

Abstract
ETS variant 1 (ETV1), also known as ETS-related protein 81, is overexpressed in prostate tumors, but whether and how this transcription factor affects tumorigenesis has remained elusive. Here, we show that ETV1 is primarily overexpressed in the most aggressive human prostate tumors. Transgenic ETV1 mice developed prostatic intraepithelial neoplasia as well as hyperplasia/neoplasia in seminal vesicles. Moreover, ETV1 cooperated with the androgen receptor (AR) to bind to the prostate-specific antigen enhancer and stimulate gene transcription. Consistent with its ability to physically interact with AR, ETV1 rendered an ETV1 binding site–driven reporter androgen inducible, and, on the other hand, ETV1 superinduced transcription from an AR binding site on androgen stimulation. In conclusion, our study substantiates that ETV1 overexpression is an underlying cause in the development of prostate and possibly also seminal vesicle cancer. Its interaction with and activation of AR provides a molecular mechanism on how ETV1 exerts its deleterious function. Thus, inhibiting ETV1 or blocking its interaction with AR may represent novel strategies in prostate cancer therapy.
Introduction
The androgen receptor (AR) plays a pivotal role in the development of prostate cancer, a leading cause of death in men in the Western hemisphere. Accordingly, androgen ablation is the cornerstone of treating prostate cancer and initially leads to the regression of the disease. However, patients generally relapse within 2 years due to the appearance of castration-resistant prostate cancer cells. Even at this advanced stage of prostate cancer, AR is believed to play the role of a dominant oncoprotein, which may be due to AR gene amplification, mutations, or changes in AR-interacting proteins, leading to its aberrant activation in an androgen-depleted environment (1).
Recently, it was discovered that chromosomal rearrangements involving ETS transcription factors occur in the majority of human prostate tumors (2). These rearrangements fuse the promoter/5′-end of various genes, which are either androgen inducible or constitutively expressed in the prostate, to the entire or truncated coding sequence of four ETS factors, namely, ETS variant 1 (ETV1), also called ETS-related protein 81 (ER81), ETV4, ETV5, or ETS-related gene (ERG). In addition, overexpression of ETV1 without evidence of any gene fusion has been observed in many prostate tumors (3–5).
Interestingly, chromosomal translocations involving ETV1, ETV4, and ERG are also causative for the development of Ewing’s sarcomas (6), suggesting that dysregulation of these ETS DNA binding proteins and their target genes is involved in the genesis of various tumors. This may include breast cancer, in which especially ETV1 and ETV4 have been implicated, possibly because they are downstream effectors of the HER2/Neu oncoprotein that is overexpressed in ~30% of all breast tumors (7–10). Of note, overexpression of the receptor tyrosine kinase HER2/Neu is also observed in prostate tumors and increases with the progression of the disease, with more than half of all castration-resistant tumors staining HER2/Neu positive in some studies (11–13). Thus, aberrant gene activation on ETV1 overexpression may be exacerbated by HER2/Neu in prostate cancer cells.
Here, we examined the ability of ETV1 to induce prostatic lesions in transgenic mice and provide mechanistic insight into how ETV1 may exert its deleterious effect in the prostate.
Materials and Methods
Analysis of tumor samples
Laser capture microdissection was performed on 122 and macrodissection on 40 fresh-frozen tissues. ETV1 expression was determined by microarray analyses (14). Formalin-fixed, paraffin-embedded tumor samples from an independent study were analyzed by quantitative reverse transcription-PCR (RT-PCR; ref. 15). Informed consent was obtained from all patients, and these studies were approved by the Mayo Clinic Institutional Review Board.
Transgenic mice
Coding sequence for full-length human ETV1 or its amino acids 132 to 477 (ΔETV1), which were preceded by a Kozak sequence and followed by an SV40 polyadenylation site and intron, was cloned downstream of the composite ARR2PB promoter (16). These gene cassettes were injected into one-cell fertilized FVB mice embryos, which were then reimplanted into pseudopregnant females. Resulting pups that tested positive for transgene insertion by PCR were crossed with C57BL/6 mice. Prostates from transgenic animals were dissected, paraffinized, cut, and stained with H&E (17). These experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Luciferase assays
LNCaP cells were seeded at 40% confluency in six-well plates and then transfected by the calcium phosphate coprecipitation method (18). One microgram of prostate-specific antigen (PSA)-luc that encompasses 5.8 kb of PSA upstream sequences or 1.5 μg of PSA-enhancer-luc reporter plasmid, in which the indicated PSA enhancer sequences were cloned into the tk80 luciferase construct, were used for transfection. As indicated, the empty vector pEV3S or ETV1 expression vectors were cotransfected, and pBluescript KS+ was used as a carrier to bring the total DNA amount up to 5 μg. The precipitate was washed away with PBS after 12 h. Cells were then incubated in medium containing 10% charcoal/dextran-treated fetal bovine serum (FBS) with or without mibolerone for 36 h, and luciferase activities were determined as described (19).
Similarly, RK13 cells were seeded at 25% confluency in 12-well plates and transfected with 0.5 μg of E743-tk80-luc (18) or the AR binding site–driven luciferase reporter ARE4-luc containing four copies of the AR binding site AGCACTTGCTGTTCT (20). Where indicated, 100 ng of ETV1 expression plasmid or empty vector pEV3S and 500 ng (in case of E743-tk80-luc) or 20 ng (in case of ARE4-luc) of pSG-AR expression vector or empty vector pSG6 were cotransfected.
