcription in murine DC (12). To test if FOXO3A is important in CD28-Ig-induced IL-6 production via CD80/CD86 engagement, we used BMDC from FOXO3A+/− or FOXO3A−/− mice that have 50 and 100% less FOXO3A mRNA expression versus WT (Fig. 2A), and we compared them with wild type DC for IL-6 induction. DC from FOXO3A+/− and FOXO3A−/− mice produced significantly higher amounts of IL-6 compared with WT DC upon CD28-Ig addition (Fig. 2B), suggesting that FOXO3A was suppressing CD80/CD86-induced IL-6 production. However, BMDC from FOXO3A+/− and FOXO3A−/− mice produced minimal IL-6 in the same 24-h period without CD28-Ig (Fig. 2B). This suggests that in the absence of CD80/CD86 activation, the reduction in FOXO3A expression, which would suppress its ability to negatively regulate IL-6 production, is not sufficient to induce DC IL-6 production and that another molecule downstream of AKT was positively inducing IL-6 production. This led us to examine whether other pathways downstream of AKT are up-regulating IL-6 expression. One pathway activated by AKT is NF-κB (21, 23), which also induces IL-6 production in murine DC (5). Cross-linking CD80/CD86 by CD28-Ig induced NF-κB signaling (as measured by electromobility gel shift assays), which was markedly decreased upon inhibiting PI3K or AKT activity (Fig. 2C, left panel). Supershift assays for canonical NF-κB signaling demonstrated increased p50 and p65 activity with CD28-Ig that was abrogated by inhibiting PI3K or AKT activity (Fig. 2C, right panel). The noncanonical NF-κB signaling pathway was not activated, as evidenced by the lack of RELB induction by CD28-Ig when compared with the positive control (the myeloma cell line U266 treated with a CD28-activating mAb (16)) (Fig. 2D). Consistent with these findings, the NF-κB inhibitor Bay-11-7082 significantly reduced CD28-Ig-mediated up-regulation of DC IL-6 production (Fig. 2E). Collectively, our data suggest that the CD80/CD86 → PI3K → p-AKT → p-FOXO3A pathway de-represses the negative regulation of IL-6 production by FOXO3A, whereas the CD80/CD86 → PI3K → p-AKT → NF-κB pathway positively induces IL-6 expression and that both are necessary for the maximal IL-6 response.
FIGURE 2.

A, cell lysates of BMDC from wild type mice (WT), FOXO3A heterozygotes (F+/−), or knock outs (F−/−) were analyzed for FOXO3A mRNA expression by RT-PCR. B, cell culture supernatants of murine BMDC from wild type, FOXO3A+/−, or FOXO3A+/+ mice cultured +/− CD28-Ig were used to perform ELISA for IL-6. C, whole cell lysates of DC cultured alone, with LY294002, or AKT II +/− CD28-Ig were analyzed by electromobility gel shift assay with primers containing consensus NF-κB-binding sites. The arrow (left panel) indicates NF-κB dimers. Supershift EMSA was performed to detect p50 and p65 subunits as represented by the arrow (right panel). Actin was used as loading control. D, whole cell lysates of Mo-DC cultured +/− CD28-Ig were used to perform EMSA to detect RELB subunit as represented by the arrow. U266 myeloma cells treated with anti-CD28 activating antibody were used as positive control for RELB expression. Actin was used as the loading control. E, cell culture supernatants of Mo-DC cultured +/− vehicle control (VC), control IgG, or the NF-κB inhibitor Bay-11-7082 were assayed for IL-6 by ELISA. Results shown are representative of three experiments. ns, not significant; **, p < 0.01; ***, p < 0.001.
Cross-talk between NOTCH and CD80/CD86 Signaling in DC IL-6 Production
In addition to playing a critical role in T cell activation, IL-6 produced by DC is an important stromal factor for plasma cell and multiple myeloma growth/survival (13, 14, 24). We have previously reported that the CD28-expressing normal PC and MM cells induced DC IL-6 production in a CD80/CD86-dependent manner (15, 16). Paralleling these observations, it has been reported that JAGGED2 on MM cells induced IL-6 in NOTCH1-expressing stromal cells (17), and we have found that MM induced DC IL-6 production in a NOTCH-dependent fashion (16).
Upon ligation of NOTCH receptors by one of its ligands (belonging to the JAGGED or Delta-like family) a conformational change is induced in the receptor negative regulatory region (NRR) that enables the first stage cleavage by a disintegrin and metalloprotease (ADAM) proteases. This is followed by a second stage cleavage of the receptor's cytoplasmic region by γ-secretase, which releases the NICD to translocate to the nucleus where it transcribes its target genes, including HES-1 (25, 26). We have found that human Mo-DC coexpress NOTCH1 and JAGGED2 (Fig. 3A), and these DC exhibit constitutive NOTCH signaling as evidenced by expression of NICD (Fig. 3B). To investigate if NOTCH signaling induces IL-6 independent of CD28-Ig-triggered CD80/CD86 signaling, we first cultured DC with soluble JAGGED2 (sJAG2) and found no further increase in NICD expression, in contrast to the effect of sJAG2 on activating NOTCH1 in the positive control macrophage cell line RAW (Fig. 3B) (27). Consistent with this, sJAG2 induced no more IL-6 than DC cultured alone, in contrast to a significant induction by CD28-Ig (Fig. 3C). This suggests that in DC, NOTCH signaling does not independently induce IL-6. To investigate if there was an interaction between NOTCH and CD80/CD86 signaling, we blocked NOTCH signaling using the γ-secretase inhibitor DAPT. Although CD28-Ig significantly up-regulated DC IL-6 production, blocking NOTCH signaling (as demonstrated by a decrease in the NOTCH target gene HES-1 (see Fig. 4C)) significantly inhibited this effect (Fig. 3D). Importantly, DAPT treatment did not cause significant alterations in Mo-DC as evidenced by the phenotype (Fig. 3E, compared with Mo-DC cultured in medium, Fig. 1A). Because DAPT is a pan-NOTCH inhibitor, we investigated the specific role for NOTCH1 by using an anti-NRR1 antibody that specifically blocks NOTCH1 signaling by binding to the NRR region and inhibiting cleavage by ADAM proteases (28), and we found that the anti-NRR1 mAb also significantly inhibited CD28-Ig-mediated IL-6 induction (Fig. 3F). Because both DAPT and anti-NRR1 antibody prevents NOTCH1 cleavage but does not block NOTCH1 binding to JAGGED2, these data also suggest that potential signaling through JAGGED2 (or other NOTCH1 ligands) is not involved. Similarly, siRNA-mediated silencing of NOTCH1 (95% knockdown of NOTCH1 signaling as measured by NICD, Fig. 3G, inset) significantly reduced CD28-Ig-mediated IL-6 production by DC (Fig. 3G). Furthermore, siRNA knockdown of JAGGED2 expression (85% knockdown of JAGGED2, Fig. 3H, inset) completely abrogated CD28-Ig-induced IL-6 production (Fig. 3H). To eliminate the possibility that these findings were limited to human DC, we inhibited NOTCH signaling using DAPT in murine BMDC and found that CD28-Ig-mediated up-regulation of IL-6 production was significantly abrogated (Fig. 3I). These data indicate that NOTCH1 and JAGGED2 play a key role in regulating CD80/CD86-induced DC IL-6 production and also suggest that other NOTCH receptor and ligand family members are not involved.
