

Background: The alternative NF-κB pathway plays important roles in osteoclastogenesis through an unknown mechanism.
Results: RelB-induced Cot expression rescues RANKL-induced osteoclastogenesis in cells isolated from aly/aly mice, which lack active NIK.
Conclusion: The overexpression of RelB rescues RANKL-induced osteoclastogenesis by Cot/IKKα-induced NF-κB2 processing in aly/aly cells.
Significance: The Akt/Cot/IKKα pathway that is induced by RelB contributes to RANKL-induced osteoclastogenesis by activating the alternative NF-κB pathway.
Keywords: Bone, NF-kappaB, Osteoclast, Phosphorylation, Signal Transduction
Abstract
The alternative nuclear factor-κB (NF-κB) pathway, mainly the RelB-p52 heterodimer, plays important roles in bone metabolism through an unknown mechanism. We have previously reported that alymphoplasia (aly/aly) mice, which lack active NF-κB-inducing kinase (NIK), show mild osteopetrosis due to the inhibition of osteoclastogenesis. p100 retains RelB in the cytoplasm and inhibits RANKL-induced osteoclastogenesis in aly/aly cells. Furthermore, the overexpression of RelB in aly/aly cells rescues RANKL-induced osteoclastogenesis by inducing p100 processing. In contrast, the overexpression of p65 in aly/aly cells has no effect. However, the overexpression of RelB fails to rescue RANKL-induced osteoclastogenesis in the presence of p100ΔGRR, which cannot be processed to p52, suggesting that p100 processing is a key step in RelB-rescued, RANKL-induced osteoclastogenesis in aly/aly cells. In this study, Cot (cancer Osaka thyroid), an MAP3K, was up-regulated by RelB overexpression. Analysis of the Cot promoter demonstrated that p65 and RelB bound to the distal NF-κB-binding site and that RelB but not p65 bound to the proximal NF-κB-binding site in the Cot promoter. The knocking down of Cot expression significantly reduced the RANKL-induced osteoclastogenesis induced by RelB overexpression. The phosphorylation of IKKα at threonine 23 and its kinase activity were indispensable for the processing of p100 and osteoclastogenesis by RelB-induced Cot. Finally, constitutively activated Akt enhanced osteoclastogenesis by RelB-induced Cot, and a dominant-negative form of Akt significantly inhibited it. Taken together, these results indicate that the overexpression of RelB restores RANKL-induced osteoclastogenesis by activation of Akt/Cot/IKKα-induced p100 processing.
Introduction
Osteoclasts are terminally differentiated multinucleated cells that are responsible for physiological and pathological bone resorption and, thereby, play an essential role in maintaining bone volume and homeostasis (1, 2). Osteoclast precursors that express RANK (receptor activator of nuclear factor-κB), which is a tumor necrosis factor (TNF) receptor family member, recognize RANKL (RANK ligand) and differentiate into osteoclasts in the presence of macrophage colony-stimulating factor (M-CSF) (1–3). The in vivo significance of the RANKL-RANK signaling pathway has been verified by observations of targeted disruption of either gene in mice, resulting in severe osteopetrosis due to a complete lack of osteoclasts (4, 5). One critical signaling mechanism that is triggered by RANKL following ligation of RANK is the activation of the inducible transcription factor NF-κB,2 which implicates this pathway as a key regulator of osteoclast differentiation (6, 7). This central role of NF-κB is further supported by the demonstration that the gene-specific deletion of the p50 and p52 NF-κB subunits causes severe osteopetrosis due to the absence of osteoclasts (8, 9). These in vitro and in vivo genetic studies strongly suggest that osteoclast differentiation is dependent upon RANKL-induced NF-κB activity in osteoclast precursors.
The transcription factor NF-κB participates in the expression of a wide variety of genes that are involved in the regulation of immune and inflammatory responses, proliferation, tumorigenesis, and survival (10, 11). Research in recent years has defined two distinct NF-κB activation pathways, termed the classical and alternative NF-κB signaling pathways (10, 11). Classical NF-κB activation is based on inducible IκB degradation. This pathway can be rapidly and transiently activated by a large variety of substances, such as mitogens, cytokines, and microbial components. This activation is dependent upon a specific IκB kinase (IKK) that is composed of two catalytic subunits, IKKα (IKK1) and IKKβ (IKK2), and the regulatory subunit NEMO (IKKγ). In contrast, the alternative NF-κB signaling pathway is activated by a select group of TNF receptors, such as CD40, lymphotoxin-β receptor (LTβR), and RANK. Among these factors, RANK is activated by RANKL and transduces signals through either the classical or alternative pathways. The activation of NF-κB-inducing kinase (NIK) results in the activation of IKKα homodimers and the processing of the p100 precursor to p52. The processing of p105 to p50 occurs constitutively, whereas the processing of p100 to p52 is stimulus-dependent. The removal of the ankyrin repeats from p100 reduces its IκB-like activity, and the processing of p100 allows for the nuclear translocation of RelB, which may heterodimerize with either p52 or p50 and result in subsequent gene transcription (10, 11).
Alymphoplasia (aly/aly) mice have a natural loss-of-function mutation in the gene encoding the NF-κB-inducing kinase Nik (12), which is an essential kinase for the processing of p100 to p52 in the alternative NF-κB pathway. The alternative NF-κB signaling pathway is inhibited downstream of NIK in these mice (13–15). Therefore, aly/aly mice are useful for understanding the physiological role of the alternative NF-κB pathway in tissue development and regeneration. We have previously shown that aly/aly mice have mild osteopetrosis with increased bone volume due to a significantly reduced number of osteoclasts (14, 15). Nfkb2lym1/lym1 mice contain a novel mutation in Nfkb2 that encodes a non-processable form of p100 and have phenotypes that are similar to those of aly/aly mice, such as abnormalities of splenic architecture and mild osteopetrosis (16). In contrast, p100-deficient mice, which carry a homozygous deletion of the COOH-terminal ankyrin repeats of p100 but still express functional p52 protein, have precisely the opposite bone phenotype of aly/aly mice: osteopenia with an increased number of osteoclasts (14). These results suggest that alternative NF-κB signaling regulates osteoclast differentiation. The number of osteoclasts that are induced by RANKL strongly correlates with the ratio of p52 to p100 expression levels (15).
The existence of highly distinct classical and alternative NF-κB pathways, which lead to the activation of p65 and RelB, respectively, suggests that these two subunits have different biological functions. The IκB-like domain of p100 retains p65 or RelB in the cytoplasm and inhibits RANKL-induced osteoclastogenesis in NIK-deficient (NIK−/−) cells (17). Furthermore, the overexpression of RelB, but not p65, in NIK−/− cells rescues RANKL-induced osteoclastogenesis (18). These results strongly indicate that RelB is required for full osteoclast differentiation. However, the molecular mechanism by which the overexpression of RelB rescues the suppression of osteoclastogenesis by inhibiting the alternative pathway remains unclear.
The purpose of this study was to define the role of the alternative pathway in osteoclast differentiation. Our results demonstrated that the overexpression of RelB rescued RANKL-induced osteoclastogenesis by inducing NF-κB2 processing in aly/aly bone marrow macrophages (BMMs). A genome-wide screen of RelB-overexpressing BMMs showed that Cot (cancer Osaka thyroid) was induced in RelB-overexpressing BMMs. Cot was initially identified in a screen for transforming genes that were expressed by a human thyroid carcinoma (19) and that activated ERK, JNK, and NF-κB (20, 21). Knocking down Cot expression significantly reduced RANKL-induced osteoclastogenesis by RelB overexpression. The phosphorylation of IKKα at threonine 23 and its kinase activity were indispensable for NF-κB2 processing and osteoclastogenesis by RelB-induced Cot. Furthermore, Akt modulated RANKL-induced osteoclastogenesis through RelB-induced Cot. Hence, overexpression of RelB restored RANKL-induced osteoclastogenesis by activating Akt/Cot/IKKα-induced NF-κB2 processing.
EXPERIMENTAL PROCEDURES
Animals
Mice (8 weeks old) that were heterozygous for aly (aly/+) were purchased from Nippon Clea (Tokyo, Japan). Heterozygous males and females were bred to obtain homozygous aly/aly mice, and the offspring were screened using PCR (22). NF-κB2-deficient (NF-κB2−/−) and RelB-deficient (RelB−/−) mice were obtained from the Leibniz-Institute for Age Research at the Fritz-Lipmann-Institute (Jena, Germany). All mice were maintained at the Animal Resource Center, and experimental procedures were approved by the Animal Care and Use Committee of Kyushu Dental University (approval number 10-021).
