

Abstract
Flavin-dependent monooxygenases and oxidoreductases are located at critical branch points in the biosynthesis and metabolism of cholesterol and vitamin D. These flavoproteins function as obligatory intermediates that accept 2 electrons from NAD(P)H with subsequent 1-electron transfers to a variety of cytochrome P450 (CYP) heme proteins within the mitochondria matrix (type I) and the (microsomal) endoplasmic reticulum (type II). The mode of electron transfer in these systems differs slightly in the number and form of the flavin prosthetic moiety. In the type I mitochondrial system, FAD-adrenodoxin reductase interfaces with adrenodoxin before electron transfer to CYP heme proteins. In the microsomal type II system, a diflavin (FAD/FMN)-dependent cytochrome P450 oxidoreductase [NAD(P)H-cytochrome P450 reductase (CPR)] donates electrons to a multitude of heme oxygenases. Both flavoenzyme complexes exhibit a commonality of function with all CYP enzymes and are crucial for maintaining a balance of cholesterol and vitamin D metabolites. Deficits in riboflavin availability, imbalances in the intracellular ratio of FAD to FMN, and mutations that affect flavin binding domains and/or interactions with client proteins result in marked structural alterations within the skeletal and central nervous systems similar to those of disorders (inborn errors) in the biosynthetic pathways that lead to cholesterol, steroid hormones, and vitamin D and their metabolites. Studies of riboflavin deficiency during embryonic development demonstrate congenital malformations similar to those associated with genetic alterations of the flavoenzymes in these pathways. Overall, a deeper understanding of the role of riboflavin in these pathways may prove essential to targeted therapeutic designs aimed at cholesterol and vitamin D metabolism.
Introduction
Riboflavin [7,8-dimethyl-(N-10-ribityl) isoalloxazine] or vitamin B-2 is synthesized by plants and bacteria and used by animal cells to form the flavin nucleotide coenzymes. Flavin nucleotides are essential for cell growth and development, and a deficiency of riboflavin can lead to clinical abnormalities that range from hemolytic anemia to growth retardation and neurologic dysfunctions. Marginal riboflavin deficiency as well as development of frank deficiency states can result from digestive and malabsorptive disorders that may involve intestinal resection or bypass, drug interactions, and alcohol abuse. In addition, rare congenital defects of riboflavin transport can cause persistent riboflavin deficiency. It is important to note that physical and clinical symptoms of riboflavin deficiency are not unique to riboflavin. Due to interactions of B vitamins and their interdependency, the classical signs of glossitis, angular stomatitis, cheilosis, and dermatitis may be observed in deficiencies of other B vitamins as well. A comprehensive review of riboflavin metabolism, including its antioxidant activities, involvement in cell signaling, role as a coenzyme, and clinical aspects of deficiency, has recently been published (1).
Flavoenzymes are capable of conducting electron transfer reactions. The underlying mechanisms that govern these reactions are based predominantly on the ability of the heterocyclic isoalloxazine ring of FMN and FAD to accept 1 or 2 electrons. Diagrammatic representations of riboflavin and its coenzymic nucleotide forms are shown in Fig. 1. The flavin nucleotides function with their client proteins as mediators between 1-electron acceptor/donor and 2-electron acceptor/donor activities that catalyze oxidation-reduction reactions involved primarily in energy, carbohydrate, lipid, and amino acid metabolism. In consideration of the major metabolic pathways that flavoenzymes influence, nutritional deficiencies of riboflavin impact primarily on lipid metabolism. Flavin coenzymes serve an eclectic array of proteins that function as electron transferases, dehydrogenases, oxidoreductases, monooxygenases, hydroxylases, and oxidases for reactions that desaturate essential FAs, form phospho- and ether lipids, and synthesize sphingosine, cholesterol, and steroids (1). Individuals who experience even marginal degrees of riboflavin deficiency can experience skin dyscrasias characteristic of those observed during essential FA deficiencies. Also, riboflavin deficiency can lead to a rapid and marked decrease in hepatic mitochondrial FA oxidation. Needle biopsies of the liver indicate fatty metamorphosis and those of muscle reveal vacuolar myopathy with lipid accumulation.
FIGURE 1.

Structures of flavin coenzymes. The isoalloxazine ring is shown relative to riboflavin and its nucleotide coenzymes, riboflavin 5′-phosphate (FMN) and FAD. The shaded area depicts the region of the isoalloxazine ring that is involved in the direct transfer of electrons from the flavoenzyme. Once transferred, electrons can migrate within the heterocyclic isoalloxazine ring.
Flavins also assist in the catalytic activation and degradation of other vitamins. For example, flavoenzymes affect de novo biosynthesis of ascorbic acid (2, 3), control the conversion of pyridoxine and vitamin K to their physiologically active forms (4–7), protect vitamins from oxidative degradation (8–10), and function conjointly with other vitamin-dependent enzymes, e.g., pyruvate dehydrogenase complex and respiratory chain complexes (11). Such interactions are the basis for the interdependency among vitamins and an underlying cause of secondary vitamin deficiencies. This interconnection ultimately results in the overlapping clinical signs and multiple sequelae that occur during vitamin deficiencies. In view of these interactions, the availability of riboflavin, its conversion to FMN and FAD, and their association with flavoenzymes can markedly affect the metabolism of folate, pyridoxine, vitamin K, niacin, and ascorbate. In light of these interactions, this review will address the critical role of flavoproteins in controlling cholesterogenesis and steroidogenesis and in regulating the biosynthesis and transformations of metabolites from these pathways into vitamin D.
Metabolically active vitamin D can be obtained from diet-derived, lipid-soluble phyto- and zoosterol prohormones, namely vitamin D2 [9,10-seco(5Z,7E)-5,7,10(19),21-ergostatetraene-3β-ol; ergocalciferol] and vitamin D3 [9,10-seco(5Z,7E)-5,7,10(19)-cholestatriene-3β-ol; cholecalciferol], respectively. In addition to its availability from diet and supplements, cholecalciferol can be synthesized by dermal keratinocytes after cutaneous exposure to UV radiation (UV-B, 270–300 nm). The nonenzymatic reaction involves a photolytic fission of the 9,10-carbon bond within the B ring of the sterol ring system, resulting in a seco (ring opened)-steroid. This results in conversion of provitamin D3 (7-dehydrocholesterol) to a previtamin D3 (6-s-cis form), which undergoes a thermally induced E/Z isomerization to form 6-s-trans, cholecalciferol (12). Both ergocalciferol and cholecalciferol must be converted into physiologically active vitamin D metabolites through 2 separate ring hydroxylations with subsequent formation of 1α,25-dihydroxyvitamin D [1α,25(OH)2D]3. Current investigations in healthy individuals with negligible UV-B exposure suggest that ergocalciferol and cholecalciferol are equipotent in their capacity to be 25-hydroxylated to form 25-hydroxyvitamin D [25(OH)D], the major circulating form of vitamin D (13, 14). For this review we will assume that the 25-hydroxylations of ergocalciferol and cholecalciferol to form their respective 25(OH)D2 and 25(OH)D3 metabolites as well as the 1α-hydroxylations of the latter 2 metabolites to form 1α,25(OH)2D derivatives, the active principal of the vitamin D hormone, are comparable.
Current Status of Knowledge
Molecular reactivity of the isoalloxazine ring
Riboflavin serves as the precursor for both FMN and FAD and is the functional moiety that accepts electrons. Both FMN and FAD coenzymes in association with their client flavoproteins participate in critical cellular processes that include energy and lipid metabolism, redox signaling, programmed cell death, growth regulation, xenobiotic defense responses, and regulation of biologic rhythms (15–19). These processes depend on the unique ability of the isoalloxazine moiety to react with NAD(P)H, molecular oxygen, electron-rich metabolites, and occasionally with UV light to catalyze 1- and 2-electron transfers. The heterocyclic nature of the isoalloxazine ring system coupled with its photodynamic properties enable flavin coenzymes to become potent oxidizing agents with a wide range of electron-donating substrates (20). In addition, the isoalloxazine ring facilitates delocalization of its pi-orbital electrons to sustain 3 stably configured oxidation states: fully oxidized, semiquinone (1-electron reduction), and hydroquinone (2-electron reduction) configurations. Fully oxidized flavins exhibit a planar ring system, which can be distorted by an apoflavoenzyme to accommodate a single nucleophilic attack at the C4a or N5 position and form a flavin semiquinone (Fig. 2).
FIGURE 2.

Nucleophilic attack at the N5 or C4a position of the isoalloxazine ring with formation of a flavosemiquinone. Flavosemiquinones exist either in a neutral radical (FAD•) or in an anionic radical (FAD•−) state. X− and Y− represent nucleophiles.
Semiquinone flavins can exist in either a neutral radical (FAD•) or an anionic radical (FAD•−) configuration. These semiquinone radicals are thermodynamically unstable within aqueous environments but are functionally stabilized by aromatic residues of tyrosine and tryptophan within client apoflavoenzymes (21). Such arrangements within enzyme active sites allow the unpaired electrons to translocate throughout the conjugated heterocyclic ring system. During reactions that require transference of electrons, flavoproteins that geometrically restrict the isoalloxazine structure between its fully oxidized (planar) and fully reduced (nonplanar or butterfly) configuration can stabilize the residence time of electrons that enter and leave the isoalloxazine ring (22). Accordingly, flavoproteins that constrain the isoalloxazine ring in a butterfly configuration at the N5–N10 axis favor electron transfer in the pico- to femtosecond range, whereas flavoenzymes that force a more planar configuration can delay electron delivery to within the nanosecond range (22).
The property of flavins to stabilize 2-electron distributions (i.e., formation of fully reduced flavohydroquinone) and to synchronize delivery of single electron transfers (formation of flavin semiquinones) is indispensable for shuttling electrons through an electron transport chain that involves heme proteins such as the cytochrome P450 (CYP) system or during oxidative phosphorylation within mitochondria. A full 2-electron reduction from NAD(P)H can result in a neutral flavohydroquinone (FADH2) or an anionic flavohydroquinone (FADH−) state, both displaying a “butterfly” conformation along the N5–N10 axis (23). The flavoenzyme is required to shift the conformational equilibrium of the flavin moiety from an electron-accepting mode (flavin shielded) toward an electron-donating (flavin deshielded) mode. This enzyme-induced maneuver would increase electron transfer toward the appropriate electron acceptor, such as the heme nucleus along the electron transport chain, or directly toward molecular oxygen, resulting in formation of superoxide anion (O2•−). Under normal conditions of electron transport, the pyridine ring of NAD(P)H (the electron or hydride ion donor) and isoalloxazine ring of flavins (the electron acceptor) assume a spatial conformation with a stable stacking arrangement between their heterocyclic moieties. This arrangement favors hydride transfer and subsequent release of its electron cargo to acceptor molecules in the electron transport chain (coenzyme Q10, ubiquinol) or directly to heme proteins CYP (24). However, unscheduled release of electrons from the reduced flavin component within mitochondrial complex I can contribute significantly to free radical damage of mitochondrial membranes. When reoxidation of the flavohydro- or semiquinone is impaired, the amount of reactive oxygen species markedly increases.
Flavoproteins and cytochrome P450 heme proteins in synthesis of cholesterol and vitamin D metabolites
The majority of mammalian flavoproteins have single binding domains for either FMN or FAD that serve as electron acceptor-donor moieties that act directly with their respective substrates. Other types of flavoproteins, known as electron transferring flavoproteins (ETFs), function as electron acceptor/donor intermediates between iron-sulfur clusters of other flavoproteins as well as between CYP heme proteins. These interfacing flavoproteins have highly mobile redox-active FAD domains that serve as the key metabolic branch points for acceptance and delivery of electrons (25). The synthesis and subsequent metabolism of cholesterol and vitamin D require a variety of oxidoreductases and hydroxylases that interface with a multitude of CYP heme proteins.
Cytochrome P450 heme proteins are classified as type I or type II on the basis of their association with 2 related NAD(P)H-dependent donor flavoproteins located within the mitochondrial matrix or endoplasmic reticulum, respectively. Although the sequence of electron transfer from the 2 flavoenzyme complexes to their recipient heme proteins bears some similarities, the donor flavoproteins exhibit 2 major differences in the number and form of the flavin moieties.
Mitochondrial (type I) cytochrome P450 system.
The type I mitochondrial enzyme system consists of 3 separate components, namely CYPs, ferredoxin, and ferredoxin reductase. Ferredoxin reductase (also called adrenodoxin reductase) is a 50-kDa flavoprotein located in the mitochondrial inner membrane. NAD(P)H generated within the matrix first interacts with adrenodoxin reductase, which passes electrons to adrenodoxin (or ferredoxin), a 12–14-kDa iron sulfur protein located within the mitochondrial matrix. Adrenodoxin is the electron shuttle between adrenodoxin reductase and CYPs. The pattern of electron shuttle involves NAD(P)H to FAD-adrenodoxin reductase, FAD-adrenodoxin reductase to adrenodoxin, and adrenodoxin to heme protein–associated exchanges and is shown in Fig. 3.
FIGURE 3.

Mitochondrial CYP electron transfer system (type I). IDH and G6PDH generate NAD(P)H, which transfers a hydride ion (H:) through π-π interaction between the nicotinamide ring of NAD(P)H and the isoalloxazine moiety of FAD-adrenodoxin reductase. A single electron transfer to heme-containing adrenodoxin generates a ferro-heme moiety that interfaces with a variety of CYP heme enzymes. For example, a CYP enzyme (CYP27B1) catalyzes the oxidation of substrate RH (e.g, 25-hydroxyvitamin D) by adding 1 oxygen atom of molecular oxygen to the substrate (RH), producing a hydroxylated product ROH (e.g., 1α,25-dihydroxyvitamin D); the other atom of oxygen is reduced to water. CYP, cytochrome P450; IDH, isocitrate dehydrogenase; G6PDH, glucose-6-phosphate dehydrogenase; ox, oxidized form of adrenodoxin reductase; red, reduced form of adrenodoxin reductase.
Thus, mitochondrial CYPs receive electrons from NAD(P)H, which is generated by isocitrate dehydrogenase and glucose-6-phosphate dehydrogenase located within the mitochondrial matrix (26). Through the intermediate flavoprotein, adrenodoxin reductase, a reducing equivalence of NAD(P)H is relayed to adrenodoxin and subsequently to the substrate to form a hydroxylated product. The diversity of the P450 redox system and the types of protein-protein arrangements with mono- and diflavin oxidoreductases required for electron transfers have recently been reviewed elsewhere (27).