FIGURE 3.
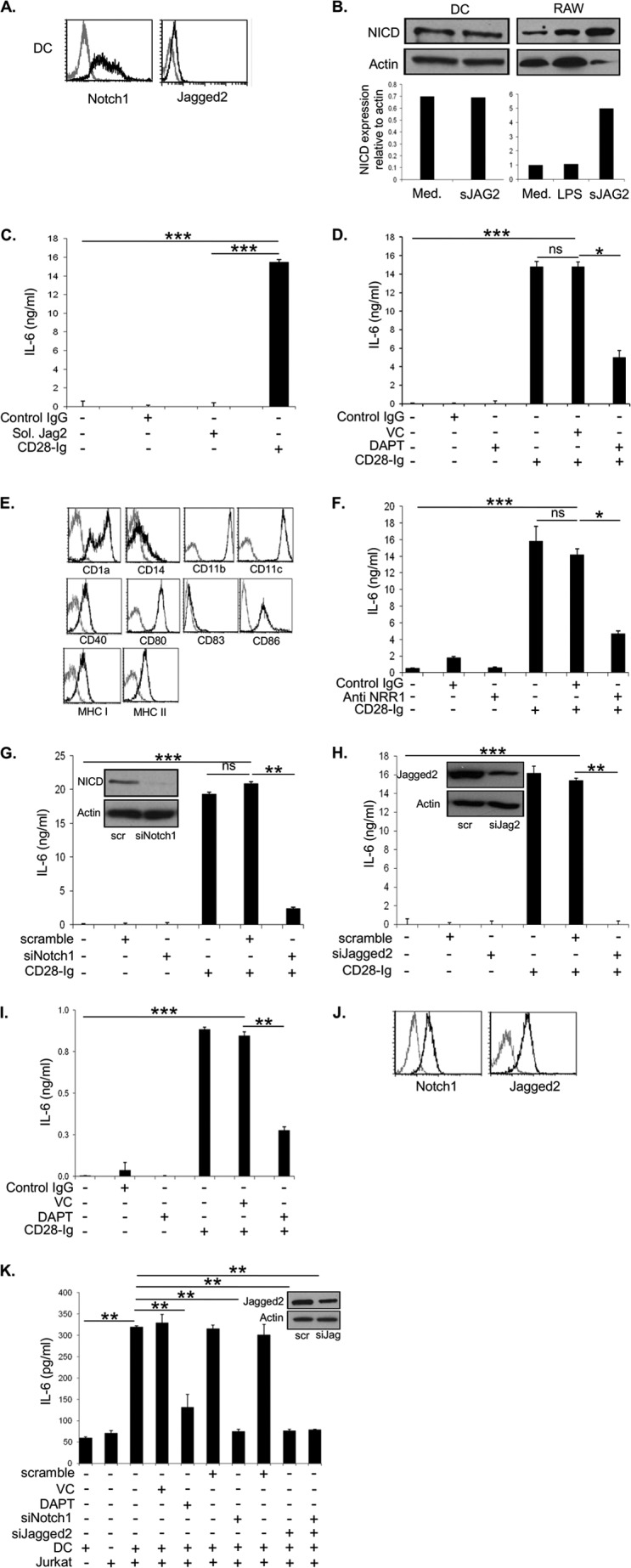
A, flow cytometric analysis of Mo-DC for expression of NOTCH1 and JAGGED2. B, cell lysates from Mo-DC cultured in medium (med) or soluble JAGGED2 (sJAG2) were used to perform Western blots for NICD. As a positive control, RAW cells were cultured in medium, with LPS or soluble JAGGED2. Densitometric analysis was performed using ImageJ software. C, ELISA for IL-6 from cell supernatants of Mo-DC was cultured in control IgG, sJAG2 +/− CD28-Ig. D, ELISA for IL-6 from cell supernatants of Mo-DC cultured alone or with vehicle control (VC), control IgG, DAPT +/− CD28-Ig. E, flow cytometric analysis of Mo-DC treated with DAPT for expression of CD1a, CD14, CD11b, CD11c, CD40, CD80, CD83, CD86, MHC I, and MHC II. F, ELISA for IL-6 from cell supernatants of Mo-DC cultured alone, with control Ig or anti-NRR1 antibody. G, Western blotting was performed using cell lysates from Mo-DC transfected with scramble/NOTCH1 siRNA (inset). ELISA for IL-6 from cell supernatants of Mo-DC cultured alone, with scramble siRNA, or NOTCH1 siRNA. H, Western blotting was performed using cell lysates of Mo-DC transfected with scramble/JAGGED2 siRNA (inset). ELISA for IL-6 from cell supernatants of Mo-DC cultured alone, with scramble siRNA, or JAGGED2 siRNA. I, ELISA for IL-6 of cell culture supernatants from murine BMDC cultured alone or with vehicle control, control IgG, DAPT +/− CD28-Ig. J, flow cytometric analysis of Jurkat for expression of NOTCH1 and JAGGED2. K, Western blots were performed using cell lysates from Jurkat cells transfected with scramble/JAGGED2 siRNA (inset). ELISA for IL-6 from cell supernatants of Mo-DC cultured with vehicle control, DAPT, scramble siRNA, siRNA NOTCH1+/− CD28-Ig, and Jurkat cells that were untransfected or transfected with JAGGED2 siRNA. Results shown are representative of three experiments. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 4.