Reagents
GST-RANKL was kindly provided by the Oriental Yeast Co., Ltd. (Shiga, Japan), and recombinant human M-CSF was purchased from PeproTech Inc. (Rocky Hill, NJ). The anti-RelB (sc-226), IκBα (sc-371), phosphorylated IKKα (sc-101706), HDAC1 (sc-7872), and Cot (sc-720) antibodies and Cot shRNA (sc-35096-SH) were purchased from Santa Cruz Biotechnology, Inc. The anti-NF-κB2 (catalog no. 4882), phosphorylated NF-κB2 (catalog no. 4810), phosphorylated IKKα/β (catalog no. 2681), Akt (catalog no. 2920), and phosphorylated Akt (catalog no. 9271) antibodies were obtained from Cell Signaling (Beverly, MA). The anti-NFATc1 (ab25916) antibodies and anti-p65 antibodies (SA-171) were purchased from Abcam (Cambridge, UK) and Biomol (Plymouth Meeting, PA), respectively. The anti-FLAG M5 and β-actin antibodies were purchased from Sigma-Aldrich.
Cell Culture
BMMs were prepared as osteoclast precursors from 5–8-week-old male WT, aly/aly, NF-κB2−/−, or RelB−/− mice. Bone marrow cells were obtained from mouse tibias and suspended in 96-well plates for 16 h in the presence of M-CSF (100 ng/ml) in α-minimal essential medium containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. Non-adherent cells were harvested and further cultured for 2 days with M-CSF (100 ng/ml). More than 90% of the adherent cells that expressed macrophage-specific antigens, such as Mac-1, Moma-2, and F4/80, were used as BMMs (23). BMMs were cultured for 3 days with RANKL (100 ng/ml). Cultures were fixed with 3.7% formaldehyde, and osteoclasts were detected by staining for tartrate-resistant acid phosphatase (TRAP). TRAP-positive (TRAP+) multinucleated cells that had more than five nuclei were observed using a microscope and counted as osteoclasts. The 293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 5% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Fractionation, Western Blot Analysis, and Immunoprecipitation
BMMs treated with RANKL for the indicated time were lysed in Tris-buffered saline (20 mm Tris-HCl and 200 mm NaCl) containing 1% Triton X-100 and protease inhibitors (aprotinin, pepstatin, dithiothreitol, and leupeptin). The cytosolic and nuclear extracts were prepared as described previously (23). For immunoprecipitation, an equal amount of protein was immunoprecipitated with anti-IKKα antibodies for 2 h; then protein A-Sepharose beads were added, and the samples were incubated for another 1 h. The immunoprecipitates were washed three times with lysis buffer and extracted in SDS sample buffer. The lysates or immunoprecipitates were resolved by 10% SDS-PAGE, transferred to Immobilon-P membranes (Millipore, Billerica, MA), and immunoblotted with individual antibodies. Then the membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). The immunoreactive proteins were visualized using ECL (Amersham Biosciences) and analyzed using a Luminescent image analyzer (Fujifilm, Tokyo, Japan).
Retroviral Transduction
The retroviral vector pMX-IRES-EGFP and Plat-E cells were kindly provided by Dr. T. Kitamura (University of Tokyo) (24, 25). The following constructs were cloned into the pMX-IRES-EGFP vector: the wild-type IKKα; a mutant form of IKKα (IKKαAA) that is not phosphorylated by NIK, with serine to alanine mutations at residues 176 and 180; a mutant form of IKKα (IKKαAAT23A) that is not phosphorylated by NIK or Akt, with serine to alanine mutations at residues 176 and 180 and a threonine to alanine mutation at residue 23; the dominant negative form of IKKα (IKKαKM), with a lysine to methionine mutation at residue 44; wild-type IKKα with a FLAG tag; wild-type p100; and p100ΔGRR, which cannot be processed to p52 (21). The constitutively activated form of Akt (AktCA), which lacks amino acids 4–129 and has an HA tag; the dominant negative form of Akt (AktDN), which has a lysine residue 179 to methionine mutation; a wild type, HA-tagged Akt; and Cot expression vectors were obtained from Addgene (Cambridge MA). Retrovirus packaging was performed by transfecting the plasmids into Plate-E cells using the Genejuice (Merck) transfection reagent. The supernatants of all retroviral vector-transfected Plat-E cells were used to infect primary BMMs in the presence of 8 μg/ml Polybrene.
Real-time PCR Analysis
Total RNA of BMMs or osteoclasts from WT or aly/aly mice was prepared using TRIzol reagent (Invitrogen). Two micrograms of total RNA was used to synthesize first-strand cDNA using SuperScript II transcriptase and random primers (Invitrogen). Real-time PCR was performed using the SYBR Green PCR Master Mix and a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The experimental results were fit to a standard curve that was generated by amplifying serially diluted products under the same PCR conditions. GAPDH expression served as an internal control. The primer sequences were as follows: Cot, 5′-cttgcatttgcaaaccatgc-3′ (forward) and 5′-ggaacaaggagaacatccga-3′ (reverse); GAPDH, 5′-aactttggcattgtggaagg-3′ (forward) and 5′-acacattggggtaggaaca-3′ (reverse).
Construction of Reporter Plasmids and Luciferase Assays
To construct the luciferase reporter plasmid, a 0.5-kb fragment upstream from the transcription initiation site of the mouse Cot gene was cloned by a standard PCR method using PrimeSTAR DNA polymerase (Takara Shuzo Co., Siga, Japan) and mouse genomic DNA as a template. It was then subcloned into the HindIII and BamHI recognition sites of the pfLuc-basic vector from Dr. Sankar Ghosh (Columbia University) (26). Genejuice was used to transfect 293 cells with the reporter plasmids. The luciferase activity was measured using a Dual-Luciferase reporter assay system (Promega, Madison, WI).
Chromatin Immunoprecipitation (ChIP)
ChIP was performed with a ChIP assay kit (Upstate Biotechnology, Waltham, MA) according to the manufacturer's instructions, using antibodies against p65, RelB, NF-κB2, and normal IgG. The purified DNA was analyzed by PCR using primers that detect sequences within the region of Cot promoter that harbors the NF-κB binding NF-κB-responsive element. The primer pairs containing the distal NF-κB-responsive element were 5′-aggatctaaaaccgcaaact-3′ (forward) and 5-cttttccatttcccttttct-3′ (reverse). The primers containing the proximal NF-κB-responsive element were 5′-ccaccgacacagaacactca-3′ (forward) and 5′-gtcttgcgcaaggtgttttc-3′ (reverse).
Data Analysis
Comparisons were made using factorial analysis of variance. When significant F values were detected, Fisher's protected least significant difference post hoc test was performed to compare each of the groups. The data were expressed as the mean ± S.D., and p values of p < 0.05 were considered significant.
RESULTS
The Translocation of RelB into the Nucleus Correlates with RANKL-induced Osteoclastogenesis
We previously reported that alymphoplasia (aly/aly) mice, in which the processing of p100 to p52 does not occur due to an inactive form of NIK, showed an osteopetrotic phenotype with significantly reduced osteoclast numbers (14, 15). In contrast, the bone phenotype of NF-κB2−/− mice, which lacks p100 and p52, was normal, and BMMs differentiated to osteoclasts, similar to WT BMMs; this result may be due to compensation for the lack of NF-κB2 by NF-κB1 (15). BMMs from WT, aly/aly, or NF-κB2−/− mice were treated with RANKL (100 ng/ml) for 6 days in the presence of M-CSF (100 ng/ml). As reported previously (15), numerous osteoclasts were formed from BMMs in WT and NF-κB2−/− mice, but few osteoclasts were formed from aly/aly BMMs (Fig. 1, A and B). Therefore, we hypothesized that the residual active p52 moiety, together with its binding partner RelB, eventually translocates into the nucleus to exert transcriptional activity. When BMMs from WT, aly/aly, or NF-κB2−/− mice were treated with RANKL for the indicated time, RANKL led to translocation of p65 to the nucleus; the maximal level of this translocation occurred at 30 min, and the degradation of IκBα coincided with the translocation of p65 in all BMMs (Fig. 1C). The translocation of RelB was observed within 6 h after RANKL stimulation in WT BMMs. RANKL transiently led to the translocation of RelB with kinetics similar to that of p65 translocation in NF-κB2−/− BMMs, which suggests that the RelB-p50 heterodimer translocates into the nucleus after RANKL stimulation in NF-κB2−/− BMMs (27). However, RelB translocation by RANKL stimulation was not observed in aly/aly BMMs (Fig. 1C). The HDAC1 expression levels were comparable in all nuclear extracts from WT, aly/aly, and NF-κB2−/− mice. These results suggested that the translocation of RelB into the nucleus correlates with RANKL-induced osteoclastogenesis.
FIGURE 1.
