

Abstract
Heparan sulphate proteoglycans are ubiquitous macromolecules of cell surfaces and extracellular matrices. Numerous extracellular matrix proteins, growth factors, morphogens, cytokines, chemokines and coagulation factors are bound and regulated by heparan sulphate. Degradation of heparan sulphate thus potentially profoundly affects cell and tissue function. Although there is evidence that several heparan sulphate-degrading endoglucuronidases (heparanases) might exist, so far only one transcript encoding a functional heparanase has been identified: heparanase-1. In the first part of this review, we discuss the current knowledge about heparan sulphate proteoglycans and the functional importance of their versatile interactions. In the second part, we summarize recent findings that have contributed to the characterization of heparanase-1, focusing on the molecular properties, working mechanism, substrate specificity, expression pattern, cellular activation and localization of this enzyme. Additionally, we review data implicating heparanase-1 in several normal and pathological processes, focusing on tumour metastasis and angiogenesis, and on evidence for a potentially direct signalling function of the molecule. In that context, we also briefly discuss heparanase-2, an intriguing close homologue of heparanase-1, for which, so far, no heparan sulphate-degrading activity could be demonstrated.
Keywords: heparan sulphate proteoglycans, heparanase, signalling, metastasis, angiogenesis
Heparan sulphate proteoglycans
Proteoglycans (PGs) form a large group of structurally complex and functionally versatile molecules that are present at the cell surface, in the extracellular matrix (ECM), in intracellular granules and even in the nucleus. They are composed of one or several polysaccharide chains of the glycosaminoglycan type (i.e. heparan sulphate (HS), chondroitin sulphate (CS), dermatan sulphate and keratan sulphate) that are covalently attached to a core protein.
Heparan sulphate
HS appeared early in metazoan evolution. Most multi-cellular organisms, from simple invertebrates like Hydra up to human beings, have the capacity to produce HS. The biosynthesis of these polysaccharide chains occurs mainly in the Golgi apparatus and involves a complex set of enzymes (Fig. 1) (reviewed by [1–3]). The first step in HS chain biosynthesis is the enzymatic transfer of a xylose (Xyl) residue, from the sugar nucleotide UDP-Xyl, to the hydroxyl group of specific serine residues of the core protein. Potential acceptor serines are localized next to a glycine residue, flanked by one or more acidic residues. Sequential attachment of two galactose (Gal) residues and one glucuronic acid (GlcA) residue to the Xyl completes the synthesis of the tetrasaccharide linkage region (GlcA-Gal-Gal-Xyl-Ser). This linkage region is identical for both HS and CS, the addition of the fifth saccharide determining the nature of the polysaccharide chain that is further made: an N-acetyl glucosamine (GlcNAc) in the case of HS and an N-acetyl galactosamine (GalNAc) in the case of CS. Amino acid determinants or structural domains lying close to the glycosaminoglycan attachment sites in the protein appear to regulate this step of the process [4, 5]. After attachment of the first GlcNAc residue, further polymerization takes place by alternate addition of GlcA and GlcNAc to the non-reducing end of the growing chain. This process is catalysed by proteins now recognized as members of the exostosin family of tumour suppressors [6]. The number of repeating GlcA-GlcNAc disaccharide units varies from 50 to 150 units per HS chain.
1.