Knockdown experiments
To down-regulate human ETV1, respective short hairpin RNAs (shRNA) were cloned into pSIREN-RetroQ. Within 2 d, LNCaP cells were thrice infected with retrovirus derived from these constructs and then incubated for 47 h with or without 0.5 nmol/L mibolerone in medium containing 10% charcoal/dextran-treated FBS (21). Protein and RNA analyses were then performed as described (22). The following sequences were targeted by shRNA: #1, GTGCCTGTACAATGT-CAGT; #4, TGTCAGTGCCTATGATCAG; and #5, TTCGATGGAGACAT-CAAAC.
Chromatin immunoprecipitation assays
These assays were performed as described (23). The following primers were used to amplify a 367-bp PSA enhancer fragment (5′-GGGGTTTGTGCCACTGGTGAG-3′ and 5′-GGGAGGCAATTCTCCATGGTT-3′), a 261-bp PSA promoter fragment (5′-TCTAGTTTCTGGTCTCAGAG-3′ and 5′-TTGCTGTTCTGCAATTACTAG-3′), and a 268-bp PSA 3′-untranslated region (3′-UTR) fragment (5′-TACTGGC-CATGCCTGGAGAC-3′ and 5′-TGGCTCACAGCCTTCTCTAG-3′). The PCR program used was as follows: 98°C for 2 min; 6 cycles of 98°C for 30 s, 64°C for 30 s (−1°C per cycle), 72°C for 25 s, followed by 32 cycles of 98°C for 30 s, 58°C for 30 s, 72°C for 25 s (+1 s per cycle); and finally by 72°C for 4 min.
Electrophoretic mobility shift assays
Reactions were set up in 10 μL of 20 mmol/L HEPES (pH 7.4), 25 mmol/L NaCl, 2 mmol/L DTT, 0.01% Tween 20, 12% glycerol, 0.1 μg/μL bovine serum albumin, and 0.05 μg/μL poly(deoxyinosinic-deoxycytidylic acid)·(deoxyinosinic-deoxycytidylic acid). Approximately 0.25 ng of 32P-labeled oligonucleotides, bacterially expressed and purified ETV1 encompassing amino acids 249 to 477, and 0.5 μL of anti-ETV1 (C-20; Santa Cruz Biotechnology) antibody were used (24).
The following oligonucleotide pairs were hybridized to generate double-stranded PSA enhancer oligonucleotides and labeled with [32P]dATP and Klenow DNA polymerase (25): ETS1, 5′-CAGTCTAGGTGGATGCTGTGCA-CACGGG-3′ and 5′-AACCCCGTGTGCACAGCATCCACCTAG-3′; ETS2/3, 5′-CTGAGATTAGGAATCCTCAATCTT-3′ and 5′-ATAAGATTGAGGATTCC-TAATCTC-3′; ETS4, 5′-GAAGATATTATCTTCATGATCTTGGATTGAAAAC-3′ and 5′-CTGTTTTCAATCCAAGATCATGAAGATAATATCTTC-3′; ETS5, 5′-CTACTCTGGAGGAACATATTGTATCGATTGTCCTTG-3′ and 5′-GTCAAG-GACAATCGATACAATATGTTCCTCCAGAGT-3′; ETS6, 5′-GACATTATCTT-TATTATCTAGGACAGTAAGCAAGCCTGGATCTGAG-3′ and 5′-CTCTCAGATCCAGGCTTGCTTACTGTCCTAGATAATAAAGATAATGTC-3′; ETS7, 5′-ATCATCTTGCAAGGATGCCTGCTTT-3′ and 5′-GTAAAGCAGG-CATCCTTGCAAGATG-3′; ETS8/9, 5′-CTTTACAAACATCCTTGAAACAA-CAATCCAGAA-3′ and 5′-TTCTGGATTGTTGTTTCAAGGATGTTTGTAA-3′; and ETS10, 5′-GTTCAGCCAGAGCCTTCCACCCTTGT-3′ and 5′-ACAAGGGTGGAAGGCTCTGGCTGA-3′.
Coimmunoprecipitations
Human embryonic kidney 293T cells grown on poly-L-lysine–coated dishes were transiently transfected by the calcium phosphate coprecipitation method (26). Cells were grown with or without 1 nmol/L mibolerone and coimmunoprecipitations were performed (22). Immunoprecipitated proteins were revealed as described (27).
Glutathione S-transferase pull-down assays
Glutathione S-transferase (GST) fusion proteins were produced in bacteria and affinity purified on glutathione agarose (28). Baculovirus encoding for AR endowed at the NH2 terminus with both a Flag-tag and 6His-tag was used to infect Sf9 insect cells, and AR was subsequently affinity purified on Ni2+-NTA agarose. Binding of AR to GST fusion proteins was assessed as described (28).
Results
ETV1 marks aggressive prostate cancer and induces PIN
Overexpression of ETV1 was reported in 6% to 16% of prostate tumors (4). To study its correlation with disease stage, we determined ETV1 expression in normal prostates, prostatic intraepithelial neoplasia (PIN), and carcinomas. Remarkably, ETV1 was found overexpressed only in Gleason pattern 4 and 5 tumors with a frequency of 3 of 46 (6.5%) and 2 of 22 (9.1%), respectively (Fig. 1A), suggesting that ETV1 expression is correlated with high-grade prostate cancer.
Figure 1.

ETV1 and prostatic lesions. A, ETV1 mRNA expression derived by microarray from normal prostates (n = 50), PINs (n = 5), Gleason pattern 3 (GP3; n = 32), Gleason pattern 4 (GP4; n = 46), Gleason pattern 5 (GP5; n = 22), and metastases (n = 7). B, ETV1 and ERG expression derived by RT-PCR from high-risk prostate cancer patients. Relapse indicates patients that developed systemic progression within 5 y after prostatectomy, whereas controls remained free of disease for at least 8 y. Dashed line, boundary of significant ETV1 or ERG overexpression. C, H&E-stained sections of the anterior (top) and ventral (bottom) prostate of transgenic ETV1 mouse 650. Right, larger magnification of PIN areas. D, similar for a 1-y-old control mouse.