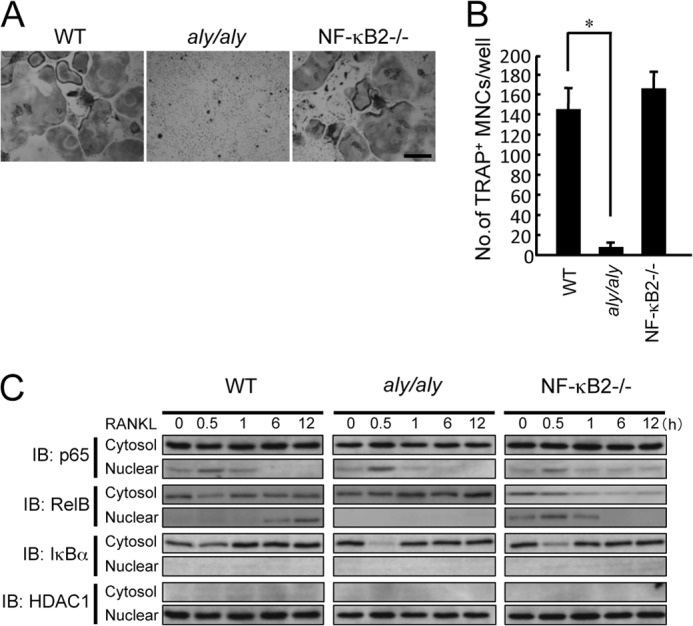
The translocation of RelB into the nucleus correlated with RANKL-induced osteoclastogenesis. BMMs from WT, aly/aly, or NF-κB2−/− mice were treated with RANKL (100 ng/ml) for 6 days in the presence of M-CSF. A, microscopic view of TRAP+ MNCs in BMMs from WT, aly/aly, or NF-κB2−/− mice. Scale bar, 100 μm. B, the data shown are the numbers of TRAP+ MNCs from WT, aly/aly, or NF-κB2−/− BMMs per culture well. The data are means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs from WT, aly/aly, or NF-κB2−/− mice were treated with RANKL (100 ng/ml) for the indicated time. The cytoplasmic and nuclear fractions were harvested from cultured cells and subjected to immunoblot analysis (IB) with specific antibodies as indicated. The expression of IκBα and HDAC1 was used to identify the cytosolic and nuclear extract, respectively. HDAC1 expression was also used as a loading control for the nuclear extracts.
The Overexpression of RelB Rescues RANKL-induced Osteoclastogenesis in aly/aly BMMs
To confirm that the translocation of RelB is important for RANKL-induced osteoclastogenesis, we transfected p65 or RelB into BMMs from aly/aly mice. The overexpression of p65 failed to rescue RANKL-induced osteoclastogenesis in aly/aly BMMs (Fig. 2, A–C). In contrast, overexpression of RelB rescued RANKL-induced osteoclastogenesis in aly/aly mice (Fig. 2, A and B). Furthermore, the overexpression of RelB induced NF-κB2 processing and NFATc1 expression (Fig. 2C).
FIGURE 2.
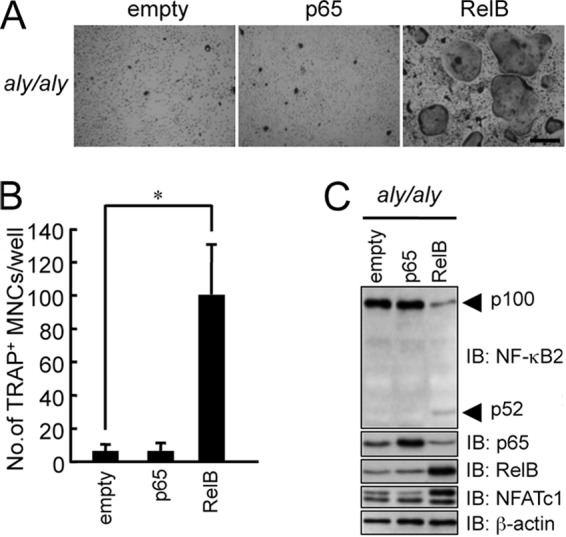
Overexpression of RelB rescued RANKL-induced osteoclastogenesis in aly/aly BMMs. BMMs from aly/aly mice were transfected with an empty vector, p65, or RelB and subsequently with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. A, microscopic view of TRAP+ MNCs in BMMs from aly/aly mice transfected with empty vector, p65, or RelB. Scale bar, 100 μm. B, the data shown are the number of TRAP+ MNCs from cultured aly/aly BMMs per culture well. The data are means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs from aly/aly mice were transfected with an empty vector, p65, or RelB, the total cell lysates were immunoblotted (IB) with specific antibodies as indicated, and β-actin was used as a loading control.
We previously reported that the osteoclastogenesis induced by RANKL correlated well with the ratio of p52 to p100 (15). As reported previously, overexpression of p100ΔGRR, which cannot be processed to p52 (28), strongly inhibited RANKL-induced osteoclastogenesis in NF-κB2−/− BMMs (Fig. 3, A and B). To examine whether NF-κB2 processing or the nuclear translocation of RelB in the nucleus is more important for RANKL-induced osteoclastogenesis, we transfected RelB into BMMs from NF-κB2−/− mice in the presence of p100ΔGRR. The overexpression of RelB failed to restore RANKL-induced osteoclastogenesis in NF-κB2−/− BMMs in the presence of p100ΔGRR, which suggested that NF-κB2 processing was more important for RANKL-induced osteoclastogenesis than RelB nuclear translocation (Fig. 3, A–C).
FIGURE 3.
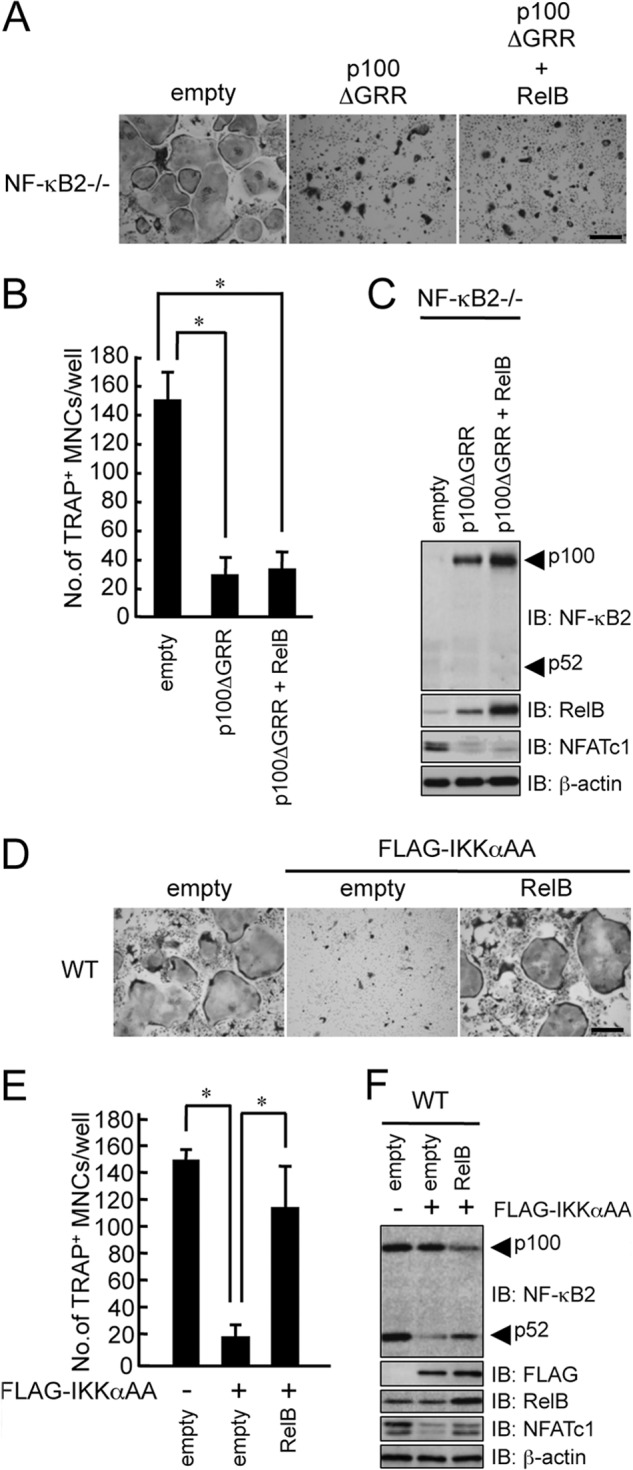
The overexpression of RelB failed to rescue RANKL-induced osteoclastogenesis in the presence of p100 in NF-κB2−/− BMMs. BMMs from NF-κB2−/− mice were transfected with an empty vector, p100ΔGRR, or p100ΔGRR with RelB and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. A, microscopic view of TRAP+ MNCs in BMMs from NF-κB2−/− mice transfected with empty vector, p100ΔGRR, or p100ΔGRR with RelB. Scale bar, 100 μm. B, the data shown are the number of TRAP+ MNCs from NF-κB2−/− BMMs per culture well and represent the means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs from NF-κB2−/− mice were transfected with an empty vector, p100ΔGRR, or p100ΔGRR with RelB, total cell lysates were immunoblotted (IB) with specific antibodies as indicated, and β-actin was used as a loading control. D, BMMs from WT mice were transfected with an empty vector, IKKαAA, or IKKαAA with RelB and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. The figure shows a microscopic view of TRAP+ MNCs in BMMs from WT mice that were transfected with empty vector, IKKαAA, or IKKαAA with RelB. Scale bar, 100 μm. E, the data shown are the number of TRAP+ MNCs from WT BMMs per culture well. The data represent the means ± S.D. (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. F, BMMs from WT mice were transfected with an empty vector, IKKαAA, or IKKαAA with RelB; total cell lysates were immunoblotted with specific antibodies as indicated; and β-actin was used as a loading control.