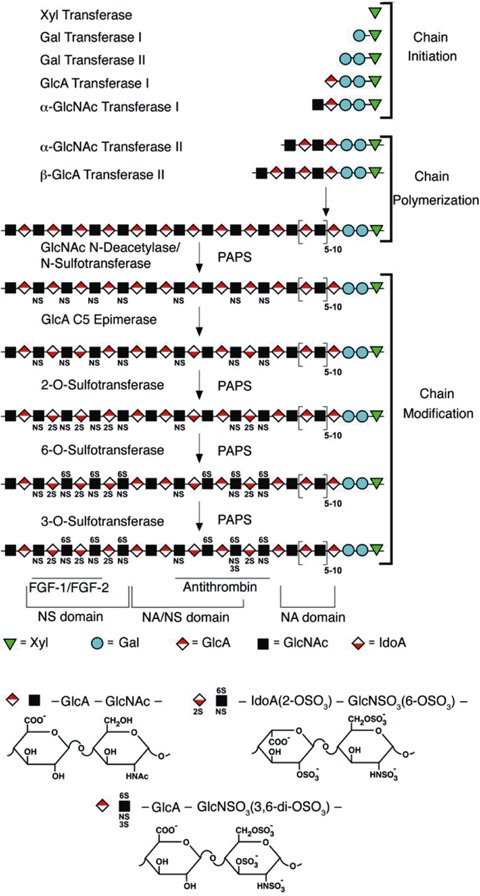
Heparan sulphate (HS) chains are covalently bound to specific serine residues of the proteoglycan core protein. After assembly of the tetrasaccharide linkage region (Xyl- Gal-Gal-GlcA) and the first GlcNAc residue, further HS polymerization takes place by alternate addition of GlcA and GlcNAc to the non-reducing end of the growing chain. While chain elongation is ongoing, the nascent polysaccharide forms a substrate for the N-deacetylase/N-sulphotransferases, which control all subsequent modifications (C5-epimerization of GlcA to IdoA by an epimerase and O-sulphations by 6-O- and 3-O-sulphotransferases). Each sugar residue is depicted by a symbol shown below the scheme. NA, NA/NS and NS domains are defined according to the degree of GlcNAc sulphation. Also shown are regions that are implicated in the binding of specific ligands, such as FGF-1/FGF-2 and antithrombin (adapted from [2]).
While chain elongation is ongoing, approximately 40–50% of the GlcNAc residues of the nascent polymer are modified into N-sulphoglucosamine (GlcNS) residues [7]. This N-deacetylation/N-sulphation step is the first of a series of modifications, and controls all subsequent modifications. Further C5-epimerization of GlcA to IdoA and O-sulphations, indeed, only occur within regions adjacent to the GlcNS residues. All modifications are catalysed by enzymes. A number of these enzymes occur as multiple isoforms, which show distinct tissue-specific distributions and substrate recognition properties. To date, four different N-deacetylase/N-sulphotransferases and various different 6-O- and 3-O-sulphotransferases have been identified in vertebrates [7]. Each modification is incomplete, as the enzymes do not modify all the available sugars and each reaction depends on previous reactions. All this results in extensive structural heterogeneity, the macroscopic organization of HS showing a typical ‘block structure’ ([8] and reviewed by [9]) whereby regions of 2–9 highly modified disaccharides (N-sulphated or NS-domain) alternate with longer regions of relatively unmodified disaccharides (N-acetylated or NA-domain). Short mixed sequences, of alternating N-acetylated/N-sulphated (NA/NS) disaccharides, may occur between the NSdomains and NA-domains. Heparin, which is pharmaceutically manufactured from mast cell rich tissues and is often used as a substitute for HS in experiments, may be considered as a single, unusually extended (∼15 kD), highly N- and O-sulphated IdoA-rich NS-domain.
Thus, the fine structure of the HS chain ultimately depends on the regulated expression and action of multiple glycosyltransferases, sulphotransferases and an epimerase, which are arrayed in the lumen of the Golgi apparatus. The processes of chain elongation and modification appear to be at least partially coupled [10]. In addition to polymerases and modifying enzymes, a series of cytoplasmic enzymes are needed to form nucleotide sugar (e.g. UDP-GlcA) and nucleotide sulphate (PAPS, which is the sulphate donor substrate), and multiple transporters are needed to import the nucleotides from the cytosol to the lumen of the Golgi apparatus. The assembly process also depends on the availability of GlcNAc or other sugars. In short, the fine structure of the HS chains on a given PG appears to be tissue [8,11,12] and cell type specific [13], and changes in HS composition occur in association with development, aging and pathology [14–16].