A, flow cytometric analysis of Mo-DC cultured alone, with control Ig, or DAPT for expression of CD80 and CD86. B, flow cytometric analysis of Mo-DC cultured alone, with control Ig, or CD28-Ig for NOTCH1 and JAGGED2 expression. C, RNA from Mo-DC cultured alone, with vehicle control (VC), DAPT, or CD28-Ig was analyzed by RT-PCR for expression of HES-1 and actin. Densitometric analysis was performed using ImageJ software. Results shown are representative of three experiments.
Because the above experiments were performed using a homogeneous population of DC, we investigated if NOTCH signaling is necessary for CD80/CD86-mediated DC IL-6 production in the more biologically relevant interaction between DC and T cells. CD28+ Jurkat human T cells also express NOTCH1 and JAGGED2, similar to Mo-DC (Fig. 3J). DC or Jurkat cultured alone in medium only produce basal amounts of IL-6 (Fig. 3K). Coculturing DC with Jurkat significantly induced IL-6 production, which was significantly reduced upon siRNA-mediated silencing of JAGGED2 (80% reduction, Fig. 3K, inset) in Jurkat or NOTCH1 in DC (90% reduction in NICD expression). Silencing both NOTCH1 on DC and JAGGED2 on T cells did not further reduce IL-6 production (Fig. 3K), suggesting that they are not interacting with other NOTCH receptors and ligands in the induction of IL-6. Similar to our previous observations in myeloma-DC cocultures, blocking NOTCH signaling in DC with DAPT significantly reduced Jurkat-induced IL-6 production (Fig. 3K). Taken together, our data suggest that DC production of IL-6 upon cross-linking CD80/CD86 during DC:T cell engagement is dependent upon NOTCH signaling.
Ligation of CD80/CD86 Does Not Affect NOTCH Signaling
To examine the mechanism of cross-talk between NOTCH signaling and CD80/CD86 signaling, we first examined the effect of inhibiting NOTCH signaling on DC CD80 and CD86 expression, and we found no marked effects (Fig. 4A). Alternatively, it is possible that CD80/CD86 signaling is modulating NOTCH1 or JAGGED2 expression. However, CD28-Ig treatment had no major effect in DC NOTCH1 or JAGGED2 expression (Fig. 4B). Furthermore, CD28-Ig treatment had no effect on constitutive NOTCH signaling as measured by expression of the NOTCH1 target gene HES-1 expression (Fig. 4C). However, inhibiting NOTCH signaling with DAPT decreased HES-1 expression irrespective of the presence of CD28-Ig, suggesting the lack of any direct effect of CD80/CD86 activation on NOTCH1 signaling in DC (Fig. 4C).
Cross-talk between NOTCH and CD80/CD86 Signaling Involves the PI3K-AKT-FOXO3A Pathway
Because of the absence of any effect on surface receptor expression or NOTCH signaling, we examined whether cross-talk between the two pathways involved molecules downstream of CD80/CD86 activation. We first evaluated if NOTCH signaling regulated CD80/CD86-induced PI3K signaling by inhibiting NOTCH signaling with DAPT. Although CD28-Ig up-regulated phosphorylation of AKT and FOXO3A, DAPT reversed this effect (Fig. 5A). Interestingly, DAPT alone inhibited basal AKT phosphorylation, suggesting there is cross-talk between NOTCH and PI3K signaling even in the absence of CD80/CD86 activation. We also found that DAPT down-regulated basal NF-κB (the second downstream target of AKT) activation (Fig. 5B, left). Although CD80/CD86 ligation up-regulated NF-κB activation, DAPT abrogated CD28-Ig-mediated induction of canonical NF-κB signaling (Fig. 5B, right). These data indicate that NOTCH signaling regulates the PI3K → p-AKT → NF-κB pathway downstream of CD80/CD86 in DC.
FIGURE 5.

Cell lysates from Mo-DC cultured alone, with vehicle control (VC), control IgG, or DAPT +/− CD28-Ig were used to perform Western blots for phosphorylated and total AKT and FOXO3A (A) and EMSA to detect NF-κB dimers as represented by the arrow (left panel) (B). Supershift EMSA was performed to detect p50 and p65 subunits as represented by the arrow (right panel). Actin was used as a loading control. Results shown are representative of three experiments.
NOTCH Signaling Modulates PI3K/AKT Activity by Regulating Casein Kinase II Activity
We next examined the mechanism by which NOTCH signaling might regulate the PI3K pathway. PTEN sequesters the PI3K substrate PIP3 and inhibits PI3K activity. PTEN phosphorylation prevents it from binding to PIP3 and allows for full PI3K activity (29). We initially hypothesized that NOTCH signaling down-regulated PTEN expression through HES-1-mediated inhibition of PTEN transcription, as reported previously in leukemic T cells (30). However, there was no change in total PTEN levels with DAPT treatment under any condition (Fig. 6A). However, compared with untreated DC, DAPT treatment with or without CD28-Ig decreased PTEN phosphorylation (which inactivates PTEN). CD28-Ig alone did not affect PTEN phosphorylation, suggesting that this was independent of CD80/CD86 signaling. Taken together, these data suggest that NOTCH signaling induces the phosphorylation/inactivation of PTEN, which permits full PI3K activity upon CD80/CD86 activation with subsequent induction of IL-6 production.
FIGURE 6.

A, Western blots were performed to detect phosphorylated and total PTEN using cell lysates from Mo-DC cultured alone or with vehicle control (VC), control IgG, or DAPT +/− CD28-Ig. B, Western blotting was performed to detect phosphorylated and total PTEN using cell lysates from Mo-DC cultured alone or with vehicle control, control IgG, or the CK II inhibitor-IV +/− CD28-Ig. C, ELISA for IL-6 from cell supernatants of Mo-DC cultured with vehicle control, control IgG, or CK II inhibitor-IV +/− addition of CD28-Ig. D, Mo-DC were cultured alone or with vehicle control, control IgG, or DAPT +/− CD28-Ig. RNA was analyzed by RT-PCR. E, Mo-DC were cultured alone or with vehicle control, control IgG, or DAPT. RNA was analyzed by quantitative-PCR for mRNA expression of CK II with actin as a loading control. F, Mo-DC were cultured alone or with vehicle control, control IgG, or DAPT +/− CD28-Ig, and cell lysates were analyzed by Western blot to detect CK II expression. Densitometric analysis was performed using ImageJ software. G, cell lysates of Mo-DC cultured alone or with vehicle control, control IgG, DAPT, or the CK II inhibitor CK II-IV +/− CD28-Ig were used to assay CK II enzyme activity. Results shown are representative of three experiments. ns, not significant; *, p < 0.05; **, p < 0.01.
