

Abstract
Protein sulfenic acids are reactive, reversibly oxidized cysteinyl residues with roles in redox catalysis and regulation. Detection and quantification of these species in proteins is accomplished through chemical modification by reagents such as 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD chloride), 2-nitro-5-thiobenzoate (TNB), dimedone, or derivatives of dimedone, followed by UV-visible spectroscopy or mass spectrometric analysis.
Keywords: sulfenic acids, cysteine modification, cysteine oxidation, UV-visible spectroscopy, mass spectrometry
INTRODUCTION
Cysteine sulfenic acids in proteins are of considerable biological interest as important players in redox catalysis and redox regulation; yet, they have been notoriously difficult to identify due to their high reactivity, particularly outside their native protein environments. Where sulfenic acids are stabilized within proteins, factors contributing to their stability include the lack of proximal thiols, presence of nearby hydrogen-bonding and/or basic side chains (to stabilize the protonated or deprotonated sulfenic acid), and restricted access to solvent (see overview, UNIT 17.1). Direct observation of sulfenic acids within proteins has been possible only with crystallography (often requiring low-temperature cryotechniques to avoid overoxidation) and NMR using 13C-cysteine-labeled protein (Claiborne et al., 1999). Electrospray ionization mass spectrometry (ESI-MS) can be successful for direct detection of the additional oxygen of the sulfenic acid (Fuangthong and Helmann, 2002), but more often it leads to detection only of the sulfonic acid form (Cys-SO3H) generated through overoxidation of the sulfenic acid of interest (Ellis and Poole, 1997a).
The protocols outlined in this unit describe four chemical modification methods useful for sulfenic acid identification. In Basic Protocol 1, the first reagent, 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD chloride), reacts with both thiol groups and sulfenic acids in proteins at pH 7, but in each case the reactions give unique products that can be distinguished by their UV-visible spectra and by their masses, with the NBD-sulfenate adduct being 16 amu larger than the NBD-thiol adduct. As an alternative (see the Alternate Protocol), NBD chloride can be replaced by 4-fluoro-7-nitrobenz-2-oxa-1,3-diazole (NBD fluoride), a less stable reagent which reacts more quickly with thiols and sulfenic acids and gives the same products. In Basic Protocol 2, a second reagent, 2-nitro-5-thiobenzoic acid (TNB)—made from 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and 1,4-dithio-DL-threitol (DTT)—reacts stoichiometrically with sulfenic acids, resulting in the loss of its bright yellow color, thereby allowing for accurate quantitation of R-SOH groups by spectroscopy as well as ESI-MS.
The third reagent, 5,5-dimethyl-1,3-cyclohexanedione (dimedone), used in Basic Protocol 3, reacts specifically with sulfenic acid—but not thiol—groups on proteins. Unfortunately, the product does not exhibit any distinguishing visible absorbance properties; therefore, proof of modification by dimedone generally relies on ESI-MS analysis, as the radiolabeled reagent is not commercially available. Thus, Basic Protocol 4 outlines the use of a set of 1,3-cyclohexanedione-based reagents (DCP-tagged reagents; Poole et al., 2007), which employ the same chemistry as dimedone for modification of sulfenic acids, but also incorporate affinity or fluorescent tags into the target protein, upon its modification.
Modifications by the reagents described in Basic Protocols 1 and 2 (NBD chloride and TNB, respectively) are readily reversed by the reductant DTT, whereas the cyclohexanedione-based reagents used in Basic Protocols 3 and 4 remain covalently attached, making the latter particularly useful reagents for subsequent tryptic digestion and peptide analysis (Support Protocol 3). If conversion of a given protein thiol group to a sulfenic acid (see Support Protocol 1) is accompanied by a spectral change, titration with buffers at different pH values may allow for the determination of the pKa of that sulfenic acid (Basic Protocol 5). Functional properties of a given protein may also be affected by sulfenic acid formation and/or modification and may be tested as well (see Support Protocol 2).
BASIC PROTOCOL 1 SULFENIC ACID TRAPPING AND DETECTION USING NBD CHLORIDE
This method is most useful in demonstrating sulfenic acid formation where this species is accessible to modification and is the only cysteine thiol or sulfenic acid present, accessible, or both. In cases where accessibility is the problem, denaturants can be used. If more than one accessible cysteine thiol and/or sulfenic acid is present per subunit, modification may still allow for the demonstration of sulfenic acid by using difference spectra.
The sulfenic acid is generated by the method of choice—typically treatment with stoichiometric hydrogen peroxide, t-butyl hydroperoxide, or cumene hydroperoxide (see Support Protocol 1)—and then trapped by reaction with NBD chloride. Once the excess free reagent is removed from the modified protein by ultrafiltration, the presence of thiol or sulfenate adducts with NBD can be assessed by UV-visible spectroscopy and/or mass spectrometry.
Materials
Putative sulfenic acid–containing protein, purified and in a neutral pH buffer (pH 6.5 to 7.5)
Neutral pH buffer: 25 mM potassium phosphate buffer, pH 7.0 (see APPENDIX 2A) or equivalent/1 mM EDTA, or other pH 7 buffer
100 mM NBD chloride (7-chloro-4-nitrobenzo-2-oxa-1,3-diazole) in DMSO (see recipe)
Acetonitrile
Formic acid
Anaerobic quartz cuvette (if required) and regular quartz cuvette
Ultrafiltration unit of appropriate MWCO (e.g., Centricon, Millipore; Apollo, Orbital Biosciences; Vivaspin, Vivascience)
UV-visible scanning or diode-array spectrophotometer
Electrospray ionization mass spectrometer (ESI-MS) or access to a fee-for-service facility
Software for comparing observed and predicted mass (e.g., Calculate pI/MW tool; http://www.expasy.ch)
Additional reagents and equipment for preparing proteins containing sulfenic acid (Support Protocol 1) and determining protein concentration by a non-NBD-affected assay (e.g., Detergent-Compatible Protein Assay Kit, Bio-Rad)
Prepare protein
-
1
Prepare at least 1 ml of a 15 μM (monomer or target thiol group concentration) solution of the putative sulfenic acid–containing protein in neutral pH buffer, using the method described in Support Protocol 1 or by other methods established by functional analyses.
For example, in a typical reaction, add 3 μl of 8 mM hydrogen peroxide to 0.5 ml of 45 μM protein (Support Protocol 1).Oxidized proteins should be prepared immediately before use.
Modify protein with NBD chloride
-
2
To the protein of interest (with or without oxidant or reductant pretreatment, as experimental and/or controls samples) add a small volume of 100 mM NBD chloride, either directly or through the sidearm of the anaerobic cuvette. Incubate 10 to 30 min at room temperature.
More reagent gives a faster reaction, allowing pseudo-first-order kinetics to be followed if any spectral changes are observed; therefore, for most experiments, an excess of at least 10-fold NBD over protein thiol and sulfenic-acid groups is desirable.Anaerobic conditions may be required to stabilize the sulfenic acid during its generation, as described in Support Protocol 1; if so, this step should also be conducted anaerobically.Denaturation of the protein may be required in this step if the sulfenic acid is not accessible (see the Troubleshooting section in the Commentary).Thus, continuing with the example from the step 1 annotation, add 3 μl NBD chloride solution and incubate 10 to 30 min at room temperature. After modification, the protein in the cuvette can be exposed to air for the ultrafiltration step that follows.
Analyze modified protein by spectroscopy
-
3
Transfer the protein-NBD solution to an ultrafiltration unit of appropriate molecular weight cutoff (MWCO). Remove the unreacted NBD chloride as follows:
Add 6 ml neutral pH buffer.
Concentrate to 50 μl according to manufacturer’s instructions.
-
Repeat two to three times.
Attention must be paid to the stated MWCO of the device so that the protein of interest will be retained. With each concentration cycle, the flow-through solution (filtrate) is removed, and fresh neutral pH buffer is added to dilute the protein again, for a total of two to three concentration/redilution cycles.Either Centricon ultrafiltration units, which allow 2 ml to be concentrated to 40 to 50 μl, or larger units offered by several other suppliers (e.g., Orbital Biosciences or Vivascience), which allow 6 to 7 ml to be concentrated to ~10 to 50 μl, can be used. In the case of the latter, the ability to concentrate more solution at once and to use swinging bucket rotors, rather than fixed-angle rotors, can speed up the washing process.Alternatively, the modified protein can be isolated (without ultrafiltration) by HPLC in tandem with injection into a mass spectrometer.
-
4
Check for the presence of free NBD in the filtrate by determining the absorbance at 343 nm. Repeat step 3 until no free reagent is detected (i.e., A343 is <0.02).
-
5
Optional: Assess the protein concentration using a protein assay that is not affected by the presence of the NBD—e.g., the Detergent-Compatible (DC) Protein Assay Kit from Bio-Rad.
The A280 (APPENDIX 3G) now has a contribution from the presence of NBD and therefore cannot be used to determine the protein concentration. Colorimetric assays monitoring wavelengths >500 nm may work (see APPENDIX 3I). -
6
Remove the concentrated protein from the ultrafiltration unit and bring to approximately the original concentration with neutral pH buffer, assuming there has been no loss of protein during the ultrafiltration step.
Protein recovery from the ultrafiltration devices can be tested independently with unmodified protein. -
7
Perform a spectral scan from ~250 to 700 nm in a quartz cuvette to assess of the nature of the modification.
Thiol adducts with NBD give a peak at 420 nm with an extinction coefficient of 13,000 M−1 cm−1 (Birkett et al., 1970). Sulfenic acid adducts with NBD give a peak at 347 nm with a similar extinction coefficient (Ellis and Poole, 1997a).
Prepare modified protein for ESI-MS
-
8
Wash the modified protein sample at least three times by complete concentration and redilution with water (as described in step 3) to remove excess reagent and buffer components.
-
9
Prepare 1 nmol modified protein in 100 μl water. Add an equal volume of acetonitrile and 2 μl formic acid before injection. Analyze by ESI-MS.
This amount of sample is far above that required for the analysis by most modern ESI-MS instruments. Optimization of sample analyses may involve changing the acetonitrile concentration and/or using a volatile buffer such as ammonium bicarbonate instead of water during the washing step (Fuangthong and Helmann, 2002).Alternatively, the modified protein can be isolated (without ultrafiltration) by HPLC in tandem with injection into the mass spectrometer. Once prepared for mass spectrometry, samples are generally stable at −20°C for weeks prior to analysis (at least in the absence of the acetonitrile and formic acid).Using this approach, the NBD adduct prepared from the thiol form of the protein will exhibit a mass 16 amu less than that prepared from the sulfenic acid form. NBD itself contributes 164 amu to the mass of the protein. -
10
Compare the obtained mass with the predicted mass using a computer program that takes into account the natural abundance of all isotopes in the protein.