NIK phosphorylation of IKKα at serine residues 176 and 180 is required for downstream signaling (29, 30). Therefore, we mutated these serine residues to alanine (IKKαAA) and then transfected BMMs from WT mice with either IKKαAA or IKKαAA and RelB. Although the overexpression of IKKαAA suppressed RANKL-induced osteoclastogenesis by suppressing NF-κB2 processing and NFATc1 induction in WT BMMs, the overexpression of RelB restored these inhibitions by IKKαAA (Fig. 3, D–F). These results strongly suggested that the overexpression of RelB induced p100 processing by a mechanism that was independent of these NIK-phosphorylated sites of IKKα.
The Overexpression of RelB Induces Expression of Cot, Which Contributes to the Processing of NF-κB2
To further examine the NIK-independent molecular mechanism by which RelB overexpression rescued RANKL-induced osteoclastogenesis in aly/aly BMMs, we performed microarray analysis using RelB-transfected BMMs. Among the several genes that were up-regulated, we focused on Cot (cancer Osaka thyroid), which is an MAP3K family member (19) that activates NF-κB. BMMs from either WT or aly/aly mice were transfected with or without RelB and then treated with RANKL. RANKL induced Cot expression in the WT BMMs but not in the aly/aly BMMs (Fig. 4A). However, the overexpression of RelB strongly induced the expression of Cot (Fig. 4A) in WT and aly/aly BMMs (Fig. 4A).
FIGURE 4.

The overexpression of RelB rescued RANKL-induced osteoclastogenesis from aly/aly BMMs by inducing Cot expression. A, BMMs from WT or aly/aly mice were transfected with or without RelB and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). Total RNA of BMMs or osteoclasts from WT or aly/aly mice was isolated, and the expression levels of Cot relative to GAPDH were measured using quantitative real-time PCR analysis. The data represent the means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. B, BMMs from WT or aly/aly mice were treated with RANKL (100 ng/ml) for the indicated time. Whole cell lysates were harvested from cultured cells and subjected to immunoblot analysis with the indicated antibodies. An antibody against β-actin was used as a loading control. C, a deletion analysis of the upstream of transcription initiation site of the mouse Cot gene using luciferase reporter constructs. The deletion constructs were generated from Cot0.5-luc. The 293 cells were individually transfected with the reporter constructs, with or without p65, RelB, or RelB and p52. The luciferase activity was measured 48 h after transfection. The data represent the means ± S.D. (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. RLU, relative luciferase units. D, BMMs isolated from WT or aly/aly mice were treated with RANKL (100 ng/ml) for the indicated time. Chromatin from individual samples was precipitated using the indicated antibodies or control immunoglobulin G. The Cot promoter containing distal or proximal NF-κB-responsive elements was amplified by PCR from precipitated DNA.
To confirm that RANKL induces Cot expression in WT and aly/aly BMMs, BMMs from either WT or aly/aly mice were treated with RANKL for the indicated time. RANKL transiently induced expression of Cot at 30 min and further induced Cot from 6 to 48 h after RANKL stimulation in WT BMMs. The induction of Cot attained a maximal level 30 min after RANKL stimulation and declined thereafter in aly/aly BMMs (Fig. 4B). As reported previously (15), NF-κB2 processing and NFATc1 induction were impaired in aly/aly BMMs compared with WT BMMs (Fig. 4B).
To examine the regulatory mechanism by which RelB induces Cot expression, we constructed a series of luciferase reporter plasmids from Cot0.5-luc, which carries a 0.5-kb fragment upstream of the transcription initiation site of the mouse Cot promoter harboring the NF-κB-responsive element. Furthermore, we generated an additional pair of deletion plasmids with 1 or no NF-κB-responsive elements, named Cot0.3-luc and Cot0.1-luc, respectively. We then transfected 293 cells with each of these luciferase reporter plasmids, with or without p65, RelB, or RelB and p52. The expression of p65 significantly stimulated Cot0.5-luc. Although RelB alone failed to stimulate Cot0.5-luc, RelB moderately stimulated Cot0.5-luc in the presence of p52 (Fig. 4C). RelB, but not p65, stimulated Cot0.3-luc. This activity was further enhanced in the presence of p52. Neither p65 nor RelB stimulated Cot0.1-luc in the presence or absence of p52 (Fig. 4C). These data indicate that the distal NF-κB-responsive element was activated mainly by the classical NF-κB pathway and that the proximal NF-κB-responsive element of the Cot promoter was activated by the alternative NF-κB pathway.
ChIP analyses confirmed that p65 was only recruited to the distal NF-κB-responsive element and not the proximal NF-κB-responsive element of the Cot promoter after 30 min of RANKL stimulation in BMMs isolated from both WT and aly/aly mice. RelB and p52 were recruited to both the distal and proximal NF-κB-responsive elements of the Cot promoter after 12 h of RANKL stimulation in BMMs isolated from WT mice. However, RelB and p52 failed to bind to the proximal NF-κB-responsive element of the Cot promoter in BMMs isolated from aly/aly or RelB−/− mice (Fig. 4D). These results suggest that the alternative NF-κB pathway is involved in the induction and/or maintenance of Cot expression.
To further examine the involvement of RelB-induced Cot expression in RANKL-induced osteoclastogenesis, we knocked down Cot expression using an shRNA that specifically targeted the expression of Cot. Although the overexpression of RelB moderately enhanced RANKL-induced osteoclastogenesis, the knockdown of Cot expression in WT BMMs suppressed RANKL-induced osteoclastogenesis in the presence of RelB (Fig. 5, A and B). In aly/aly BMMs, knocking down of Cot expression significantly suppressed the RANKL-induced osteoclastogenesis that was induced by RelB overexpression (Fig. 5, A and B). It has been reported that IKKα phosphorylates NF-κB2 at serine residues 865 and 869 (Ser-865/869) (13, 32). In our study, RelB overexpression induced phosphorylation and processing of NF-κB2 and expression of NFATc1 in aly/aly BMMs, and knocking down Cot completely inhibited these effects, suggesting that RelB-induced Cot expression triggers the phosphorylation and processing of NF-κB2 and restores RANKL-induced osteoclastogenesis in aly/aly BMMs (Fig. 5).
FIGURE 5.
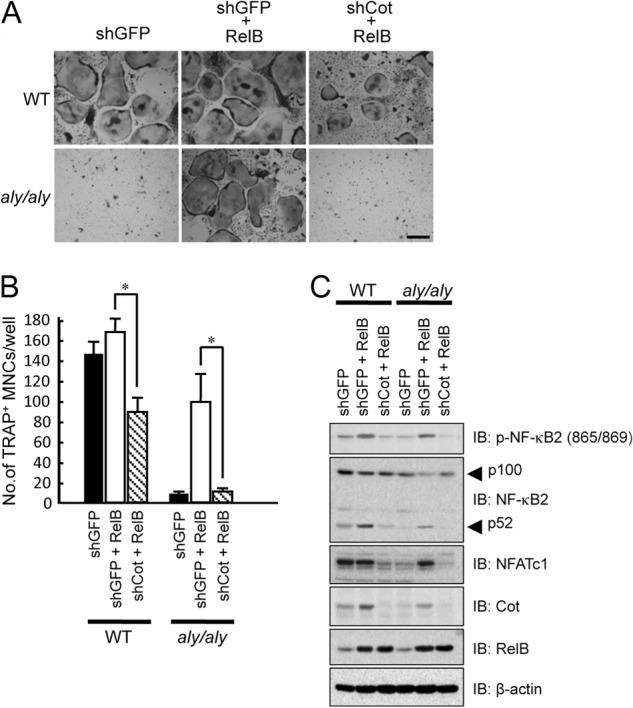
The overexpression of RelB failed to rescue RANKL-induced osteoclastogenesis in the absence of Cot. BMMs isolated from WT or aly/aly mice were transfected with or without RelB in the presence of shGFP or shCot and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. A, the figure shows a microscopic view of TRAP+ MNCs in WT or aly/aly BMMs transfected with or without RelB in the presence of shGFP or shCot. Scale bar, 100 μm. B, the data shown are the number of TRAP+ MNCs per well in cultured WT or aly/aly BMMs and represent the means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs isolated from WT or aly/aly mice were transfected with or without RelB in the presence of shGFP or shCot, and total cell lysates were immunoblotted (IB) with the indicated antibodies. β-Actin was used as a loading control.