Heparan sulphate interactions
Given the high content of charged groups in HS, it is not surprising that many molecules bind to HS through electrostatic interactions. To date, more than 100 different proteins have been reported to interact with HS/heparin, including ECM proteins, growth factors, morphogens, cytokines, chemokines, coagulation factors, proteases and their inhibitors. Detailed information on the structural requirements for binding is available in only a few cases. Two types of binding sites are known: (1) sites that depend on relatively rare modifications, exemplified by the minimal antithrombin-binding sequence (a pentasaccharide that contains a central 3-O-sulphated GlcN residue, adjacent to a GlcA residue and located to the nonreducing side of a fully modified disaccharide, as shown in Figure 1 and thoroughly reviewed by [3]) and (2) sites that consist of relatively common disaccharides. The arrangement of these common substituents in a sequence-specific manner [10], their precise spacing and the specific spacing between different (sulphated) domains (e.g. SAS-arrangement, where short NS-domains are separated by N-acetylated disaccharide units) may provide selective protein binding (reviewed by [17]). An important example of this second type of binding sites is provided by the HS structures that bind and approximate basic fibroblast growth factor (FGF2) and FGF receptor-1 (FGFR1). FGF2 is a key regulator of angiogenesis and is involved in a number of pathophysiological processes such as wound repair and tumour growth [18]. Initially, the association of FGF2 with HS had been proposed to protect this growth factor from proteolysis and thermal denaturation and to serve as a reservoir of growth factor that can be released by enzymes that degrade HSPGs [19]. However, since 1991, it has become obvious that HSPGs are essential co-receptors for FGF2, which strongly promote FGF2-FGFR1 binding and subsequent activation of the receptor [20, 21]. The minimal structure required for activating FGF2-signalling consists of a deca- to dodecasaccharide, containing N-, 2-O- and 6-O-sulphations. While an IdoA(2S) residue in an N-sulphated pentasaccharide-domain is sufficient for FGF2 binding (Fig. 1) [22, 23], 6-O-sulphation is needed for FGF2-dependent dimerization and activation of FGFR1 [24–27]. Recent surface plasmon resonance studies suggest that FGF2 initially binds to HS/heparin (Kd = 39 nM), with FGFR1 showing nearly 22-fold higher affinity for FGF2-heparin complexes than for FGF2 (Kd = 2.7 nM as compared to Kd = 62 nM) [28]. In addition to promoting FGF2FGFR1 interaction, HS/heparin plays an important second role in signalling, by promoting the dimerization of two FGF2-FGFR1 complexes. Two crystallographic models have been proposed for FGF-FGFR-heparin signalling complexes. Pellegrini et al.[29], studying the FGF1–FGFR2 interaction, found that two FGF1 ligands are dimerized by a central single heparin molecule, bringing two FGFR2 molecules together (2:2:1 heteropentameric assembly). Significant features of this model are that very few protein–protein contacts occur between the two FGF-FGFR dimers, and that one FGFR molecule is not interacting directly with the (asymmetric) heparin molecule. Schlessinger et al.[27], in contrast, but investigating the FGF2–FGFR1 interaction, propose signalling results from two 1:1:1 FGF2–FGFR1-heparin ternary complexes associating to form a symmetrical dimer. Within each 1:1:1 ternary complex, heparin interacts extensively with FGF2 and FGFR1. Moreover, the heparin structure from one 1:1:1 ternary complex also interacts with FGFR1 of the adjoining 1:1:1 ternary complex, promoting the dimerization of two ternary complexes. Yet, sterical restrictions to that approximation require that ternary complexes are assembled at or near the non-reducing end of the heparin structures (two-end model). Unlike heparin, HS-chains are composed of both NA- and (heparinlike) NS-domains (see above). Whereas heparin stimulates FGF2–FGFR1 signalling in HS-negative/ receptor-positive cells, our laboratory has shown that most of the cell surface HS, as it can be recovered from cells by proteolytic treatment, has little activity and is even inhibitory (for heparin) in this sense [30]. Bacterial heparitinase (which selectively cleaves the glycosidic bonds to GlcA, locating mainly in NA-domains), in contrast to heparinase (which specifically cleaves N-sulphated GlcN in linkage to O-sulphated IdoA, occurring in NS-domains), converts these inactive/inhibitory HS-species into potent activators of FGF2-dependent FGFR1 autophosphorylation, interpreted as the generation of heparin-like ‘end-structures’ that can cooperate in receptor dimerization. As will be explained further, mammalian heparanase-1 is surmized to be also able to liberate these ‘dormant’ HSfragments from their inhibitory contexts, with ensuing biological consequences.