In addition, we studied ETV1 expression in a cohort of patients with high risk of systemic cancer progression (Gleason score 7–10) that underwent radical retropubic prostatectomy. In this cohort, slight ETV1 overexpression was observed in 3 of 77 (3.9%) of the cases remaining cancer-free for at least 8 years after prostatectomy, whereas 10 of 80 (14.7%) of the relapsing patients showed, in part dramatic, ETV1 overexpression (Fig. 1B, left). Although not quite statistically significant (P = 0.0797, Fisher’s exact test), this further supports the notion that ETV1 overexpression is indicative of aggressive prostate cancer. In contrast, an equal percentage of cancer-free (34 of 81, 42.0%) and relapsing patients (36 of 84, 42.9%) showed ERG overexpression (Fig. 1B, right).
To assess whether overexpression of full-length ETV1 leads to neoplasia, we generated transgenic mice in which human ETV1 cDNA was expressed under the control of a composite rat probasin promoter (ARR2PB) that is androgen inducible and prostate specific (16). We analyzed prostates at 12 months of age in four mice that were derived from four independent transgenic ETV1 lines. Based on nuclear atypia, architectural changes such as cribriforming, and focality of the lesions, PIN was identified in all four transgenic ETV1 mice analyzed (Fig. 1C; Supplementary Table S1); please note that age-matched wild-type mice do not display PIN or hyperplasia (Fig. 1D). Further, RT-PCR of whole prostates indicated that the level of transgene expression (human ETV1) was similar to the level of endogenous murine ETV1/ER81 expression (Supplementary Table S2). Because ETV1 expression is elevated from 2-fold to >10-fold in human prostate tumors (4, 29), the level of ETV1 transgene expression is not unreasonably high. In conclusion, these results indicate that overexpression of full-length ETV1 is causally involved in prostate carcinogenesis.
Overexpression of ETV1 and the ΔETV1 truncation, which lacks the first 131 amino acids of full-length ETV1, occurs with roughly equal frequency in prostate tumors (3–5). Therefore, we also generated transgenic mice expressing ΔETV1 and analyzed four mice derived from three independent ΔETV1 transgenic lines. We observed the development of PIN in one and hyperplasia in the other three ΔETV1 mice (Supplementary Table S1); moreover, ΔETV1 and ETV1 transgene expression were not drastically different as determined by RT-PCR (Supplementary Table S2). These data indicate that both truncated and full-length ETV1 are capable of initiating prostate cancer formation. While this study was in progress, another report confirmed that overexpression of ΔETV1 induces PIN (4).
The only other organ apart from prostate displaying activity, albeit at a lower level, of the ARR2PB promoter driving expression of the ETV1 and ΔETV1 transgenes is the seminal vesicle (16). Notably, we observed macroscopic differences in the seminal vesicles in one of four ETV1 and two of three ΔETV1 transgenic lines (Supplementary Table S1; Supplementary Fig. S1), including different color, enlargement, or anatomically different location in the abdomen. Moreover, pathologic examination of these abnormal seminal vesicles revealed hyperplasia or neoplasia, suggesting that ETV1/ΔETV1 overexpression may cause neoplastic transformation in the seminal vesicles.
Stimulation of PSA expression by ETV1
AR and ETS binding sites reside frequently close together in the genome (30), suggesting that the ETS protein ETV1 may cooperate with AR in regulating gene transcription. We noted the presence of 10 putative ETS sites in the enhancer of the PSA, a serum marker for prostate cancer whose expression is regulated by multiple AR sites (31, 32). Thus, we wondered if overexpression of ETV1 might affect PSA transcription. We overexpressed ETV1 in AR-positive LNCaP prostate cancer cells and assessed the activity of a luciferase reporter construct driven by 5.8 kb of PSA upstream sequences, including its enhancer and proximal promoter. As shown in Fig. 2A, a synthetic AR agonist, mibolerone, induced PSA luciferase activity by 14.7-fold. Further, overexpression of ETV1 stimulated PSA luciferase activity by 2.3-fold in hormone-depleted medium. Importantly, ETV1 and hormone synergized to affect a 41.3-fold stimulation of luciferase activity. These data suggest that ETV1 cooperates with AR in the regulation of PSA transcription.
Figure 2.

Activation of the PSA gene by ETV1. A, ETV1 or its 6xA mutant (300 ng) was cotransfected with the 5.8-kb PSA luciferase reporter construct into LNCaP cells stimulated with or without 0.1 nmol/L mibolerone. B, anti-Myc Western blot showing comparable expression of Myc-tagged wild-type ETV1 and its 6xA mutant in LNCaP cells. C, ETV1 down-regulation by three different shRNAs inhibits expression of endogenous PSA mRNA and protein in LNCaP cells. Top two rows, agarose gels revealing reverse-transcribed mRNA; bottom three rows, Western blots. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and actin protein levels were determined as loading controls. D, top, scheme of the PSA enhancer and proximal promoter with its AR binding sites in Roman numerals and potential ETS sites in Arabic numerals. AR site III is the most important AR binding site. Numbering is according to ref. 31. Bottom, chromatin immunoprecipitation assays assessing the binding of ETV1 and AR to the PSA enhancer, proximal promoter, and 3′-UTR in LNCaP cells before and after 1 or 8 h of stimulation with 1 nmol/L mibolerone.