We then confirmed that the RelB-dependent expression of Cot was critical for RANKL-induced osteoclastogenesis using RelB−/− BMMs. BMMs from WT or RelB−/− mice were treated with RANKL for the indicated time, and the expression levels of Cot were examined. Similar to aly/aly BMMs, RANKL transiently induced the expression of Cot at 30 min, but not between 6 and 48 h, after RANKL stimulation in RelB−/− BMMs, suggesting that the second wave of Cot induction depends on RelB (Fig. 6A). NF-κB2 processing and NFATc1 induction were also impaired in RelB−/− BMMs compared with in WT BMMs (Fig. 6A).
FIGURE 6.
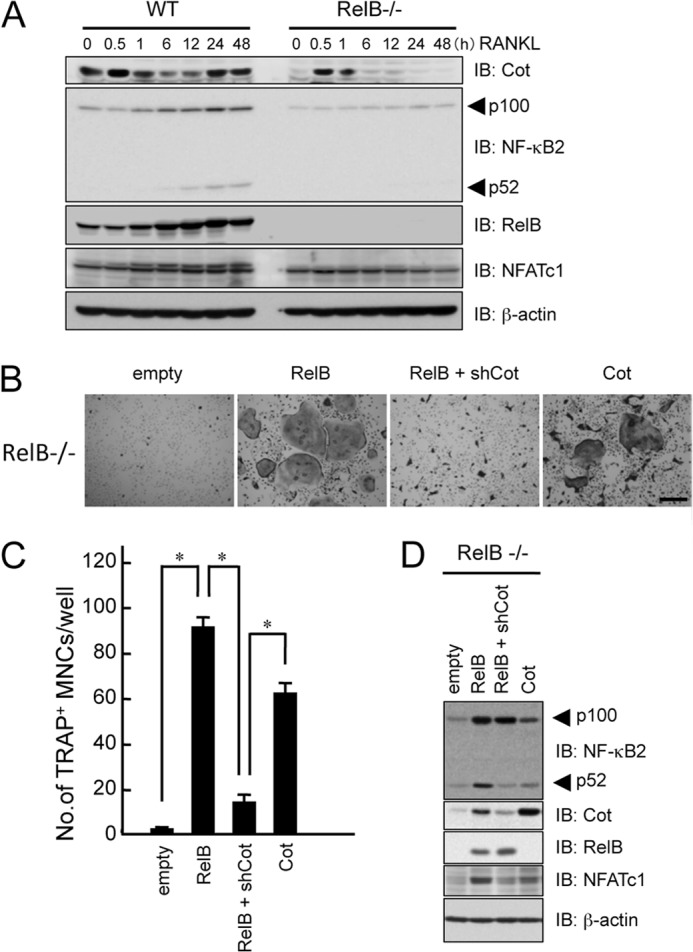
RelB-induced Cot expression is critical for RANKL-induced osteoclasts. A, BMMs from WT or RelB−/− mice were treated with RANKL (100 ng/ml) for the indicated time. Whole-cell lysates were harvested from cultured cells and subjected to immunoblot analysis (IB) with the indicated antibodies. An antibody for β-actin was used as a loading control. B, BMMs from RelB−/− mice were transfected with empty or RelB vectors in the presence or absence of shCot or Cot and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs in BMMs from RelB−/− mice that were transfected with empty or RelB vectors in the presence or absence of shCot or Cot. Scale bar, 100 μm. C, data shown are the number of TRAP+ MNCs from RelB−/− BMMs per culture well. The data are means ± S.D. (error bars) (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. D, BMMs from RelB−/− mice were transfected with empty or RelB vectors in the presence or absence of shCot or Cot, and total cell lysates were immunoblotted with the indicated antibodies. β-Actin was used as a loading control.
Few osteoclasts were formed in the RelB−/− BMMs after RANKL stimulation, and the processing of NF-κB2 was barely detectable (Fig. 6, B and C). The overexpression of RelB in RelB−/− BMMs fully rescued RANKL-induced osteoclastogenesis (Fig. 6, B and C). The induction of Cot expression and NF-κB2 processing was also observed in RelB-overexpressing RelB−/− BMMs (Fig. 6D). In contrast, knocking down Cot suppressed RANKL-induced osteoclastogenesis and the processing of NF-κB2, even in the presence of RelB (Fig. 6). In addition, overexpression of Cot partially rescued RANKL-induced osteoclastogenesis and the processing of NF-κB2 (Fig. 6, B–D). These results strongly indicated that the RelB-dependent expression of Cot was critical for RANKL-induced osteoclastogenesis by the alternative NF-κB pathway.
RelB-induced Cot Phosphorylates Threonine 23 of IKKα, Which Induces Processing of NF-κB2
We then examined whether the Cot-induced processing of NF-κB2 depended on IKKα kinase activity. Knocking down IKKα expression failed to restore the RANKL-induced osteoclastogenesis that was induced by RelB overexpression as well as the inhibition of processing of NF-κB2 and NFATc1 expression in aly/aly BMMs (Fig. 7, A–C).
FIGURE 7.

The overexpression of Cot rescued RANKL-induced osteoclastogenesis from IKKαAA-transfected BMMs but not IKKαKM-transfected BMMs. BMMs from aly/aly mice were transfected with or without RelB in the presence or absence of shCot or shIKKα and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. A, a microscopic view of TRAP+ MNCs with BMMs from aly/aly mice that were transfected with or without RelB in the presence or absence of shCot or shIKKα. Scale bar, 100 μm. B, the data shown are the number of TRAP+ MNCs from aly/aly BMMs per culture well. The data are means ± S.D. (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs from aly/aly mice were transfected with or without RelB in the presence or absence of shCot or shIKKα, and total cell lysates were immunoblotted (IB) with the indicated antibodies. β-Actin was used as a loading control. D, BMMs from WT mice were transfected with or without Cot in the presence of empty vector, IKKαWT, IKKαAA, or IKKαKM and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs with BMMs from WT mice transfected with or without Cot in the presence of empty vector, IKKαWT, IKKαAA, or IKKαKM. Scale bar, 100 μm. E, the data shown are the number of TRAP+ MNCs from WT BMMs per culture well and are represented as the means ± S.D. (n = 3); *, p < 0.01 versus RelB-transfected cells. Similar results were obtained in three independent experiments. F, BMMs from WT mice were transfected with or without Cot in the presence of empty vector, IKKαWT, IKKαAA, or IKKαKM, and total cell lysates were immunoblotted with the indicated antibodies. β-Actin was used as a loading control. IP, immunoprecipitation. G, BMMs isolated from WT mice were transfected with or without Cot in the presence of empty vector, IKKαAA, or IKKαAAT23A and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). The cells were then fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs in WT BMMs transfected with or without Cot in the presence of empty vector, IKKαAA, or IKKαAAT23A. Scale bar, 100 μm. H, the data shown represent the number of TRAP+ MNCs per well in cultured WT BMMs. The data represent the mean ± S.D. (n = 3); *, p < 0.01 versus RelB-transfected cells. Similar results were obtained in three independent experiments. I, BMMs isolated from WT mice were transfected with or without Cot in the presence of empty vector, IKKαAA, or IKKαAAT23A, and total cell lysates were immunoblotted with the indicated antibodies. β-Actin was used as a loading control.
WT BMMs transfected with empty, IKKαWT, IKKαAA, or IKKαKM (kinase-dead) and with or without Cot were treated with RANKL and M-CSF for 6 days. Overexpression of IKKα WT enhanced RANKL-induced osteoclastogenesis by autophosphorylating IKKα at serine 180, compared with control, and overexpression of IKKαWT with Cot further enhanced RANKL-induced osteoclastogenesis by further phosphorylating Thr-23 of IKKα (Fig. 7, D and E). Overexpression of IKKαAA alone markedly suppressed osteoclastogenesis compared with control or IKKαWT (Fig. 7, D and E). In contrast, overexpression of Cot in the presence of IKKαAA recovered osteoclastogenesis compared with IKKαAA alone. Furthermore, overexpression of Cot strongly induced the phosphorylation of IKKα at Thr-23 and the phosphorylation (Ser-865/869) and processing of NF-κB2 (Fig. 7F). To further examine the importance of Cot-induced IKKα phosphorylation at Thr-23, we generated an additional construct, IKKαAAT23A. The overexpression of Cot failed to rescue RANKL-induced osteoclastogenesis because the phosphorylation (Ser-865/869) and processing of NF-κB2 was inhibited (Fig. 7, G–I). Although overexpression of Cot resulted in phosphorylation of IKKα at Thr-23, it failed to induce phosphorylation and processing of NF-κB2 and osteoclastogenesis in the presence of IKKαKM (Fig. 7, D–F). These results strongly indicate that phosphorylation of IKKα at Thr-23 and its kinase activity are important for NF-κB2 processing and osteoclastogenesis by the overexpression of Cot.