ALTERNATE PROTOCOL SULFENIC ACID TRAPPING AND DETECTION USING NBD FLUORIDE
This method can be utilized to generate an NBD adduct with protein cysteine sulfenic acids, which can then be followed by UV-visible spectroscopy and/or ESI-MS as described in Basic Protocol 1. The only difference between this and Basic Protocol 1 is that 100 mM 4-fluoro-7-nitrobenz-2-oxa-1,3-diazole (NBD fluoride; see recipe), rather than NBD chloride, is used as the chemical modification agent. The reactivity of NBD fluoride is approximately ten times greater than that of NBD chloride (described in the handbook from Molecular Probes), allowing for more rapid modification. Note that NBD fluoride should be freshly made and used as soon as possible, given its reactivity and instability. The incubation time with this reagent can be decreased relative to that needed for NBD chloride (Basic Protocol 1, step 2), and the ultrafiltration step should be performed very quickly after modification (Basic Protocol 1, steps 3 and 4).
BASIC PROTOCOL 2 QUANTITATION OF SULFENIC ACID FORMATION BY REACTION WITH TNB
In order to quantify cysteine sulfenic acids in proteins, this method takes advantage of the thiol reactivity of sulfenic acids and the resulting formation of mixed disulfide bonds with TNB, a chromophoric thiol-containing reagent generated by reduction of DTNB. The TNB reagent must be tested just before use to ensure that there is no excess of DTT or DTNB (see recipe for TNB solution). Assessment of the extent of reaction can be made immediately by monitoring the decrease in A412. Further proof of disulfide bond formation is obtained by isolation of the TNB-labeled protein through ultrafiltration, analysis of spectral properties of the modified protein, and release of the protein-associated TNB by DTT treatment. TNB-labeled protein can also be digested by trypsin and the peptides separated by HPLC and monitored for the presence of the TNB label.
Materials
Putative sulfenic acid–containing protein, purified and in a neutral pH buffer (pH 6.5 to 7.5)
Neutral pH buffer: 25 mM potassium phosphate buffer, pH 7.0 to 7.5 (see APPENDIX 2A)/1 mM EDTA, or other pH 7.0 to 7.5 buffer
4 mM 2-nitro-5-thiobenzoic acid (TNB) solution (see recipe)
100 mM dithiothreitol (DTT; see recipe)
Anaerobic quartz cuvette (if required) and regular quartz cuvette
UV-visible scanning or diode-array spectrophotometer
Ultrafiltration unit of appropriate MWCO (e.g., Centricon, Millipore; Apollo, Orbital Biosciences; Vivaspin, Vivascience)
Additional reagents and equipment for preparing proteins containing sulfenic acid (Support Protocol 1), determining protein concentration by a non-TNB-affected assay (e.g., Detergent-Compatible Protein Assay Kit, Bio-Rad; optional), and performing functional assays (Support Protocol 2; optional)
Prepare protein
-
1
Prepare at least 1 ml of a 15 μM (monomer concentration) solution of the putative sulfenic acid–containing protein in neutral pH buffer, using the method described in Support Protocol 1 or by other methods established by functional analyses.
Oxidized proteins should be prepared immediately before use.
Modify protein with TNB
-
2
To the protein of interest (with or without oxidant or reductant pretreatment as experimental and/or controls samples) add a small volume of 4 mM TNB either directly or through the sidearm of the anaerobic cuvette. Incubate at room temperature and monitor the decrease in A412 until the spectral changes are complete (usually within 2 to 5 min, if accessible). Include controls containing peroxide and other oxidants (without protein) to assess the contribution of their reactions.
Anaerobic conditions may be required to stabilize the sulfenic acid during its generation, as described in Support Protocol 1; if so, this step should also be conducted anaerobically.At long incubation times (>20 to 30 min), TNB can air oxidize, a process that must be taken into account using appropriate control reactions.A 2- to 3-fold excess of reagent can be used, keeping in mind that the quantitation of sulfenic acids will be most accurate where the total A412 of TNB added is 1.0 or slightly less (i.e., within the linear range of the spectrophotometer) and the absorbance change upon adding the reagent to the protein is >0.1.Excess peroxide and at least some other oxidants also react with TNB over time; if present, these oxidants should be added to control reactions without protein to assess the contribution of this reaction.Denaturation of the protein may be required in this step if the sulfenic acid is not accessible (see the Troubleshooting section in the Commentary). -
3
Read the A412 of a blank consisting of neutral pH buffer containing the same concentration of TNB as the sample and incubated for the same amount of time.
-
4
Calculate the sulfenic-acid content of the protein as follows:
Subtract the final A412 value of the modified sample (step 2) from the A412 of the blank (step 3).
Convert this value to concentration of sulfenic acid–containing residues, using the extinction coefficient 14,150 M−1 cm−1 in buffers of pH 7 or higher (Riddles et al., 1979).
-
Divide by the protein concentration to obtain the sulfenic acid content of the protein.
The contribution of excess peroxide and other oxidants that react with TNB over time should be assessed based on the controls.
Isolate TNB-labeled protein
-
5
Transfer the reaction mixture to an ultrafiltration unit of appropriate molecular weight cutoff (MWCO). Remove unreacted TNB as follows:
Add 6 ml neutral pH buffer.
Concentrate to 50 μl according to manufacturer’s instructions.
-
Repeat one to three times.
Attention must be paid to the stated MWCO of the device so that the protein of interest will be retained. With each concentration cycle, the flow-through solution (filtrate) is removed, and fresh neutral pH buffer is added to dilute the protein again, for a total of one to three concentration/redilution cycles.Either Centricon ultrafiltration units, which allow 2 ml to be concentrated to 40 to 50 μl, or larger units offered by several other suppliers (e.g., Orbital Biosciences or Vivascience) which allow 6 to 7 ml to be concentrated to ~10 to 50 μl, can be used. In the case of the latter, the ability to concentrate more solution at once and to use swinging bucket rotors, rather than fixed-angle rotors, can speed up the washing process.
-
6
Check for the presence of free TNB in the filtrate by determining the absorbance at 412 nm. Repeat step 5 until no free reagent is detected (i.e., A412 is <0.02).
-
7
Remove the concentrated protein from the ultrafiltraton unit and bring to approximately the original concentration with neutral buffer. Transfer the solution to a quartz cuvette.
-
8
Optional: Assess the protein concentration using a protein assay that is not affected by the presence of the TNB—e.g., the Detergent-Compatible (DC) Protein Assay Kit from Bio-Rad.
-
9
Optional: If identification of the labeled peptide is important, perform a tryptic digest and analyze, as described in Support Protocol 3.
Release TNB from the modified protein
-
10
Add a 10- to 100-fold excess of 100 mM DTT (up to 5 mM final concentration for 45 μM protein), monitoring the spectral changes from 280 to 600 nm until complete (i.e., until no change is detected).
The increase in A412 is used to assess the amount of TNB released from the isolated, modified protein. The calculation is basically the same as in step 4 except that the A412 after DTT treatment is higher than before. -
11
Optional: If regeneration of the thiol group(s) in the protein is expected to restore or alter the functional properties of the protein, perform an appropriate assay to determine this (see Support Protocol 2), with or without an additional ultrafiltration step (see step 5), depending mostly on the potential for the excess DTT to interfere with the assay.
BASIC PROTOCOL 3 MODIFICATION AND DETECTION OF PROTEIN SULFENIC ACIDS WITH DIMEDONE
Reactivity of proteins with dimedone is diagnostic for the presence of cysteine sulfenic acds. This reaction is not monitored by spectral changes, but rather by the mass increase observed upon modification of the sulfenic acid. When localization of the incorporated dimedone is of interest, tryptic digests followed by HPLC and mass spectrometric analyses can be used to identify modified peptides and/or residues (see Support Protocol 3).
Materials
Putative sulfenic acid–containing protein sample, purified and in a neutral pH buffer (pH 6.5 to 7.5)
Neutral pH buffer: 25 mM potassium phosphate buffer, pH 7.0 (see APPENDIX 2A)/ 1 mM EDTA, or other pH 7 buffer
100 mM 5,5-dimethyl-1,3-cyclohexanedione (dimedone): prepared by adding 14 mg dimedone (Sigma-Aldrich) to 1 ml DMSO; store in aliquots up to several months at −20°C
Acetonitrile
Formic acid
Anaerobic cuvette
Ultrafiltration unit of appropriate MWCO (e.g., Centricon, Millipore; Apollo, Orbital Biosciences; Vivaspin, Vivascience)
Electrospray ionization mass spectrometer (ESI-MS) or access to a fee-for-service facility
Software for comparing observed and predicted mass (e.g., Calculate pI/MW tool; http://www.expasy.ch)
Additional reagents and equipment for preparing proteins containing sulfenic acid (Support Protocol 1) and determining the identity of the labeled peptide (Support Protocol 3; optional)
Prepare protein
-
1
Prepare at least 200 μl of a 15 μM (monomer concentration) solution of the putative sulfenic acid–containing protein in neutral pH buffer, using the method described in Support Protocol 1 or by other methods established by functional analyses.
Oxidized proteins should be prepared immediately before use.
Modify with dimedone
-
2
To the protein of interest (with or without oxidant or reductant pretreatment as experimental and/or controls samples) add at least a 100-fold excess (or up to 1 mM) of 100 mM dimedone either directly or through the sidearm of the anaerobic cuvette. Incubate 1 to 2 hr or overnight at 25°C.
Anaerobic conditions maybe required to stabilize the sulfenic acid during its generation, as described in Support Protocol 1; if so, this step should also be conducted anaerobically.Depending on accessibility of the sulfenic acid, denaturation of the protein may be required to promote the labeling reaction. This can be accomplished by adding urea or guanidine hydrochloride to the dimedone, preferably under anaerobic conditions, as denaturation will decrease the stability of the sulfenic acid.
Prepare modified protein for ESI-MS
-
3
Transfer the reaction mixture to an ultrafiltration unit of appropriate molecular weight cutoff (MWCO). Remove the unreacted dimedone and buffer components as follows:
Add 6 ml deionized water.
Concentrate to 50 μl according to manufacturer’s instructions.
-
Repeat at least three times.