Candidate molecules regulating PTEN phosphorylation in DC include casein kinase II (CK II), as it plays a predominant role in phosphorylating/inactivating PTEN in a number of other systems (31, 32). Consistent with this, inhibiting CK II activity (see Fig. 6G) with the chemical inhibitor CK II-IV decreased PTEN phosphorylation versus untreated DC (Fig. 6B). As seen in Fig. 6A, CD28-Ig alone had no effect on PTEN phosphorylation and also did not affect CK II-mediated inhibition of PTEN phosphorylation (Fig. 6B). However, CD28-Ig-induced up-regulation of IL-6 was completely abrogated by inhibiting CK II activity (Fig. 6C).
To investigate how NOTCH signaling might be regulating CK II, we first examined the effect of inhibiting NOTCH signaling on CK II expression, and we found that CK II mRNA as assessed by RT-PCR (Fig. 6D), quantitative-PCR (Fig. 6E), or protein as analyzed by Western blots (Fig. 6F) remained unchanged. However, DC treated with DAPT exhibit a significantly lower CK II enzymatic activity versus DC treated with vehicle control (Fig. 6G). Unexpectedly, given our findings that CD28-Ig does not affect the (in)activation of PTEN, CD28-Ig significantly up-regulated CK II activity. However, DAPT completely blocked CD28-Ig-mediated up-regulation of CK II activity to levels similar to the CK II inhibitor (Fig. 6G). This suggests that although CD80/CD86-mediated signaling can up-regulate CK II activity (and thus may sustain PI3K signaling), the initiation of CD80/CD86 signaling cannot occur without a prior NOTCH1-mediated alleviation of the signaling blockade by PTEN.
NOTCH1 Target Gene PIN1 Regulates Casein Kinase II Enzyme Activity
To elucidate how NOTCH1 up-regulates CK II activity, we investigated the role of the prolyl isomerase PIN1. PIN1 has been previously reported to be a NOTCH1 gene target and up-regulates CK II activity in human breast cancer cells, but it has not been studied in DC (33). First, we tested if PIN1 is a target of NOTCH1 in human DC and found that siRNA-mediated inhibition of NOTCH1 signaling (95% knockdown of NICD, Fig. 7A) markedly reduced PIN1 mRNA compared with scramble siRNA control (Fig. 7B). This observation was reflected in PIN1 protein levels as well (Fig. 7C). We next examined if PIN1 is involved in CD28-Ig-mediated production of DC IL-6, and we found that siRNA-mediated silencing of PIN1 (90% knockdown of PIN1, Fig. 7D, inset) significantly reduced IL-6 production by DC compared with the scrambled siRNA control (Fig. 7D). This was consistent with a significant reduction in CK II activity upon silencing PIN1 (Fig. 7E). Altogether, these data suggest that NOTCH1 signaling induces transcription of PIN1, which in turn up-regulates casein kinase II activity.
FIGURE 7.
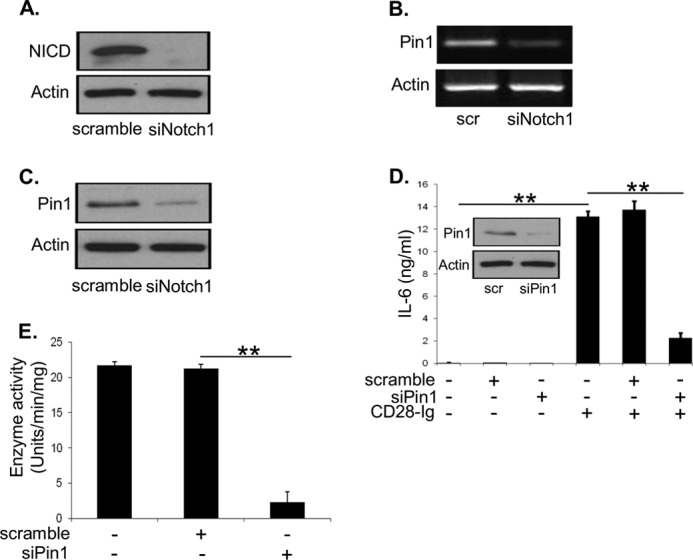
A, Western blots were performed to detect NICD and actin using cell lysates from DC transfected with scramble siRNA or NOTCH1 siRNA. B, RT-PCR was performed to detect PIN1 and actin mRNA using RNA extracted from DC transfected with scramble siRNA or NOTCH1 siRNA. C, Western blots were performed to detect PIN1 and actin using cell lysates from DC transfected with scramble siRNA or NOTCH1 siRNA. D, Western blots were performed to detect PIN1 and actin using cell lysates from DC transfected with scramble siRNA or PIN1 siRNA (inset). ELISA for IL-6 from cell supernatants of Mo-DC cultured alone, with scramble siRNA, or PIN1 siRNA +/− CD28-Ig. E, Mo-DC were cultured alone, with scramble siRNA, or PIN1 siRNA +/− addition of CD28-Ig. Cell lysates were analyzed for enzyme activity assay for CK II. Results shown are representative of three experiments. ns, not significant; **, p < 0.01.