Cot and Akt Cooperatively Induce the Processing of NF-κB2 and RANKL-induced Osteoclastogenesis
Previous reports showed that Akt phosphorylates IKKα at Thr-23 (33) and that Akt and Cot physiologically associate and functionally cooperate (34). Thus, we examined the relationship between Akt and Cot during RelB-induced osteoclastogenesis in WT or aly/aly BMMs. First, BMMs isolated from WT or aly/aly mice were transfected with RelB with or without a constitutively activated form of Akt (AktCA) or a dominant-negative form of Akt (AktDN) and were then treated with RANKL and M-CSF for 6 days. In WT BMMs, RelB overexpression slightly enhanced RANKL-induced osteoclastogenesis. The co-expression of AktCA and RelB further enhanced RANKL-induced osteoclastogenesis. As described above, RelB overexpression in aly/aly BMMs rescued RANKL-induced osteoclastogenesis. The co-expression of AktCA and RelB further enhanced RANKL-induced osteoclastogenesis in these cells (Fig. 8, A and B). However, Cot knockdown moderately suppressed these effects in WT BMMs and significantly suppressed the AktCA and RelB-dependent rescue of RANKL-induced osteoclastogenesis in aly/aly BMMs (Fig. 8, A and B). The phosphorylation and processing of NF-κB2 induced by RelB overexpression in both WT and aly/aly BMMs was further enhanced by the co-expression of AktCA and RelB (Fig. 8C). In contrast, AktDN expression strongly inhibited RelB-induced osteoclastogenesis and the phosphorylation and processing of NF-κB2 in WT and aly/aly BMMs (Fig. 8, A–C).
FIGURE 8.

Cot and Akt cooperatively induced the processing of p100 and RANKL-induced osteoclastogenesis. BMMs from WT or aly/aly mice were transfected with empty vector, RelB, or RelB together with AktCA or AktDN in the presence or absence of shCot and subsequently cultures with M-CSF (100 ng/ml) and RANKL (100 ng/ml). A, cells were fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs in BMMs from WT or aly/aly mice transfected with empty vector, RelB, or RelB together with AktCA or AktDN in the presence or absence of shCot. Scale bar, 100 μm. B, data shown are the number of TRAP+ MNCs from WT or aly/aly BMMs per culture well. The data are means ± S.D. (error bars) (n = 3). *, p < 0.01. Similar results were obtained in three independent experiments. C, BMMs from WT or aly/aly mice were transfected with empty vector RelB or RelB together with AktCA or AktDN in the presence or absence of shCot, and then total cell lysates were immunoblotted with specific antibodies as indicated, and β-actin was used as a loading control. D, BMMs from WT or aly/aly mice were transfected with or without AktCA in the presence or absence of shCot and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). Cells were fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs in BMMs from WT or aly/aly mice transfected with or without AktCA in the presence or absence of shCot. Scale bar, 100 μm. E, data shown are the number of TRAP+ MNCs from WT or aly/aly BMMs per culture well. The data are means ± S.D. (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. F, BMMs from WT or aly/aly mice were transfected with empty vector, AktCA in the presence or absence of shCot, and then total cell lysates were immunoblotted (IB) with specific antibodies as indicated, and β-actin was used as a loading control. G, BMMs from WT or aly/aly mice were transfected with or without Cot in the presence or absence of AktDN and subsequently cultured with M-CSF (100 ng/ml) and RANKL (100 ng/ml). Cells were fixed and stained for TRAP. Shown is a microscopic view of TRAP+ MNCs in BMMs from WT mice transfected with or without Cot in the presence or absence of AktDN. Scale bar, 100 μm. H, data shown are the number of TRAP+ MNCs from WT or aly/aly BMMs per culture well. The data are means ± S.D. (n = 3); *, p < 0.01. Similar results were obtained in three independent experiments. I, BMMs from WT or aly/aly mice were transfected with or without Cot in the presence or absence of AktDN, and then total cell lysates were immunoblotted with specific antibodies as indicated, and β-actin was used as a loading control.
To further investigate the relationship between Cot and Akt, BMMs isolated from WT or aly/aly mice were transfected with shCot, with or without AktCA, and then treated with RANKL and M-CSF for 6 days. AktCA moderately enhanced RANKL-induced osteoclastogenesis by enriching the phosphorylation (Ser-865/869) and processing of NF-κB2, whereas Cot knockdown inhibited RANKL-induced osteoclastogenesis in WT BMMs, even in the presence of AktCA (Fig. 8, D–F). The overexpression of AktCA alone failed to promote RANKL-induced osteoclastogenesis in aly/aly cells, possibly due to the markedly reduced expression of Cot in aly/aly BMMs compared with WT BMMs (Fig. 8, D and E).
The slightly enhanced osteoclastogenesis caused by Cot overexpression in WT BMMs was suppressed in the presence AktDN (Fig. 8, G and H). Cot overexpression promoted RANKL-induced osteoclastogenesis by enhancing the phosphorylation (Ser-865/869) and processing of NF-κB2 and increasing NFATc1 expression, whereas co-transfection of Cot and AktDN negated these effects (Fig. 8, G–I). These results suggest that RelB-induced Cot collaborates with Akt to regulate NF-κB2 processing and RANKL-induced osteoclastogenesis.
DISCUSSION
We previously reported that aly/aly mice showed an osteopetrotic phenotype and a reduced number of osteoclasts (14, 15). We also showed a strong correlation between the number of osteoclasts that were induced by RANKL and the ratio of p52 to p100 expression (15). Furthermore, previous reports showed that the overexpression of RelB, but not p65, in NIK-deficient cells rescued RANKL-induced osteoclastogenesis (18). However, the molecular mechanism by which the overexpression of RelB rescued RANKL-induced osteoclastogenesis has not yet been examined. Therefore, the goal of this study was to define how the overexpression of RelB rescues RANKL-induced osteoclastogenesis in aly/aly BMMs. We have shown here that Cot induction by RelB overexpression is a critical factor for RANKL-induced osteoclastogenesis in aly/aly BMMs.
In the alternative pathway, NIK and IKKα are required for NF-κB2 processing and allow for the nuclear translocation of RelB, which may, in turn, heterodimerize with either p52 or p50 (10, 11). We showed that the accumulation of p100 acts as an inhibitor by binding to RelB, but not p65, and preventing its nuclear translocation, thereby inhibiting its DNA binding activity in aly/aly BMMs and aly/aly lymphocytes (35). Thus, the overexpression of RelB forces nuclear translocation and restores RANKL-induced osteoclastogenesis in aly/aly BMMs by inducing NF-κB2 processing. Furthermore, RelB overexpression failed to rescue RANKL-induced osteoclastogenesis in the presence of p100ΔGRR. Indeed, RelB overexpression rescued RANKL-induced osteoclastogenesis in IKKαAA-transfected BMMs. These results strongly indicate that NF-κB2 processing is more important than RelB translocation into the nucleus for inducing osteoclastogenesis. Furthermore, the overexpression of RelB activates IKKα by a NIK-independent mechanism to induce the processing of NF-κB2.
Using a genome-wide screen, we observed that Cot is induced in RelB-overexpressing aly/aly BMMs. We confirmed that the protein expression of Cot is markedly increased during osteoclastogenesis in WT BMMs but not in aly/aly or RelB−/− BMMs. We also found that there are two NF-κB-responsive elements from upstream of the transcription initiation site of the Cot promoter, one in the distal and one in the proximal region. Although p65 and RelB, together with p52, bound the distal NF-κB-responsive element, RelB, together with p52 but not p65, bound the proximal NF-κB-responsive element in the Cot promoter. The overexpression of RelB also induced the phosphorylation of NF-κB2 at Ser-865/869, which are phosphorylated by IKKα (13, 32), suggesting that the overexpression of RelB enhanced IKKα kinase activity. The loss of Cot function via an shRNA that targeted Cot significantly suppressed the RANKL-dependent osteoclastogenesis that was induced by RelB overexpression in WT and aly/aly BMMs. Indeed, the overexpression of Cot directly or indirectly resulted in the phosphorylation of IKKα at Thr-23 and rescued the RANKL-induced osteoclastogenesis in IKKαAA-transfected BMMs but not in IKKαAAT23A- or IKKαKM-transfected BMMs.