Attention must be paid to the stated MWCO of the device so that the protein of interest will be retained. With each concentration cycle, the flow-through solution (filtrate) is removed, and deionized water is added to dilute the protein again, for a total of at least three concentration/redilution cycles.Either Centricon ultrafiltration units, which allow 2 ml to be concentrated to 40 to 50 μl, or larger units offered by several other suppliers (e.g., Orbital Biosciences or Vivascience), which allow 6 to 7 ml to be concentrated to ~10 to 50 μl, can be used. In the case of the latter, the ability to concentrate more solution at once and to use swing-out bucket rotors, rather than fixed-angle rotors, can speed up the washing process.
-
4
Prepare 1 nmol modified protein in 100 μl water. Add an equal volume of acetonitrile and 2 μl formic acid before injection. Analyze by ESI-MS
This amount of sample is far above that required for the analysis by most modern ESI-MS instruments. Optimization of sample analyses may involve changing the acetonitrile concentration and/or using a volatile buffer such as 10 or 50 mM ammonium bicarbonate instead of water during the washing step (Fuangthong and Helmann, 2002).Alternatively, the modified protein can be isolated (without ultrafiltration) by HPLC in tandem with injection into the mass spectrometer, skipping step 3 and the sample preparation above. Once prepared for mass spectrometry, samples are generally stable at −20°C for weeks prior to analysis (at least in the absence of the acetonitrile and formic acid).Using this approach, only peptides containing a cysteine sulfenic acid will form adducts with dimedone. Dimedone itself contributes an additional 138 amu to the mass of the protein. -
5
Optional: If identification of the labeled peptide is important, perform a tryptic digest of the isolated modified protein and analyze, as described in Support Protocol 3.
BASIC PROTOCOL 4 MODIFICATION AND DETECTION OF PROTEIN SULFENIC ACIDS WITH BIOTIN-LINKED OR FLUOROPHORE-LINKED DIMEDONE DERIVATIVES
Reactivity of proteins with the 1,3-cyclohexanedione-based compounds like dimedone is diagnostic for the presence of cysteine sulfenic acids. This reaction can be monitored by the mass increase observed upon modification of the sulfenic acid and/or by other appropriate means when conjugated compounds (probes) are used, e.g., immunoblotting (biotinylated probes) or fluorescence detection in gels (fluorphore-conjugated probes). Where localization of the incorporated probe is of interest, tryptic digests followed by HPLC and mass spectrometric analyses can be used to identify modified peptides and/or residues (Support Protocol 3). Use of a biotinylated probe allows for enrichment of mixtures of protein in samples by affinity capture on avidin-conjugated beads.
Materials
Putative sulfenic acid–containing protein sample, purified and in a neutral pH buffer (pH 6.5 to 7.5)
Neutral pH buffer: 25 mM potassium phosphate buffer, pH 6.5 to 7.5 (see APPENDIX 2A)/1 mM EDTA, or other pH 7 buffer
-
250 mM DCP-based affinity and fluorescent reagents (see recipe; see Fig. 17.2.3 and Poole et al., 2007 for structures):
Biotinylated DCP-linked probe (DCP-Bio1, DCP-Bio2, DCP-Bio3)
Fluorescent-labeled DCP-linked probe (DCP-FL1, DCP-FL2, DCP-Rho1, DCP-Rho2)
50 mM and 0.1 M ammonium bicarbonate buffer (not pH adjusted)
Acetonitrile
Formic acid
Acetone or trichloroacetic acid (optional)
Avidin-conjugated (e.g., streptavidin, monoavidin, or neutravidin) Sepharose or agarose beads
Phosphate-buffered saline (APPENDIX 2A)
SDS sample buffer (containing 2-mercaptoethanol; e.g., see APPENDIX 3F)
Figure 17.2.3.

Electrospray ionization mass spectrometry analysis of adducts with the sulfenic form (Cys46-SOH) of C165S AhpC from Salmonella typhimurium. Shown are the transformed data from electrospray ionization mass spectrometric analyses of protein conjugates generated with the DCP-linked reagents (B, C, D), or vehicle only (A), and the chemical structures of the modifying reagents used. The peaks observed at 20,632 and 20,648 Da represent the protein with the active site Cys46 in the sulfinic and sulfonic acid states, respectively. For more details, see Figure 3 in Poole et al. (2007).
Anaerobic cuvette or pear-shaped flask
Ultrafiltration unit of appropriate MWCO (e.g., Centricon, Millipore; Apollo, Orbital Biosciences; Vivaspin, Vivascience)
Electrospray ionization mass spectrometer (ESI-MS) or access to a fee-for-service facility
PD-10 columns (GE Healthcare) or Bio-Gel P6 spin columns (Bio-Rad), optional Software for comparing observed and predicted mass (e.g., Calculate pI/MW tool; http://www.expasy.ch)
Gel documentation system with filters for fluorescein or rhodamine B
Additional reagents and materials for preparing proteins containing sulfenic acid (Support Protocol 1), determining the identity of the labeled peptide (optional; Support Protocol 3), purifying proteins (Bollag and Edelstein, 1991), performing SDS-PAGE (APPENDIX 3F or Bollag and Edelstein, 1991), and performing immunoblotting (see UNIT 2.3)
Prepare protein
-
1
Prepare at least 200 μl of a 15 μM (monomer concentration) solution of the putative sulfenic acid–containing protein in neutral pH buffer, using the method described in Support Protocol 1 or by other methods established by functional analyses.
Oxidized proteins should be prepared immediately before use.
Modify with DCP-based probe
-
2
To the protein of interest (with or without oxidant or reductant pretreatment as experimental and/or controls samples) add at least a 100-fold molar excess (or up to 5 mM) of a 250 mM stock of the probe (in DMSO) either directly or through the sidearm of the anaerobic cuvette. Incubate 1 to 2 hr or overnight at 25°C.
Anaerobic conditions maybe required to stabilize the sulfenic acid during its generation, as described in Support Protocol 1; if so, this step should also be conducted anaerobically.Depending on accessibility, denaturation of the protein may be required to promote the labeling reaction. Denaturation of the protein may be required in this step if the sulfenic acid is not accessible (see the Troubleshooting section in the Commentary).Some proteins may be sensitive to and precipitate out of solution in the presence of 1% to 2% DMSO, so lower concentrations of the probe stock should be used in those cases. -
3
Optional: If identification of the labeled peptide is important, perform a tryptic digest of the isolated modified protein and analyze, as described in Support Protocol 3.
Prepare modified protein for ESI-MS
-
4a
Transfer the reaction mixture to an ultrafiltration unit of appropriate molecular weight cutoff (MWCO). Remove the unreacted reagent and buffer components as follows:
Add 6 ml deionized water or volatile buffer (e.g., 10 M ammonium bicarbonate).
Concentrate to 50 μl according to the manufacturer’s instructions.
-
Repeat three to four times (or more).
Attention must be paid to the stated MWCO of the device so that the protein of interest will be retained. With each concentration cycle, the flow-through solution (filtrate) is removed, and deionized water or buffer is added to dilute the protein again, for a total of at least three concentration/redilution cycles. More washes may be required for sticky fluorescein- and rhodamine-linked reagents.Either Centricon ultrafiltration units, which allow 2 ml to be concentrated to 40 to 50 μl, or larger units offered by several other suppliers (e.g., Orbital Biosciences or Vivascience), which allow 6 to 7 ml to be concentrated to ~10 to 50 μl, can be used. In the case of the latter, the ability to concentrate more solution at once and to use swing-out bucket rotors, rather than fixed-angle rotors, can speed up the washing process.
-
5a
Prepare 1 nmol modified protein in 100 μl water. Add an equal volume of acetonitrile and 2 μl formic acid before injection. Analyze by ESI-MS.
This amount of sample is far above that required for the analysis by most modern ESI-MS instruments. Optimization of sample analyses may involve changing the acetonitrile concentration and/or using a volatile buffer such as 10 or 50 mM ammonium bicarbonate instead of water during the washing step (Fuangthong and Helmann, 2002).Alternatively, the modified protein (without ultrafiltration) can be isolated by HPLC in tandem with injection into a mass spectrometer, skipping steps 4a and 5a. Once prepared for mass spectrometry, samples are generally stable at −20°C for weeks prior to analysis (at least in the absence of the acetonitrile and formic acid).Using this approach, only peptides containing a cysteine sulfenic acid will form adducts with the dimedone derivatives (Poole et al., 2005). Each of the derivatives contributes a unique additional mass to the protein (see Poole et al., 2007).
Affinity capture and detect biotin-labeled proteins
-
4b
Remove excess biotin-containing reagents from the protein sample by ultrafiltration as in step 4a, by gel filtration with PD-10 columns from GE Healthcare or Bio-Gel P6 spin columns to remove small molecules, or by precipitation of proteins with acetone or TCA by standard methods (see Bollag and Edelstein, 1991).
These methods are compatible with the use of lysis buffer to disrupt cells in tissue culture as a first step; in such cases, the DCP-based biotin reagent can be added directly to the lysis buffer to label cellular proteins. For such samples, subsequent steps can be carried out with phosphate-buffered saline or ammonium bicarbonate buffers.Affinity capture of biotinylated peptides may also be performed instead of or in addition to affinity capture of the intact proteins, following proteolytic digestion of the protein sample as described in Support Protocol 3. -
5b
Incubate the sample with commercially available avidin-conjugated Sepharose or agarose beads (amounts recommended by manufacturer) for 0.5 to 2 hr (or overnight) at room temperature or 4°C.
To control for nonspecific binding of “sticky” proteins (e.g., where cellular extracts are being used), samples can be pretreated with chemically similar beads without the avidin (e.g., Sepharose CL-4B beads) prior to application of the sample to the avidin beads. -
6b
Wash the beads sequentially two times each with column volumes of:
Buffer (e.g., 0.1 M ammonium bicarbonate or phosphate-buffered saline)
1 M NaCl
Buffer
Water.
Centrifuge between washes 5 min at 5,000 × g, 4°C and remove the supernatant.
The stringency of the wash step is of great importance for determining the degree of enrichment obtained for biotinylated versus nonbiotinylated proteins. With a low stringency wash procedure (as described above), one may retain more of the biotinylated proteins, as well as more nonspecifically bound proteins or proteins bound in complexes with those that are biotinylated. For intact proteins, higher stringency washing includes successive incubations with 1% SDS (30 min), 4 M urea (30 min), 1 M NaCl (10 min), 0.1 M ammonium bicarbonate buffer, and then water (Vila et al., 2008). -
7b
Elute the biotinylated protein from the beads using one or both (sequentially) of the following (see Vila et al., 2008):
70% acetonitrile with 5% formic acid
Boiling in SDS sample buffer (for SDS-PAGE).