IDO Production by DC upon Cross-linking CD80/CD86
Although engagement of CD80/CD86 by CD28-Ig induces IL-6 in murine DC, CTLA4-Ig induces IDO, an enzyme that catabolizes tryptophan to l-kynurenine and blocks T cell activation (8). This raised the previously unexplored question of whether the pathways downstream of CD80/CD86 that are inducing DC IL-6 production are also inducing IDO production in human DC. We found that CTLA4-Ig induced IDO protein in Mo-DC, at a later time point (24 h) than seen for IL-6 (4 h), and that maximal expression occurs in combination with IFN-γ treatment (Fig. 8A). Cross-linking CD80/CD86 by CTLA4-Ig, anti-CD80, or anti-CD86 mAb also significantly increases IDO activity (as measured by production of the l-kynurenine metabolite) at 48 h in the presence of IFN-γ (Fig. 8B). We next examined if CTLA4-Ig-mediated IDO production is dependent upon the downstream PI3K/AKT pathway. CTLA4-Ig increased IDO activity, which was significantly inhibited by blocking PI3K signaling with LY294002 (Fig. 8C). As predicted, inhibition of the activity of the downstream targets of PI3K, AKT (Fig. 8D), and NF-κB (Fig. 8E) also significantly decreased CTLA4-Ig-induced IDO activity. Similar to IL-6 production, we found that inhibiting NOTCH signaling with DAPT or the anti-NRR1 blocking antibody significantly decreased CD80/CD86-induced IDO activity (Fig. 8F). The cross-talk between NOTCH and CD80/CD86 signaling in DC IDO production also involves CK II, as CK II inhibition significantly decreased CTLA4-Ig-induced IDO activity (Fig. 8G). Interestingly, we found that cross-linking CD80/CD86 with CD28-Ig also induces active IDO in the presence of IFN-γ (Fig. 8H) and that CTLA4-Ig can also induce DC IL-6 production (Fig. 8I), which is in contrast to what has been reported in murine DC (5, 8). These data suggest that DC IDO production is dependent on the PI3K signaling pathway downstream of CD80/CD86, which cross-talks with NOTCH signaling. Additionally, regardless of its binding partner, CD80/CD86 can induce both IL-6 and IDO in Mo-DC.
FIGURE 8.

A, Western blots were performed to detect IDO and actin using cell lysates from Mo-DC cultured with IFN-γ +/− CTLA4-Ig for 18, 24, or 48 h. B, Mo-DC were cultured with IFN-γ followed by addition of CTLA4-Ig, anti-CD80, or anti-CD86. Cell culture supernatants were assayed for l-kynurenine levels to estimate IDO activity. Mo-DC cultured with IFN-γ and treated with the PI3K inhibitor LY294002 (C), AKT inhibitor AKT II (D), NF-κB inhibitor Bay-11-7062 (E), NOTCH signaling inhibitor DAPT or anti-NRR1 blocking antibody (F), or CK II inhibitor CK II-IV +/− CTLA4-Ig (G). Cell culture supernatants were assayed for l-kynurenine levels to estimate IDO activity. Mo-DC were cultured with or without IFN-γ followed by addition of CTLA4-Ig or CD28-Ig. Cell culture supernatants were used to assay l-kynurenine levels to estimate IDO activity (H) or perform ELISA for IL-6 (I).
DISCUSSION
Antigen-specific activation of T cells is critically dependent upon the costimulatory signals provided by DC involving the CD28-CD80/CD86 interaction. T cell activation is regulated not only by the T cell-intrinsic signals provided by CD28 but also by cytokines such as IL-6 produced by DC upon CD80 and CD86 activation (6, 7, 34). In the B cell lineage, we have previously demonstrated that these same molecular interactions between PC and DC induce DC IL-6 production, which enhances normal PC immunoglobulin production and malignant PC survival (15, 16). However, a thorough understanding of the molecular pathways involved in CD80/CD86-induced DC IL-6 production is lacking. To decipher signaling downstream of CD80/CD86, we used CD28-Ig, which binds both the ligands specifically and induces DC IL-6 production (5, 20). IL-6 induced by anti-CD80 and anti-CD86 antibodies in DC was comparable with the effect of CD28-Ig, which supports the conclusion that CD28-Ig is functioning by activating CD80/CD86 and argues against off-target effects. We also found that engagement of either CD80 or CD86 was sufficient to induce human DC IL-6 production, which is in contrast to findings by us (15) and others (5) in murine DC that both CD80 and CD86 are essential. However, our findings are consistent with other reports that describe the redundancy of CD80 and CD86 in human DC production of IDO (10). Why there is a difference between murine and human CD80 and CD86 is not known, but alignment of the human versus murine protein sequence shows that CD80 is homologous across species but CD86 is not.4 We have also found by in silico analysis that the murine and human CD80/CD86 cytoplasmic tails contain potential binding motifs for different kinases (data not shown), suggesting that they may be distinctly phosphorylated and initiate different signaling pathways upon activation. This is consistent with our observation that although PI3K plays a dominant role in human DC IL-6 production (blocking PI3K activity completely abrogates IL-6 production), in murine DC blocking PI3K activity results in significant yet incomplete down-regulation of IL-6 production, suggesting that a second pathway involving p38 MAPK (activated by MAPK kinases) activation may be regulating IL-6 production in murine DC (5).
The most proximal signaling molecules that activate PI3K downstream of CD80/CD86 activation in human DC are currently unknown. In B cells, it has been reported that CD86 engagement activates the membrane adaptor proteins prohibitins (PHB) (11). Although PHB interacts with PIP3 (36) and AKT (37), it is unknown if PHB interacts with PI3K. Because PHB are expressed by Mo-DC (38) and they up-regulate NF-κB signaling upon CD86 engagement in B cells (11) (similar to our findings reported here), we speculate that PHB may play a role in PI3K activation in DC. Downstream of this, we have found that PI3K activates AKT with subsequent phosphorylation of FOXO3A preventing it from suppressing IL-6 transcription. Simultaneously, activation of NF-κB by AKT induces DC IL-6 production.
In addition to IL-6 production, CD80/CD86 activation by CTLA4-Ig induces production of immunosuppressive tryptophan catabolizing IDO (8). The literature is inconclusive whether CD80/CD86-induced IDO induction utilizes the same signaling pathways as CD80/CD86-induced IL-6 production. In CD8α+ splenic DC from nonobese diabetic mice, CD80/CD86 engagement by CTLA4-Ig down-regulated PI3K signaling by increasing PTEN expression, which enhanced FOXO3A-mediated production of IDO (39). How CTLA4-Ig up-regulates PTEN expression was not determined, however. One possibility is that CD80/CD86 ligation induces IFN-γ (39), which produces nitric oxide (NO) in murine DC (40). Nonobese diabetic mice express high amounts of nitric oxide (NO) (41) that transcribes p53 (42) and EGR-1 (43). Separately, it has been reported that p53 and EGR-1 induce PTEN transcription (44, 45). Although these data have not been reported in DC, they suggest a potential role for CD80/CD86-induced NO in PTEN up-regulation in nonobese diabetic mice. However, in DC under homeostatic conditions, NO is nearly absent (46), suggesting that direct regulation of PTEN expression by activated CD80/CD86 via NO may be lacking.