ETV1 is activated through phosphorylation by mitogen-activated protein (MAP) and MAPKAP kinases on six serine/threonine residues (8, 33, 34), and LNCaP cells express high levels of the MAP kinase stimulator HER2/Neu (35). Accordingly, mutation of all six ETV1 phosphorylation sites to alanine suppressed the ability of ETV1 to induce PSA luciferase activity on hormone treatment in LNCaP cells (6×A mutant in Fig. 2A). Western blotting revealed that this effect was not due to different protein levels of the 6×A mutant compared with wild-type ETV1 (Fig. 2B). Thus, activation of ETV1 by MAP kinases is required for maximal PSA transcription. Interestingly, in the absence of hormone treatment, the 6×A mutant was slightly more active than wild-type ETV1, suggesting that ETV1 phosphorylation may suppress basal PSA expression.
To show that endogenous ETV1 affects PSA transcription, we knocked down ETV1 with three different shRNAs in LNCaP cells. All of these shRNAs specifically and efficiently down-regulated ETV1 expression both in the presence or in the absence of mibolerone (Fig. 2C); please note that ETV1 expression is up-regulated by mibolerone because the entire ETV1 gene locus is translocated into an androgen-responsive region in LNCaP cells (4). Importantly, both PSA mRNA and protein levels were reduced on ETV1 down-regulation in the presence and absence of mibolerone (Fig. 2C). Further, down-regulation of ETV1 did not significantly affect AR levels (Supplementary Fig. S2). In conclusion, ETV1 enhances endogenous PSA expression.
To assess if the PSA gene is targeted by ETV1, we performed chromatin immunoprecipitation assays in LNCaP cells. As expected, AR binding to the PSA enhancer dramatically increased after 1 or 8 hours of hormone stimulation (Fig. 2D). Consistent with a previous report (36), AR binding to the proximal PSA promoter was negligible, and none was observed at the 3′-UTR of the PSA gene. Neither an irrelevant antibody (anti-Rcl) nor omission of antibody led to the precipitation of PSA enhancer, promoter, or 3′-UTR DNA. Like AR, some ETV1 interacted with the PSA enhancer before hormone stimulation (Fig. 2D). On hormone stimulation, strongly increased binding of ETV1 to the PSA enhancer was evident, whereas negligible or no binding of ETV1 to the PSA proximal promoter and 3′-UTR was observed. Collectively, these data indicate that PSA is a target gene of ETV1 and that ETV1 and AR coordinately bind to the PSA enhancer on androgen stimulation.
Binding of ETV1 to the PSA enhancer
To assess whether the PSA enhancer is regulated directly by ETV1, we cloned it in front of a thymidine kinase basal promoter and the luciferase gene and transfected this reporter construct with and without ETV1 into LNCaP cells. Indeed, ETV1 enhanced luciferase activity from 0.02 to 0.12 in the absence of hormone and from 21.7 to 43.14 on hormone stimulation (Fig. 3A, −4324/−3875 construct). We then divided the enhancer into two parts: one containing the pivotal AR binding site III (−4324/−4119 construct) and the other encompassing ETS sites 6 to 10 (−4127/−3875 construct). Both constructs displayed a dramatically decreased response to hormone stimulation. Importantly, the effect of ETV1 overexpression was curtailed (Fig. 3A). These data suggest that ETS sites 6 to 10 cooperate with the AR binding site III in regulating the PSA enhancer.
Figure 3.

Binding of ETV1 to the PSA enhancer. A, activation of the PSA enhancer by ETV1. Left, scheme of the PSA enhancer and two truncations thereof; right, luciferase reporter assays with these enhancer fragments in LNCaP cells transfected with or without ETV1 (250 ng). Where indicated, cells were stimulated with 1 nmol/L mibolerone. B, binding of ETV1 to the 10 ETS sites in the PSA enhancer was assessed in electrophoretic mobility shift assays. Where indicated, anti-ETV1 antibody (Ab) was added to the binding reaction. C, comparison of the activities of the wild-type and mETS#8 (mutation of ETS site 8) −4324/−3875 PSA enhancer in LNCaP cells. Where indicated, ETV1 (600 ng) was cotransfected and 0.25 nmol/L mibolerone was used.
To elucidate which ETS sites in the PSA enhancer might be bound by ETV1, we performed electrophoretic mobility shift assays. Oligonucleotides spanning 1 or 2 of the 10 ETS sites within the PSA enhancer were generated and incubated with recombinant ETV1 in vitro. We observed weak binding of ETV1 to ETS sites 8/9 (Fig. 3B, white arrow) and the known ETV1 site within the matrix metalloproteinase-1 (MMP-1) promoter. However, inclusion of an anti-ETV1 antibody, which stabilizes the binding of ETV1 to selected ETS sites such as in the MMP-1 or Smad7 promoters (8, 37), resulted in much more pronounced DNA binding to both ETS sites 8/9 and the MMP-1 target site (Fig. 3B, black arrow). Moreover, DNA binding was now also found at other ETS sites in the following order of decreasing strength: ETS8/9 > ETS7 > ETS5 > ETS1,6 > ETS10. Furthermore, mutation of ETS site 8 but not site 9 abrogated binding of ETV1 to the ETS8/9 oligonucleotide (Supplementary Fig. S3A), indicating that ETS site 8 but not ETS site 9 is targeted by ETV1; this result is also consistent with the fact that ETS site 8 matches the ETV1 consensus DNA binding site much better than ETS site 9 (Supplementary Fig. S3B). In conclusion, ETV1 can bind to several sites within the PSA enhancer, with ETS site 8 being the strongest binder.