-
8b
Separate biotinylated proteins using standard one-dimensional or two-dimensional polyacrylamide gel electrophoresis methods (APPENDIX 3F or Bollag and Edelstein, 1991), followed by electroblotting and immunodetection with anti-biotin antibody (generally described in UNIT 2.3).
Alternatively, the primary and secondary antibodies typically used in such detection methods can be replaced with commercially available biotin detection reagents like HRP-conjugated streptavidin.
Detect fluorescein- or rhodamine-labeled proteins
-
4c
Wash fluorescein- or rhodamine-labeled proteins free of the unreacted reagent as described in step 4a.
-
5c
Evaluate fluorophore incorporation spectroscopically by the absorbance of the labeled protein in pH 7 phosphate buffer at the appropriate wavelength:
493 nm for protein labeled with DCP-FL1 or DCP-FL2 (extinction coefficient ~ 67,000 M−1 cm−1)
570 nm for protein labeled with DCP-Rho1 or DCP-Rho2 (extinction coefficients ~70,000 or 61,000 M−1 cm−1, respectively).
-
6c
Separate fluorescent proteins using standard one-dimensional or two-dimensional polyacrylamide gel electrophoresis methods (see APPENDIX 3F or Bollag and Edelstein, 1991), first soaking the gels in water or 50 mM ammonium bicarbonate buffer for 30 min to 1 hr.
-
7c
Detect fluorescence using appropriate settings and filters for fluorescein or rhodamine B using a gel documentation system.
Such systems can detect these DCP-linked fluorophores at amounts as low as 0.1 to 0.5 pmol (Poole et al., 2007).Because these detection methods do not discriminate between covalently linked fluo-rophores and those binding nonspecifically to proteins of interest, specificity must be demonstrated with proper controls (e.g., for the sulfenic acid modification) and/or by demonstrating covalent modification of the protein of interest by mass spectrometry (step 5a, above).
BASIC PROTOCOL 5 DETERMINATION OF THE PROTEIN SULFENIC ACID DISSOCIATION CONSTANT (pKa)
In some cases, proteins containing sulfenic acids may exhibit a band (εmax of ~320 to 370 nm) due to the presence of the deprotonated sulfenate species (Poole and Ellis, 2002), as has been observed previously in small molecules (Tripolt et al., 1993). In these cases, lowering of the pH to protonate this group will result in the disappearance of this absorbance band. Titration of this absorbance can be used to determine the pKa value of the cysteine sulfenic acid within the protein. This knowledge could be useful for understanding the effects of the protein microenvironment in stabilizing the sulfenic acid and/or the function of sulfenic acid formation in a given protein. Depending on the extinction coefficient for this absorbance and on the pH stability properties of the protein, large amounts of the protein containing the stable sulfenic acid and a stopped-flow spectrophotometer (for enhanced reproducibility and rapid mixing) are optimal for this determination.
Materials
Citrate/phosphate buffer (pH from 3 through 7.6)/1 mM EDTA (see recipe)
Protein, purified and in 10 mM neutral pH buffer (e.g., 10 mM potassium phosphate buffer pH ~7; see APPENDIX 2A)
UV-visible spectrophotometer or stopped-flow spectrophotometer (recommended) 3- to 5-ml syringes
Additional reagents and equipment for preparing proteins containing sulfenic acid (Support Protocol 1) and determining the identity of the labeled peptide (optional; Support Protocol 3)
Prepare sample
-
1
Prepare at least a 150 μM (monomer concentration) solution of the (putative) sulfenic acid–containing protein in 10 mM neutral pH buffer, using the method described in Support Protocol 1 or other methods established by functional analyses.
Oxidized proteins should be prepared immediately before use. -
2
To observe the expected absorbance change and determine the feasibility of this experiment, mix protein samples with an equal volume of high and low pH citrate/phosphate buffers (e.g., pH 4 and 7.6) and record the UV-visible spectral change from 250 to 400 nm.
The spectral change should be immediate.If the near UV absorbance band is lost at low pH, the following experiment is feasible.In the case of the sulfenate anion of the C165S mutant of AhpC, maximal absorbance was at 367 nm (Poole and Ellis, 2002).
Conduct rapid mixing (pH jump) experiment
-
3
Set the monochromator wavelength of the spectrophotometer at the λmax for the protein sulfenate peak (e.g., 367 nm in the example above).
-
4a
For a stopped-flow spectrophotometer: Load the sulfenic acid–containing protein into one 5-ml syringe and one of the buffers (i.e., high or low pH) into another. Rapidly mix the protein and buffer solutions and acquire data at the peak wavelength for 10 sec. Repeat ten times or more for each pH point (minimum of eight suggested), controlling for instrument drift by frequently checking the absorbance of buffer alone.
For accurate results, this data acquisition should be carried out at a minimum of eight different pH values bracketing the pKa.Keep the sample volume as low as possible to minimize protein consumption (although protein may be recovered from this nondestructive procedure at pH values where the protein is stable). Any formation of precipitated protein or any conformational changes resulting from the pH change may, however, confound the results. The use of the stopped-flow spectrophotometer allows for the absorbance changes that ensue after the rapid pH change to be measured before any slower pH-dependent denaturation occurs. -
4b
For a standard spectrophotometer: Rapidly mix the buffer and protein by adding the phosphate/citrate buffer to the protein in the cuvette, holding a piece of Parafilm tightly over the cuvette with the index finger, and gently inverting the cuvette several times. Measure the absorbance at the peak wavelength as rapidly as possible. Repeat measurements at each pH value at least three times for a minimum of eight different pH values, as above.
-
5Plot absorbance versus pH, averaging all data collected at a given pH. If the expected sigmoidal curve is observed, fit data to the following equation:
where A is the absorbance at high pH;
B is the absorbance at low pH;
y is the absorbance at pH x.
SUPPORT PROTOCOL 1 PREPARATION OF SULFENIC ACID–CONTAINING PROTEIN
Susceptible protein thiol groups are oxidized to sulfenic acids by the addition of even mild oxidants including hydrogen peroxide, organic hydroperoxides, hypochlorous acid, peroxynitrite, S-nitrosoglutathione, and other NO-generating or NO-derived signaling molecules. This oxidation may be stoichiometric or may require an excess of the oxidant, and anaerobiosis may or may not be necessary to stabilize the sulfenic acid formed. This protocol describes the use of peroxides as the oxidant but is generally applicable to use of the other oxidants as well.
Materials
Protein solution
Argon or oxygen-free nitrogen (optional)
Neutral pH buffer: 25 mM potassium phosphate (pH 7.0)/1 mM EDTA (or other buffer at pH 7 or lower)
Peroxide of choice, e.g., 8 mM hydrogen peroxide (H2O2) or cumene hydroperoxide (see recipes)
100 mM dithiothreitol (DTT; see recipe)
Anaerobic cuvette assembly (Williams et al., 1979) or small pear-shaped flask with stopcock
-
1
Determine if anaerobic conditions are necessary for the protein solution of interest, by using one of the methods described in Basic Protocols 1 to 4 for detecting the stabilized protein generated under anaerobic conditions, then use the same method under aerobic conditions.
If the protein sulfenic acid can be detected under anaerobic conditions, but the sulfenic acid content of the protein decreases markedly when using aerobic conditions, then preparation and initial modification steps of the sulfenic acid–containing protein should be conducted anaerobically.Preparation of the sulfenic acid form of the target cysteinyl residue(s) on the protein may or may not require anaerobic conditions for stabilization. -
2
Optional: If anaerobic conditions are necessary:
Place the protein solution in an anaerobic cuvette assembly or small pear-shaped flask with stopcock.
While gently rocking, flush with argon or nitrogen for 1 to 2 min, followed by gentle vacuum for 1 to 2 min. Repeat for a total of ten to twenty cycles (20 to 30 min total).
-
3
Add a stoichiometric amount of the peroxide of choice (e.g., 8 mM H2O2 or cumene hydroperoxide) to the protein solution.
Excess peroxide may also be added, although there is a risk of thiol overoxidation beyond the sulfenic acid oxidation state.For cysteine-based peroxidases (e.g., NADH peroxidase and peroxiredoxins) and at least one peroxide-sensitive transcriptional regulator (OxyR), this reaction is very fast (≥105 M−1 sec−1 second-order rate constant); however, in general, the reaction is much slower and varies greatly for other protein thiol or thiolate groups. As a result, no clear statement can be made as to the length of time required for the incubation of the protein with the peroxide (this should be treated as a variable). For example, continue treatment for 2 min to 24 hr at 4° or 25°C. -
4
Perform a wavelength scan from 250 to 400 nm to determine if any spectral changes due to addition of peroxide are present.
Any spectral signature can provide both a way to monitor sulfenic acid formation and a way to discriminate between protonated and deprotonated forms of the sulfenic acid for pKa determination (see Basic Protocol 5). However, for observations of such low-extinction coefficient spectral changes, the protein needs to be at very high concentration (≥80 μM).
SUPPORT PROTOCOL 2 FUNCTIONAL ANALYSES OF THE MODIFIED PROTEIN
If a free thiol group is required for the functional activity of the protein under investigation, either the modification of that group with NBD chloride or the oxidation of the thiol group to sulfenic acid followed by NBD chloride, TNB, or dimedone modification should block its activity. Whether or not oxidation to the sulfenic acid itself alters the protein’s activity depends on the function of this modification:
In cysteine-based peroxidases, the sulfenic acid is a naturally occurring intermediate during turnover and is therefore catalytically active as long as the reductant being used will reduce this species, and overoxidation to inactive sulfinic and sulfonic acids forms does not occur preferentially.
In at least some peroxide-sensitive transcriptional regulators, the sulfenic acid form may be an important functional form, either activating or derepressing transcription from genes related to oxidative stress protection (see UNIT 17.1). In these cases an assay for transcriptional activation is appropriate.
In protein tyrosine phosphatases, the active site cysteine is reversibly inhibited by conversion to the sulfenic acid form. Therefore, an assay for phosphatase activity using a model phosphorylated peptide can be used for these proteins. It is of great interest to test the consequences of modification and/or oxidation of target cysteines in active sites of enzymes or binding sites of transcriptional regulators.