Instead, we have found in human DC that PI3K signaling downstream of CD80/CD86 activation is being modulated by NOTCH1 regulation of PTEN. Although total PTEN expression is unaffected, PTEN is phosphorylated/inactivated by NOTCH1 signaling (initiated upon ligation of DC NOTCH1 by JAGGED2 expressed on a neighboring DC), which up-regulates PIN1 expression resulting in increased CK II activity. This allows for full PI3K activation downstream of CD80/CD86 ligation (summarized in Fig. 9). NOTCH1 has been reported to regulate PI3K signaling, but the mechanisms are distinct in various cell types. In activated T cells, NOTCH1 forms a complex with p56lck and PI3K that activates downstream AKT signaling (47). DC, however, do not express p56lck (48), and such a molecular interaction seems unlikely in our system. In T cell acute lymphoblastic leukemia, activating mutations of NOTCH1 up-regulate HES-1 expression that subsequently inhibits PTEN expression (30). PTEN phosphorylation by CK II was also previously reported in primary T cell acute lymphoblastic leukemia (49); however, this was independent of NOTCH signaling. In pancreatic cancer cells, RhoA/ROCK1 activation downstream of NOTCH signaling results in PTEN phosphorylation (50), but it is not known whether CK II is also involved or whether this pathway also occurs in nontransformed cells. Here, in human DC we propose that NOTCH1 → PIN1 → CK II → phospho-PTEN allows for very rapid signaling cross-talk with the PI3K pathway, which may be essential to initiate the very early events required for DC-mediated activation of T cells.
FIGURE 9.

Engagement of NOTCH1 on DC by its ligand JAGGED2 expressed on a neighboring DC initiates NOTCH1 signaling resulting in the transcription of the prolyl isomerase PIN1. Subsequently, casein kinase II activity is up-regulated, which inactivates PTEN by phosphorylation. This allows for full activation of PI3K downstream of CD80/CD86 ligation. Downstream of PI3K, AKT is activated, which in turn phosphorylates/inactivates FOXO3A and inhibits IL-6 suppression. Simultaneous activation of NF-κB downstream of AKT allows for IL-6 production by DC.
Consistent with this idea, we found that DC IL-6 and IDO production (which is dependent upon the same signaling pathways downstream of CD80/CD86 ligation in the presence of IFN-γ) is temporally regulated. CD80/CD86-induced IL-6 production occurs early (4 h), although IDO production/activation occurs late (24 h). This suggests that CD80/CD86 activation initially induces DC IL-6 production to promote T cell activation, but the same pathway later induces IDO production (in conjunction with evidence of a successfully activated T cell, namely IFN-γ production) to self-limit this activation.
Because the DC in our study have constitutive NOTCH signaling that is not inducibly regulated, this raises the question of the relevance of NOTCH in modulating physiological CD80/CD86 signaling. However, NOTCH1-CD80/CD86 cross-talk may be particularly relevant in vivo, where unlike in our in vitro experiments there is not a high density of DC interacting with one another to drive NOTCH1 signaling. One such setting as demonstrated by us is the T cell/DC interaction, where CD28+ T cells expressing JAGGED2 engage NOTCH1 on CD80/CD86+ DC inducing IL-6 production. The second setting may be in the PC/MM-DC interaction, where CD28+ PC/MM cells expressing JAGGED2 engage NOTCH1 on CD80/CD86+ DC, inducing production of IL-6 to support PC/MM survival (16). Intriguingly, our data also demonstrate that blocking NOTCH signaling inhibits basal AKT and FOXO3A phosphorylation, implying that NOTCH signaling can regulate the PI3K pathway independent of CD80/CD86 activation. In DC, such regulation of PI3K signaling by NOTCH may modulate PI3K-mediated regulation of DC maturation, antigen presenting capability, and blockade of DC IL-12 production (35, 51). Thus, the modulation of PI3K signaling by NOTCH1, regardless of its initiating source, may be a general regulatory pathway for many different biological responses.
Acknowledgments
We thank Dr. Arthur A. Hurwitz (NCI-Frederick, National Institutes of Health) for helping us with experiments with the FOXO3A heterozygous and FOXO3A homozygous knock-out mice (originally generated by Dr. Stephen M. Hedrick, University of California, San Diego, La Jolla, CA).
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA140622, R01 CA121044, R01 AI10015, and T32 CA085183. This work was also supported by the Multiple Myeloma Research Foundation.
This article was selected as a Paper of the Week.
L. Boise, unpublished data.
- DC
- dendritic cell
- PC
- plasma cell
- MM
- multiple myeloma
- PIP3
- phosphatidylinositol 1,4,5-phosphate
- BMDC
- bone marrow derived dendritic cell
- FOXO3A
- Forkhead Box O3a
- NF-κB
- nuclear factor κB
- PTEN
- phosphatase and tensin homolog
- NICD
- NOTCH intracellular domain
- NRR
- negative regulatory region
- PIN1
- peptidyl-prolyl isomerase 1
- CK
- casein kinase
- IDO
- indoleamine 2,3-dioxygenase
- PHB
- prohibitin
- Mo-DC
- monocyte-derived DC; N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester.