Core proteins
The core proteins of HSPGs dictate when, where and to what extent the HS-chains are expressed. In the ECM, HSPGs are mostly found in the basement membranes (BM), which separate endothelial cells (ECs) and epithelial cells from their subjacent connective tissues. Three main BM HSPGs have been well characterized: perlecan, collagen XVIII (both reviewed by [31]) and agrin (reviewed by [32]). Most HS-chains at the cell surface are attached to transmembrane syndecan (reviewed by [33, 34]) and GPIanchored glypican (reviewed by [35]) core proteins, the number of HSPG-molecules at the cell surface varying between 105 and 106 per cell [36]. All these core proteins show differential and highly regulated expressions during development. Some domains of these proteins are remarkably conserved throughout evolution, implying that not only the HS-chains but also the core proteins of the HSPGs engage in highly specific functional interactions. Indeed, the syndecan transmembrane domain is believed to be important for localizing these HSPGs to distinct membrane compartments [37], for syndecan oligomerization [38] and, together with the cytoplasmic domain, for internalization of bound ligands [39]. Although quite small (ca. 30 amino acids), the syndecan cytoplasmic domain interacts with several cytoskeletal and cytoplasmic proteins. One of these proteins is syntenin, a prominent component of focal adhesions and cell–cell junctions [40]. Recently, it has been shown that syntenin can also bind to PtdIns(4,5)P2 [41], and that this interaction controls stimulusinduced and Arf6-mediated syndecan recycling through endosomal compartments [42]. Importantly, cargo for the HS-chains of syndecans (i.e. FGF2 and FGFR1) accompanies syndecan along the syntenin recycling pathway, where they might encounter heparanase-1, as will be explained further
Similarly, an increasing number of reports shows that also the core proteins of the glypicans support essential functions, other than just the anchorage of HS to the cell surface. While first reports suggested that glypican-3 binds to insulin-like growth factor-II via its core protein and modulates its action [43, 44], others were unable to confirm this claim [45]. Yet from genetic studies, mostly obtained in Drosophila, it is becoming increasingly clear that the different glypicans (i.e. Dally and Dally-like protein) have distinct, non redundant functions in morphogen gradient formation (e.g. Wingless/Wnt) and signal transduction during development (reviewed by [46, 47]). Their differential roles can be partially explained by distinct expression patterns [48], but also implicate the different core proteins of these PGs. Our laboratory found that the glypican-3 core protein is processed by a pro-protein convertase, and that this processing, along with the HS, is essential for glypican-3 modulating Wnt signalling and cell survival in vitro and for regulation of embryonic cell movements in zebrafish [49]. One possible scheme could be that the processing uncovers a cryptic site in the core protein that is necessary, together with the HS chains, for optimal binding of the Wnt ligand. In that sense and context, heparanases are also potential modulators of Wnt-signalling.
Heparanase
Mammalian heparanases are endoglycosidases that cleave heparin/HS chains at specific intrachain sites. The first report on heparanases goes back to 1975, when Ögren and Lindahl described an endoglucuronidase from mouse mast cells, capable of cleaving macromolecular heparin at a limited number of sites [50]. A few months later, Höök et al. reported an endoglycosidase from rat liver tissue that degrades HS-polymers into oligosaccharides [51]. Since then, heparanase activity has been identified in a wide variety of normal and malignant cells and tissues. The purification and further characterization of heparanase(s) was initially hampered by limited supply and unstable enzyme activity, as well as by the lack of a simple assay for monitoring the degradation of heparin/HS chains. This has lead to conflicting reports on the physicochemical properties and substrate specificity of the enzyme(s) [52]. More recently, in 1999, four different groups independently reported the cloning of a single human gene encoding a functional heparanase [53–56]. Since then, identical or highly homologous cDNA sequences were derived from different types of normal and malignant cells and from different species respectively. So far, no other cDNA sequence encoding an active heparanase enzyme has been identified, indicating that mammalian cells express primarily one single dominant heparanase enzyme: heparanase-1.