If the protein sulfenic acid is known to be stable toward air (or at least stable over the 10 to 30 min during which the functional analyses is performed; see Support Protocol 1), the assay of interest can be carried out aerobically. The air stability of the chemically modified proteins is not a problem, although in these cases, an exogenous chemical has been added to the protein to give a non-native modification. For NBD- or TNB-modified protein, the thiol(ate) group is restored by DTT treatment (excess reagents may need to be removed by ultrafiltration prior to carrying out the functional assay). Dimedone modification of sulfenic acids is not reversed by DTT treatment.
SUPPORT PROTOCOL 3 TRYPTIC DIGESTION OF MODIFIED PROTEINS
To determine the peptide (or even the residue) of the protein modified by one of the reagents described in Basic Protocols 1 to 4, chemical modification of the protein can be followed by tryptic digestion and isolation, and analysis of the peptides. The dimedone or DCP-based reagent modifications (Basic Protocols 3 and 4) are best for this analysis, due to their irreversibility.
Materials
Modified protein (see Basic Protocols 1, 2, 3, or 4) and unmodified protein (optional)
50 mM N-ethylmaleimide (NEM; optional; see recipe)
10 M urea or 8 M guanidine hydrochloride (see recipes)
100 mM Tris·Cl buffer, pH 8.0 (APPENDIX 2A) or 100 mM HEPES, pH 7.6 (see recipe)
100 mM calcium chloride (1.11 g anhydrous CaCl2 in 100 ml water; store up to several weeks at room temperature)
TPCK-treated trypsin solution (see recipe)
Acetic acid
10 mM EGTA
100 mM DTT
95° or 60°C water bath (optional)
HPLC equipped with a C18 reversed-phase column, solvents for peptide isolation: e.g., 0.1% (v/v) trifluoroacetic acid, 70% (v/v) acetonitrile with 0.08% trifluoroacetic acid
Electrospray ionization mass spectrometer (ESI-MS) or access to a fee-for-service facility
Additional reagents and equipment for performing MALDI-TOF mass spectrometry (optional), SDS-PAGE (APPENDIX 3F; optional), or Tris-tricine-PAGE (APPENDIX 3F; optional)
-
1
Optional: To block additional cysteine thiol groups, add 50 mM NEM to a 100-fold excess and incubate 1 hr at room temperature to alkylate the additional cysteine thiol groups.
-
2
Prepare a small volume (≤200 μl) containing ~1 mg isolated, modified, blocked (if necessary) protein and 8 M urea or 6 M guanidinium hydrochloride. Optionally, incubate 15 to 20 min at 95°C, or 45 to 60 min at 60°C to ensure denaturation.
Protein eluted from avidin or antibody-linked beads into acetonitrile and reduced in volume in vacuo can also be used directly for digestion. -
3
Once the sample has cooled, add sufficient 100 mM Tris·Cl, pH 8.0 (or if the sample is NBD-labeled, add 100 mM HEPES, pH 7.6) to lower the denaturant concentration to ≤1 M. Add 1/100 vol of 100 mM calcium chloride (1 mM final).
Alternatively, ammonium bicarbonate buffer (pH 7.4) can be used as the digestion buffer.If NBD- or TNB-labeled protein is used, the conditions for trypsin digestion can be pretested to ensure that the label remains covalently attached to the protein (reductants remove both labels and cannot be used, and amine groups can react with and remove the NBD moiety). -
4
Add TPCK-treated trypsin to a protease/protein ratio of 1:100 to 1:40 (w/w). Incubate 12 hr at 37°C. Add a second aliquot of TPCK-treated trypsin solution and incubate another 12 hr.
Shorter incubation times may also suffice. -
5
Optional: To monitor the progress of the digestion, remove small aliquots of the reaction mixture and analyze using reversed-phased HPLC, MALDI-TOF mass spectrometry, SDS-PAGE, or Tris-tricine-PAGE.
-
6a
For storage: Freeze samples at −20°C and store up to 2 weeks.
-
6b
For immediate use: Inactivate the trypsin by adding sufficient acetic acid to lower the pH to <4, or by adding 10 mM EGTA to chelate calcium.
-
7a
To analyze NBD- and TNB-labeled proteins (Basic Protocols 1 and 2) by absorbance: Use the following procedure:
To an aliquot of digested peptide, add 1/10 vol of 100 mM DTT.
-
Separate the digested peptide samples with and without DTT treatment by HPLC, using an appropriate column and gradient.
For example, use a C18 or C8 column and a linear gradient over 60 to 100 min from 0% to 60% solvent B (70% acetonitrile/0.08% trifluoracetic acid) with the balance being solvent A (0.1% trifluoracetic acid).Monitor the absorbance at 347 or 420 nm for NBD-treated samples, or 325 nm for TNB-treated samples. Peptide elution is typically monitored at 215 nm where the peptide bonds absorb strongly and the contributions of solvents A and B are appropriately balanced.For further analysis of these peptides, additional purification using a more shallow gradient should be carried out. -
Identify labeled peptides by comparing HPLC chromatograms of the tryptic digests with and without DTT treatment.
DTT treatment will remove the chromophore and will likely shift the retention time of the peptide.
-
7b
To analyze fluorophore-labeled proteins (Basic Protocol 4) by fluorescence: Perform reversed-phase HPLC (see step 7a, ii) and identify labeled peptides with excitation and emission wavelengths appropriate to the fluorescent tag used.
For DCP-FL1 and DCP-FL2: λex,max = 493 nm and λem,max ~517 nm.
For DCP-Rho1 and DCP-Rho2: λex,max = 570 nm and λem,max ~588 nm.
See Poole et al. (2007).DTT does not reverse the alkylation with these 1,3-cyclohexanedione-based labeling agents. -
7c
To analyze labeled samples by ESI-MS: Perform reversed-phase HPLC (step 7a, ii) in tandem with ESI-MS on digests. Identify labeled residues by the added mass.
-
8
Optional: Repeat steps 1 to 6 with unlabeled protein and analyze by HPLC in tandem with ESI-MS to verify labeling.
Where only a single cysteine is encoded within that peptide, further localization is unnecessary. Additional analysis by MS-MS using collision-induced fragmentation can also be used to determine the sequence and location of the labeled cysteine. Alternatively, the peptide can be subjected to Edman degradation to determine the position of the blocked cysteine, although its identification may rely only on the absence of an identifiable peak at the position of the modified cysteine.
REAGENTS AND SOLUTIONS
Use Milli-Q-purified water or equivalent for all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Citrate/phosphate buffer
Combine the following:
245 ml water
9.61 g citric acid (~200 mM final)
7.10 g dibasic sodium phosphate (~200 mM Na2HPO4 final)
93 mg disodium EDTA
-
Measure the pH (should be ~3). Slowly drip in a solution of 1 M sodium hydroxide (4.0 g in 100 ml) to raise the pH, and remove ~10-ml aliquots at each desired pH value (about every 0.2 pH units). Store the aliquots up to 1 month at room temperature, but recheck pH of each before each use.
These solutions allow for mixing of the sulfenic acid–containing protein with strong buffers at various pH values to determine the pKa of the protein-associated sulfenic acid (Basic Protocol 5).
Cumene hydroperoxide, 8 mM
Prepare a stock solution containing 20 μl cumene hydroperoxide solution (80% stock from Sigma; store aliquots up to 1 year at −20°C) and 980 μl dimethyl sulfoxide (DMSO). Prepare a 15× dilution of the stock solution in water to give a final concentration of ~8 mM. Prepare fresh daily.
DCP-based affinity and fluorescent reagents, 250 mM
-
Synthesize 1,3-cyclohexanedione linked reagents—including three biotin-linked (DCP-Bio1, DCP-Bio2, DCP-Bio3), two fluorescein-linked (DCP-FL1, DCP-FL2), and two rhodamine-linked reagents (DCP-Rho1, DCP-Rho2)—according to published procedures (Poole et al., 2007). Add 5 mg of one of the compounds to DMSO to give a stock concentration of 250 mM (using DMSO volumes of 50.5, 42.0, 39.2, 36.8, 32.1, 28.3, and 25.2 μl for the different compounds listed respectively). Dispense into aliquots and store up to several months or more at −20°C.
Each reagent has a unique molecular weight (see Table 1 of Poole et al., 2007).
DTNB, 5 mM
Prepare a 100 mM stock solution by dissolving 0.198 g of 5,5′-dithiobis(2-nitrobenzoic acid) in 5 ml DMSO. Dispense in aliquots and store up to several months at −20°C. Before use, dilute the stock DTNB solution 20-fold into 25 mM phosphate buffer, pH 7 (see APPENDIX 2A).
To standardize the working solution: Dilute 3 μl in 0.5 ml 25 mM phosphate buffer. While monitoring the A412, add 8-μl aliquots of 0.5 mM DTT, pausing between each addition until the spectral changes are complete. When the A412 no longer increases with additional DTT, use the maximal A412 to calculate the DTNB concentration, using an ε412 value of 14,150 M−1 cm−1 for TNB (the solution resulting from the combination of DTNB and DTT) and the fact that 2 moles of TNB are released per mole DTNB (Riddles et al., 1979).
DTT, 100 mM
Dissolve 154 mg of 1,4-dithio-DL-threitol (DTT) in 10 ml water. Store in aliquots up to 2 weeks at −20°C.
-
To standardize the DTT: First perform the standardization of DTNB (see recipe). Calculate the DTT concentration, using the titration breakpoint from a plot of A412 versus volume of DTT added to determine the volume of DTT required to titrate the known amount of DTNB.
DTT has two thiols per molecule; therefore, its concentration is equivalent to the concentration of DTNB with which it reacts.
Guanidine hydrochloride, 8 M
To ~4 ml water add 7.64 g guanidine hydrochloride (ultrapure; Sigma-Aldrich). Warm the solution and add water to fully dissolve. Bring the volume to 10 ml total. Store up to several months at room temperature in a tightly sealed bottle.
HEPES, 100 mM (pH 7.6)
To 90 ml of water, add 2.38 g HEPES and dissolve. Add 1 M sodium hydroxide dropwise to bring the pH to 7.6, and then add water to bring the final volume to 100 ml. Store up to several weeks at room temperature.
Hydrogen peroxide, 8 mM
Dilute ~182 μl of 30% (v/v) H2O2 solution (stored at 4°C) into 250 ml water. Prepare fresh daily.