REFERENCES
- 1. Lanier L. L., O'Fallon S., Somoza C., Phillips J. H., Linsley P. S., Okumura K., Ito D., Azuma M. (1995) CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J. Immunol. 154, 97–105 [PubMed] [Google Scholar]
- 2. Sharpe A. H., Freeman G. J. (2002) The B7-CD28 superfamily. Nat. Rev. Immunol. 2, 116–126 [DOI] [PubMed] [Google Scholar]
- 3. Frauwirth K. A., Riley J. L., Harris M. H., Parry R. V., Rathmell J. C., Plas D. R., Elstrom R. L., June C. H., Thompson C. B. (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 [DOI] [PubMed] [Google Scholar]
- 4. Bour-Jordan H., Blueston J. A. (2002) CD28 function: a balance of costimulatory and regulatory signals. J. Clin. Immunol. 22, 1–7 [DOI] [PubMed] [Google Scholar]
- 5. Orabona C., Grohmann U., Belladonna M. L., Fallarino F., Vacca C., Bianchi R., Bozza S., Volpi C., Salomon B. L., Fioretti M. C., Romani L., Puccetti P. (2004) CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat. Immunol. 5, 1134–1142 [DOI] [PubMed] [Google Scholar]
- 6. Gajewski T. F., Renauld J. C., Van Pel A., Boon T. (1995) Costimulation with B7-1, IL-6, and IL-12 is sufficient for primary generation of murine antitumor cytolytic T lymphocytes in vitro. J. Immunol. 154, 5637–5648 [PubMed] [Google Scholar]
- 7. Holsti M. A., McArthur J., Allison J. P., Raulet D. H. (1994) Role of IL-6, IL-1, and CD28 signaling in responses of mouse CD4+ T cells to immobilized anti-TCR monoclonal antibody. J. Immunol. 152, 1618–1628 [PubMed] [Google Scholar]
- 8. Grohmann U., Orabona C., Fallarino F., Vacca C., Calcinaro F., Falorni A., Candeloro P., Belladonna M. L., Bianchi R., Fioretti M. C., Puccetti P. (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101 [DOI] [PubMed] [Google Scholar]
- 9. Fallarino F., Grohmann U., Hwang K. W., Orabona C., Vacca C., Bianchi R., Belladonna M. L., Fioretti M. C., Alegre M. L., Puccetti P. (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212 [DOI] [PubMed] [Google Scholar]
- 10. Munn D. H., Sharma M. D., Mellor A. L. (2004) Ligation of B7–1/B7–2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol. 172, 4100–4110 [DOI] [PubMed] [Google Scholar]
- 11. Lucas C. R., Cordero-Nieves H. M., Erbe R. S., McAlees J. W., Bhatia S., Hodes R. J., Campbell K. S., Sanders V. M. (2013) Prohibitins and the cytoplasmic domain of CD86 cooperate to mediate CD86 signaling in B lymphocytes. J. Immunol. 190, 723–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dejean A. S., Beisner D. R., Ch'en I. L., Kerdiles Y. M., Babour A., Arden K. C., Castrillon D. H., DePinho R. A., Hedrick S. M. (2009) Transcription factor FOXO3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat. Immunol. 10, 504–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uchiyama H., Barut B. A., Mohrbacher A. F., Chauhan D., Anderson K. C. (1993) Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood 82, 3712–3720 [PubMed] [Google Scholar]
- 14. Kawano M., Hirano T., Matsuda T., Taga T., Horii Y., Iwato K., Asaoku H., Tang B., Tanabe O., Tanaka H. (1988) Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature 332, 83–85 [DOI] [PubMed] [Google Scholar]
- 15. Rozanski C. H., Arens R., Carlson L. M., Nair J., Boise L. H., Chanan-Khan A. A., Schoenberger S. P., Lee K. P. (2011) Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med. 208, 1435–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nair J. R., Carlson L. M., Koorella C., Rozanski C. H., Byrne G. E., Bergsagel P. L., Shaughnessy J. P., Jr., Boise L. H., Chanan-Khan A., Lee K. P. (2011) CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J. Immunol. 187, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Houde C., Li Y., Song L., Barton K., Zhang Q., Godwin J., Nand S., Toor A., Alkan S., Smadja N. V., Avet-Loiseau H., Lima C. S., Miele L., Coignet L. J. (2004) Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 104, 3697–3704 [DOI] [PubMed] [Google Scholar]
- 18. Cejas P. J., Carlson L. M., Zhang J., Padmanabhan S., Kolonias D., Lindner I., Haley S., Boise L. H., Lee K. P. (2005) Protein kinase C βII plays an essential role in dendritic cell differentiation and autoregulates its own expression. J. Biol. Chem. 280, 28412–28423 [DOI] [PubMed] [Google Scholar]
- 19. Bahlis N. J., King A. M., Kolonias D., Carlson L. M., Liu H. Y., Hussein M. A., Terebelo H. R., Byrne G. E., Jr., Levine B. L., Boise L. H., Lee K. P. (2007) CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 109, 5002–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linsley P. S., Brady W., Grosmaire L., Aruffo A., Damle N. K., Ledbetter J. A. (1991) Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 173, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kin N. W., Sanders V. M. (2006) CD86 stimulation on a B cell activates the phosphatidylinositol 3-kinase/AKT and phospholipase Cγ2/protein kinase Cαβ signaling pathways. J. Immunol. 176, 6727–6735 [DOI] [PubMed] [Google Scholar]
- 22. Cantley L. C. (2002) The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 23. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) AKT promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 24. Minges Wols H. A., Underhill G. H., Kansas G. S., Witte P. L. (2002) The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol. 169, 4213–4221 [DOI] [PubMed] [Google Scholar]
- 25. Kidd S., Lieber T., Young M. W. (1998) Ligand-induced cleavage and regulation of nuclear entry of NOTCH in Drosophila melanogaster embryos. Genes Dev. 12, 3728–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schroeter E. H., Kisslinger J. A., Kopan R. (1998) NOTCH-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 [DOI] [PubMed] [Google Scholar]
- 27. Tsao P. N., Wei S. C., Huang M. T., Lee M. C., Chou H. C., Chen C. Y., Hsieh W. S. (2011) Lipopolysaccharide-induced NOTCH signaling activation through JNK-dependent pathway regulates inflammatory response. J. Biomed. Sci. 18, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y., Cain-Hom C., Choy L., Hagenbeek T. J., de Leon G. P., Chen Y., Finkle D., Venook R., Wu X., Ridgway J., Schahin-Reed D., Dow G. J., Shelton A., Stawicki S., Watts R. J., Zhang J., Choy R., Howard P., Kadyk L., Yan M., Zha J., Callahan C. A., Hymowitz S. G., Siebel C. W. (2010) Therapeutic antibody targeting of individual NOTCH receptors. Nature 464, 1052–1057 [DOI] [PubMed] [Google Scholar]