Type I electron transfers exhibit 1 FAD-binding domain, whereas type II electron transfers with NAD(P)H-cytochrome P450 reductase (CPR) [see NAD(P)H cytochrome P450 oxidoreductase (CPR) below] use both FAD and FMN. Type II transfers are described below (see NADPH-cytochrome P450 reductase, CPR) and differ from type I in that CPR has evolved an FMN-binding domain that functions exclusively within the microsomal compartment. Some type II reactions will also recruit cytochrome b5 for electron transfers but only after the first electron has been delivered by the FMN moiety of CPR.
Microsomal (type II) cytochrome P450 system.
A small but biologically important subfamily of flavoenzymes contains both FAD and FMN coenzymes and constitutes the family of diflavin reductases. The crystallographic and amino acid structures of the diflavin reductases suggest that these proteins evolved by a fusion of 2 ancestral genes that encoded the N-terminal portion of the protein, an FMN-containing flavodoxin, and the C-terminal portion, an FAD-containing ferredoxin-NADP(H)-reductase (28). The prototypic members of mammalian diflavin reductases are NO synthases (EC 1.14.13.39), methionine synthase reductase (EC 1.16.1.8), NAD(P)H sulfite reductase (EC 1.8.1.2), cytoplasmic NR1 protein, and CPR (9, 29, 30). It is the last member of these diflavoproteins that is a membrane-bound component of the endoplasmic reticulum. CPR is the obligate partner protein for all microsomal P450 enzymes involved in metabolism of cholesterol, steroids, and vitamin D.
NAD(P)H cytochrome P450 oxidoreductase (CPR).
CPR was the first member of the diflavin family to be isolated and the most extensively characterized. In mammals, a single CPR is responsible for the electron transport to a diverse array of P450s that are involved in the synthesis of biologically important endogenous compounds such as steroids, FAs, and prostaglandins. Thus, knockout mouse models are embryonically lethal (31). In addition to its physiologic functions, CPR plays a vital role in the reductive degradation and sometimes activation of therapeutic drugs and xenobiotic substances. CPR has been extensively studied (32). It is an 82-kDa, membrane-bound protein that serves as an integral component of the microsomal monooxygenase system that transfers electrons from NAD(P)H to ~57 CYPs via its FAD and FMN component coenzymes.
Studies of the CPR active site show that the proton status of a highly conserved γ-carboxylic acid moiety from glutamate favors binding of NAD(P)H over that of NADP+ (33). The resident flavin moieties are located in domains distinct from each other and are separated by a flexible hinge region. After transfer of a hydride ion carrying an electron pair from the reduced nicotinamide ring of NAD(P)H, FAD aligns its isoalloxazine ring with that of FMN and discharges its cargo electrons. The redox environment of FMN allows sequential electron flow to a variety of partner proteins that include both heme and other flavoproteins (34). The electron transfer usually follows the pathway:
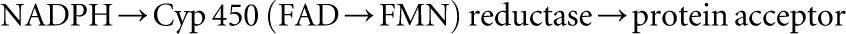 |
Thus, each catalytic cycle of CPR restores the reduced status of CYP with an electron flow from NAD(P)H to the flavin electron acceptors before reacting with its target acceptor proteins.
Two factors that markedly influence the activity of CPR and thus affect cholesterogenesis are dietary riboflavin status and genetic mutations. Studies in riboflavin-deficient animals showed marked decreases in the concentrations of CPR and cytochrome b5 proteins. Subsequent repletion of animals with riboflavin resulted in complete recovery (35, 36). In microsomes of riboflavin-deficient rats, the amount of FMN bound to CPR is lower than that of FAD. A 1:1 ratio of FMN to FAD is critical for maximum activity of CPR (37). The altered FMN:FAD ratio is consistent with the observation that riboflavin deficiency causes a decrease in flavokinase activity and an increase in FMN pyrophosphorylase (FAD synthetase) activity, the enzymes that synthesize FMN and FAD, respectively (38). The addition of FAD to liver microsomes isolated from riboflavin-deficient animals failed to replete enzymatic activity, which illustrates the selectivity for the flavin moieties; however, the FAD that binds to the FMN domain can be readily replaced by FMN, which restores CPR activity. Thus, CPR activity illustrates the critical need to maintain correct balance of the flavin nucleotides and proper coenzymic form within the flavin-binding domains.
Although several polymorphisms and missense mutations have been identified in the CPR gene, 2 missense mutations, in particular V492E and R457H, result in instability and misfolding of CPR that involve FAD binding (39). The binding affinity of V492E is lower than that of R457H for FAD; however, it does cause the protein to have an activity as an electron donor that is more easily reconstituted with the addition of exogenous FAD. V492E is located at the base of the β-flap and influences stacking and arrangement of the molecules between the FAD/NAD(P)H domain and the connecting domain. By contrast, the amino acid substitution, R457H, displays a more tightly bound FAD, but reestablishment of activity is somewhat more resistant to FAD reconstitution. Regions of polypeptide unfolding within CPR differ among these CPR variants. V492E is less stable due to its low binding affinity for FAD and demonstrates a gradual, localized unfolding in the absence of FAD as demonstrated by partial trypsin digestion (40). R457H, on the other hand, binds FAD more tightly, but unfolding is observed more globally throughout the CPR variant. Both mutants are stable to partial trypsinization in the presence of FAD (40). The presence of FAD markedly stabilizes CPR function and suggests a preventive role for riboflavin supplementation in women whose fetuses may harbor these mutations. Mutations in CPR manifest in utero as ambiguous genitalia, abnormal patterns of steroid hormones, and/or skeletal deformities. Thus, patients identified prenatally or with less severe cases of CYP reductase deficiency may be rescued postnatally with supplemental riboflavin (40).
Given the obligatory role of CPR as the primary electron donor for a variety of CYP enzymes, deficits in flavin availability other than those caused by dietary deficiency would also have far-reaching metabolic and developmental consequences. Riboflavin deficiencies can be caused by endocrine abnormalities such as adrenal or thyroid hormone insufficiencies, overexposure to certain tri- and tetracyclic drugs, or excessive ethanol consumption. These deficits in riboflavin would manifest with deformities similar to those occurring in patients with CPR missense variants, V492E and R457H. Such abnormalities would impair riboflavin status by blocking conversion of the vitamin to its physiologically active forms, by interfering with intestinal absorption and transport, and by enhancing renal excretion. Reviews of factors that adversely affect riboflavin metabolism have been published elsewhere (1, 35, 38). The teratogenic effects of specific drugs that interfere with flavin metabolism will be discussed later in this review. In particular, tri- and tetracyclic drugs that bear structural relations with riboflavin and interfere with its metabolism can cause abnormalities similar to those in individuals with defects in FAD-dependent oxidoreductase, 24-dehydrocholesterol reductase (DHCR24), CYP51, and the microsomal 25-hydroxylase, CYP2R1. The pattern of abnormalities observed in individuals with mutations in these enzymes is similar to the teratogenic effects observed with drugs that induce alterations in flavin metabolism. Many of these abnormalities include cleft palate formation, hydrocephalus, and skeletal deformities as well as shortening of distal extremities of fetuses.
Flavoenzymes in cholesterogenesis and steroidogenesis
Several mitochondrial and microsomal flavin-dependent oxidoreductases that function within the cholesterol biosynthetic pathway produce essential branch point metabolites. These metabolites are immediate precursors in the pathways that lead to steroid hormones, bile acids, and vitamin D analogs. As described previously, the CYP heme proteins involved in these processes are classified on the basis of their association with 2 NAD(P)H-dependent donor flavoproteins (41) located in either the mitochondria (type I) or endoplasmic reticulum (type II). A combination of 57 type I and type II enzymes have been identified that play key roles in steroidogenesis, eicosanoid metabolism, and in metabolism of drugs and xenobiotic compounds. Approximately 25% of these proteins exhibit unspecified function and are considered orphan P450 enzymes (42).
CYPs involved with metabolism of cholesterol and steroid hormones receive their electrons primarily from CPR in the endoplasmic reticulum. CPR is a key regulatory flavoenzyme that donates electrons to a number of acceptor proteins in the pathway, which include the squalene monooxygenase, microsomal cytochrome CYP51A1, and DHCR24. Studies using mouse knockout models for CPR indicate that the flavoenzyme is indispensable for cholesterogenesis and steroidogenesis because no other electron donor is capable of providing reducing equivalents to the microsomal CYP heme proteins (43). Thus, the NAD(P)H-supported catalytic activity of microsomal monooxygenases and reductases requires an interaction between CPR and the enzyme or particular heme CYP protein before reacting with substrate.
With regard to cholesterol biosynthesis, flavoproteins have an obligate role within the postsqualene pathway, which is divided into the Bloch and the Kandutsch-Russell branches (44, 45). Squalene monooxygenase and DHCR24 exhibit FAD-binding domains that are the intermediate electron recipients reacting with NAD(P)H. CYP51A1, the sole P450 participant in cholesterogenesis, receives reducing equivalents directly from CPR. These reactions occur before formation of 7-dehydrocholesterol, a branch point metabolite in the cholesterol pathway which functions as the previtamin D.
Squalene monooxygenase.
Squalene monooxygenase (EC 1.14.99.7), an FAD-dependent enzyme, is a potential regulatory site in cholesterogenesis that relies on CPR for reducing equivalents. This 90-kDa enzyme catalyzes oxidation of squalene to squalene-2,3-epoxide [(3S)-2,3-oxidosqualene]. This epoxide undergoes a rapid cyclization catalyzed by lanosterol synthase to form lanosterol, the first C30 steroidal intermediate (46). In this remarkable reaction, a series of 1,2-methyl group and hydride transfers occur along the squalene chain to cause ring closures and formation of the tetracyclic structure of cholesterol, the cyclopentanophenanthrene ring (47). This process is considered one of the most complex single enzymatic reactions thus far identified for an enzyme of such a small molecular size (48). FAD binding to the protein occurs within a moderately hydrophobic pocket bearing a conserved amino acid sequence motif, GxGxxG, characteristic of Rossmann folds, which recognize the ADP moiety of FAD (49). The isoalloxazine ring of FAD interacts with conserved aromatic amino acid residues, thus forming π-π interacting ring structures, which help stabilize the enzyme (50).
Within the endoplasmic reticulum, sterol carrier protein 1 [also known as supernatant protein factor (SPF)] (51) binds squalene in the presence of phosphatidylserine, FAD, NAD(P)H, and oxygen and promotes squalene epoxidation (52). In the squalene monooxygenase reaction, NAD(P)H forms a transient complex with the enzyme and dissociates immediately after transferring its hydride equivalence to FAD (53). Purified SPF stimulates squalene monooxygenase and squalene-2,3-oxide lanosterol cyclase [also known as lanosterol synthase (LSS)]. The latter enzyme also conducts a side reaction with squalene-2,3-epoxide to produce a potentially useful 2,3;22,23-squalene dioxide, a reaction that has an absolute requirement for exogenous FAD (54, 55). These studies show that 2,3;22,23-squalene dioxide has a half-maximal inhibitory concentration (IC50) of 16 μmol/L for liver oxidosqualene cyclase (LSS), the enzyme that generates lanosterol, the tetracyclic precursor of cholesterol. Partial inhibition of LSS by squalene dioxide favors synthesis of 24(S),25-epoxycholesterol over that of cholesterol. 24(S),25-Epoxycholesterol has been shown to repress 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase activity and enhance its degradation. Thus, supplemental dietary riboflavin may have the potential to favor formation of 24(S),25-epoxycholesterol. Overproduction of this cholesterol derivative would result in the generation of a negative regulatory feedback loop with HMG-CoA reductase that would limit cholesterol biosynthesis. Epoxidations of the terminal double bonds of squalene are shown in Fig. 4.
FIGURE 4.

Formation of squalene epoxides. Squalene monooxygenase catalyzes oxidation of squalene to form 2,3-oxidosqualene. This mixed-function oxidase requires NAD(P)H as the reductant and molecular O2 as oxidant. One oxygen atom of molecular oxygen is incorporated into the substrate (as the epoxide), and the other oxygen atom is reduced to water. An additional side reaction in the presence of excess FAD (bold letters) can occur with 2,3-oxidosqualene to produce 2,3;22,23-dioxidosqualene. R represents the internal isoprene section of squalene from carbon 3 to 22.
Squalene monooxygenase appears to play a central role in overall regulation of cholesterol biosynthesis. Studies show that addition of cholesterol to hepatocytes in culture suppresses mRNA levels of squalene monooxygenase, subsequently diminishing the activity of squalene monooxygenase and resulting in accumulation of squalene (56). Thus, squalene monooxygenase is an attractive therapeutic target for drugs to lower serum cholesterol (57). In view of its flavin requirement, the apparent Michaelis constant (Km) of 0.3 μmol/L is consistent with a freely dissociable FAD moiety, and the Km for its protein partner, CPR, the primary reductant flavoprotein, is in the low nmol/L range (46). The associated Km of squalene monooxygenase with CPR is lower than that compared with most of the other CPR client proteins (including heme partner enzymes). In view of the high-affinity protein-protein interactions, targeting CPR may potentially limit monooxygenase activity (58). A blockade at the site of squalene monooxygenase would result in accumulation of squalene, which is considered nontoxic and thus would be expected to spare needed isoprenyl units for other vital cell metabolic processes (57). By contrast, inhibitors of enzymes involved in the initial phase of cholesterogenesis, such as HMG-CoA reductase, would limit production of dolichol, farnesyl, and geranylgeranyl, key isoprenoid metabolites essential for protein signaling and cell proliferation.
Several dietary constituents have been shown to interfere with squalene monooxygenase, which may explain their marginal effects on lowering serum cholesterol and improving cardiovascular function. Of interest are the green tea polyphenolics (epigallocatechin-3-O-gallates), red wine stilbenoids (resveratrol, trans-3,4′,5-trihydroxystilbene), and allylsulfides (including allylcysteines) from garlic. These derivatives act as reversible, noncompetitive inhibitors by scavenging the reactive oxygen species formed at the active site of the flavin moiety, namely the flavin C4a hydroperoxide (59, 60). A review of nonstatin strategies for treatment of hypercholesterolemia has been published elsewhere and offers a perspective for the use of inhibitors that start at the site of squalene synthase in the cholesterol biosynthetic pathway (61).
Cytochrome P450, family 51, subfamily A, polypeptide 1.