To determine the functional importance of ETS site 8, we mutated it in the PSA enhancer. Both in the presence and in the absence of hormone, this led to a reduction of enhancer activity (Fig. 3C). Whereas ETV1 was still able to enhance the activity of the mutated PSA enhancer in the absence of hormone, albeit the degree of activation was less than with the wild-type enhancer, ETV1 over-expression no longer enhanced PSA transcription in the presence of hormone (Fig. 3C). Thus, ETS site 8 seems to be especially crucial for the androgen-inducible expression of the PSA gene.
ETV1 forms complexes with AR
The apparent in vivo corecruitment of ETV1 and AR to the PSA enhancer after hormone stimulation suggested that these factors may be part of one complex. To explore this possibility, we expressed Flag-tagged AR with or without Myc-tagged ETV1 and performed anti-Myc immunoprecipitations. Probing the precipitate with anti-Flag antibodies revealed that AR coprecipitated with ETV1 (Fig. 4A). Moreover, this interaction of ETV1 and AR was independent of the presence or absence of hormone. To confirm this interaction, we also performed coimmunoprecipitation experiments with endogenous AR and ETV1 in LNCaP cells. Anti-ETV1 antibodies, but not control antibodies, led to the coprecipitation of AR (Fig. 4B), indicating that endogenous AR and ETV1 form complexes in vivo.
Figure 4.

Interaction of ETV1 and ERG with AR. A, binding of Flag-tagged AR to Myc-tagged ETV1 was assessed by coimmunoprecipitation in the presence and absence of mibolerone. B, mibolerone-treated LNCaP cells were lysed and ETV1 was immunoprecipitated. Coprecipitated AR was revealed by anti-AR Western blotting. C, GST pull-down experiments in the presence or absence of 1 nmol/L mibolerone. Indicated GST fusion proteins or GST was used to pull down Flag-tagged, baculovirus-expressed AR. Bottom, amounts of GST fusion proteins used by Coomassie staining. D, coimmunoprecipitation of AR with Myc-tagged ERG in the presence of mibolerone.
To assess whether the interaction between ETV1 and AR is direct, we produced ETV1 as a GST fusion protein in bacteria, affinity purified it, and bound it to glutathione agarose beads. Incubation of these beads with affinity-purified, baculovirus-expressed AR revealed that AR bound to GST-ETV1, but not to GST or ETV1 amino acids 182 to 334, in a hormone-independent manner (Fig. 4C, left). Thus, ETV1 and AR are capable of binding directly to each other. Similarly, ERG and AR interacted with each other in vitro and in vivo (Fig. 4C (right) and D).
Next, we sought to determine the interaction domain(s) within AR. Thus, we expressed full-length, Flag-tagged AR or various truncations thereof and probed for coprecipitation of Myc-tagged ETV1 after anti-Flag immunoprecipitation. Only amino acids 650 to 919, which encompass the ligand binding domain of AR, coprecipitated ETV1 with a similar efficiency as full-length AR1–919 (Fig. 5A). Conversely, we probed which domain(s) in ETV1 is responsible for the interaction with AR (Fig. 5B). The NH2-terminal activation domain of ETV1 encompassing amino acids 1 to 182 did not bind to AR, but ETV1 lacking these amino acids (ETV1182–477) did, indicating that the NH2-terminal activation domain of ETV1 is not involved in the interaction with AR. Moreover, ETV1249–477 was unable to bind to AR. Thus, amino acids 182 to 249 are crucial for the interaction with AR, yet not sufficient as ETV11–249 did not bind to AR. Further, ETV11–334 was unable to bind to AR (Fig. 5B), indicating that amino acids 334 to 477 are also required for the interaction with AR. However, these amino acids alone were not sufficient because ETV1333–477 did not bind to AR. In conclusion, ETV1 amino acids in the center domain cooperate with the COOH-terminal 144 amino acids in AR binding.
Figure 5.

Mapping of interaction domains. A, Flag-tagged fragments of AR were coexpressed with Myc-tagged ETV1. After anti-Flag immunoprecipitation, coprecipitated ETV1 was revealed by anti-Myc Western blotting. A/B, activation domain; C, DNA binding domain; D, hinge region; E/F, ligand binding domain of AR. B, coprecipitation of AR with Myc-tagged fragments of ETV1. Sketch of ETV1 at the top shows its activation domains and DNA binding ETS domain.
Reciprocal coactivation by ETV1 and AR
We next assessed if ETV1 and AR may recruit each other as cofactors, meaning that only one of these transcription factors binds to DNA, whereas the other is recruited by protein-protein interactions. To this end, we used the E74 site that is derived from the Drosophila E74 gene and is tightly bound by ETV1 (18) but does not contain an AR binding site. Moreover, we used rabbit kidney RK13 cells that do not contain AR but have been often used to study ETV1/ER81 (8, 18, 34). AR expression or mibolerone stimulation, either alone or in combination, did not activate E74-dependent transcription in the absence of ectopic ETV1 (Fig. 6A). Similarly, neither AR expression nor mibolerone stimulation alone raised luciferase activities in the presence of ectopic ETV1. However, AR activated by mibolerone stimulated E74-dependent transcription by 2.1-fold in the presence of ectopic ETV1. This suggests that ETV1 recruits AR to the E74 site.
Figure 6.