- 29. Das S., Dixon J. E., Cho W. (2003) Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. U.S.A. 100, 7491–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palomero T., Sulis M. L., Cortina M., Real P. J., Barnes K., Ciofani M., Caparros E., Buteau J., Brown K., Perkins S. L., Bhagat G., Agarwal A. M., Basso G., Castillo M., Nagase S., Cordon-Cardo C., Parsons R., Zúñiga-Pflücker J. C., Dominguez M., Ferrando A. A. (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 13, 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torres J., Pulido R. (2001) The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 276, 993–998 [DOI] [PubMed] [Google Scholar]
- 32. Kim K. Y., Shin H. K., Lee J. H., Kim C. D., Lee W. S., Rhim B. Y., Shin Y. W., Hong K. W. (2004) Cilostazol enhances casein kinase 2 phosphorylation and suppresses tumor necrosis factor-α-induced increased phosphatase and tensin homolog deleted from chromosome 10 phosphorylation and apoptotic cell death in SK-N-SH cells. J. Pharmacol. Exp. Ther. 308, 97–104 [DOI] [PubMed] [Google Scholar]
- 33. Rustighi A., Tiberi L., Soldano A., Napoli M., Nuciforo P., Rosato A., Kaplan F., Capobianco A., Pece S., Di Fiore P. P., Del Sal G. (2009) The prolyl-isomerase PIN1 is a NOTCH1 target that enhances NOTCH1 activation in cancer. Nat. Cell Biol. 11, 133–142 [DOI] [PubMed] [Google Scholar]
- 34. June C. H., Bluestone J. A., Nadler L. M., Thompson C. B. (1994) The B7 and CD28 receptor families. Immunol. Today 15, 321–331 [DOI] [PubMed] [Google Scholar]
- 35. Fukao T., Tanabe M., Terauchi Y., Ota T., Matsuda S., Asano T., Kadowaki T., Takeuchi T., Koyasu S. (2002) PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881 [DOI] [PubMed] [Google Scholar]
- 36. Ande S. R., Mishra S. (2009) Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem. Biophys. Res. Commun. 390, 1023–1028 [DOI] [PubMed] [Google Scholar]
- 37. Han E. K., Mcgonigal T., Butler C., Giranda V. L., Luo Y. (2008) Characterization of AKT overexpression in MiaPaCa-2 cells: prohibitin is an AKT substrate both in vitro and in cells. Anticancer Res. 28, 957–963 [PubMed] [Google Scholar]
- 38. Liu K., Li Y., Prabhu V., Young L., Becker K. G., Munson P. J., Weng N. (2001) Augmentation in expression of activation-induced genes differentiates memory from naive CD4+ T cells and is a molecular mechanism for enhanced cellular response of memory CD4+ T cells. J. Immunol. 166, 7335–7344 [DOI] [PubMed] [Google Scholar]
- 39. Fallarino F., Bianchi R., Orabona C., Vacca C., Belladonna M. L., Fioretti M. C., Serreze D. V., Grohmann U., Puccetti P. (2004) CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J. Exp. Med. 200, 1051–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu L., Bonham C. A., Chambers F. G., Watkins S. C., Hoffman R. A., Simmons R. L., Thomson A. W. (1996) Induction of nitric oxide synthase in mouse dendritic cells by IFN-γ, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J. Immunol. 157, 3577–3586 [PubMed] [Google Scholar]
- 41. Weinberg J. B., Granger D. L., Pisetsky D. S., Seldin M. F., Misukonis M. A., Mason S. N., Pippen A. M., Ruiz P., Wood E. R., Gilkeson G. S. (1994) The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-l-arginine. J. Exp. Med. 179, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Forrester K., Ambs S., Lupold S. E., Kapust R. B., Spillare E. A., Weinberg W. C., Felley-Bosco E., Wang X. W., Geller D. A., Tzeng E., Billiar T. R., Harris C. C. (1996) Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. U.S.A. 93, 2442–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cibelli G., Policastro V., Rössler O. G., Thiel G. (2002) Nitric oxide-induced programmed cell death in human neuroblastoma cells is accompanied by the synthesis of Egr-1, a zinc finger transcription factor. J. Neurosci. Res. 67, 450–460 [DOI] [PubMed] [Google Scholar]
- 44. Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T. W. (2001) Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325 [DOI] [PubMed] [Google Scholar]
- 45. Virolle T., Adamson E. D., Baron V., Birle D., Mercola D., Mustelin T., de Belle I. (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 3, 1124–1128 [DOI] [PubMed] [Google Scholar]
- 46. Paolucci C., Burastero S. E., Rovere-Querini P., De Palma C., Falcone S., Perrotta C., Capobianco A., Manfredi A. A., Clementi E. (2003) Synergism of nitric oxide and maturation signals on human dendritic cells occurs through a cyclic GMP-dependent pathway. J. Leukocyte Biol. 73, 253–262 [DOI] [PubMed] [Google Scholar]
- 47. Sade H., Krishna S., Sarin A. (2004) The anti-apoptotic effect of NOTCH-1 requires p56lck-dependent, AKT/PKB-mediated signaling in T cells. J. Biol. Chem. 279, 2937–2944 [DOI] [PubMed] [Google Scholar]
- 48. Liu P., Aitken K., Kong Y. Y., Opavsky M. A., Martino T., Dawood F., Wen W. H., Kozieradzki I., Bachmaier K., Straus D., Mak T. W., Penninger J. M. (2000) The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat. Med. 6, 429–434 [DOI] [PubMed] [Google Scholar]
- 49. Silva A., Jotta P. Y., Silveira A. B., Ribeiro D., Brandalise S. R., Yunes J. A., Barata J. T. (2010) Regulation of PTEN by CK2 and NOTCH1 in primary T-cell acute lymphoblastic leukemia: rationale for combined use of CK2- and γ-secretase inhibitors. Haematologica 95, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vo K., Amarasinghe B., Washington K., Gonzalez A., Berlin J., Dang T. P. (2011) Targeting NOTCH pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol. Cancer 10, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bhattacharyya S., Sen P., Wallet M., Long B., Baldwin A. S., Jr., Tisch R. (2004) Immunoregulation of dendritic cells by IL-10 is mediated through suppression of the PI3K/AKT pathway and of IκB kinase activity. Blood 104, 1100–1109 [DOI] [PubMed] [Google Scholar]