After formation of lanosterol, the first sterol in the cholesterogenic pathway, the 14α-methyl moiety is oxidatively removed by lanosterol 14α-demethylase to produce 4,4-dimethyl-5α-cholest-8(9),14,24-triene-3β-ol. This enzyme also demethylates 24,25-dihydrolanosterol, which is the reaction product of the flavoenzyme, sterol-Δ24-reductase, on lanosterol (see 24-dehydrocholesterol reductase below). Removal of the C14-methyl moiety is critical because sterols with a methyl in this position cannot replace membrane cholesterol or any of the steroid hormones (61). Lanosterol 14α-demethylase is a 53-kDa CYP51A1 protein located in the endoplasmic reticulum that catalyzes a 3-step reaction each requiring 1 molecule of oxygen and 2 NAD(P)H-derived reducing equivalents supplied by CPR (62). Figure 5 shows the transfer of reducing equivalence required in 3 consecutive reactions resulting in the C14-demethylation of lanosterol to 4,4-dimethyl-5α-cholest-8(9),14,24-triene-3β-ol.
FIGURE 5.

Oxidative demethylation of lanosterol. Lanosterol 14α-demethylase CYP51A1 catalyzes a 3-step reaction requiring molecule oxygen and reducing equivalents from NAD(P)H. The methyl moiety at the 14α position is oxidized to a hydroxymethyl, which undergoes subsequent oxidations and then final elimination from the sterol ring. CPR (type II reaction) functions as the interfacing transfer protein between NAD(P)H and CYP51A1 to form the demethylated product, 4,4-dimethylcholest-8(9),14-diene-3β-ol (also 4,4-dimethyl-5α-cholest-8(9),14,24-triene-3β-ol). CPR, NAD(P)H-cytochrome P450 reductase; e•, represents the electron transfer from CPR to CYP51A1; S, indicates the sulfhydryl moiety on the CYP51A1 protein to which heme iron is bound.
CPR plays a critical role in this reaction in that a mutation in FAD binding in this flavoenzyme or a diminished association with CYP51A1 is linked to a skeletal dysmorphology syndrome (Antley-Bixler syndrome) (63, 64). Interference in the ability of these 2 partner proteins to interact appears to be the major underlying mechanism that leads to Antley-Bixler syndrome. This view is supported by the fact that, to date, no genetic mutation has been identified that directly disrupts or abolishes CYP51A1 enzymic activity (65). However, a V13E amino acid variant in the N-terminal portion of CYP51A1 has been reported to affect its ability to associate with the endoplasmic membrane and perhaps the membrane-bound CPR (42, 62). As mentioned previously, mutations identified in CPR could also account for decreased CYP51A1. A decrease in the activity of this enzyme is observed in patients with Antley-Bixler syndrome.
Of the 14 cytochrome proteins involved in cholesterol homeostasis, CYP51A1 is the only P450 enzyme that participates in cholesterol biosynthesis. Thirteen other CYPs are involved solely in cholesterol degradation, subsequent steroidogenesis, and bile acid formation (66). Thus, interfering with the ability of the CYPs to interact with CPR or disrupting the stability of protein-protein complexes may provide a viable target to control cholesterogenesis. A blockade of CYP51A1 (lanosterol demethylase) activity results in accumulation of lanosterol and 24-dihydrolanosterol (67). In studies using CPR-null mouse livers, the accumulation of these methylated sterols stimulated ubiquitination of HMG-CoA reductase causing degradation of the enzyme and blocking the initial phase of cholesterogenesis (68).
24-Dehydrocholesterol reductase.
The FAD-dependent oxidoreductase DHCR24 (EC 1.3.1.72) catalyzes reduction of the Δ24 double bond in the side chain of several obligatory intermediates in the cholesterol pathway. Due to the broad substrate specificity of this flavoenzyme for metabolites between lanosterol and cholesterol, the precise sequence of reactions that lead directly to cholesterol appears to follow a circuitous route. For convenience, the postsqualene pathway is divided into the Bloch and the Kandutsch-Russell pathways. In the Kandutsch-Russell division, DHCR24 acts initially on lanosterol. Thus, subsequent metabolites from 24,25-dihydrolanosterol to 7-dehydrocholesterol exhibit a saturated side chain. Among the other key precholesterol metabolites in the Bloch division, zymosterol and desmosterol are excellent substrates for DHCR24, whereas 24-dehydrolathosterol exhibits the highest affinity for DHCR24 and is the most reactive substrate (69). In contrast to metabolites in the Kandutsch-Russell pathway, Bloch division metabolites from lanosterol to desmosterol exhibit the Δ24 double bond. Figure 6 shows the separate but interactive pathways of postlanosterol metabolites possessing a fully saturated side chain (Kandutsch-Russell pathway) and a Δ24 double bond (Bloch pathway). The molar ratios of the sister metabolites in each pathway are governed by the catalytic capacity of the flavoenzyme, DHCR24, the bioavailability of dietary riboflavin, or drugs known to block conversion of riboflavin to its physiologically active coenzymes.
FIGURE 6.

Postsqualene cholesterol biosynthetic pathway. The figure depicts oxidation of squalene by the flavoenzyme squalene monooxygenase and subsequent cyclization by LSS to form lanosterol, the first sterol (cyclopentanophenanthrene ring) in the cholesterogenic pathway. Lanosterol is a major branch point metabolite between 2 proposed biosynthetic pathways to cholesterol: the Bloch and the Kandutsch-Russell pathways. The metabolites in these pathways differ in that Bloch metabolites exhibit a Δ24 double bond in the aliphatic side chain as opposed to having a fully saturated side chain (Kandutsch-Russell metabolites). Both pathways depend on 2 flavin-dependent enzymes: CYP51A1 and DHCR24. The former enzyme conducts demethylations of lanosterol and 24,25-dihydrolanosterol–generating sterols: 14-demethyllanosterol and 14-demethyl dihydrolanosterol, respectively. These lanosterol derivatives are precursors to a series of “sister” metabolites that exhibit the Δ24 double bond (Bloch metabolites) or 24,25 dihydro side chain (Kandutsch-Russell metabolites). The latter pathway is predominant in most tissues and has cholesterol as its final metabolite. In contrast to cholesterol, incorporation of Bloch pathway sterols (especially desmosterol) into cell membranes increases their permeability and fluidity. Multiple arrows indicate reactions not shown in each metabolic pathway. Other enzymes shared by both pathways are EBP, SC5D, and DHCR7. DHCR7, 7-dehydrocholesterol reductase; EBP, emopamil binding protein (3-β-hydroxysteroid-Δ8,Δ7-isomerase); LSS, lanosterol synthase; SC5D, sterol-C5-desaturase-like.
Desmosterolosis, a rare disorder of cholesterol biosynthesis, is caused by mutations in the gene that encodes for DHCR24 (70). To date, desmosterolosis has been described in only 3 patients (71). DHCR24 contains a leader sequence that directs it to the endoplasmic reticulum membrane. A missense mutation in this flavoprotein causes desmosterolosis, which presents biochemically with elevated desmosterol and phenotypically with spasticity, microcephaly, and micrognathia in affected patients (72). In 1 patient, the mutation of R94H, for which the moiety R94 resides at the FAD binding site, resulted in desmosterolosis (71).
The syndrome resulting from genetic mutations of DHCR24 is similar to those observed in animals during severe maternal riboflavin deficiency, namely micrognathia, cleft palate, hydrocephalus, and skeletal anomalies as well as reduction-type defects in the distal extremities of fetuses (73, 74). It is also intriguing to consider that drugs that bear structural similarities to those of riboflavin [e.g., tricyclic antidepressants and phenothiazine derivatives (75–77)] and interfere with its conversion to FMN and FAD exhibit comparable teratogenic manifestations (78).
Enzyme analyses of DHCR24 that follow the reduction of the Δ24 double bond in desmosterol to form cholesterol show 2-fold activity increases in the presence of exogenous FAD. This indicates the responsiveness of the conserved domain to exogenous flavins, which is characteristic of the FAD-dependent oxidoreductases found in the DHCR24 amino acid sequence (72). Conversely, in constructs containing mutant DHCR24 alleles from patients with desmosterolosis, the conversion from desmosterol into cholesterol is absent or markedly reduced (72).
Importance of Bloch and Kandutsch-Russell metabolites in membrane stability, fluidity, and flexibility
Changes in the membrane content of Bloch- and Kandutsch-Russell–type sterols, coupled with selective compartmentalization of these sterols throughout the membrane, can influence ciliary activity and flagella motility. Using filipin (a histochemical stain for cholesterol) as a probe for the presence of membrane cholesterol, differential distribution of cholesterol has been observed along the distal cilial regions and regions of the ciliary necklace (79, 80). The necklace region of cilia is the specialized structural feature of ciliary membranes thought to serve as a timing mechanism for their wave motion or beat. Filipin staining demonstrates that cholesterol is abundant in distal portions of the cilium but is virtually absent from regions of the ciliary necklace. A subcellular-selective decrease in cholesterol deposition or perhaps an increase deposition of Bloch pathway metabolites would account for these differences. In similar fashion, controlled expression of DHCR24 activity or alterations in flavin binding or availability would increase the ratio of Bloch to Kandutsch-Russell sterols and provide a more “flexible” arrangement within the membrane segment to accommodate ciliary locomotion and wave action. Studies have shown that the addition of cholesterol to membranes can amplify the average membrane thickness by 0.3–0.6 nm and thus decrease flexibility and fluidity of membranes (81).
The development of specialized cells such as sperm cells and oocytes requires modeling and remodeling of their membrane compartments to enable specific changes to occur in membrane permeability and fluidity (82). To this end, desmosterol rather than cholesterol preferentially accumulates in sperm flagella where greater membrane fluidity and motility are required. Studies show that ablation of the Dhcr24 gene, which would suspend flavoenzyme-induced conversion of desmosterol to cholesterol, results in animals that are unable to reproduce (83). However, studies in rats with marginal riboflavin deficiency alone displayed minimal changes in sperm motility within the caudal epididymis (84). By contrast, in these same studies, rats fed a riboflavin-deficient diet in combination with a metabolic stress such as infection demonstrated a marked decrease in sperm motility compared with that observed in control rats with infection alone.
DHCR24 was first identified as Seladin-1 (selective Alzheimer disease indicator 1) and has since been ascribed diverse functions that involve regulation of cell growth and senescence as well as contributing to defenses against oncogenic and oxidative stress (85). The downregulation of DHCR24 in brains of patients with Alzheimer disease may explain the diminished formation of cholesterol in affected patients. The neuroprotective effect of this highly regulated flavoprotein may be partly associated with its essential role in converting Bloch pathway sterols to cholesterol. DHCR24 is a key regulator and stabilizer of lipid rafts, particularly within the central nervous system (CNS). Rafts are organized centers for assembly of signal proteins and provide for membrane trafficking of proteins, receptors, neurotransmitters, and nutrient fluxes. As a component of the Bloch pathway, desmosterol as well as other Δ24 double-bond metabolites cannot fulfill the domain-promoting role assigned for cholesterol in raft-dependent functions (86). Understanding the ability of cells to regulate DHCR24 activity and make differential use of these cholesterogenic metabolites may provide novel insights into age-related impairments of cells and the pathophysiology of multiple CNS disorders that include Smith-Lemli-Opitz syndrome (see “7-Dehydrocholesterol reductase” below) and Alzheimer diseases (87–89).
With regard to Alzheimer disease, strong evidence supports a relation between disease progression and dysfunction in neuronal sterol metabolism. Cholesterol turnover in the brain is ~20-fold lower than that in the periphery (90). Although hypercholesterolemia strongly correlates with Alzheimer disease, the exchange of brain cholesterol with that in plasma is negligible. Thus, the adult brain needs to efficiently recycle its cholesterol in order to preserve constant concentrations of cholesterol. Both neuronal and glial cells are major contributors to de novo biosynthesis of cholesterol, which declines as a function of age and brain development (91). Thus, neuronal membranes must be viewed as highly dynamic structures that transiently trap diffusing sterol metabolites. In this regard, DHCR24 may be a critical regulatory protein that balances the formation of the raft-promoting Kandutsch-Russell metabolites and the less optimal raft components of the Bloch pathway.
In view of the dependency of DHCR24 on FAD, riboflavin nutriture can likewise play an essential role in maintaining a critical balance between metabolites of the Bloch and Kandutsch-Russell pathways. Members of these pathways differ primarily in the extent of saturation between carbons 24 and 25. For example, an increased membrane concentration of lanosterol, which has a double bond at the Δ24 carbon, over that of cholesterol, which lacks the double bond, enhances permeability and fluidity of the cell membrane (92). By contrast, a high membrane content of cholesterol increases specific condensation of membrane phospholipids and thereby decreases membrane fluidity. Various cells maintain strict control of the cholesterol content of their membranes. For example, the age of disc membranes and their spatial distribution in the outer segment of retinal rods correlate with their cholesterol content. Even modest changes in the ratio of cholesterol to other sterols within cell membranes result in significant alterations in structural properties of membranes that would affect their physiologic homeostasis (93, 94).
Deficits in riboflavin intake and metabolism as well as defects in flavoenzyme activity result in marked structural alterations within the skeletal and central nervous systems similar to those of disorders (inborn errors) in the biosynthetic pathways that lead to cholesterol, steroid hormone, vitamin D, and their metabolites. Such disorders include desmosterolosis, Antley-Bixler syndrome, and Smith-Lemli-Opitz syndrome (95). The multiple congenital malformations associated with these disorders share craniofacial traits similar to those of animals born to riboflavin-deficient mothers, namely microcephaly, bitemporal narrowing, ptosis, short nasal root, anteverted nares, cleft palate, and micrognathia. In the majority of cases 2- to 3-digit syndactyly has also been observed. Abnormalities in the skull, limbs, and fingers have been traced to a missense mutation in CPR and/or defects in DHCR24. Defects in these flavoenzymes coupled with availability of riboflavin can influence the ratio of cholesterol (Kandutsch-Russell pathway metabolite) and Bloch division metabolites.
In studies related to cholesterol metabolite imbalances in membranes, offspring from riboflavin-deficient animals exhibit distinctive encephalic malformations, in particular, cystic expansion of the third and predominantly fourth ventricle and reduced amounts of cerebellum, medulla oblongata, pons, and tegmentum mesencephali (96). These teratogenic features due to maternal riboflavin deficiency are characteristic of those observed in a rare congenital brain defect, Dandy-Walker syndrome (97). In this syndrome, ventricular extensions and other CNS abnormalities have been attributed to deficits in the structural integrity of the surrounding nervous tissues. Thus, further investigations are required to ascribe distortions of the shape and structure of ganglionic regions and the weakening of the tectonic arrangement of the adjoining tissues to cholesterol deficits of neuronal tissues.
7-Dehydrocholesterol reductase.