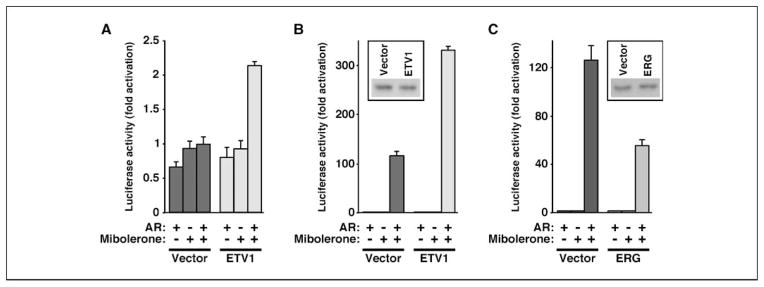
A, activation of a luciferase gene driven by the ETS site E74. Where indicated, AR and/or ETV1 were transfected into RK13 cells and stimulated with 0.5 nmol/L mibolerone. B, analogous stimulation of an AR binding site–driven luciferase reporter in the presence or absence of 1 nmol/L mibolerone. Western blotting (inset) shows that AR levels were unaffected by ETV1 expression in the presence of mibolerone. C, as in B with ERG instead of ETV1.
Conversely, we tested whether AR could use ETV1 as a coactivator. To this end, we used an AR binding site–dependent luciferase reporter that was stimulated 116-fold by AR and mibolerone in the absence of ectopic ETV1 (Fig. 6B). In the presence of ectopic ETV1, which did not affect AR protein levels (Fig. 6B, inset), AR and mibolerone stimulated luciferase activity by 330-fold, implicating that ETV1 is recruited by AR to the AR binding site.
Finally, we also tested whether ERG may cooperate with AR. Surprisingly, ERG expression led to a more than 2-fold decrease of the ability of AR to induce the AR binding site–dependent luciferase reporter in the presence of mibolerone (Fig. 6C). Likewise, ERG inhibited the induction of the PSA promoter by hormone in LNCaP cells (Supplementary Fig. S4). Together, these data suggest that ERG and ETV1 exert opposing effects on their common binding partner, AR.
Discussion
Recurrent chromosomal translocations involving ETS transcription factors, including ETV1, are present in most prostate tumors, leading to the overexpression of truncated or full-length ETS proteins (2). Moreover, many prostate tumors do not display chromosomal translocations involving ETV1, ETV4, ETV5, or ERG transcription factors yet display an overexpression of these factors in a nontruncated form (3–5). Our study shows that overexpression of full-length ETV1 results in the development of PIN, the precursor lesion of malignant prostate cancer. In addition, our results with transgenic mice expressing truncated ETV1 complement other reports showing that ΔETV1 and ΔERG overexpression induces PIN (4, 38, 39). Altogether, these transgenic animal studies support the concept that overexpression of ETS proteins is causative for the initiation of prostate cancer.
Transgenic ETV1/ΔETV1 mice analyzed in this report were at most 15 months of age. It remains to be determined whether older mice not only display PIN but also progress to develop adenocarcinomas. Alternatively, ETV1/ΔETV1 overexpression may cooperate with other genetic lesions in the development of malignant prostate cancer; indeed, two recent studies show that ERG overexpression cooperates with loss of the tumor suppressor PTEN to induce prostate carcinogenesis (40, 41).
Our data on human prostate tumors suggest that ETV1 overexpression is associated with a higher Gleason pattern and thus support a previous study showing that ETV1 gene alterations are correlated with a higher Gleason score (42). In contrast, the evidence for a correlation between ERG overexpression and Gleason score is controversial (2). Furthermore, our data suggest that overexpression of ETV1, but not ERG, is correlated with a higher frequency of relapse after radical retropubic prostatectomy. Collectively, these data implicate that ETV1 overexpression may mark the most aggressive prostate tumors.
The composite ARR2PB promoter driving the expression of ETV1/ΔETV1 transgenes is active not only in the prostate but also in the seminal vesicle (16), most likely due to the fact that seminal vesicles also express high levels of AR (43). This explains why seminal vesicles displayed various defects in ETV1/ΔETV1 transgenic mice, including the development of neoplasia in one case. Notably, this neoplasia is not a consequence of invading prostate cancer cells because the prostate of the affected animal exhibited only PIN. Chromosomal translocations involving the ETV1 locus or other genetic defects leading to the overexpression of ETV1 should occur in the seminal vesicle with a probability that is similar to prostate. Thus, ETV1 overexpression may indeed occur in human seminal vesicles and cause cancer. In fact, seminal vesicle cancer is observed in a significant proportion of prostate cancer patients (5–23%) and interpreted as spreading of the prostate cancer to the seminal vesicles (44, 45). Our data challenge this view because it is also possible that the cancer arises in the seminal vesicles and then spreads into the prostate.
Although ETV1, ETV4, ETV5, or ERG overexpression is thought to contribute to prostate carcinogenesis, it has remained unknown by which molecular mechanism these transcription factors achieve their deleterious effects. Our study provides a mechanistic explanation in case of ETV1, namely, through its cooperation with AR in inducing transcription as exemplified for the PSA gene. Analysis of the PSA enhancer revealed that several binding sites for ETV1 are present in close proximity to AR binding sites, and mutation of the most avidly binding ETV1 site significantly reduced the activity of the PSA enhancer. Moreover, ETV1 and AR occupancy of the PSA enhancer was coordinately increased within 1 hour of hormone induction, suggesting that these two proteins bind the PSA enhancer as a complex due to their physical association. Because AR and ETS binding sites are frequently close together in the genome (30), such a scenario may be widespread and could account for how ETV1 overexpression augments the function of AR, thereby leading to the aberrant activation of AR and its target genes that is critical for the development of prostate cancer.
In a second scenario, ETV1 may render genes androgen inducible by recruiting AR to gene promoters that have no AR binding site, as indicated by our results with the E74 ETS site. Again, recruitment of AR to gene promoters by ETV1 may be widespread and provide a new paradigm of androgen inducibility. Reciprocally, our data with the AR binding site–driven reporter construct indicated that AR might recruit ETV1 to many gene promoters with ETV1 serving the function of a coactivator.