Molecular properties of human heparanase-1
The heparanase-1 gene (∼50kb) is located on human chromosome 4q22 [57]. The single-copy gene is expressed as 5 and 1.7 kb mRNA species, generated by alternative splicing [54, 57]. Both forms contain the same open reading frame of 1629bp that encodes a polypeptide of 543 amino acids, with a calculated Mr of ∼61.2 kD. Based on the predicted amino acid sequence, heparanase-1 contains a putative N-terminal signal sequence (Met1-Ala35), a C-terminal hydrophobic region (Pro515-Ile534), 5 cysteine residues and 6 N-glycosylation sites [58] (Fig. 2). The mature, active enzyme purified from various cells and tissues was found to have a Mr of ∼50 kD, and have its N-terminus situated 157 amino acids downstream from the initiation codon [53, 54, 56]. Moreover, western blot analysis of lysates of CHO cells transfected with human full-length heparanase-1 revealed a major band of ∼50 kD and a minor band of ∼65 kD [53], suggesting some form of post-translational processing. Heparanase activity was readily obtained in mammalian cells after transfection with the full-length heparanase-1 cDNA [53–56], but not after transfection with cDNA encoding the N-truncated ∼50 kD protein, suggesting that the N-terminal region is essential for the expression of heparanase-1 activity [54]. Active heparanase-1 was subsequently shown to be a heterodimer, consisting of a ∼50 kD subunit (Lys158-Ile543) associated non-covalently with an ∼8 kD peptide (Gln36-Glu109), generated by the excision of a ∼6 kD peptide (Ser110-Gln157) from the precursor [59] (Fig. 2). Co-expressing the ∼8 and ∼50 kD heparanase-1 subunits in insect cells [60] or mammalian cells [61] results in an active enzyme, indicating that the heterodimer is necessary and sufficient for heparanase-1 enzymatic activity. Coexpressions in HEK293-T cells, investigating the interaction of the ∼50 kD subunit with the ∼8 kD subunit, revealed that a middle fragment (Glu228-Lys417) can be co-precipitated with the ∼8 kD peptide [61]. Consistently, a deletion construct of the ∼50 kD subunit lacking Lys411-Arg432 fails to interact with the ∼8 kD subunit [62]. It is worth noting that combining (culture media of insect cells containing) individually expressed subunits fails to produce active protein [60], suggesting that the subunits need to intimately fold together within a cellular environment (do not fold independently during synthesis) to produce active protein. Recently it has been shown that a bulky hydrophobic amino acid at position 156 (Tyr156) is required for proper processing and activation of pro-heparanase-1 by a cathepsin L-like protease. In complementary experiments, incubation of purified recombinant ∼65 kD pro-heparanase-1 with a purified preparation of cathepsin L resulted in processing of the precursor into ∼8 and ∼50 kD subunits and in heparanase-activity [63]. Similarly, by engineering tobacco etch virus protease cleavage sites at the Nand C-terminal junctions of the ∼6 kD fragment, the proteolytic activation of heparanase-1 could be reproduced in vitro using purified components [64]. Therefore, it can be concluded that the activation switch is solely mediated by the presence/removal of the ∼6 kD fragment (see next section) and does not require additional components.
2.
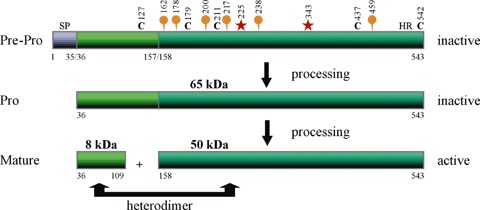
Predicted structure and processing of human heparanase-1. Heparanase-1 is synthesized as an inactive precursor of ∼65 kD that subsequently undergoes proteolytic cleavage, yielding ∼8 and ∼50 kD protein subunits that heterodimerize to form the active enzyme. The protein contains a putative N-terminal signal peptide (SP, Met1-Ala35), a C-terminal hydrophobic region (HR, Pro515-Ile534), five cysteine residues (black C's) and six N-linked oligosaccharides (orange balloons). Heparanase-1 uses a general acid catalysis mechanism for the hydrolysis of the HS-chains, requiring two critical residues, a proton donor (Glu225) and a nucleophile (Glu343) (red asterisks).
Working mechanism and substrate specificity of human heparanase-1
Sequence homology studies and secondary structure predictions demonstrate that mammalian heparanase-1 is related to members of the ‘clan A’ glycosylhydrolases, especially in the active-site regions of these enzymes [65]. This family of enzymes uses a general acid catalysis mechanism for the hydrolysis of glycosidic bonds, requiring two critical residues, a proton donor and a nucleophile, both of which appear to be conserved in heparanase-1 at Glu225 and Glu343, respectively (Fig. 2) [65].