7-Dehydrocholesterol reductase (DHCR7) catalyzes the terminal reaction in cholesterol biosynthesis, which involves reducing 7-dehydrocholesterol to form cholesterol. Functional DHCR7 is a 55.5-kDa NAD(P)H-requiring protein located in the microsomal membrane. Previous studies had suggested that DHCR7 required CPR for catalyzing the reduction of 7-dehydrocholesterol (98). However, recent studies show that the activity of DHCR7 is not enhanced in the presence of FAD, nor does it form a partner complex with the flavoprotein CPR (99). Further studies showed that in microsomes from livers of CPR-null mice, a modest increase in levels of DHCR7 mRNA occurs.
It is important to consider here that 7-dehydrocholesterol is the immediate precursor of both cholesterol and vitamin D3 (cholecalciferol) and thus represents a critical branch point in the cholesterol biosynthetic pathway (56, 100). Most important, cholesterol cannot be oxidized back to 7-dehydrocholesterol and therefore cholesterol is not a precursor of vitamin D3. Rather, both vitamin D3 and cholesterol share 7-dehydrocholesterol as a common precursor (see Fig. 7).
FIGURE 7.

Flavin-dependent enzymes in postsqualene synthesis of 7-dehydrocholesterol. The diagram depicts flavoenzymes that use FAD directly (squalene monooxygenase and DHCR24) or indirectly CYP51A1 using FMN/FAD-dependent cytochrome P450 reductase. 7-Dehydrocholesterol is the critical branch point metabolite required for the synthesis of both cholesterol and cholecalciferol (vitamin D). DHCR24, 24-dehydrocholesterol reductase; LSS, lanosterol synthase (squalene-2,3-oxide lanosterol cyclase).
In view of the regulatory function that several flavoenzymes have within the postsqualene section of the cholesterol biosynthetic pathway, riboflavin status potentially would have an impact on controlling vitamin D3 formation from 7-dehydrocholesterol. Studies show that the intracellular concentrations of cholesterol per se influence vitamin D3 status at 2 sites controlled by FAD-dependent enzymes, namely at squalene monooxygenase and CYP51A1 (101). In studies using cell culture models for cholesterogenesis, both the expression of mRNA and the activity of squalene monooxygenase were suppressed by the addition of cholesterol to the media (68, 102). This inhibition at the squalene level would decrease formation of 7-dehydrocholesterol and limit available substrate for the photosynthesis of vitamin D3. At the second site, inhibition of the CYP51 (lanosterol demethylase) would result in accumulation of lanosterol and 24-dihydrolanosterol, both of which block 7-dehydrocholesterol formation by accelerating ubiquitination and proteasomal degradation of HMG-CoA reductase (67, 103). Thus, decreased dermal formation of 7-dehydrocholesterol would be expected to diminish its conversion to cholecalciferol (previtamin D3) when exposed to UV-B sunlight. By contrast, in patients with Smith-Lemli-Opitz syndrome (104, 105), which is characterized by marked increases in 7-dehydrocholesterol, cutaneous synthesis of vitamin D and circulating concentrations of vitamin D metabolites were not found to differ from normal controls who were matched for age and season of blood collection (106).
Cholesterol and transmembrane riboflavin efflux protein
A direct relation between riboflavin and cholesterol exists through their interaction with the ATP-binding cassette (ABC) transporter G2 (ABCG2) located in membrane rafts of endothelial cells (107–109). ABCG2, which is evolutionarily conserved among mammals, protects cells from the potential toxicity of xenobiotic compounds and accumulation of some nutrients by transporting these substances from the hepatocyte into bile, from intestinal epithelia back to the lumen, and from brain or retinal endothelium to the capillary lumen (110). It is an integral membrane protein both in normal and cancer cells and has been identified as the breast cancer–resistance protein. In the brain, this transport protein is a major component of the blood-brain barrier located on endothelial cells near tight junctions. Studies in Abcg2-null mice suggest a gatekeeper function for the protein, particularly in patients with Alzheimer disease where it may limit access of β-amyloid peptides to the brain or increase clearance or extrusion from the brain (111).
With regard to flavin nutriture, ABCG2 controls efflux of riboflavin into extracellular fluids such as blood plasma, cerebral spinal fluid, bile, semen, and breast milk as well as protects cells from excessive accumulation of riboflavin. Cholesterol is an essential activator of ABCG2 function and has a major impact on its capacity to efflux metabolites (112, 113). Cholesterol loading of membrane rafts significantly improves ABCG2-ATPase activity and vesicular transport (114). Thus, the concentration of cholesterol within lipid rafts has a paradoxical dual function of potentiating toxicant elimination and of enhancing efflux of riboflavin into extracellular fluids such as breast milk, semen, and cerebral spinal fluids (109). The ability of cholesterol to control riboflavin efflux and the influence that riboflavin and flavoproteins has on cholesterogenesis and its regulatory branch points require further investigation and may lead to novel therapeutic options for metabolic control of cholesterol and its metabolites.
Flavoproteins and cytochrome P450 heme proteins in synthesis of vitamin D metabolites
The activation of vitamin D to its major circulating form, 25(OH)D, and its hormonally active form, 1α,25(OH)2D, requires both type I and type II CYP heme proteins. The reliance on flavin-dependent heme proteins in this process supports further the association between riboflavin and vitamin D status (115).
Formation of 25-hydroxycholecalciferol: role of type II cytochrome P450 heme proteins.
Cholecalciferol, either biosynthesized in skin or derived from dietary sources, is converted to 25(OH)D in a variety of tissues. This vitamin D metabolite is the major circulating form of vitamin D and a key biomarker of vitamin D status. The formation of 25(OH)D is catalyzed by 5 microsomal CYP hydroxylases (CYP2R1, CYP2J2/3, CYP3A4, CYP2D25, and CYP2C11) (type II) and 1 mitochondrial hydroxylase (CYP27A1) (type I) (116, 117). Each of the CYP proteins exhibits different catalytic properties that are not exclusive in their formation of 25(OH)D from cholecalciferol because they also hydroxylate other vitamin D analogs, other sterols, and bile acid metabolites. An in-depth summary of vitamin D 25-hydroxylases covering 4 decades of research has been published (117).
Cytochrome P450 2R1.
Of the microsomal 25-hydroxylases, CYP2R1 appears to play a prominent role in maintaining physiologic concentrations of 25(OH)D in plasma (118). Unlike the other microsomal 25-hydroxylases, kinetic studies using recombinant Cyp2R1 indicate that the enzyme can conduct 25-hydroxylation by using physiologic concentrations of cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2) as well as with their respective 1α(OH)-derivatives (119). To further suggest that CYP2R1 plays a physiologically important role in the vitamin D 25-hydroxylation in humans, genetic defects in the other microsomal enzymes show no or only marginal decreases in circulating 25(OH)D and present minimal risk for formation of rickets or osteoporotic lesions. By contrast, studies using Cyp2R1−/− mice indicate that serum concentrations of 25(OH)D were reduced by 50% of those in control animals, whereas 1α,25-dihydroxyvitamin D [1α,25(OH)2D] concentrations in serum remained unchanged (120). Nonetheless, the importance of the enzymatic redundancy and promiscuity among the 25-hydroxylases for sterol metabolites requires further study (117).
As discussed previously with the cholesterogenic (type II) microsomal enzymes, vitamin D 25-hydroxylases require reducing equivalence from the FAD/FMN oxidoreductase, CPR, which is tethered to the membrane of the endoplasmic reticulum. CPR is the exclusive flavoprotein that donates electrons to all 5 of the microsomal hydroxylases, and thus mutations in CPR would be expected to present clinically with overlapping symptoms such as those observed with apparent pregnene hydroxylation deficiency, Antley-Bixler skeletal malformation syndrome, Shprintzen-Goldberg syndrome, and DHCR7 deficiency, which manifests phenotypically as Smith-Lemli-Opitz syndrome (121–126).
In similar fashion, deficits in riboflavin availability or imbalances in the intracellular ratio of FAD and FMN, particularly during fetal development, would be expected to elicit clinical manifestations similar to those caused by mutations of CPR. This flavoenzyme exhibits a commonality of function with all microsomal CYP enzymes. Early studies of acute riboflavin deficiency during embryonic development suggested that specific pathways governed by the action of flavoenzymes were essential for normal differentiation. According to this concept, if the flavin concentration of the embryo, or within certain areas of the embryo, falls below a critical nadir, anomalies will result (127). Subsequent investigations showed that the total riboflavin and FAD concentrations of differentiating rat pup embryos can be reduced by as much as 30% without ill effects. By contrast, a 60% reduction in FAD is teratogenic (128).
Congenital malformations due to riboflavin deficiency and those associated with the genetic alteration of CPR and vitamin D deficiencies manifest as cleft palate, micrognathia, micromelia, absent or fused vertebrae, short ribs, and failed closure of the neurotube (craniorachischisis). As mentioned previously, these same teratogenic manifestations have also been related to use of phenothiazine drugs, which bear structural analogies to the isoalloxazine moiety of flavins. An increased risk of congenital malformations in humans occurs when the human fetus is exposed to phenothiazines during weeks 4 to 10 of gestation (129). In addition, the side chain of tricyclic compounds plays an important role in their inhibitory action on flavoenzymes and conversion of riboflavin to FMN and FAD (130). Accordingly, in rats, riboflavin administration can lower prenatal death rates caused by single doses of phenothiazines given on the 14th day of gestation and can provide varying degrees of protection against certain other drug-induced teratogenicities (131). The experimental congenital malformations induced in the offspring of pregnant riboflavin-deficient rats and in those administered drugs structurally similar to riboflavin have features in common with those induced by defects in vitamin D and sterol metabolism. The commonality in these defects appears to converge on a central alteration associated with diminished activity of CPR, due possibly to decreased riboflavin availability, displacement of the FMN and FAD coenzymes from their respective binding sites, or alterations in the FMN:FAD ratio of the enzyme within the endoplasmic reticulum (121).
Clinical investigations that examine metabolism of micronutrients often find that deficiency symptoms of vitamins tend to be multiple. The impact of a single vitamin deficiency on the efficacy of another is best examined by using animal models where pair-feeding regiments as well as eucaloric and control diets can be maintained. Aside from rare genetic defects in riboflavin transport such as the Brown-Vialetto-Van Laere syndrome, which is characterized by progressive sensorineuropathies, paralysis, and eventual respiratory failure, isolated deficiencies of riboflavin are not widely prevalent in the general population.
Several physical and clinical symptoms that manifest during riboflavin deficiency are not pathognomonic or exclusive to deficits in riboflavin. In this regard, only 1 study was found in the literature that addressed the impact of riboflavin deficiency directly on vitamin D metabolism (132). In this animal study, riboflavin deficiency caused a moderate decline in serum calcium as well as a decrease in the active transport of calcium through the small intestine. Measurements of 25(OH)D in the blood serum, the formation of 24,25-dihydroxyvitamin D [24,25(OH)2D] in kidney slices and the content of nuclear receptors for 1α,25(OH)2D in intestinal mucosa were significantly lower than those of control animals. Administration of vitamin D to riboflavin-deficient rats 6 d before death restored calcium homeostasis.
In conclusion, the 5 microsomal P450 isoforms capable of synthesizing 25(OH)D have different catalytic properties toward sterols and secosterols, exhibit different tissue distributions, and contribute diversely to sterol homeostasis. Despite their apparent variability as well as redundancy toward substrates, microsomal P450 enzymes draw electrons from the same diflavin oxidoreductase (CPR) within the endoplasmic reticulum and thus can be influenced by the extent of riboflavin trafficking and the concentration and ratio of flavin coenzymes within subcellular compartments.
Formation of 25(OH)D, 1α,25(OH)2D, and their 24-hydroxy metabolites: role of type I oxidoreductases.
The other enzymes reliant on flavoproteins and critical for vitamin D metabolism are located exclusively in mitochondria. Accordingly, CYP27A1, CYP27B1, and CYP24 thus rely on type I oxidoreductase reactions within the mitochondrial matrix.
CYP27A1 (sterol 27-hydroxylase).
CYP27A1 is the only ubiquitously expressed mitochondrial enzyme that conducts a 25-hydroxylation with cholecalciferol. Thus, as a mitochondrial enzyme, its electron transfer partners are FAD-dependent adrenodoxin reductase and 2Fe-2S-adrenodoxin (133). CYP27A1 is located primarily in the liver, kidney, intestine, ovary, lung, and skin and the human CYP27A1 gene is under modest regulatory control by growth hormone, insulin-like growth factor-1, thyroid hormones, and dexamethasone (134).
Human CYP27A1 can hydroxylate many metabolites of cholecalciferol (but not ergocalciferol) to form C-25, C-26, and C-27 monohydroxy and C-24,25, C-1α,25 and C-25,26 dihydroxy derivatives (135, 136). Human CYP27A1 can also catalyze multiple reactions with cholesterol and bile acids (137). In addition, hydroxylations of vitamin D metabolites exhibit smaller maximum velocity (Vmax):Km ratio value (i.e., catalytic efficiency) than those of cholesterol metabolites (138). Thus, although CYP27A1 has the ability to activate vitamin D, it nonetheless lacks specificity and catalyzes multiple hydroxylations in addition to that of a 25-hydroxylase, a property it shares with the microsomal CYP hydroxylase CYP2D25 (139).
CYP27A1 exhibits a greater affinity toward cholesterol metabolites than toward vitamin D metabolites. Thus, high concentrations of cholesterol can diminish 25(OH)D synthesis. This effect is compatible with the observations that vitamin D insufficiency is often associated with increased blood cholesterol concentrations (140) and that obese children and adults with hypercholesterolemia usually express low circulating concentrations of 25(OH)D (141–143). More detailed analysis showed that the low concentrations of circulating 25(OH)D correlate with high concentrations of total cholesterol, LDL cholesterol, and TGs, whereas the low concentrations of active 1α,25(OH)2D are associated with low HDL cholesterol (144).
Genetic mutations in CYP27A1 can completely inactivate the enzyme and lead to a reduced production of bile acids, in particular chenodeoxycholic acid, with a concomitant increase in bile acid alcohols. To date, >50 genetic mutations have been implicated that affect the amino acid domains critical for enzymic activity. These changes in amino acid profile of the enzyme markedly affect substrate binding, heme binding, interfacing with its mitochondrial redox partner (ferredoxin/adrenodoxin), membrane binding, and protein folding (41).
Patients with inborn errors in CYP27A1 manifest with cerebrotendinous xanthomatosis (CTX), a rare lipid-storage disease that is characterized by bile acid deficiency, neurologic dysfunctions, premature atherosclerosis, and sometimes osteoporosis. The clinical manifestations of CYP27A1 mutations also mimic features of amyotrophic lateral sclerosis (ALS) with progressive upper and lower motor neuron loss (145). It is interesting to consider that a relation between CTX and Brown-Vialetto-Van Laere syndrome may exist in that both neurodegenerative disorders are characterized by progressive multiple neurologic abnormalities in combination with ALS. Brown-Vialetto-Van Laere syndrome has been identified with mutations in 2 riboflavin transporter genes (146). An extension of these studies to ALS patients in general revealed decreases in expression of FAD synthetase, riboflavin kinase, cytochrome c1, and succinate dehydrogenase complex subunit B, a mitochondrial enzyme bearing a covalently linked FAD moiety (147). These results suggest that specific electron transport chain abnormalities associated with mitochondrial riboflavin metabolism or interference with a flavoenzyme may underlie or contribute to these neurodegenerative disorders.