We predict that ETV4 and ETV5, which are highly homologous to ETV1, will also cooperate with AR in activating gene transcription. However, ERG, which is different from ETV1 outside its DNA binding domain, seems to behave differently: although capable of binding to AR like ETV1, ERG suppresses AR-dependent transcription. This functional difference between ERG and ETV1 might be the underlying cause why overexpression of the latter apparently leads to more aggressive prostate cancer.
The oncoprotein HER2/Neu has been implicated in prostate carcinogenesis (11–13). Our previous results have shown that ETV1 is a downstream target of HER2/Neu and MAP kinases (8, 9, 19). Accordingly, mutating the six HER2/Neu-inducible phosphorylation sites in ETV1 reduced the ability of ETV1 to synergize with AR in inducing the PSA promoter. Notably, activation of MAP kinases not only is limited to HER2/Neu overexpression but also can result from the overexpression of various growth factors (e.g., epidermal growth factor, transforming growth factor-α, basic fibroblast growth factor, and insulin-like growth factor-I) or their cognate receptors in prostate tumors. Remarkably, activation of extracellular signal-regulated kinase MAP kinases is correlated with increasing Gleason score and tumor stage (46), suggesting that this exacerbates the tumor-promoting effects of ETV1.
p300 is a coactivator endowed with acetyltransferase activity that binds to ETV1 and stimulates its function (19, 27). Similarly, the SRC-3/AIB1/ACTR oncoprotein is a coactivator of ETV1 (47). Of note, p300 as well as SRC-3 are overexpressed in prostate cancer and may stimulate cancer cell proliferation (48–50). Possibly, p300 and/or SRC-3 and/or HER2/Neu overexpression represents secondary genetic lesions that exacerbate ETV1 overexpression and may be required for the progression of ETV1-induced PIN to malignancy.
In conclusion, this study has revealed that overexpression of ETV1 can be an underlying cause of prostate cancer and may also contribute to the formation of seminal vesicle cancer. Furthermore, our data suggest that ETV1 exerts its deleterious effects on the prostate through interacting and cooperating with AR. Thus, developing strategies to inhibit ETV1 activation or its interaction with AR may be useful to combat prostate cancer.
Supplementary Material
Acknowledgments
Grant support: Department of Defense Prostate Cancer Research Program grant W81XWH-08-1-0177, Fraternal Order of Eagles, and Mayo Clinic Specialized Program of Research Excellence in Prostate Cancer.
We thank Drs. Robert Matusik and Michael Carey for generously providing the ARR2PB and ARE4-luc construct, respectively.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–71. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 2.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai C, Hsieh CL, Omwancha J, et al. ETV1 is a novel androgen receptor-regulated gene that mediates prostate cancer cell invasion. Mol Endocrinol. 2007;21:1835–46. doi: 10.1210/me.2006-0480. [DOI] [PubMed] [Google Scholar]
- 4.Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 5.Hermans KG, van der Korput HA, van Marion R, et al. Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res. 2008;68:7541–9. doi: 10.1158/0008-5472.CAN-07-5930. [DOI] [PubMed] [Google Scholar]
- 6.Janknecht R. EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene. 2005;363:1–14. doi: 10.1016/j.gene.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Benz CC, O’Hagan RC, Richter B, et al. HER2/Neu and the Ets transcription activator PEA3 are coordinately upregulated in human breast cancer. Oncogene. 1997;15:1513–25. doi: 10.1038/sj.onc.1201331. [DOI] [PubMed] [Google Scholar]
- 8.Bosc DG, Goueli BS, Janknecht R. HER2/Neu-mediated activation of the ETS transcription factor ER81 and its target gene MMP-1. Oncogene. 2001;20:6215–24. doi: 10.1038/sj.onc.1204820. [DOI] [PubMed] [Google Scholar]
- 9.Goueli BS, Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 2004;24:25–35. doi: 10.1128/MCB.24.1.25-35.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin S, Bosc DG, Ingle JN, Spelsberg TC, Janknecht R. Rcl is a novel ETV1/ER81 target gene upregulated in breast tumors. J Cell Biochem. 2008;105:866–74. doi: 10.1002/jcb.21884. [DOI] [PubMed] [Google Scholar]
- 11.Signoretti S, Montironi R, Manola J, et al. Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92:1918–25. doi: 10.1093/jnci/92.23.1918. [DOI] [PubMed] [Google Scholar]
- 12.Osman I, Mikhail M, Shuch B, et al. Serum levels of shed Her2/neu protein in men with prostate cancer correlate with disease progression. J Urol. 2005;174:2174–7. doi: 10.1097/01.ju.0000181205.23233.65. [DOI] [PubMed] [Google Scholar]
- 13.Nishio Y, Yamada Y, Kokubo H, et al. Prognostic significance of immunohistochemical expression of the HER-2/neu oncoprotein in bone metastatic prostate cancer. Urology. 2006;68:110–5. doi: 10.1016/j.urology.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 14.Cheville JC, Karnes RJ, Therneau TM, et al. Gene panel model predictive of outcome in men at high-risk of systemic progression and death from prostate cancer after radical retropubic prostatectomy. J Clin Oncol. 2008;26:3930–6. doi: 10.1200/JCO.2007.15.6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosari F, Munz JM, Savci-Heijink CD, et al. Identification of prognostic biomarkers for prostate cancer. Clin Cancer Res. 2008;14:1734–43. doi: 10.1158/1078-0432.CCR-07-1494. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141:4698–710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs B, Inwards CY, Janknecht R. Vascular endothelial growth factor expression is up-regulated by EWS-ETS oncoproteins and Sp1 and may represent an independent predictor of survival in Ewing’s sarcoma. Clin Cancer Res. 2004;10:1344–53. doi: 10.1158/1078-0432.ccr-03-0038. [DOI] [PubMed] [Google Scholar]