To standardize hydrogen peroxide: Prepare solutions of 10 mg/ml o-dianisidine in methanol and 1 mg/ml horseradish peroxidase (HRP) in an appropriate buffer—e.g., 25 mM potassium phosphate (pH 7.0)/1 mM EDTA or other buffer at pH 7; store these solutions up to 1 month or more at 4°C in the dark. Add 10 μl of the 10 mg/ml o-dianisidine solution and 2 to 10 μl of the 8 mM (putative) peroxide solution to 0.9 ml 25 mM phosphate buffer, pH 7 (APPENDIX 2A)/0.1% Triton X-100. Bring to 0.99 ml with deionized water. Use this solution to blank a spectrophotometer at 460 nm, then add 10 μl of the 1 mg/ml HRP solution to the cuvette. Monitor the A460 change which is complete within a few seconds. Use ε460 = 11,300 M−1 cm−1 for oxidized o-dianisidine and dilution factors to compute peroxide concentration. Use three to four different volumes of the hydrogen peroxide solution to be tested. Perform linear regression to determine the change in A460 per microliter (the slope of the line) to obtain an accurate concentration: slope/11,300 × cuvette path length in centimeters = millimolar concentration of hydrogen peroxide.
NBD chloride, 100 mM
To 1 ml DMSO, add 20 mg 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD chloride). Store in aliquots up to several weeks at −20°C, protected from light.
-
To determine the concentration of the solution: First, make a 20-fold dilution of the stock in 25 mM phosphate buffer, pH 7 (APPENDIX 2A), and then add 16 μl of this diluted NBD chloride solution to 1.0 ml methanol, using a glass pipet. Measure the A336. Use an ε336 of 9800 M−1 cm−1 and the dilution factor to calculate the concentration of the stock.
Use a glass pipet rather than micropipettor to measure out the methanol and avoid leakage.
NBD chloride is called 4-chloro-7-nitrobenz-2-oxa-1,3-diazole by Molecular Probes. This reagent is also available from other suppliers (e.g., Sigma-Aldrich). Another abbreviation for the compound is Nbf-Cl.
NBD fluoride, 100 mM
-
To 100 μl DMSO, add 1.8 mg of 4-fluoro-7-nitrobenz-2-oxa-1,3-diazole (NBD fluoride; Molecular Probes). Keep protected from light at room temperature, and use within several hours after it is prepared.
Do not store at −20°C as for NBD chloride.
-
To determine the concentration of the solution: First, make a 20-fold dilution of the stock into 25 mM phosphate buffer, pH 7 (APPENDIX 2A), and then add 16 μl of this diluted NBD fluoride solution to 1.0 ml methanol, using a glass pipet. Measure the A328. Use an ε328 of 8000 M−1 cm−1 and the dilution factor to calculate the concentration of the stock.
Use a glass pipet rather than a micropipettor to measure the methanol and avoid leakage.
NEM, 50 mM
To 1 ml DMSO, add 6.3 mg N-ethylmaleimide (NEM). Dispense into aliquots and store up to a few weeks at −20°C.
-
To standardize the exact concentration (if required): First, prepare a 1:20 dilution of 100 mM DTT (see recipe) and then add 6 μl of this dilution to 1 ml of 25 mM phosphate buffer, pH 7 (APPENDIX 2A). Add 14 μl of a 1:20 dilution of the NEM stock. Incubate 30 min at room temperature, and then add 15 μl of 0.25 mM DTNB, diluted from the 5 mM working solution (see recipe).
The loss of thiol groups corresponds to the amount of NEM added, allowing for calculation of the concentration of the stock solution. Therefore, the concentration of NEM in the final test solution is equal to the A412 of the control (without NEM) minus the A412 in the presence of NEM divided by the ε412 value of 14,150 M−1 sec−1 for TNB.
TNB solution, 16 mM
To a dark-colored microcentrifuge tube (or a tube wrapped with aluminum foil) containing 0.84 ml of 25 mM phosphate buffer, pH 7 (APPENDIX 2A), add 80 μl of 100 mM stock DTNB (see recipe) and 80 μl of 100 mM DTT (see recipe). The solution will turn dark orange. Store up to 1 week at −20°C.
-
To determine the concentration of TNB: Add 7.5 μl of the TNB solution to 0.5 ml phosphate buffer, measure the A412, and calculate the concentration using an ε412 of 14,150 M−1 cm−1 for TNB (Riddles et al., 1979). Add 1 μl of 100 mM or 2 μl of 5 mM DTNB (see recipe) and observe any increase in A412. If there is an increase, calculate the amount of DTNB that should be added to the stock TNB solution to bring it to the maximum A412 but not beyond. This amount will be equal to 1/2 the concentration of TNB generated (according to the increased A412 divided by the ε412 of 14,150 M−1 sec−1), taking into account the relevant dilutions factions and the volume of the TNB stock. Perform the same test using 100 mM DTT instead of DTNB, adding more DTT to the TNB stock solutions if necessary, as calculated for the needed addition above.
The reagent is properly prepared if addition of neither dithiothreitol nor DTNB leads to an increase in A412. This reagent tends to air oxidize and should be tested every few hours for DTNB formation (readjust with additional DTT as needed).
TPCK-treated trypsin solution
-
Prepare a stock solution of 1 mg TPCK-treated trypsin (Worthington Biochemical) in 100 μl of 1 mM HCl (8.5 μl concentrated HCl in 100 ml water), or as indicated by the manufacturer. Dispense into aliquots and store up to several months at −70°C, if necessary, but do not subject to multiple freeze/thaw cycles.
The best grades of trypsin available (e.g., sequencing-grade trypsin from Promega) include modified forms of trypsin that do not undergo self-digestion and are therefore active longer during the digestion (and do not generate contaminating peptides from the trypsin, itself). Immobilized trypsin (Pierce Biotechnology) can also be used so that the digestion can be stopped by centrifugation and removal of the supernatant to a separate tube.
Note that for digestion of 1 mg of the target protein using a protease/protein ratio of 1:50, 2 μl of the prepared trypsin solution is added.
Urea, 10 M
To ~4 ml of water add 6.01 g urea (ultrapure; Sigma-Aldrich). Warm the solution and add water to fully dissolve and bring to 10 ml final volume. Store 1 to 2 days at room temperature.
COMMENTARY
Background Information
The procedures described herein allow for the detection and measurement of sulfenic acids in proteins by chemical means and do not require the specialized approaches of crystallography or NMR. The intrinsic instability of sulfenic acids makes them inherently diffi-cult to work with; mass spectrometry can theoretically be used to demonstrate the single oxygen added to the cysteine and/or protein, but without use of a trapping agent, only the further oxidized sulfinic (R-SO2H) and/or sulfonic (R-SO3H) acid species are typically observed. With NBD modification, the sulfenate oxygen is incorporated into the product, and subsequent mass spectrometry allows for its demonstration. With the other covalent modi-fication methods, the oxygen is lost upon reaction, but the presence of the sulfenic acid in the protein is indicated simply by the ability to react with it.
Modification with NBD chloride
As described above, NBD modification may be the simplest way to trap and demonstrate sulfenic acids in proteins. In fact, access to a mass spectrometer is not required, as the product of NBD reaction with sulfenic acids has a distinctive spectral signature with the peak shifted some 73 nm with respect to that generated on reaction with thiol groups. However, because this peak is shifted only ~4 nm from that of the free reagent, the protein must be washed free of excess reagent before the spectral signature of the covalently attached NBD can be discerned. This very slight spectral perturbation on modification of sulfenic acids by NBD limits the ability to follow the course of the reaction spectroscopically, although with sufficient protein and only a small excess of reagent, this may be possible. On the other hand, modification of thiols by NBD chloride is readily monitored spectroscopically and can be used to determine the rate of modification and the extent of the reaction without subsequent washing steps. Note that rates of modification for thiol versus sulfenic acid groups by NBD chloride appear to be similar, as reported earlier by Ellis and Poole (1997a). NBD chloride may not be the reagent of choice if there are additional accessible thiol groups in the protein that may obscure the spectral signature of the R-S(O)-NBD adduct. Reactivity of NBD chloride toward other amino acid side chains can occasionally cause problems in these analyses, as well; however, the adducts with amino or tyrosyl groups are typically formed only at higher pH and have distinct spectral properties (i.e., λmax of 382 and 480 nm for NBD adducts with tyrosines and amines, respectively; Ghosh and Whitehouse, 1968; Aboderin and Boedefeld, 1976; Miki, 1985). In some cases, NBD adducts with thiol groups have been shown to migrate to proximal amine groups of lysine (e.g., Senior et al., 1998), and this process may occur with the NBD adducts of sulfenic acids, as well.
Modification with NBD fluoride
The Alternate Protocol describes the use of NBD fluoride as a replacement for NBD chloride. Since the product formed is identical, all the advantages and disadvantages described above for NBD chloride modification are relevant to this reagent as well. NBD fluoride has the advantage of reacting more quickly with thiol and sulfenic acid groups but has the related disadvantage of being rather unstable in solution and considerably more expensive than NBD chloride. Additionally, uncharacterized spectral changes may also occur during the modification reaction, but the modified protein, once washed free of unbound reagent, still has the same appearance as that generated with NBD chloride.
Modification with TNB
Reactivity with TNB is very useful, as the spectral changes that ensue allow for immediate quantitation of sulfenic acid content. Subsequent to this reaction, covalent modification of the protein with TNB can be confirmed upon washing, spectral analysis, and release of the bound TNB by DTT treatment. The major disadvantage of using this reagent is its tendency to air oxidize, and any DTNB present in the solution can react with other thiol groups in the protein and cause an increase in the absorbance at 412 nm. TNB can also react with excess peroxides or other oxidants that may remain in the solution, and this reactivity may obscure the reaction of interest. This must therefore be accounted for by control reactions conducted in the absence of protein.
Modification with dimedone
Dimedone reacts only with sulfenic acids in proteins, a reaction considered diagnostic for these species. It is also not reversed by DTT treatment and is therefore particularly useful for peptide analysis following digestion of the modified protein by trypsin. Unfortunately, however, dimedone as a label has no distinguishing spectral features and thus requires the use of mass spectrometry to establish its incorporation. Because dimedone does not react with thiols, it offers the advantage that it can be added before or at the same time as the oxidant is added and may therefore allow for trapping of the sulfenic acid as it is formed and before it becomes further oxidized to the sulfinic and sulfonic acids. This could be particularly useful for systems where the sulfenic acid is formed rather slowly, and further oxidation of this species occurs readily.
Modification with DCP-linked fluorescent and affinity reagents
The DCP-linked reagents, based on the reactive 1,3-cyclohexanedione core of dime-done, have all the advantages (as with dime-done) of irreversible alkylation diagnostic for sulfenic acid modification on proteins. In addition, they include linked fluorescent or biotin groups that allow for sensitive gel-based detection and/or affinity capture of the proteins or peptides after labeling. This enables use of proteomic approaches to analyzing complex mixtures of oxidized proteins and peptides.