In a similar fashion, interference with mitochondrial riboflavin metabolism after alcohol exposure in utero may underlie the cardiac pathologies and other deformities associated with the fetal alcohol syndrome. Ethanol markedly decreases bioavailability of dietary flavins (148) and has been shown to reduce activities of 2 mitochondrial inner membrane enzymes, cytochrome c oxidase and succinate dehydrogenase (149). When flavin-linked substrates were used to assess mitochondrial function in prenatal mice, respiration was depressed. By contrast, ADP-stimulated respiration was not compromised (149).
Earlier investigations had considered that CYP27A1 is a major contributor to the circulating pool of 25(OH)D (138). However, knockout mouse models of CYP27A1 and monoclonal antibodies raised against CYP27A1 did not show reductions in 25-hydroxylation of cholecalciferol (150). Even patients with CTX demonstrate only modest depressions in serum concentrations of 25(OH)D and exhibit only marginal risk of development of bone lesions (151). In view of the importance of CYP27A1 in the synthesis of bile acids, determinants that affect development of osteoporosis are probably associated more with malabsorption of fat-soluble vitamin D coupled with secondary production deficits in bile acids rather than with reduced formation of 25(OH)D (152). In this regard, it is important to consider that the gene promoter region of CYP27A1 does not exhibit a vitamin D–response element but is controlled by bile acids. In addition, in terms of activation of vitamin D metabolites and its utility for vitamin D metabolism, CYP27A1 exhibits specificity for cholecalciferol (vitamin D3) and not ergocalciferol (vitamin D2). This selectivity for vitamin D analogs is in contrast to that of the microsomal CYP2R1, which hydroxylates both vitamin D2 and vitamin D3 (135).
CYP27B1 (sterol 1α-hydroxylase).
The conversion of 25(OH)D to the active hormone 1α,25(OH)2D or calcitriol is under strict regulatory control by CYP27B1, which occurs primarily within kidney mitochondria (153). In addition to renal production of calcitriol, real-time PCR and specific immunoprecipitation reactions have confirmed the extrarenal presence of 1α-hydroxylase (CYP27B1). Thus, mitochondria in placenta, macrophages, astrocytes, skin, intestine, as well as breast and prostate cells can also synthesize calcitriol (154). The activity of CYP27B1 in kidney is highly regulated. Renal CYP27B1 is upregulated by parathyroid hormone and downregulated by calcitonin, major hormones of the mammalian calciotropic homeostatic loop. In addition, renal CYP27B1 is downregulated by fibroblast growth factor-23 (FGF-23), a component of the phosphotropic homeostatic loop. Renal mitochondria are important for systemic control of calcitriol formation. By contrast, 1α-hydroxylase activity in extrarenal mitochondria functions in a paracrine/autocrine manner and is influenced by cytokines such as interleukins and interferon-γ.
CYP24A1 hydroxylase (sterol 24-hydroxylase).
Another critical mitochondrial enzyme that controls the vitamin D metabolic machinery is CYP24A1 hydroxylase. This enzyme is responsible for converting 25(OH)D and 1α,25(OH)2D to less bioactive metabolites, 24R,25(OH)2D and 1α,24,25(OH)3D, respectively, and is responsible for the 26-hydroxylation of these metabolites (155). The former metabolite, 24R,25(OH)2D, has been reported to counteract the hypercalcemic activity of calcitriol and regulate calcium homeostasis for bone formation, whereas the latter trihydroxy metabolite is further catabolized via a 6-step monooxygenation to form calcitroic acid, a urinary metabolite of vitamin D (156). An overview of the microsomal and mitochondrial CYP enzymes involved in vitamin D metabolism is shown in Fig. 8.
FIGURE 8.

CYP enzymes involved in the synthesis and degradation of vitamin D. The pathway shows an overview of the multifunctional capacity of microsomal and mitochondrial CYP enzymes to hydroxylate vitamin D analogs. Type I mitochondrial enzymes are indicated with an asterisk (*). CYP, cytochrome P450; 1α,24,25(OH)3D, 1α,24,25-trihydroxyvitamin D; 1α,24,26(OH)3D, 1α,24,26-trihydroxyvitamin D; 1α,25(OH)2D, 1α,25-dihydroxyvitamin D; 24,25(OH)2D, 24,25-dihydroxyvitamin D; 25,26(OH)2D, 25,26-dihydroxyvitamin D; 25(OH)D, 25-hydroxyvitamin D.
With regard to understanding the role of riboflavin and the importance of flavoenzymes in mitochondrial vitamin D metabolism, consideration must be made of the bioavailability and delivery of riboflavin into mitochondria and the activation of the nascent apoflavoenzyme within the matrix. In this regard, CYP27A1, CYP27B1, and CYP24A1 are dependent on the reducing equivalents delivered by adrenodoxin reductase and adrenodoxin to the P450 heme complex. Thus, a coordination of protein import and cofactor supply to the mitochondrial matrix is indispensable for mitochondrial function. Although more in-depth information is required, recent studies have focused attention on the subcellular distribution of riboflavin and its coenzymic forms between the cytosolic, mitochondrial, and peroxisomal compartments (157).
Mitochondrial flavin homeostasis and associations with the cytochrome P450 system
Biogenesis and molecular reactivities of the mitochondrial CYP enzyme system in mammals have been intensively investigated over the past 30 y (158, 159). All 3 protein families (adrenodoxin reductase, adrenodoxin, and CYP heme proteins) of the mitochondrial P450 system are located within the matrix side of the inner membrane and are of nuclear origin. To penetrate the mitochondria, each P450 enzyme bears a canonical mitochondrial NH2-terminal targeting sequence (160). The exact processing of the heme- and FAD-dependent proteins in this complex has not been completely defined. It is assumed that an unfolded version of these proteins has the mitochondrial targeting presequence removed by a mitochondrial processing peptidase before being imported into the matrix (161). Refolding of the heme- and the FAD-dependent proteins is thought to occur after they encounter their respective prosthetic groups with the aid of another flavin-dependent enzyme, sulfhydryl oxidase Erv1 (essential for respiratory and vegetative growth 1). This flavoenzyme is mandatory for folding of the secondary and tertiary structures of incoming proteins via formation of crosslinking disulfide bonds (162–164). For example, mitochondrial medium-chain acyl-CoA dehydrogenase requires folding in the presence of an FAD isoalloxazine ring to enable conformational competence of the holoenzyme (165). In a similar fashion, adrenodoxin reductase has an FAD folding requirement and exhibits an FAD-binding motif, glycine-X-glycine-X-X-glycine sequence (166), similar to that of a Rossmann fold to which nucleotide cofactors such as FAD and NAD(P) become associated (49).
In contrast to the multiple P450 isoforms, the FAD-dependent adrenodoxin reductase and the 2Fe-2S heme-dependent adrenodoxin exist as single isoforms and are expressed in concentrations lower than those of the CYPs. Studies in adrenal cortex mitochondria have shown that the average relative molar ratio of adrenodoxin reductase, adrenodoxin, and CYP is 1:3:8 (167). In view of this molar ratio and interaction among these components, activation of vitamin D and its subsequent degradation appear to be influenced primarily by the availability of substrate within the mitochondrial matrix and the mobile independence of the CYPs. Further analyses show that the process of electron transfer in the mitochondrial metabolism of vitamin D is most dependent on the formation of transient partner protein complexes with adrenodoxin reductase, which rely critically on the proximity of the redox domains and the availability of FAD within the mitochondrial matrix (168–171).
Availability of FAD within the mitochondrial matrix.
Recent studies in trafficking of riboflavin between subcellular compartments that include mitochondria and peroxisomes have focused on the existence of cytosolic and mitochondrial isoforms of FAD synthetase within eukaryotic cells (172, 173). In the cytosol, FMN is pyrophosphorylated with ATP to form FAD by FAD synthetase (FADS; ATP:FMN adenylyl transferase, FMNAT; or FAD pyrophosphorylase, EC 2.7.7.2) (174). The mitochondrial isoform is distinct in size and intracellular location from that in the cytosol (175). The mitochondrial isoform is referred to as FAD synthetase-1, whereas the cytosolic form is referred to as FAD synthetase-2. Mitochondrial FAD synthetase-1 displays an additional 97-amino acid domain at the N-terminus portion and localizes to the mitochondrial matrix. Isoform-1 also displays a 17-amino acid region, which represents a cleavable mitochondrial targeting sequence similar to that found in other proteins synthesized via nuclear DNA (176).
Investigations of the 2 isoforms have called special attention to a riboflavin/FAD cycle between the cytosolic compartment and mitochondria (177). The formation of intramitochondrial FAD requires transport of riboflavin from the cytosol into mitochondria and subsequent phosphorylation to FMN via a putative mitochondrial flavokinase (178). FAD synthetase-1 transforms FMN into FAD, which then combines with imported client apoflavoproteins inside the mitochondrial matrix. In yeast mitochondria, unbound FAD is quickly exported from the mitochondria to the cytosol via a flavin exchange protein (FLX1), which belongs to a family of mitochondrial transport proteins (179). In yeast, FLX1 plays a crucial role in maintaining normal levels of FAD-binding enzyme activity within the mitochondria and serves as a key regulator for unbound FAD within the mitochondria (180). Because FAD itself does not traverse membranes, FAD requires degradation back to riboflavin. Mitochondria display a pair of FAD dephosphorylating enzymes that differ from the cytosolic isoforms. Namely, mitochondrial FAD pyrophosphatase and FMN phosphohydrolase differ from the extramitochondrial enzymes in their pH maxima and sensitivity to inhibitors (178). Thus, in view of the complete complement of both flavin biosynthetic and dephosphorylating enzymes, as well as the presence of a flavin exchange protein, FLX1, it has been postulated that a riboflavin-FAD cycle occurs within mitochondria and further that it plays a major role in controlling mitochondrial flavoprotein turnover.
On the basis of sequence similarities to FLX1, a human candidate gene described as a mitochondrial folate transporter (MFT) was able to functionally complement the yeast FLX1 (181). In addition to the MFT, the ABCG2 transporter previously described to export riboflavin from the cell has also been detected on the inner membrane of the mitochondria (182). It is interesting to speculate whether MFT and ABCG2 may share a dual function because they both export riboflavin and folate from the mitochondria. Thus, FAD unaffiliated with client proteins is dephosphorylated to its parent form, riboflavin. Riboflavin is quickly “escorted” from the mitochondrial matrix by either the MFT or ABCG2 to prevent unscheduled electron transfer and avert formation of reactive oxygen species. Understanding the homeostatic control and turnover of the flavin coenzymes within the mitochondrial compartment is crucial to clarifying the regulatory functions of flavoenzymes in maintenance of normal cellular metabolism and bioenergetics.
It is important to note that the mitochondrial CYP system in the presence of unbound FAD or in the presence of excess riboflavin can function as a nonproductive electron transport system and react with molecular oxygen (183). As noted previously, unscheduled electron transfer via P450s and molecular oxygen can produce superoxide and other reactive oxygen species (184). Thus, a coordination among imported proteins, coenzymes, and cofactors within the mitochondrial matrix is indispensable for “correct” CYP function. Flavins unaffiliated with their client proteins or not confined within their proper domains in the electron transport chain may force unscheduled electron shunting within the chain and result in inappropriate formation of reactive oxygen species (185). Leakage of electrons within the matrix is the major source of reactive oxygen in the mitochondria. The generation of superoxide occurs at both the matrix side of the inner mitochondrial membrane as well as the cytosolic side (186). Riboflavin transported into mitochondria requires immediate transformation to FMN and FAD by the mitochondrial flavin biosynthetic enzymes.
A compromised reduction in activities of the flavin biosynthetic enzymes (flavokinase and FAD synthetase-1), an altered FMN:FAD ratio, or a defect in an export protein could result in accumulation of free riboflavin that may contribute to formation of excessive reactive oxygen species (187). FMN and FAD serve as coenzymes and are stabilized against photoreactivity and electron transfer while buried within their protein domains. Unlike its coenzymic derivatives, FMN and FAD, the parent compound, riboflavin, does not share the same extent of privileged binding domains and thus can interfere with scheduled electron transfers. Human motor neurons are particularly susceptible to injury if marked changes occur in components involved with mitochondria electron transfers (188). Thus, unattended riboflavin present within the inner compartments of mitochondrial membranes may short circuit the tightly controlled flow of electrons through heme transport and would be expected to interfere with vitamin D metabolism.
Conclusions
This review article focused attention on the subcellular compartmentalization of flavoproteins and their ability to interface with CYP proteins as well as with other electron transferring flavoproteins required for cholesterogenesis and vitamin D synthesis/metabolism. Another feature addressed in this review is the mechanism by which different flavoenzymes participate in cholesterogenesis and vitamin D metabolism and the overlapping clinical, cellular, and biochemical aspects of various diseases and inborn errors of metabolism (172). Several key P450s have been identified to date in mammalian mitochondria and endoplasmic reticulum that are involved in cholesterogenesis and vitamin D activation and degradation. The eukaryotic members of this gene superfamily of proteins exhibit dynamic interactions among their substrates, their various redox partners, and membrane associations. Numerous studies have examined the structure-function relation of P450s, factors that determine substrate specificity and selectivity, factors that establish redox-partner binding, and which regions are involved in membrane binding. Mammalian cells contain 2 major CYP systems located within the mitochondria (type I) and the endoplasmic reticulum (type II).
In the latter system, NAD(P)H cytochrome P450 reductase, which binds both FAD and FMN nucleotide coenzymes, catalyzes an electron pair transfer from NAD(P)H onto the isoalloxazine ring of FAD, which, in turn, engages in a single electron transfer to FMN followed by its release to CYP during the P450 catalytic cycle. This enzyme appears to have evolved from 2 separate flavoprotein families: ferredoxin nucleotide reductase and flavodoxin. The subdomains can be reconstituted to perform the monooxygenase activity of the P450 system. The mitochondrial CYP system comprises the adrenodoxin reductase, adrenodoxin, and several CYP enzymes. Studies of the factors that control the molecular recognition and interaction among these 3 proteins are complicated by their protein-protein contact and dynamic changes in the electrostatic strength and redox status of each of the components. The primary importance of dietary riboflavin in cell biology is associated with its conversion into FMN and FAD, coenzymes for a large number of dehydrogenases, reductases, monooxygenase, and oxidases involved in energy metabolism, redox homeostasis, protein folding, and a diverse number of regulatory, biosynthetic and degradative pathways.