Secondary structure predictions suggest that heparanase-1 is likely to contain an (α/β)8 TIM-barrel fold, which is another characteristic of clan A glycosylhydrolases [65]. However, the region Lys158-Lys409 within the ∼50 kD fragment corresponds to only one α-helix plus six β/α units, suggesting either a departure from the classical (α/β)8 TIM-barrel fold or that other parts of the protein contribute the missing units [65]. Recently, on the basis of multiple-sequence alignments, it has been suggested that the first β/α/β elements are contributed by the ∼8 kD subunit and that the intermediate ∼6 kD fragment connects the second β-strand and the second α-helix of the barrel [64]. Since adding extra flanking regions of the ∼6 kD fragment to the N-terminus of the ∼50 kD subunit prevents the formation of an active enzyme from ∼8 and ∼50 kD heparanase-1 subunits that are coexpressed in insect cells [60], and since substitution of the ∼6 kD fragment with a shorter loop leads to a constitutively active, single-chain heparanase-1 in insect cells [64], it is postulated that the ∼6 kD fragment impedes substrate binding and/or obstructs access to the active site. Indeed, using the homology- modelling approach, the critical glutamate residues (Glu225 and Glu343) are proposed to be localized inside the barrel (active-site cleft) [65, 66]. As for many other glycosylhydrolases in which β/α units 1 and 2 seem to participate in substrate binding, the region Lys158-Asp171 (corresponding to α- helix 2) has been reported to be responsible for the interaction with the HS-substrate [62]. Consistent with such a role for the Lys158-Asp171 peptide, polyclonal antibodies raised in rabbits against this peptide significantly inhibit (in a dose-dependent manner) the HS-degrading activity of purified, active recombinant human heparanase-1 as well as they inhibit the activity of heparanase-1 transfected cells and the activity of mammalian cells that express high levels of endogenous heparanase-1 [67]. Similarly, oligomannurarate sulphate (JG3), a novel marinederived oligosaccharide that inhibits heparanaseactivity seems to act via binding to the Lys158-Asp171 (and to a lesser extent to the Gln270-Lys280) domain of the heparanase-1 molecule [68].
Mammalian heparanase-1 cleaves the HS chains at only a few sites, resulting in HS fragments of still appreciable size (5–10 kD, 10-20 sugar units). Recently, it was made clear that heparanase-1 is also responsible for the intracellular processing of macromolecular heparin [69]. Macromolecular heparin is synthesized in mast cells as part of a unique proteoglycan, serglycin, and has a Mr of 60–100 kD. Heparanase-1 cuts macromolecular heparin into fragments of 5–20 kD (which can be recovered from most animal tissues and correspond to ‘commercial heparin’) [69], suggesting that it corresponds to the heparin-degrading enzyme originally identified by Ögren and Lindahl [50]. Infrequent enzyme cleavage sites may be due to either the recognition of a single, unusual modification in the heparin/HS chains or the requirement of a specific extended carbohydrate sequence for the cleavage. All studies examining the substrate specificity of heparanase-1 agree that the enzyme is an endo-β-D-glucuronidase (producing a GlcA at the newly formed reducing terminus), but no unified picture of the heparin/HS-sequence recognized and cleaved by heparanase-1 can yet be provided [70–73]. For example, Pikas et al. reported that a heparin-derived octasaccharide, which binds to antithrombin, is cleaved at a single site (by heparanase-1 partially purified from human hepatoma or platelets) (Fig. 3A) [70]. No NS-groups or IdoA units are required, but a 2-O-sulphated hexuronic acid residue located 2 monosaccharides away from the target GlcA residue, towards the reducing end, is essential for substrate recognition. On the contrary, Okada et al. reported that this 2-O-sulphated hexuronic acid residue is not an absolute requirement for cleavage by human heparanase-1, highly purified from transfected human melanoma cells, and that the 3-O-sulphate group on the GlcN at the reducing side of the cleavage, which is required for antithrombin binding, is dispensable and even has an inhibitory effect when located in a NS-region [73]. Instead, the GlcNS structure on the reducing site and GlcN(6S) structure on the nonreducing side of the target GlcA residue are considerably important for substrate recognition (Fig. 3B). Notably, all studies examining the substrate specificity of heparanase-1 not only use different cell-lines and methods to purify heparanase-1, but also different approaches (assays and non-uniform substrates) to define the substrate recognition properties.