Reversibility of modification
As noted above, NBD and TNB modifications of sulfenic acids are both reversible by addition of DTT, returning the target cysteine to the reduced (thiol) state. This can allow for restoration of function, which may be shown in subsequent experiments. At the same time, the reversibility of these modifications may complicate the use of tryptic digests to verify the site of modification, although TNB- and NBD-modified peptides have in some cases been isolated successfully (Chae et al., 1994; Jeong et al., 2000).
Determination of pKa
If spectral properties of the sulfenic acid–containing protein allow for the determination of the pKa as described in Basic Protocol 5, this relatively simple method of assessing this parameter can be a major advance, as only a few pKa values for sulfenic acids, particularly within proteins, have been reported (Claiborne et al., 1999; Poole and Ellis, 2002). Unfortunately, such a spectral feature may not be observed, especially if other chromophores are present in the protein. The procedure requires large quantities of protein in which the sulfenic acid has been stably generated, but it is nondestructive as long as the protein is stable in the buffers at various pHs.
Critical Parameters
Before any of the spectral analyses can be performed, it must be clear that there is enough protein available to give absorbance changes that are sufficient for quantitative analysis (i.e., >0.1). In the case of the dimedone or 1,3-cyclohexadione-based reagent treatments, which do not necessarily rely on spectral analyses, the requirement for protein may be less, as the amount used must be sufficient for identification of the modification by ESI-MS, or gel-based blotting or fluorescence detection methods.
The chemical modification reagents, if used in excess, do not necessarily have to be standardized by spectral titrations with model reagents unless there are problems with the modification or exact amounts are required. However, the oxidant levels used to generate the sulfenic acid in the first place may be quite critical. For addition of a stoichiometric amount of hydrogen peroxide, for example, exact quantitation of the hydrogen peroxide concentration by the HRP assay is an important first step. As has been mentioned above, the TNB reagent is particularly prone to air-oxidation and must be checked for the presence of excess DTNB or DTT every 2 hr or so.
Loss of protein during ultrafiltration can be a major source of error, so it is advantageous to test in advance for the recovery of a given protein with the specific ultrafitration device to be used.
It is helpful, particularly with the NBD modification methods, to know in advance how many accessible thiols are in the protein. This can be done by adding a 10-fold excess of DTNB and observing the rise in A412 over time, until completion of the reaction. The amount of TNB released is directly proportional to the number of reactive thiols present in the protein. However, reactivity and accessibility of given protein thiols to particular reagents can be different.
For pKa determination, the pH values of all phosphate/citrate buffer solutions used should be measured on the day the analysis is performed.
Troubleshooting
Choice of modifying reagent
Where multiple thiol groups are present, only one of which is oxidant sensitive, the demonstration of sulfenic acid formation by NBD chloride modification may require the use of difference spectroscopy to clearly identify the 347-nm peak of the R-S(O)-NBD adduct. In these cases, quantitation will likely suffer as well. Other possibilities are to compare the modified spectra with those obtained when the cysteine of interest has been mutated to a serine.
Alternatively, the cysteinyl thiols that obfuscate the analysis can themselves be removed by mutagenesis to serines (assuming that such changes do not affect the normal function of the protein). If there is a way to reversibly block only the cysteine of interest (e.g., by sulfenic-acid formation, then reaction with 1 mole reduced glutathione or TNB for each mole cysteine), this can be done initially, followed by alkylation of the remaining cysteine thiols by a reagent such as N-ethylmaleimide. If the rereduced enzyme is fully functional, then effects and detection of sulfenic acid formation at the target cysteine can be analyzed for the preblocked protein using the NBD chloride method without interference by the other cysteine thiols.
Another approach could be to use the sulfenic acid–containing protein preblocked with dimedone, then reacted with NBD chloride to generate the control spectrum with only the interfering cysteine thiols modified for comparison with the spectrum of the modified protein containing the sulfenic acid as well as the additional thiols. If the problem is too great, then dimedone or the 1,3-cyclohexandione-based reagents rather than NBD chloride should be used.
Use of denaturants
With all of the chemical modification methods for detecting sulfenic acids, the presumption is that the target sulfenic acids are accessible to reaction by these reagents. Where modification reactions are unsuccessful, lack of accessibility may be the problem (even if the corresponding thiol group is accessible toward DTNB and NBD chloride modification, the sulfenic acid moiety may not be accessible due to conformational changes or other differences). In these cases, addition of denaturants may be necessary for trapping of the sulfenic acid to take place (e.g., see modification of NADH peroxidase as reported in Ellis and Poole, 1997a).
Denaturants can include guanidine hydrochloride (GuHCl), urea, SDS, or acetonitrile, although urea may itself react with NBD chloride. Low amounts (0.5 to 2 M) of urea or GuHCl can be tried first, or, if necessary, higher amounts (4 to 6 M) can be used. In these cases, it is best if the modifying reagent is already present when the denaturant is added, since denaturation of the protein can also promote autoxidation of the sulfenic acid (the denaturant solution can also be bubbled with nitrogen or argon for 20 min before addition to remove oxygen). During ultrafiltration to remove excess reagent, the continued presence of some amount of the denaturant (e.g., 2 M GuHCl), as is compatible with the ultrafiltration device being used, may be necessary to avoid protein precipitation.
One adverse effect of denaturation could be to expose additional cysteinyl thiol groups if the protein has them; in this case, use of dimedone or the 1,3-cyclohexandione-based reagents may be preferred. Proteins can be analyzed by spectroscopy in the presence of the denaturant but must be freed of denaturant for ESI-MS analysis and may be best analyzed using reversed-phase HPLC linked to ESI-MS analysis.
Interfering lysines
Where modification of the sulfenic acid by NBD chloride is successful, but the NBD group migrates to a proximal lysine side chain over time (as noted by the decrease in A347 and increase in A480), one approach may be to preblock accessible amino groups of lysine using amine-specific reagents such as succinimidyl esters (see the handbook from Molecular Probes). Then, once the NBD modification of the sulfenic acid is complete, there should be no further migration of the NBD group and the full absorbance increase at 347 nm can be observed. Again, if the identity of the lysine group is known or suspected, mutagenesis can be used to change this lysine group to a different amino acid, provided this mutation does not also result in a change in the functional properties of the protein.
Poor ESI-MS resolution
The possibility also exists that, if there is insufficient resolution due to the large size of the target protein and/or the quality of data obtained from the mass spectrometer, a mass difference will be obtained for the NBD-modified sulfenic acid product that is less that the full 16 amu expected (relative to the NBD-thiol adduct). This may indicate only partial conversion of the protein to the sulfenic acid prior to modification and an inability to resolve the R-S-NBD and R-S(O)-NBD products by mass spectrometry. One solution is to allow the oxidation of the thiol group to proceed for a longer time or with more oxidant prior to NBD chloride modification. Alternatively, dimedone or the 1,3-cyclohexandione-based reagents could be the preferred modification agents, as they give much greater mass shifts on modification.
Anticipated Results
Modification with NBD
For modification reactions with NBD chloride (or NBD fluoride), the thiol form of the protein will generate a peak for the NBD adduct with a maximum at 420 nm, while the sulfenic acid form, once washed free of excess reagent, will exhibit a maximal absorbance at 347 nm, with the 347-nm peak rising concomitant with a decrease at 420 nm (Fig. 17.2.2). If multiple cysteine residues are present, demonstration of the 347-nm peak may be improved by using difference spectroscopy (Fig. 17.2.1), although quantitation will likely suffer under these conditions. ESI-MS analyses will indicate that the R-S(O)-NBD adduct is 16 amu larger than the R-S-NBD adduct (Fig. 17.2.3), and the R-S(O)-NBD adduct will be nonfluorescent, unlike the R-S-NBD adduct (λex,max = 422 nm, λem,max = 527 nm). NBD itself accounts for an additional mass of 164 amu.
Figure 17.2.2.
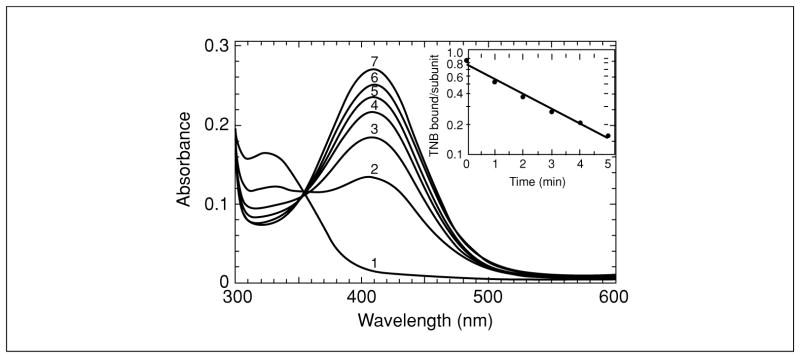
Reduction of TNB-labeled C165S AhpC by DTT. The TNB-labeled enzyme generated by TNB treatment of the protein oxidized by a stoichiometric amount of hydrogen peroxide was isolated by ultrafiltration with Centricon CM-30 concentrators, then treated with a 10-fold excess of DTT. Shown are spectra before (spectrum 1), and after addition of DTT for 1, 2, 3, 4, 5 and 30 min (spectra 2 to 7, respectively). The inset represents a semilogarithmic plot of the change in absorbance, converted into units of TNB/subunit, versus time. Reprinted with permission from Ellis, H.R., and Poole, L.B., Biochemistry 36:13349–13356. Copyright 1997, American Chemical Society.
Figure 17.2.1.
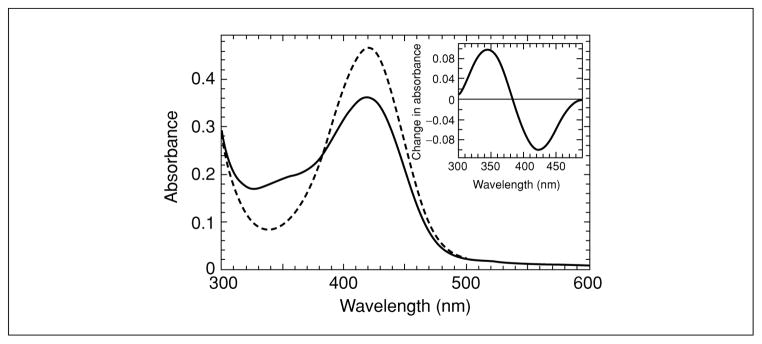
Hypothetical UV-visible spectra for NBD adducts of reduced and oxidized forms of a protein with three additional reactive cysteine thiol groups. Spectra correspond to the R-S-NBD product (dotted line) and the products, including R-S(O)-NBD, after NBD chloride modification of the protein in which one of the four cysteine residues has been oxidized to a sulfenic acid (solid line). The inset represents the difference spectrum calculated by subtracting the spectrum of the fully reduced and modified form of the protein from that of the oxidized, modified form.