Understanding the homeostatic control and turnover of the flavin coenzymes within the mitochondrial compartment and endoplasmic reticulum is crucial to clarifying the regulatory functions of flavoenzymes in the maintenance of normal cellular metabolism that controls cholesterogenesis and vitamin D formation. A number of recent studies that shed new light on the cellular processes and biologic effects of riboflavin in metabolic disease have been discussed.The deficiency of riboflavin in humans and in experimental animal models has been linked to several diseases, including neuromuscular and neurologic disorders and even cancer. Overall, a deeper understanding of the role of riboflavin may prove essential to targeted therapeutic designs aimed at cholesterol and vitamin D metabolism.
Acknowledgments
Both authors read and approved the final manuscript.
Footnotes
Abbreviations used: ABCG2, ATP-binding cassette transporter G2; ALS, amyotrophic lateral sclerosis; CNS, central nervous system; CPR, NAD(P)H-cytochrome P450 reductase; CTX, cerebrotendinous xanthomatosis; CYP, cytochrome P450; DHCR7, 7-dehydrocholesterol reductase; DHCR24, 24-dehydrocholesterol reductase; ETF, electron transferring flavoprotein; FGF-23, fibroblast growth factor-23; FLX1, flavin exchange protein; HMG-CoA, 3-hydroxy-3-methylglutaryl–coenzyme A; Km, Michaelis constant; LSS, lanosterol synthase (squalene-2,3-oxide lanosterol cyclase); MFT, mitochondrial folate transporter; 1α,25(OH)2D, 1α,25-dihydroxyvitamin D; 25(OH)D, 25-hydroxyvitamin D.
Literature Cited
- 1.Pinto JT, Rivlin RS. Riboflavin In:Zempleni J, Suttie JW, Gregory JF, Stover PJ. editors. 5th ed. New York: Taylor and Francis; 2013. p. 191–265 [Google Scholar]
- 2.Nakagawa H, Asano A, Sato R. Ascorbate-synthesizing system in rat liver microsomes. II. A peptide-bound flavin as the prosthetic group of L-gulono-gamma-lactone oxidase. J Biochem. 1975;77:221–32 [PubMed] [Google Scholar]
- 3.Kiuchi K, Nishikimi M, Yagi K. L-gulonolactone oxidase activity and vitamin C status in riboflavin-deficient rats. Biochim Biophys Acta. 1980;630:330–7 [DOI] [PubMed] [Google Scholar]
- 4.Mushtaq S, Su H, Hill MH, Powers HJ. Erythrocyte pyridoxamine phosphate oxidase activity: a potential biomarker of riboflavin status? Am J Clin Nutr. 2009;90:1151–9 [DOI] [PubMed] [Google Scholar]
- 5.Adelekan DA, Adekile AD, Thurnham DI. Dependence of pyridoxine metabolism on riboflavin status in sickle cell patients. Am J Clin Nutr. 1987;46:86–90 [DOI] [PubMed] [Google Scholar]
- 6.Zhou Z, Fisher D, Spidel J, Greenfield J, Patson B, Fazal A, Wigal C, Moe OA, Madura JD. Kinetic and docking studies of the interaction of quinones with the quinone reductase active site. Biochemistry. 2003;42:1985–94 [DOI] [PubMed] [Google Scholar]
- 7.Preusch PC, Suttie JW. Vitamin K-dependent reactions in rat liver: role of flavoproteins. J Nutr. 1981;111:2087–97 [DOI] [PubMed] [Google Scholar]
- 8.Seekamp A, Hultquist DE, Till GO. Protection by vitamin B2 against oxidant-mediated acute lung injury. Inflammation. 1999;23:449–60 [DOI] [PubMed] [Google Scholar]
- 9.Gherasim CG, Zaman U, Raza A, Banerjee R. Impeded electron transfer from a pathogenic FMN domain mutant of methionine synthase reductase and its responsiveness to flavin supplementation. Biochemistry. 2008;47:12515–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNulty H, McKinley MC, Wilson B, McPartlin J, Strain JJ, Weir DG, Scott JM. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am J Clin Nutr. 2002;76:436–41 [DOI] [PubMed] [Google Scholar]
- 11.Depeint F, Bruce WR, Shangari N, Mehta R, O'Brien PJ. Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem Biol Interact. 2006;163:94–112 [DOI] [PubMed] [Google Scholar]
- 12.Bikle DD. Vitamin D metabolism and function in the skin. Mol Cell Endocrinol. 2011;347:80–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisk CM, Theobald HE, Sanders TA. Fortified malted milk drinks containing low-dose ergocalciferol and cholecalciferol do not differ in their capacity to raise serum 25-hydroxyvitamin D concentrations in healthy men and women not exposed to UV-B. J Nutr. 2012;142:1286–90 [DOI] [PubMed] [Google Scholar]
- 14.Tripkovic L, Lambert H, Hart K, Smith CP, Bucca G, Penson S, Chope G, Hyppönen E, Berry J, Vieth R, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95:1357–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massey V. The chemical and biological versatility of riboflavin. Biochem Soc Trans. 2000;28:283–96 [PubMed] [Google Scholar]
- 16.Green J, Paget MS. Bacterial redox sensors. Nat Rev Microbiol. 2004;2:954–66 [DOI] [PubMed] [Google Scholar]
- 17.Dong H, Beer SV. Riboflavin induces disease resistance in plants by activating a novel signal transduction pathway. Phytopathology. 2000;90:801–11 [DOI] [PubMed] [Google Scholar]
- 18.Macheroux P, Kappes B, Ealick SE. Flavogenomics—a genomic and structural view of flavin-dependent proteins. FEBS J. 2011;278:2625–34 [DOI] [PubMed] [Google Scholar]
- 19.Tokutomi S, Matsuoka D, Zikihara K. Molecular structure and regulation of phototropin kinase by blue light. Biochim Biophys Acta. 2008;1784:133–42 [DOI] [PubMed] [Google Scholar]
- 20.Silva E, Edwards AM. editors. Flavins: photochemistry and photobiology. Cambridge (UK): RSC Publishing; 2006 [Google Scholar]
- 21.Lostao A, Gómez-Moreno C, Mayhew SG, Sancho J. Differential stabilization of the three FMN redox forms by tyrosine 94 and tryptophan 57 in flavodoxin from Anabaena and its influence on the redox potentials. Biochemistry. 1997;36:14334–44 [DOI] [PubMed] [Google Scholar]
- 22.Kao YT, Saxena C, He TF, Guo L, Wang L, Sancar A, Zhong D. Ultrafast dynamics of flavins in five redox states. J Am Chem Soc. 2008;130:13132–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beinert WD, Rüterjans H, Müller F, Bacher A. Nuclear magnetic resonance studies of the old yellow enzyme. 2. 13C NMR of the enzyme recombined with 13C-labeled flavin mononucleotides. Eur J Biochem. 1985;152:581–7 [DOI] [PubMed] [Google Scholar]
- 24.Berrisford JM, Sazanov LA. Structural basis for the mechanism of respiratory complex I. J Biol Chem. 2009;284:29773–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toogood HS, Leys D, Scrutton NS. Dynamics driving function: new insights from electron transferring flavoproteins and partner complexes. FEBS J. 2007;274:5481–504 [DOI] [PubMed] [Google Scholar]
- 26.Mailloux RJ, Harper ME. Glucose regulates enzymatic sources of mitochondrial NADPH in skeletal muscle cells; a novel role for glucose-6-phosphate dehydrogenase. FASEB J. 2010;24:2495–506 [DOI] [PubMed] [Google Scholar]
- 27.Guengerich FP, Munro AW. Unusual cytochrome p450 enzymes and reactions. J Biol Chem. 2013;288:17065–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter TD, Kasper CB. NADPH-cytochrome P-450 oxidoreductase: flavin mononucleotide and flavin adenine dinucleotide domains evolved from different flavoproteins. Biochemistry. 1986;25:1682–7 [DOI] [PubMed] [Google Scholar]
- 29.Aigrain L, Pompon D, Truan G. Role of the interface between the FMN and FAD domains in the control of redox potential and electronic transfer of NADPH-cytochrome P450 reductase. Biochem J. 2011;435:197–206 [DOI] [PubMed] [Google Scholar]
- 30.Mowat CG, Gazur B, Campbell LP, Chapman SK. Flavin-containing heme enzymes. Arch Biochem Biophys. 2010;493:37–52 [DOI] [PubMed] [Google Scholar]
- 31.Shen AL, O'Leary KA, Kasper CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002;277:6536–41 [DOI] [PubMed] [Google Scholar]
- 32.Iyanagi T, Xia C, Kim JJ. NADPH-cytochrome P450 oxidoreductase: prototypic member of the diflavin reductase family. Arch Biochem Biophys. 2012;528:72–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dumit VI, Cortez N, Matthias Ullmann G. Distinguishing two groups of flavin reductases by analyzing the protonation state of an active site carboxylic acid. Proteins. 2011;79:2076–85 [DOI] [PubMed] [Google Scholar]
- 34.Murataliev MB, Feyereisen R, Walker FA. Electron transfer by diflavin reductases. Biochim Biophys Acta. 2004;1698:1–26 [DOI] [PubMed] [Google Scholar]
- 35.Patel JM, Pawar SS. Riboflavin and drug metabolism in adult male and female rats. Biochem Pharmacol. 1974;23:1467–77 [DOI] [PubMed] [Google Scholar]
- 36.Hara T, Taniguchi M. Relationship between changes in properties and contents of riboflavin derivatives of NADPH-cytochrome P-450 reductase in the liver microsomes of riboflavin-deficient rats. J Biochem. 1985;97:473–82 [DOI] [PubMed] [Google Scholar]
- 37.Vermilion JL, Ballou DP, Massey V, Coon MJ. Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. J Biol Chem. 1981;256:266–77 [PubMed] [Google Scholar]
- 38.Fass S, Rivlin RS. Regulation of riboflavin-metabolizing enzymes in riboflavin deficiency. Am J Physiol. 1969;217:988–91 [DOI] [PubMed] [Google Scholar]
- 39.Kranendonk M, Marohnic CC, Panda SP, Duarte MP, Oliveira JS, Masters BS, Rueff J. Impairment of human CYP1A2-mediated xenobiotic metabolism by Antley-Bixler syndrome variants of cytochrome P450 oxidoreductase. Arch Biochem Biophys. 2008;475:93–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia C, Panda SP, Marohnic CC, Martásek P, Masters BS, Kim JJ. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc Natl Acad Sci USA. 2011;108:13486–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pikuleva IA, Waterman MR. Cytochromes p450: roles in diseases. J Biol Chem. 2013;288:17091–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller WL. Role of mitochondria in steroidogenesis. Endocr Dev. 2011;20:1–19 [DOI] [PubMed] [Google Scholar]
- 43.Gu J, Weng Y, Zhang QY, Cui H, Behr M, Wu L, Yang W, Zhang L, Ding X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–901 [DOI] [PubMed] [Google Scholar]
- 44.Chen HW, Kandutsch AA, Waymouth C. Inhibition of cell growth by oxygenated derivatives of cholesterol. Nature. 1974;251:419–21 [DOI] [PubMed] [Google Scholar]
- 45.Bloch KE. Sterol structure and membrane function. CRC Crit Rev Biochem. 1983;14:47–92 [DOI] [PubMed] [Google Scholar]
- 46.Laden BP, Tang Y, Porter TD. Cloning, heterologous expression, and enzymological characterization of human squalene monooxygenase. Arch Biochem Biophys. 2000;374:381–8 [DOI] [PubMed] [Google Scholar]
- 47.Wendt KU. Enzyme mechanisms for triterpene cyclization: new pieces of the puzzle. Angew Chem Int Ed Engl. 2005;44:3966–71 [DOI] [PubMed] [Google Scholar]
- 48.Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem. 2009;284:11374–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleiger G, Eisenberg D. GXXXG and GXXXA motifs stabilize FAD and NAD(P)-binding Rossmann folds through C(alpha)-H...O hydrogen bonds and Van der Waals interactions. J Mol Biol. 2002;323:69–76 [DOI] [PubMed] [Google Scholar]
- 50.Abe I, Abe T, Lou W, Masuoka T, Noguchi H. Site-directed mutagenesis of conserved aromatic residues in rat squalene epoxidase. Biochem Biophys Res Commun. 2007;352:259–63 [DOI] [PubMed] [Google Scholar]
- 51.Mokashi V, Singh DK, Porter TD. Rat supernatant protein factor-like protein stimulates squalene monooxygenase and is activated by protein kinase A. Biochem Biophys Res Commun. 2004;316:688–92 [DOI] [PubMed] [Google Scholar]
- 52.Lin LF. Roles of phospholipid and detergent in soluble protein activation of squalene epoxidase. Biochemistry. 1980;19:5135–40 [DOI] [PubMed] [Google Scholar]
- 53.van Berkel WJ, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–89 [DOI] [PubMed] [Google Scholar]
- 54.Bai M, Xiao XY, Prestwich GD. Epoxidation of 2,3-oxidosqualene to 2,3;22,23-squalene dioxide by squalene epoxidase. Biochem Biophys Res Commun. 1992;185:323–9 [DOI] [PubMed] [Google Scholar]
- 55.Nagumo A, Kamei T, Sakakibara J, Ono T. Purification and characterization of recombinant squalene epoxidase. J Lipid Res. 1995;36:1489–97 [PubMed] [Google Scholar]
- 56.Bogh MK, Schmedes AV, Philipsen PA, Thieden E, Wulf HC. Vitamin D production after UVB exposure depends on baseline vitamin D and total cholesterol but not on skin pigmentation. J Invest Dermatol. 2010;130:546–53 [DOI] [PubMed] [Google Scholar]
- 57.Belter A, Skupinska M, Giel-Pietraszuk M, Grabarkiewicz T, Rychlewski L, Barciszewski J. Squalene monooxygenase—a target for hypercholesterolemic therapy. Biol Chem. 2011;392:1053–75 [DOI] [PubMed] [Google Scholar]
- 58.Feidt DM, Klein K, Nüssler A, Zanger UM. RNA-interference approach to study functions of NADPH: cytochrome P450 oxidoreductase in human hepatocytes. Chem Biodivers. 2009;6:2084–91 [DOI] [PubMed] [Google Scholar]
- 59.Abe I, Seki T, Umehara K, Miyase T, Noguchi H, Sakakibara J, Ono T. Green tea polyphenols: novel and potent inhibitors of squalene epoxidase. Biochem Biophys Res Commun. 2000;268:767–71 [DOI] [PubMed] [Google Scholar]
- 60.Gupta N, Porter TD. Garlic and garlic-derived compounds inhibit human squalene monooxygenase. J Nutr. 2001;131:1662–7 [DOI] [PubMed] [Google Scholar]