- 18.Janknecht R. Analysis of the ERK-stimulated ETS transcription factor ER81. Mol Cell Biol. 1996;16:1550–6. doi: 10.1128/mcb.16.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goel A, Janknecht R. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 2003;23:6243–54. doi: 10.1128/MCB.23.17.6243-6254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Matherly J, Smallwood A, et al. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Ther. 2001;8:1416–26. doi: 10.1038/sj.gt.3301549. [DOI] [PubMed] [Google Scholar]
- 21.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–6. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 22.Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–8. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
- 23.Goueli BS, Janknecht R. Regulation of telomerase reverse transcriptase gene activity by upstream stimulatory factor. Oncogene. 2003;22:8042–7. doi: 10.1038/sj.onc.1206847. [DOI] [PubMed] [Google Scholar]
- 24.De Haro L, Janknecht R. Cloning of the murine ER71 gene (Etsrp71) and initial characterization of its promoter. Genomics. 2005;85:493–502. doi: 10.1016/j.ygeno.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 25.De Haro L, Janknecht R. Functional analysis of the transcription factor ER71 and its activation of the matrix metalloproteinase-1 promoter. Nucleic Acids Res. 2002;30:2972–9. doi: 10.1093/nar/gkf390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TD, Shin S, Janknecht R. Repression of Smad3 activity by histone demethylase SMCX/JARID1C. Biochem Biophys Res Commun. 2008;366:563–7. doi: 10.1016/j.bbrc.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Papoutsopoulou S, Janknecht R. Phosphorylation of ETS transcription factor ER81 in a complex with its coactivators CREB-binding protein and p300. Mol Cell Biol. 2000;20:7300–10. doi: 10.1128/mcb.20.19.7300-7310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knebel J, De Haro L, Janknecht R. Repression of transcription by TSGA/Jmjd1a, a novel interaction partner of the ETS protein ER71. J Cell Biochem. 2006;99:319–29. doi: 10.1002/jcb.20945. [DOI] [PubMed] [Google Scholar]
- 29.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 30.Massie CE, Adryan B, Barbosa-Morais NL, et al. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007;8:871–8. doi: 10.1038/sj.embor.7401046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang W, Shostak Y, Tarr P, Sawyers C, Carey M. Cooperative assembly of androgen receptor into a nucleoprotein complex that regulates the prostate-specific antigen enhancer. J Biol Chem. 1999;274:25756–68. doi: 10.1074/jbc.274.36.25756. [DOI] [PubMed] [Google Scholar]
- 32.Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. J Clin Oncol. 2003;21:383–91. doi: 10.1200/JCO.2003.02.083. [DOI] [PubMed] [Google Scholar]
- 33.Wu J, Janknecht R. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J Biol Chem. 2002;277:42669–79. doi: 10.1074/jbc.M205501200. [DOI] [PubMed] [Google Scholar]
- 34.Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1) Oncogene. 2003;22:746–55. doi: 10.1038/sj.onc.1206185. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Liu B, Schmidt M, Lu Y, Wels W, Fan Z. Antitumor effect of an HER2-specific antibody-toxin fusion protein on human prostate cancer cells. Prostate. 2001;47:21–8. doi: 10.1002/pros.1043. [DOI] [PubMed] [Google Scholar]
- 36.Louie MC, Yang HQ, Ma AH, et al. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci U S A. 2003;100:2226–30. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dowdy SC, Mariani A, Janknecht R. HER2/Neu- and TAK1-mediated up-regulation of the transforming growth factor β inhibitor Smad7 via the ETS protein ER81. J Biol Chem. 2003;278:44377–84. doi: 10.1074/jbc.M307202200. [DOI] [PubMed] [Google Scholar]
- 38.Klezovitch O, Risk M, Coleman I, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105–10. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomlins SA, Laxman B, Varambally S, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King JC, Xu J, Wongvipat J, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Attard G, Clark J, Ambroisine L, et al. Heterogeneity and clinical significance of ETV1 translocations in human prostate cancer. Br J Cancer. 2008;99:314–20. doi: 10.1038/sj.bjc.6604472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruizeveld de Winter JA, Trapman J, Vermey M, Mulder E, Zegers ND, van der Kwast TH. Androgen receptor expression in human tissues: an immunohistochemical study. J Histochem Cytochem. 1991;39:927–36. doi: 10.1177/39.7.1865110. [DOI] [PubMed] [Google Scholar]
- 44.Bostwick DG, Qian J, Bergstralh E, et al. Prediction of capsular perforation and seminal vesicle invasion in prostate cancer. J Urol. 1996;155:1361–7. [PubMed] [Google Scholar]
- 45.Wang L, Hricak H, Kattan MW, et al. Prediction of seminal vesicle invasion in prostate cancer: incremental value of adding endorectal MR imaging to the Kattan nomogram. Radiology. 2007;242:182–8. doi: 10.1148/radiol.2421051254. [DOI] [PubMed] [Google Scholar]
- 46.Gioeli D. Signal transduction in prostate cancer progression. Clin Sci (Lond) 2005;108:293–308. doi: 10.1042/CS20040329. [DOI] [PubMed] [Google Scholar]
- 47.Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem. 2004;279:14909–16. doi: 10.1074/jbc.M400036200. [DOI] [PubMed] [Google Scholar]
- 48.Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen de AL, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer Res. 2003;63:7638–40. [PubMed] [Google Scholar]
- 49.Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer. 2001;85:1928–36. doi: 10.1054/bjoc.2001.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou HJ, Yan J, Luo W, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–83. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.