Modification with TNB
Modification of sulfenic acid–containing proteins with TNB will result in the stoichiometric incorporation of the TNB into the protein, as judged by the decrease in the A412 relative to that expected from the dilution of the TNB reagent into buffer lacking protein. Conversion of this A412 decrease to the concentration of TNB consumed using the extinction coefficient for TNB and dividing that result by the protein concentration will yield the sulfenic acid content of the protein. On isolation of the TNB-modified protein by ultra-filtration, the spectrum of the modified protein will have a peak at ~325 nm corresponding to the TNB-mixed disulfide bond formed at the cysteine of interest (Ellis and Poole, 1997b). Addition of DTT to this solution will then result in the isosbestic decrease in this peak and increase in the TNB peak, with its absorbance maximum at 412 nm (Fig. 17.2.2). If no protein is lost during ultrafiltration, this is a second opportunity to quantitate the TNB modification of the protein. The quantitative decrease in A412 upon TNB modification can be used as an assay method for sulfenic acid generation during oxidant titration of the protein (e.g., as in Figure 4 of Poole and Ellis, 2002) or for sulfenic acid generation over time in the presence of excess oxidant, although reactivity of the TNB with the oxidant in the absence of protein must be considered in the latter case.
Modification with dimedone and DCP-linked labeling agents
Reaction of the sulfenic acid in the protein with dimedone will result in a 138-amu increase in mass. Dimedone modification should also eliminate the functionality of the cys-teine residue and will block any additional attempts to modify this group with NBD chloride (as a cross-confirmation of the target of these reagents as the sulfenic acid).
In a parallel manner, modification at sulfenic acid groups in proteins by all of the reagents based on the 1,3-cyclohexanedione core of dimedone leads to increases in molecular weight in the protein and labeled peptide of 396 to 792 amu depending on the specific reagent used (Fig. 17.2.3; Poole et al., 2007). In addition, fluorescently labeled reagents (DCP-FL1, DCP-FL2, DCP-Rho1 and DCP-Rho2) detected spectroscopically can be evaluated for extent of labeling based on approximate extinction coefficients in pH 7 phosphate buffer (ε493 ~67,000 M−1 cm−1 for protein labeled with DCP-FL1 or DCP-FL2; ε570 ~70,000 or 61,000 M−1 cm−1 for protein labeled with DCP-Rho1 or DCP-Rho2, respectively).
Fluorescence imaging of one-dimensional and two-dimensional gels after soaking will detect proteins labeled with the fluorescein-or rhodamine-linked reagents in amounts as low as 0.1 to 0.5 pmol. DCP-based biotin reagents are detectable through electrotrans-fer and blotting approaches (with anti-biotin antibodies or HRP-streptavidin conjugates) as are the fluorescein-linked reagents if commercially available anti-fluorescein antibodies are used. Proteins modified with the biotin-linked reagents are also readily captured from solution using avidin-conjugated beads prior to analyses by gel- or MS-based methods.
Determination of pKa
If there are observable differences between the absorbance of the deprotonated sulfenate base at higher pH values and the absorbance of the protonated sulfenic acid at lower pH values (Fig. 17.2.4), then a fit of the absorbance versus pH data to the equation given in Basic Protocol 5 will allow for the determination of the pKa of the sulfenic acid (Fig. 17.2.5). In several small molecules, pKa values for the stabilized sulfenic acids have ranged from 4.8 to 6.3 (Claiborne et al., 2001), while a pKa of ~6.1 has been determined for one protein sulfenic acid so far (Poole and Ellis, 2002).
Figure 17.2.4.
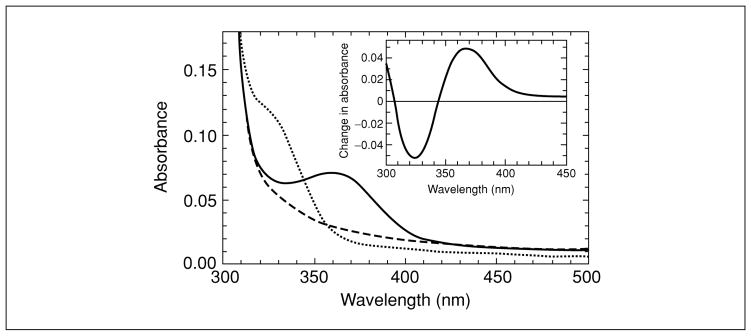
Spectral properties of thiol(ate) (Cys46-SH or Cys46-S-), sulfenate (Cys46-SO-), and sulfenic acid (Cys46-SOH) forms of C165S AhpC from Salmonella typhimurium. Spectra of the mutant enzyme (200 μM) at pH 7.0 in the absence (dotted line) or presence (solid line) of 1.1 equivalent of H2O2 are shown. The dashed spectrum represents the Cys46-SOH form of the enzyme at pH 4.6. The inset depicts the difference spectrum between the Cys46-S(H) and Cys46-SO− forms of C165S AhpC. Reprinted with permission from Poole, L.B., and Ellis, H.R. 2002. Methods Enzymol. 348:122–136. Copyright 2002, Academic Press.
Figure 17.2.5.

Hypothetical determination of pKa value for a protein sulfenic acid given absorbance values determined in various pH buffers. Given an experiment similar to that described in Basic Protocol 5, representative data are shown for absorbance values at 367 nm versus pH of a sulfenic acid–containing protein (~50 μM) with a pKa of ~6.1. The fitted curve is obtained using the equation given in the protocol.
Time Considerations
For most of the chemical modification procedures, once the reagents are prepared and the protein concentration is determined, the generation of the sulfenic acid and reaction with the modifying agent is complete within < 1 to 2 hr. Ultrafiltration may take 20 min to 3 hr, depending on the particular device and the molecular weight cutoff (lower cutoff, slower filtration). The pKa determination itself takes about 1 to 3 hr, with preparation and/or pH measurement of the citrate/phosphate buffers requiring 1 to 2 additional hr. Tryptic digests are carried out for 12 to 24 hr, and each HPLC run takes ~90 to 120 min.
Literature Cited
- Aboderin AA, Boedefeld E. Reaction of chicken egg white lysozyme with 7-chloro-4-nitrobenz-2-oxa-1,3-diazole. II. Sites of modification. Biochim Biophys Acta. 1976;420:177–186. doi: 10.1016/0005-2795(76)90356-1. [DOI] [PubMed] [Google Scholar]
- Birkett DJ, Price NC, Radda GK, Salmon AG. The reactivity of SH groups with a fluorogenic reagent. FEBS Lett. 1970;6:346–348. doi: 10.1016/0014-5793(70)80095-3. [DOI] [PubMed] [Google Scholar]
- Bollag DM, Edelstein SJ. Protein Methods. Wiley-Liss, Inc; New York: 1991. [Google Scholar]
- Chae HZ, Uhm TB, Rhee SG. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc Natl Acad Sci USA. 1994;91:7022–7026. doi: 10.1073/pnas.91.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, III, Charrier V, Parsonage D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Prot Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine-sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997a;36:15013–15018. doi: 10.1021/bi972191x. Describes, for the first time, the use of the NBD chloride method for identification of the cysteine sulfenic acid of the C165S mutant of AhpC. [DOI] [PubMed] [Google Scholar]
- Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997b;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine–sulfenic acid derivative. Proc Natl Acad Sci USA. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh PB, Whitehouse MW. 7-chloro-4-nitrobenz-2-oxa-1,3-diazole: A new fluorigenic reagent for amino acids and other amines. Biochem J. 1968;108:155–156. doi: 10.1042/bj1080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W, Cha MK, Kim IH. Thioredoxin-dependent hydroperoxide peroxidase activity of bacterioferritin comigratory protein (BCP) as a new member of the thiol-specific antioxidant protein (TSA)/alkyl hydroperoxide peroxidase C (AhpC) family. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- Miki M. Chemical modification of tropomysosin with NBD-chloride. J Biochem (Tokyo) 1985;97:1067–1072. doi: 10.1093/oxfordjournals.jbchem.a135149. [DOI] [PubMed] [Google Scholar]
- Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Meth Enzymol. 2002;348:122–136. doi: 10.1016/s0076-6879(02)48632-6. Gives practical details about the use of several protocols, including NBD chloride, TNB, and dimedone-based methods for identification of sulfenic acids. [DOI] [PubMed] [Google Scholar]
- Poole LB, Zeng BB, Knaggs SA, Yakubu M, King SB. Synthesis of chemical probes to map sulfenic acid modifications on proteins. Bioconjug Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. Describes the synthesis and use of seven fluorescent or affinity-tagged reagents based on the 1,3-cyclohexanedione core of dimedone that can be used for detection and identification of sites of sulfenic acid modification on proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddles PW, Blakeley RL, Zerner B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—A reexamination. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- Senior AE, Gros P, Urbatsch IL. Residues in P-glycoprotein catalytic sites that react with the inhibitor 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole. Arch Biochem Biophys. 1998;357:121–125. doi: 10.1006/abbi.1998.0778. [DOI] [PubMed] [Google Scholar]
- Tripolt R, Belaj F, Nachbaur E. Unexpectedly stable sulfenic acid: 4,6-Dimethoxy-1,3,5-triazine-2-sulfenic acid; synthesis, properties, molecular and crystal structure. Z Naturforsch. 1993;48B:1212–1222. [Google Scholar]
- Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CH, Jr, Arscott LD, Matthews RG, Thorpe C, Wilkinson KD. Methodology employed for anaerobic spectrophotometric titrations and for computer-assisted data analysis. Meth Enzymol. 1979;62:185–198. doi: 10.1016/0076-6879(79)62217-6. [DOI] [PubMed] [Google Scholar]
- Willett WS, Copley SD. Identification and localization of a stable sulfenic acid in peroxide-treated tetrachlorohydroquinone dehalogenase using electrospray mass spectrometry. Chem Biol. 1996;3:851–857. doi: 10.1016/s1074-5521(96)90071-x. Describes the use of dimedone modification and LC/MS/MS to identify and localize sulfenic acids within proteins. [DOI] [PubMed] [Google Scholar]