- 61.Rozman D, Monostory K. Perspectives of the non-statin hypolipidemic agents. Pharmacol Ther. 2010;127:19–40 [DOI] [PubMed] [Google Scholar]
- 62.Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta. 2007;1770:467–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kelley RI, Kratz LE, Glaser RL, Netzloff ML, Wolf LM, Jabs EW. Abnormal sterol metabolism in a patient with Antley-Bixler syndrome and ambiguous genitalia. Am J Med Genet. 2002;110:95–102 [DOI] [PubMed] [Google Scholar]
- 64.Marohnic CC, Panda SP, Martásek P, Masters BS. Diminished FAD binding in the Y459H and V492E Antley-Bixler syndrome mutants of human cytochrome P450 reductase. J Biol Chem. 2006;281:35975–82 [DOI] [PubMed] [Google Scholar]
- 65.Keber R, Motaln H, Wagner KD, Debeljak N, Rassoulzadegan M, Ačimovič J, Rozman D, Horvat S. Mouse knockout of the cholesterogenic cytochrome P450 lanosterol 14α-demethylase (Cyp51) resembles Antley-Bixler syndrome. J Biol Chem. 2011;286:29086–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pikuleva IA. Cytochrome P450s and cholesterol homeostasis. Pharmacol Ther. 2006;112:761–73 [DOI] [PubMed] [Google Scholar]
- 67.Song BL, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005;1:179–89 [DOI] [PubMed] [Google Scholar]
- 68.Li L, Porter TD. Hepatic cytochrome P450 reductase-null mice reveal a second microsomal reductase for squalene monooxygenase. Arch Biochem Biophys. 2007;461:76–84 [DOI] [PubMed] [Google Scholar]
- 69.Bae SH, Paik YK. Cholesterol biosynthesis from lanosterol: development of a novel assay method and characterization of rat liver microsomal lanosterol delta 24-reductase. Biochem J. 1997;326:609–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zolotushko J, Flusser H, Markus B, Shelef I, Langer Y, Heverin M, Björkhem I, Sivan S, Birk OS. The desmosterolosis phenotype: spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter. Eur J Hum Genet. 2011;19:942–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schaaf CP, Koster J, Katsonis P, Kratz L, Shchelochkov OA, Scaglia F, Kelley RI, Lichtarge O, Waterham HR, Shinawi M. Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet A. 2011;155A:1597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI, Wanders RJ. Mutations in the 3-beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nelson MM, Bated CDC, Weight HV, Evans HM. Multiple congenital abnormalities in the rat resulting from riboflavin deficiency induced by the antimetabolite galactoflavin. J Nutr. 1956;58:125–34 [DOI] [PubMed] [Google Scholar]
- 74.Mackler B. Studies of the molecular basis of congenital malformations. Pediatrics. 1969;43:915–26 [PubMed] [Google Scholar]
- 75.Pinto J, Huang YP, Rivlin RS. Inhibition of riboflavin metabolism in rat tissues by chlorpromazine, imipramine and amitriptyline. J Clin Invest. 1981;67:1500–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinto J, Huang YP, Rivlin RS. Inhibition by chlorpromazine of thyroxine modulation of flavin metabolism in liver, cerebrum and cerebellum. Biochem Pharmacol. 1985;34:93–5 [DOI] [PubMed] [Google Scholar]
- 77.Dutta P, Raiczyk GB, Pinto J. Inhibition of riboflavin metabolism in cardiac and skeletal muscle of rats by quinacrine and tetracycline. J Clin Biochem Nutr. 1988;4:203–8 [Google Scholar]
- 78.Gabay S, Harris SR. Inhibition of flavoenzymes by phenothiazines. Agressologie. 1968;9:79–89 [PubMed] [Google Scholar]
- 79.Cuevas P, Gutierrez Diaz JA. Absence of filipin-sterol complexes from the ciliary necklace of ependymal cells. Anat Embryol (Berl). 1985;172:97–9 [DOI] [PubMed] [Google Scholar]
- 80.Chailley B, Boisvieux-Ulrich E. Detection of plasma membrane cholesterol by filipin during microvillogenesis and ciliogenesis in quail oviduct. J Histochem Cytochem. 1985;33:1–10 [DOI] [PubMed] [Google Scholar]
- 81.Martinez-Seara H, Róg T, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M, Reigada R. Interplay of unsaturated phospholipids and cholesterol in membranes: effect of the double-bond position. Biophys J. 2008;95:3295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keber R, Rozman D, Horvat S. Sterols in spermatogenesis and sperm maturation. J Lipid Res. 2013;54:20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res. 2006;47:1535–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tumkiratiwong P, Sukkachart H, Buapuan T, Tungtrongchitr R, Tungtrongchitr A. Riboflavin-deficient and Trichinella spiralis-induced stresses on plasma corticosterone associated with spermatogenesis in male Wistar rats. Southeast Asian J Trop Med Public Health. 2006;37:250–6 [PubMed] [Google Scholar]
- 85.Kuehnle K, Crameri A, Kälin RE, Luciani P, Benvenuti S, Peri A, Ratti F, Rodolfo M, Kulic L, Heppner FL, et al. Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress. Mol Cell Biol. 2008;28:539–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vainio S, Jansen M, Koivusalo M, Rog T, Karttunen M, Vattulainen I, Ikonen E. Significance of sterol structural specificity: desmosterol cannot replace cholesterol in lipid rafts. J Biol Chem. 2006;281:348–55 [DOI] [PubMed] [Google Scholar]
- 87.Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55:1265–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peri A, Serio M. Neuroprotective effects of the Alzheimer's disease-related gene seladin-1. J Mol Endocrinol. 2008;41:251–61 [DOI] [PubMed] [Google Scholar]
- 89.Reid PC, Urano Y, Kodama T, Hamakubo T. Alzheimer's disease: cholesterol, membrane rafts, isoprenoids and statins. J Cell Mol Med. 2007;11:383–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–97 [DOI] [PubMed] [Google Scholar]
- 91.Hung YH, Bush AI, La Fontaine S. Links between copper and cholesterol in Alzheimer's disease. Front Physiol. 2013;4:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Connor WE, Lin DS, Wolf DP, Alexander M. Uneven distribution of desmosterol and docosahexaenoic acid in the heads and tails of monkey sperm. J Lipid Res. 1998;39:1404–11 [PubMed] [Google Scholar]
- 93.Huster D, Scheidt HA, Arnold K, Herrmann A, Müller P. Desmosterol may replace cholesterol in lipid membranes. Biophys J. 2005;88:1838–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hitzemann RJ, Johnson DA. Developmental changes in synaptic membrane lipid composition and fluidity. Neurochem Res. 1983;8:121–31 [DOI] [PubMed] [Google Scholar]
- 95.Shackleton CH. Role of a disordered steroid metabolome in the elucidation of sterol and steroid biosynthesis. Lipids. 2012;47:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kalter H. Congenital malformations induced by riboflavin deficiency in strains of inbred mice. Pediatrics. 1959;23:222–30 [PubMed] [Google Scholar]
- 97.Aluclu MU, Bahceci S, Bahceci M. A rare embryological malformation of brain - Dandy-Walker syndrome—and its association with Kallmann's syndrome. Neuroendocrinol Lett. 2007;28:255–8 [PubMed] [Google Scholar]
- 98.Nishino H, Ishibashi T. Evidence for requirement of NADPH cytochrome P450 oxidoreductase in the microsomal NADPH Sterol delta 7-reductase system. Arch Biochem Biophys. 2000;374:293–8 [DOI] [PubMed] [Google Scholar]
- 99.Zou L, Li L, Porter TD. 7-Dehydrocholesterol reductase activity is independent of cytochrome P450 reductase. J Steroid Biochem Mol Biol. 2011;127:435–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hafner M, Rezen T, Rozman D. Regulation of hepatic cytochromes p450 by lipids and cholesterol. Curr Drug Metab. 2011;12:173–85 [DOI] [PubMed] [Google Scholar]
- 101.Glossmann HH. Origin of 7-dehydrocholesterol (provitamin D) in the skin. J Invest Dermatol. 2010;130:2139–41 [DOI] [PubMed] [Google Scholar]
- 102.Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011;13:260–73 [DOI] [PubMed] [Google Scholar]
- 103.Doolman R, Leichner GS, Avner R, Roitelman J. Ubiquitin is conjugated by membrane ubiquitin ligase to three sites, including the N terminus, in transmembrane region of mammalian 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for sterol-regulated enzyme degradation. J Biol Chem. 2004;279:38184–93 [DOI] [PubMed] [Google Scholar]
- 104.DeBarber AE, Eroglu Y, Merkens LS, Pappu AS, Steiner RD. Smith-Lemli-Opitz syndrome. Expert Rev Mol Med. 2011;13:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rossi M, Federico G, Corso G, Parenti G, Battagliese A, Frascogna AR, Della Casa R, Dello Russo A, Strisciuglio P, Saggese G, et al. Vitamin D status in patients affected by Smith-Lemli-Opitz syndrome. J Inherit Metab Dis. 2005;28:69–80 [DOI] [PubMed] [Google Scholar]
- 107.Ifergan I, Goler-Baron V, Assaraf YG. Riboflavin concentration within ABCG2-rich extracellular vesicles is a novel marker for multidrug resistance in malignant cells. Biochem Biophys Res Commun. 2009;380:5–10 [DOI] [PubMed] [Google Scholar]
- 108.van Herwaarden AE, Schinkel AH. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol Sci. 2006;27:10–6 [DOI] [PubMed] [Google Scholar]
- 109.van Herwaarden AE, Wagenaar E, Merino G, Jonker JW, Rosing H, Beijnen JH, Schinkel AH. Multidrug transporter ABCG2/breast cancer resistance protein secretes riboflavin (vitamin B2) into milk. Mol Cell Biol. 2007;27:1247–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jonker JW, Freeman J, Bolscher E, Musters S, Alvi AJ, Titley I, Schinkel AH, Dale TC. Contribution of the ABC transporters Bcrp1 and Mdr1a/1b to the side population phenotype in mammary gland and bone marrow of mice. Stem Cells. 2005;23:1059–65 [DOI] [PubMed] [Google Scholar]
- 111.Shen S, Callaghan D, Juzwik C, Xiong H, Huang P, Zhang W. ABCG2 reduces ROS-mediated toxicity and inflammation: a potential role in Alzheimer’s disease. J Neurochem. 2010;114:1590–604 [DOI] [PubMed] [Google Scholar]
- 112.Telbisz Á, Özvegy-Laczka C, Hegedűs T, Váradi A, Sarkadi B. Effects of the lipid environment, cholesterol and bile acids on the function of the purified and reconstituted human ABCG2 protein. Biochem J. 2013;450:387–95 [DOI] [PubMed] [Google Scholar]
- 113.Storch CH, Ehehalt R, Haefeli WE, Weiss J. Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J Pharmacol Exp Ther. 2007;323:257–64 [DOI] [PubMed] [Google Scholar]
- 114.Pál A, Méhn D, Molnár E, Gedey S, Mészáros P, Nagy T, Glavinas H, Janáky T, von Richter O, Báthori G, et al. Cholesterol potentiates ABCG2 activity in a heterologous expression system: improved in vitro model to study function of human ABCG2. J Pharmacol Exp Ther. 2007;321:1085–94 [DOI] [PubMed] [Google Scholar]
- 115.Miller WL. Steroidogenic enzymes. Endocr Dev. 2008;13:1–18 [DOI] [PubMed] [Google Scholar]
- 116.Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29:664–73 [DOI] [PubMed] [Google Scholar]
- 117.Zhu J, DeLuca HF. Vitamin D 25-hydroxylase—four decades of searching, are we there yet? Arch Biochem Biophys. 2012;523:30–6 [DOI] [PubMed] [Google Scholar]
- 118.Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. 2004;101:7711–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shinkyo R, Sakaki T, Kamakura M, Ohta M, Inouye K. Metabolism of vitamin D by human microsomal CYP2R1. Biochem Biophys Res Commun. 2004;324:451–7 [DOI] [PubMed] [Google Scholar]
- 120.Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci USA. 2013;110:15650–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miller WL. P450 oxidoreductase deficiency: a new disorder of steroidogenesis with multiple clinical manifestations. Trends Endocrinol Metab. 2004;15:311–5 [DOI] [PubMed] [Google Scholar]
- 122.Wudy SA, Hartmann MF, Draper N, Stewart PM, Arlt W. A male twin infant with skull deformity and elevated neonatal 17-hydroxyprogesterone: a prismatic case of P450 oxidoreductase deficiency. Endocr Res. 2004;30:957–64 [DOI] [PubMed] [Google Scholar]
- 123.Shackleton C, Malunowicz E. Apparent pregnene hydroxylation deficiency (APHD): seeking the parentage of an orphan metabolome. Steroids. 2003;68:707–17 [DOI] [PubMed] [Google Scholar]
- 124.Adachi M, Tachibana K, Asakura Y, Suwa S, Nishimura G. A male patient presenting with major clinical symptoms of glucocorticoid deficiency and skeletal dysplasia, showing a steroid pattern compatible with 17alpha-hydroxylase/17,20-lyase deficiency, but without obvious CYP17 gene mutations. Endocr J. 1999;46:285–92 [DOI] [PubMed] [Google Scholar]
- 125.Kelley RI, Robinson D, Puffenberger EG, Strauss KA, Morton DH. Amish lethal microcephaly: a new metabolic disorder with severe congenital microcephaly and 2-ketoglutaric aciduria. Am J Med Genet. 2002;112:318–26 [DOI] [PubMed] [Google Scholar]
- 126.Robinson PN, Neumann LM, Demuth S, Enders H, Jung U, König R, Mitulla B, Müller D, Muschke P, Pfeiffer L, et al. Shprintzen-Goldberg syndrome: fourteen new patients and a clinical analysis. Am J Med Genet A. 2005;135:251–62 [DOI] [PubMed] [Google Scholar]
- 127.Warkany J, Schraffenberger E. Congenital malformations induced in rats by maternal nutritional deficiency. J Nutr. 1944;27:477–84 [Google Scholar]