Consistent with previous reports that showed that Cot induces NF-κB1 processing by activating IKK (36, 37), RelB-induced Cot induced NF-κB2 processing via IKKα activation. Taken together, these data suggest that RelB-induced Cot activates IKKα kinase activity by phosphorylating Thr-23, which ultimately activates NF-κB2 processing.
A previous study showed that Cot-deficient (Cot−/−) mice exhibited normal bone density and that Cot−/− BMMs differentiated normally into osteoclasts in the presence of RANKL with M-CSF (38). Cot−/− BMMs exhibit severely impaired osteoclastogenesis when co-cultured with mouse ST2 bone marrow stromal cells in the presence of FK506, which is a calcineurin inhibitor. Although osteoclastogenesis by IP3R2/Cot double knock-out BMMs was substantially impaired, IP3R2/Cot double knock-out mice had normal bone density. One possible explanation for the discrepancy between these in vitro and in vivo phenotypes is the suppression of bone formation in the IP3R2/Cot double knock-out mice. In contrast, 1,7-naphthyridine-3-carbonitrile, which is a selective inhibitor of Cot, suppresses RANKL-induced osteoclastogenesis in RAW264.7 cells by inhibiting ERK but not JNK or p38 activities (39). These discrepancies are probably due to differences in experimental strategies, such as the use of genetic versus pharmacological inhibition. Although the role of Cot in osteoclastogenesis is still controversial, our results clearly provide another example of RelB-induced Cot expression rescuing RANKL-induced osteoclastogenesis by inducing NF-κB2 processing independently of NIK (Fig. 9).
FIGURE 9.
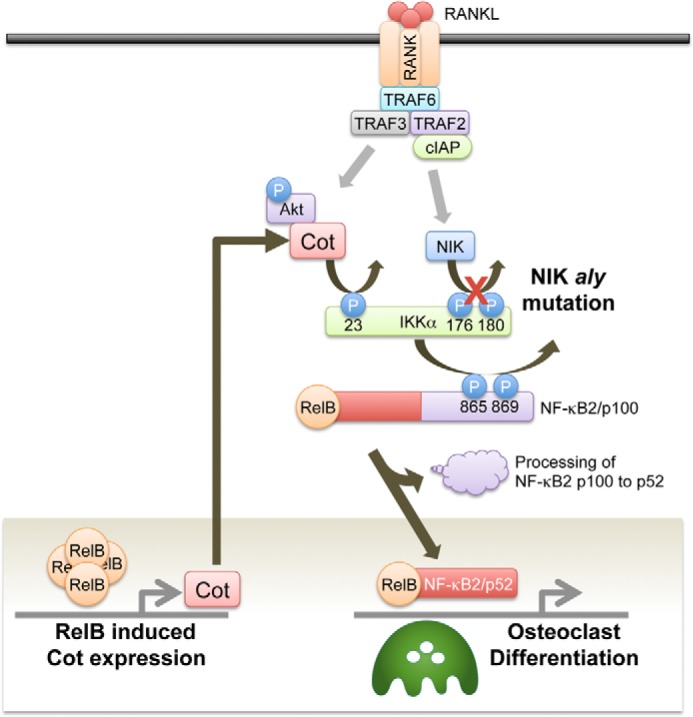
A schema of the molecular mechanism by which the overexpression of RelB rescued RANKL-induced osteoclastogenesis from aly/aly BMMs. RelB-induced Cot activates IKKα kinase activity by phosphorylating Thr-23, which ultimately activates NF-κB2 processing and induces RANKL-induced osteoclastogenesis in aly/aly BMMs.
There are three Akt family members: Akt1, Akt2, and Akt3 (40, 41). Akt1 and Akt2, but not Akt3, are abundantly expressed in both osteoblasts and osteoclasts (42). Although mice lacking a single Akt isoform exhibited a mild phenotype, Akt1/Akt2 double KO mice displayed severely impaired bone development and dwarfism (43). Akt1−/− mice display impaired bone resorption via cell autonomous effects on differentiation and survival (42). Ablation of both Akt1 and Akt2 inhibited osteoclastogenesis by down-regulating the RANKL-induced DNA binding of NF-κB (44). Furthermore, LY29400, a specific inhibitor of PI3K, strongly inhibited RANKL-induced Akt activation and osteoclastogenesis, whereas other signaling pathways, such as JNK, p38, and NF-κB, were not affected (45). The overexpression of constitutively activated Akt strongly enhanced RANKL-induced osteoclastogenesis via the NFATc1 signaling cascade (45). Thus, Akt activation regulates RANKL-induced osteoclastogenesis by modulating NF-κB signaling.
Akt directly phosphorylates IKKα at Thr-23 (13, 31). Furthermore, Akt and Cot can physically associate through the amino terminus of Cot, and Akt mediates the phosphorylation of Cot (34). Cot binding to Akt is critical for Cot function because Akt phosphorylates Cot at serine 400, and this phosphorylation is necessary for Cot induction of NF-κB-dependent transcription (35). Although AktCA alone failed to induce NF-κB2 processing and RANKL-induced osteoclastogenesis in aly/aly BMMs, AktCA induced NF-κB2 processing and RANKL-induced osteoclastogenesis in aly/aly BMMs in the presence of RelB. Moreover, knocking down Cot by shCot suppressed the NF-κB2 processing and RANKL-induced osteoclastogenesis in aly/aly BMMs that were induced by AktCA and RelB. These results suggest that Akt and Cot cooperatively form a complex with IKKα and that the complex activates the NF-κB alternative pathway and RANKL-induced osteoclastogenesis in aly/aly BMMs.
In conclusion, we have identified, at least in part, a previously unappreciated role for the alternative NF-κB pathway in osteoclast differentiation. Although the activation of NIK/IKKα may be important for RANKL-induced osteoclastogenesis, the Akt/Cot/IKKα pathway induced by RelB also contributes, in part, to RANKL-induced osteoclastogenesis. An in depth understanding of the molecular mechanism of osteoclast differentiation by the alternative NF-κB pathway should help to provide a molecular basis for future therapeutic strategies targeting bone disease.
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology of Japan Grants 23390424 (to E. J.), 23593039 (to M. K.), and 24890214 (to K. O.).
- NF-κB
- nuclear factor-κB
- NIK
- NF-κB-inducing kinase
- IKK
- IκB kinase
- TRAP
- tartrate-resistant acid phosphatase
- BMM
- bone marrow macrophage
- MNC
- multinucleated cell.
REFERENCES
- 1. Matsuo K., Otaki N. (2012) Bone cell interactions through Eph/ephrin. Bone modeling, remodeling and associated diseases. Cell Adh. Migr. 6, 148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanaka S. (2013) Regulation of bone destruction in rheumatoid arthritis through RANKL-RANK pathways. World J. Orthop. 4, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakashima T., Hayashi M., Takayanagi H. (2012) New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 23, 582–590 [DOI] [PubMed] [Google Scholar]
- 4. Kong Y. Y., Yoshida H., Sarosi I., Tan H. L., Timms E., Capparelli C., Morony S., Oliveira-dos-Santos A. J., Van G., Itie A., Khoo W., Wakeham A., Dunstan C. R., Lacey D. L., Mak T. W., Boyle W. J., Penninger J. M. (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323 [DOI] [PubMed] [Google Scholar]
- 5. Dougall W. C., Glaccum M., Charrier K., Rohrbach K., Brasel K., De Smedt T., Daro E., Smith J., Tometsko M. E., Maliszewski C. R., Armstrong A., Shen V., Bain S., Cosman D., Anderson D., Morrissey P. J., Peschon J. J., Schuh J. (1999) RANK is essential for osteoclast and lymph node development. Genes Dev. 13, 2412–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson D. M., Maraskovsky E., Billingsley W. L., Dougall W. C., Tometsko M. E., Roux E. R., Teepe M. C., DuBose R. F., Cosman D., Galibert L. (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390, 175–179 [DOI] [PubMed] [Google Scholar]
- 7. Jimi E., Akiyama S., Tsurukai T., Okahashi N., Kobayashi K., Udagawa N., Nishihara T., Takahashi N., Suda T. (1999) Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J. Immunol. 163, 434–442 [PubMed] [Google Scholar]
- 8. Iotsova V., Caamaño J., Loy J., Yang Y., Lewin A., Bravo R. (1997) Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 3, 1285–1289 [DOI] [PubMed] [Google Scholar]
- 9. Franzoso G., Carlson L., Xing L., Poljak L., Shores E. W., Brown K. D., Leonardi A., Tran T., Boyce B. F., Siebenlist U. (1997) Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 11, 3482–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DiDonato J. A., Mercurio F., Karin M. (2012) NF-κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400 [DOI] [PubMed] [Google Scholar]
- 11. Ghosh S., Hayden M. S. (2012) Celebrating 25 years of NF-κB research. Immunol. Rev. 246, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shinkura R., Kitada K., Matsuda F., Tashiro K., Ikuta K., Suzuki M., Kogishi K., Serikawa T., Honjo T. (1999) Alymphoplasia is caused by a point mutation in the mouse gene encoding NF-κB-inducing kinase. Nat. Genet. 22, 74–77 [DOI] [PubMed] [Google Scholar]
- 13. Xiao G., Harhaj E. W., Sun S. C. (2001) NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 7, 401–409 [DOI] [PubMed] [Google Scholar]
- 14. Soysa N. S., Alles N., Weih D., Lovas A., Mian A. H., Shimokawa H., Yasuda H., Weih F., Jimi E., Ohya K., Aoki K. (2010) The pivotal role of the alternative NF-κB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J. Bone Miner. Res. 25, 809–818 [DOI] [PubMed] [Google Scholar]
- 15. Maruyama T., Fukushima H., Nakao K., Shin M., Yasuda H., Weih F., Doi T., Aoki K., Alles N., Ohya K., Hosokawa R., Jimi E. (2010) Processing of the NF-κB2 precursor, p100, to p52 is critical for RANKL-induced osteoclast differentiation. J. Bone Miner. Res. 25, 1058–1067 [DOI] [PubMed] [Google Scholar]
- 16. Tucker E., O'Donnell K., Fuchsberger M., Hilton A. A., Metcalf D., Greig K., Sims N. A., Quinn J. M., Alexander W. S., Hilton D. J, Kile B. T., Tarlinton D. M., Starr R. (2007) A novel mutation in the Nfkb2 gene generates an NF-κB2 “super repressor”. J. Immunol. 179, 7514–7522 [DOI] [PubMed] [Google Scholar]
- 17. Novack D. V., Yin L., Hagen-Stapleton A., Schreiber R. D., Goeddel D. V., Ross F. P., Teitelbaum S. L. (2003) The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 198, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaira S., Johnson T., Hirbe A. C., Alhawagri M., Anwisye I., Sammut B., O'Neal J., Zou W., Weilbaecher K. N., Faccio R., Novack D. V. (2008) RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 3897–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyoshi J., Higashi T., Mukai H., Ohuchi T., Kakunaga T. (1991) Structure and transforming potential of the human cot oncogene encoding a putative protein kinase. Mol. Cell Biol. 11, 4088–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiariello M., Marinissen M. J., Gutkind J. S. (2000) Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol. Cell Biol. 20, 1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin X., Cunningham E. T., Jr., Mu Y., Geleziunas R., Greene W. C. (1999) The proto-oncogene Cot kinase participates in CD3/CD28 induction of NF-κB acting through the NF-κB-inducing kinase and IκB kinases. Immunity 10, 271–280 [DOI] [PubMed] [Google Scholar]
- 22. Macpherson A. J., Uhr T. (2003) The donor splice site mutation in NF-κB-inducing kinase of alymphoplasia (aly/aly) mice. Immunogenetics 54, 693–698 [DOI] [PubMed] [Google Scholar]
- 23. Fukushima H., Nakao A., Okamoto F., Shin M., Kajiya H., Sakano S., Bigas A., Jimi E., Okabe K. (2008) The association of Notch2 and NF-κB accelerates RANKL-induced osteoclastogenesis. Mol. Cell Biol. 28, 6402–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning. Powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 25. Nosaka T., Kawashima T., Misawa K., Ikuta K., Mui A. L., Kitamura T. (1999) STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 18, 4754–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong H., May M. J., Jimi E., Ghosh S. (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol. Cell 9, 625–636 [DOI] [PubMed] [Google Scholar]
- 27. Fusco A. J., Savinova O. V., Talwar R., Kearns J. D., Hoffmann A., Ghosh G. (2008) Stabilization of RelB requires multidomain interactions with p100/p52. J. Biol. Chem. 283, 12324–12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heusch M., Lin L., Geleziunas R., Greene W. C. (1999) The generation of nfkb2 p52. Mechanism and efficiency. Oncogene 18, 6201–6208 [DOI] [PubMed] [Google Scholar]
- 29. Ling L., Cao Z., Goeddel D. V. (1998) NF-κB-inducing kinase activates IKKα by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U.S.A. 95, 3792–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delhase M., Hayakawa M., Chen Y., Karin M. (1999) Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284, 309–313 [DOI] [PubMed] [Google Scholar]
- 31. Ozes O. N., Mayo L. D., Gustin J. A., Pfeffer S. R., Pfeffer L. M., Donner D. B. (1999) NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401, 82–85 [DOI] [PubMed] [Google Scholar]
- 32. Xiao G., Fong A., Sun S. C. (2004) Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 279, 30099–30105 [DOI] [PubMed] [Google Scholar]
- 33. Yuan Z. Q., Feldman R. I., Sun M., Olashaw N. E., Coppola D., Sussman G. E., Shelley S. A., Nicosia S. V., Cheng J. Q. (2002) Inhibition of JNK by cellular stress- and tumor necrosis factor α-induced AKT2 through activation of the NF-κB pathway in human epithelial Cells. J. Biol. Chem. 277, 29973–29982 [DOI] [PubMed] [Google Scholar]
- 34. Kane L. P., Mollenauer M. N., Xu Z., Turck C. W., Weiss A. (2002) Akt-dependent phosphorylation specifically regulates Cot induction of NF-κB-dependent transcription. Mol. Cell Biol. 22, 5962–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramakrishnan P., Wang W., Wallach D. (2004) Receptor-specific signaling for both the alternative and the canonical NF-κB activation pathways by NF-κB-inducing kinase. Immunity 21, 477–489 [DOI] [PubMed] [Google Scholar]
- 36. Waterfield M. R., Zhang M., Norman L. P., Sun S. C. (2003) NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell 11, 685–694 [DOI] [PubMed] [Google Scholar]
- 37. Beinke S., Robinson M. J., Hugunin M., Ley S. C. (2004) Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol. Cell. Biol. 24, 9658–9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuroda Y., Hisatsune C., Mizutani A., Ogawa N., Matsuo K., Mikoshiba K. (2012) Cot kinase promotes Ca2+ oscillation/calcineurin-independent osteoclastogenesis by stabilizing NFATc1 protein. Mol. Cell Biol. 32, 2954–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hirata K., Taki H., Shinoda K., Hounoki H., Miyahara T., Tobe K., Ogawa H., Mori H., Sugiyama E. (2010) Inhibition of tumor progression locus 2 protein kinase suppresses receptor activator of nuclear factor-κB ligand-induced osteoclastogenesis through down-regulation of the c-Fos and nuclear factor of activated T cells c1 genes. Biol. Pharm. Bull. 33, 133–137 [DOI] [PubMed] [Google Scholar]
- 40. Altomare D. A., Guo K., Cheng J. Q., Sonoda G., Walsh K., Testa J. R. (1995) Cloning, chromosomal localization and expression analysis of the mouse Akt2 oncogene. Oncogene 11, 1055–1060 [PubMed] [Google Scholar]
- 41. Altomare D. A., Lyons G. E., Mitsuuchi Y., Cheng J. Q., Testa J. R. (1998) Akt2 mRNA is highly expressed in embryonic brown fat and the AKT2 kinase is activated by insulin. Oncogene 16, 2407–2411 [DOI] [PubMed] [Google Scholar]
- 42. Kawamura N., Kugimiya F., Oshima Y., Ohba S., Ikeda T., Saito T., Shinoda Y., Kawasaki Y., Ogata N., Hoshi K., Akiyama T., Chen W.S., Hay N., Tobe K., Kadowaki T., Azuma Y., Tanaka S., Nakamura K., Chung U. I., Kawaguchi H. (2007) Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS One 2, e1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng X. D., Xu P. Z., Chen M. L., Hahn-Windgassen A., Skeen J., Jacobs J., Sundararajan D., Chen W. S., Crawford S. E., Coleman K. G., Hay N. (2003) Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 17, 1352–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sugatani T., Hruska K. A. (2005) Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J. Biol. Chem. 280, 3583–3589 [DOI] [PubMed] [Google Scholar]
- 45. Moon J. B., Kim J. H., Kim K., Youn B. U., Ko A., Lee S. Y., Kim N. (2012) Akt induces osteoclast differentiation through regulating the GSK3β/NFATc1 signaling cascade. J. Immunol. 188, 163–169 [DOI] [PubMed] [Google Scholar]