- 128.Miller Z, Poncet I, Takacs E. Biochemical studies on experimental congenital malformations: flavin nucleotides and folic acid in fetuses and livers from normal and riboflavin-deficient rats. J Biol Chem. 1962;237:968–73 [PubMed] [Google Scholar]
- 129.Patton SW, Misri S, Corral MR, Perry KF, Kuan AJ. Antipsychotic medication during pregnancy and lactation in women with schizophrenia: evaluating the risk. Can J Psychiatry. 2002;47:959–65 [DOI] [PubMed] [Google Scholar]
- 130.Pinto J, Huang YP, Pelliccione N, Rivlin RS. Cardiac sensitivity to the inhibitory effects of chlorpromazine, imipramine, and amitriptyline upon formation of flavins. Biochem Pharmac. 1982;31:3495–9 [DOI] [PubMed] [Google Scholar]
- 131.Horvath C, Druga A. Action of the phenothiazine derivative methophenazine on prenatal development in rats. Teratology. 1975;11:325–9 [DOI] [PubMed] [Google Scholar]
- 132.Sergeev IN, Kim RH, Arkhapchev IuP, Kodentsova VM, Alekseeva IA. [Metabolism of 25-hydroxyvitamin D3 in the kidney and nuclear receptors of 1,25-dihydroxyvitamin D3 in small intestine mucosa of rats with vitamin B2 deficiency.] Vopr Med Khim. 1987;33:96–103. [PubMed] [Google Scholar]
- 133.Ziegler GA, Vonrhein C, Hanukoglu I, Schulz GE. The structure of adrenodoxin reductase of mitochondrial P450 systems: electron transfer for steroid biosynthesis. J Mol Biol. 1999;289:981–90 [DOI] [PubMed] [Google Scholar]
- 134.Araya Z, Tang W, Wikvall K. Hormonal regulation of the human sterol 27-hydroxylase gene CYP27A1. Biochem J. 2003;372:529–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–231 [DOI] [PubMed] [Google Scholar]
- 136.Gupta RP, Patrick K, Bell NH. Mutational analysis of CYP27A1: assessment of 27-hydroxylation of cholesterol and 25-hydroxylation of vitamin D. Metabolism. 2007;56:1248–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schwarz M, Russell DW, Dietschy JM, Turley SD. Alternate pathways of bile acid synthesis in the cholesterol 7-alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J Lipid Res. 2001;42:1594–603 [PubMed] [Google Scholar]
- 138.Ohyama Y, Masumoto O, Usui E, Okuda K. Multi-functional property of rat liver mitochondrial cytochrome P-450. J Biochem. 1991;109:389–93 [DOI] [PubMed] [Google Scholar]
- 139.Araya Z, Hosseinpour F, Bodin K, Wikvall K. Metabolism of 25-hydroxyvitamin D3 by microsomal and mitochondrial vitamin D3 25-hydroxylases (CYP2D25 and CYP27A1): a novel reaction by CYP27A1. Biochim Biophys Acta. 2003;1632:40–7 [DOI] [PubMed] [Google Scholar]
- 140.Forrest KY, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. 2011;31:48–54 [DOI] [PubMed] [Google Scholar]
- 141.Sacheck J, Goodman E, Chui K, Chomitz V, Must A, Economos C. Vitamin D deficiency, adiposity, and cardiometabolic risk in urban schoolchildren. J Pediatr. 2011;159:945–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumar J, Muntner P, Kaskel FJ, Hailpern SM, Melamed ML. Prevalence and associations of 25-hydroxyvitamin D deficiency in US children: NHANES 2001–2004. Pediatrics. 2009;124:e362–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gagnon C, Lu ZX, Magliano DJ, Dunstan DW, Shaw JE, Zimmet PZ, Sikaris K, Ebeling PR, Daly RM. Low serum 25-hydroxyvitamin D is associated with increased risk of the development of the metabolic syndrome at five years: results from a national, population-based prospective study (the Australian Diabetes, Obesity and Lifestyle Study: AusDiab). J Clin Endocrinol Metab. 2012;97:1953–61 [DOI] [PubMed] [Google Scholar]
- 144.Karhapää P, Pihlajamäki J, Pörsti I, Kastarinen M, Mustonen J, Niemelä O, Kuusisto J. Diverse associations of 25-hydroxyvitamin D and 1,25-dihydroxy-vitamin D with dyslipidaemias. J Intern Med. 2010;268:604–10 [DOI] [PubMed] [Google Scholar]
- 145.Diekstra FP, Saris CG, van Rheenen W, Franke L, Jansen RC, van Es MA, van Vught PW, Blauw HM, Groen EJ, Horvath S, et al. Mapping of gene expression reveals CYP27A1 as a susceptibility gene for sporadic ALS. PLoS ONE. 2012;7:e35333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Johnson JO, Gibbs JR, Megarbane A, Urtizberea JA, Hernandez DG, Foley AR, Arepalli S, Pandraud A, Simón-Sánchez J, Clayton P, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain. 2012;135:2875–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lin J, Diamanduros A, Chowdhury SA, Scelsa S, Latov N, Sadiq SA. Specific electron transport chain abnormalities in amyotrophic lateral sclerosis. J Neurol. 2009;256:774–82 [DOI] [PubMed] [Google Scholar]
- 148.Pinto J, Huang YP, Rivlin RS. Mechanisms underlying the differential effects of ethanol upon the bioavailability of riboflavin and flavin adenine dinucleotide. J Clin Invest. 1987;79:1343–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nyquist-Battie C, Freter M. Cardiac mitochondrial abnormalities in a mouse model of the fetal alcohol syndrome. Alcohol Clin Exp Res. 1988;12:264–7 [DOI] [PubMed] [Google Scholar]
- 150.Rosen H, Reshef A, Maeda N, Lippoldt A, Shpizen S, Triger L, Eggertsen G, Björkhem I, Leitersdorf E. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem. 1998;273:14805–12 [DOI] [PubMed] [Google Scholar]
- 151.Berginer VM, Shany S, Alkalay D, Berginer J, Dekel S, Salen G, Tint GS, Gazit D. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism. 1993;42:69–74 [DOI] [PubMed] [Google Scholar]
- 152.Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, Turley SD. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia: reversal by cholic acid feeding. J Biol Chem. 2000;275:39685–92 [DOI] [PubMed] [Google Scholar]
- 153.Urushino N, Yamamoto K, Kagawa N, Ikushiro S, Kamakura M, Yamada S, Kato S, Inouye K, Sakaki T. Interaction between mitochondrial CYP27B1 and adrenodoxin: role of arginine 458 of mouse CYP27B1. Biochemistry. 2006;45:4405–12 [DOI] [PubMed] [Google Scholar]
- 154.Jones G. Extrarenal vitamin D activation and interactions between vitamin D2, vitamin D3, and vitamin D analogs. Annu Rev Nutr. 2013;33:23–44 [DOI] [PubMed] [Google Scholar]
- 155.Beckman MJ, Tadikonda P, Werner E, Prahl J, Yamada S, DeLuca HF. Human 25-hydroxyvitamin D3–24-hydroxylase, a multicatalytic enzyme. Biochemistry. 1996;35:8465–72 [DOI] [PubMed] [Google Scholar]
- 156.Inouye K, Sakaki T. Enzymatic studies on the key enzymes of vitamin D metabolism; 1 alpha-hydroxylase (CYP27B1) and 24-hydroxylase (CYP24). Biotechnol Annu Rev. 2001;7:179–94 [DOI] [PubMed] [Google Scholar]
- 157.Olsen RK, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford MW, Dean JC, Cornelius N, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–54 [DOI] [PubMed] [Google Scholar]
- 158.Estabrook RW. A passion for p450s (remembrances of the early history of research on cytochrome p450). Drug Metab Dispos. 2003;31:1461–73 [DOI] [PubMed] [Google Scholar]
- 159.Peterson JA, Sevrioukova I, Truan G, Graham-Lorence SE. P450BM-3; a tale of two domains—or is it three? Steroids. 1997;62:117–23 [DOI] [PubMed] [Google Scholar]
- 160.Addya S, Anandatheerthavarada HK, Biswas G, Bhagwat SV, Mullick J, Avadhani NG. Targeting of NH2-terminal-processed microsomal protein to mitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2. J Cell Biol. 1997;139:589–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Omura T. Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J Biochem. 1998;123:1010–6 [DOI] [PubMed] [Google Scholar]
- 162.Endo T, Yamano K, Kawano S. Structural basis for the disulfide relay system in the mitochondrial intermembrane space. Antioxid Redox Signal. 2010;13:1359–73 [DOI] [PubMed] [Google Scholar]
- 163.Riemer J, Fischer M, Herrmann JM. doi: 10.1016/j.bbamem.2010.06.003. Oxidation-driven protein import into mitochondria: Insights and blind spots. Biochim Biophys Acta. 2011;1808:981–9. [DOI] [PubMed] [Google Scholar]
- 164.Araki K, Inaba K. Structure, mechanism, and evolution of Ero1 family enzymes. Antioxid Redox Signal. 2012;16:790–9 [DOI] [PubMed] [Google Scholar]
- 165.Saijo T, Tanaka K. Isoalloxazine ring of FAD is required for the formation of the core in the Hsp60-assisted folding of medium chain acyl-CoA dehydrogenase subunit into the assembly competent conformation in mitochondria. J Biol Chem. 1995;270:1899–907 [DOI] [PubMed] [Google Scholar]
- 166.Yamazaki M, Ohnishi T, Ichikawa Y. Selective chemical modification of amino acid residues in the flavin adenine dinucleotide binding site of NADPH-ferredoxin reductase. Int J Biochem. 1992;24:223–8 [DOI] [PubMed] [Google Scholar]
- 167.Hanukoglu I, Hanukoglu Z. Stoichiometry of mitochondrial cytochromes P-450, adrenodoxin and adrenodoxin reductase in adrenal cortex and corpus luteum: implications for membrane organization and gene regulation. Eur J Biochem. 1986;157:27–31 [DOI] [PubMed] [Google Scholar]
- 168.Lambeth JD, Seybert DW, Lancaster JR, Jr, Salerno JC, Kamin H. Steroidogenic electron transport in adrenal cortex mitochondria. Mol Cell Biochem. 1982;45:13–31 [DOI] [PubMed] [Google Scholar]
- 169.Kowluru R, Yamazaki T, McNamara BC, Jefcoate CR. Metabolism of exogenous cholesterol by rat adrenal mitochondria is stimulated equally by physiological levels of free Ca2+ and by GTP. Mol Cell Endocrinol. 1995;107:181–8 [DOI] [PubMed] [Google Scholar]
- 170.Müller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U. Adrenodoxin reductase-adrenodoxin complex structure suggests electron transfer path in steroid biosynthesis. J Biol Chem. 2001;276:2786–9 [DOI] [PubMed] [Google Scholar]
- 171.Cornelius N, Frerman FE, Corydon TJ, Palmfeldt J, Bross P, Gregersen N, Olsen RK. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum Mol Genet. 2012;21:3435–48 [DOI] [PubMed] [Google Scholar]
- 172.Barile M, Giancaspero TA, Brizio C, Panebianco C, Indiveri C, Galluccio M, Vergani L, Eberini I, Gianazza E. Biosynthesis of flavin cofactors in man: implications in health and disease. Curr Pharm Des. 2013;19:2649–75 [DOI] [PubMed] [Google Scholar]
- 173.Agrimi G, Russo A, Scarcia P, Palmieri F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem J. 2012;443:241–7 [DOI] [PubMed] [Google Scholar]
- 174.Oka M, McCormick DB. Complete purification and general characterization of FAD synthetase from rat liver. J Biol Chem. 1987;262:7418–22 [PubMed] [Google Scholar]
- 175.Brizio C, Galluccio M, Wait R, Torchetti EM, Bafunno V, Accardi R, Gianazza E, Indiveri C, Barile M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem Biophys Res Commun. 2006;344:1008–16 [DOI] [PubMed] [Google Scholar]
- 176.Torchetti EM, Brizio C, Colella M, Galluccio M, Giancaspero TA, Indiveri C, Roberti M, Barile M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion. 2010;10:263–73 [DOI] [PubMed] [Google Scholar]
- 177.Barile M, Brizio C, Valenti D, De Virgilio C, Passarella S. The riboflavin/FAD cycle in rat liver mitochondria. Eur J Biochem. 2000;267:4888–900 [DOI] [PubMed] [Google Scholar]
- 178.Barile M, Brizio C, De Virgilio C, Delfine S, Quagliariello E, Passarella S. Flavin adenine dinucleotide and flavin mononucleotide metabolism in rat liver–the occurrence of FAD pyrophosphatase and FMN phosphohydrolase in isolated mitochondria. Eur J Biochem. 1997;249:777–85 [DOI] [PubMed] [Google Scholar]
- 179.Tzagoloff A, Jang J, Glerum DM, Wu M. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J Biol Chem. 1996;271:7392–7 [DOI] [PubMed] [Google Scholar]
- 180.Bafunno V, Giancaspero TA, Brizio C, Bufano D, Passarella S, Boles E, Barile M. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. J Biol Chem. 2004;279:95–102 [DOI] [PubMed] [Google Scholar]
- 181.Spaan AN, Ijlst L, van Roermund CW, Wijburg FA, Wanders RJ, Waterham HR. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2005;86:441–7 [DOI] [PubMed] [Google Scholar]
- 182.Kobuchi H, Moriya K, Ogino T, Fujita H, Inoue K, Shuin T, Yasuda T, Utsumi K, Utsumi T. doi: 10.1371/journal.pone.0050082. Mitochondrial localization of ABC transporter ABCG2 and its function in 5-aminolevulinic acid-mediated protoporphyrin IX accumulation. PLoS One. 2012;7(11):e50082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hanukoglu I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab Rev. 2006;38:171–96 [DOI] [PubMed] [Google Scholar]
- 185.Henriques BJ, Olsen RK, Bross P, Gomes CM. Emerging roles for riboflavin in functional rescue of mitochondrial β-oxidation flavoenzymes. Curr Med Chem. 2010;17:3842–54 [DOI] [PubMed] [Google Scholar]
- 186.Ghiasi P, Hosseinkhani S, Noori A, Nafissi S, Khajeh K. Mitochondrial complex I deficiency and ATP/ADP ratio in lymphocytes of amyotrophic lateral sclerosis patients. Neurol Res. 2012;34:297–303 [DOI] [PubMed] [Google Scholar]
- 187.Vergani L, Barile M, Angelini C, Burlina AB, Nijtmans L, Freda MP, Brizio C, Zerbetto E, Dabbeni-Sala F. Riboflavin therapy: biochemical heterogeneity in two adult lipid storage myopathies. Brain. 1999;122:2401–11 [DOI] [PubMed] [Google Scholar]
- 188.Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A, Sciacco M, Bordoni A, Fassone E, Fortunato F, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. 2010;67:849–54 [DOI] [PubMed] [Google Scholar]