3.
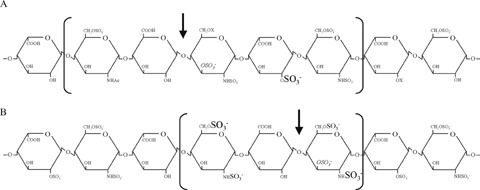
HS-sequences recognized and cleaved by human heparanase-1. (A) One candidate human heparanase-1 cleavage site, as identified by Pikas et al.[70]. The heparin-derived octasaccharide, which binds antithrombin (binding sequence corresponds to units 2–6, between brackets), is cleaved by human heparanase-1 at a single site (arrow), yielding a GlcA at the newly formed reducing terminus (endo-β-D-glucuronidase activity). The 2-O-sulphate group on a hexuronic acid residue located two monosaccharide units away from the cleavage site is essential for substrate recognition (shown in the largest font size). The 3-O-sulphate group on the GlcN residue, required for high affinity binding of antithrombin, but of no particular relevance in the present context, is shown in italics. (B) A target octasaccharide sequence for human heparanase-1, as identified by Okada et al.[73]. Although longer oligosaccharides are most likely better substrates for the cleavage by human heparanase-1, the minimum size required for recognition is only a trisaccharide (indicated between brackets). A highly sulphated structure is crucial for enzyme action, and the GlcNS structure on the reducing side and GlcN(6S) structure on the non-reducing side of the cleavage site are considered as important for the substrate recognition (shown in the largest font size). The additional 2-N-sulphate group on the non-reducing GlcN or 6-O-sulphate group on the reducing GlcN appear to promote cleavage by heparanase-1 (shown in the middle font size). The GlcN 3-O-sulphate structure (shown in italics) promotes cleavage when it resides in a relatively low sulphated sequence but inhibits cleavage when located in a highly sulphated sequence.
Expression of human heparanase-1
Under normal conditions, heparanase-1 expression is restricted primarily to placental trophoblasts, blood-borne cells and keratinocytes [53, 54, 74]. However, as will be addressed further, highly metastatic cells and tumours often show elevated expression of heparanase-1 in comparison with their non-invasive counterparts and normal tissues. Elevated expression of human heparanase-1 protein also occurs in ECs of sprouting capillaries and small vessels in the vicinity of a tumour, but not of mature, quiescent blood vessels [75]. Moreover, human ECs that are stimulated with FGF2 [75], inflammatory cytokines, oxidized LDL or fatty acids [76, 77] demonstrate a high expression of heparanase-1 mRNA and protein, in contrast to non-stimulated ECs or stimulated smooth muscle cells (SMCs). Increased heparanase-1 expression has also been noted in (diabetic) kidney disorders [78, 79].
The heparanase-1 promoter is GC-rich [80], and appears to be subjected to multiple, both positive and negative controls (schematically presented in Fig. 4). First of all, several transcription factors have been shown to regulate heparanase-1 transcription (trans-regulation). The transcription factor Sp1 and the Ets family member GA-binding protein cooperate in binding to a number of sites in the minimal promoter region (which was mapped to the 0.3-kb region upstream of the ATG initiation site), controlling the expression of heparanase-1 in thyroid tumour cells [80]. Sp1 was suggested to be the fatty acid regulatory target in the 0.3-kb promoter region in ECs [76]. Two Ets sites located upstream of the 0.3-kb minimal promoter are functionally important in binding Ets1 and Ets2, and in regulating heparanase-1 mRNA expression in metastatic breast tumour cells [81]. Moreover, the expression of early growth response-1 (EGR1, a member of the zinc-finger family of transcription factors that is rapidly induced in response to a variety of signals, including growth factors, cytokines, vascular injury and hypoxia) correlates with the levels of heparanase-1 mRNA in human prostate and bladder cancer cells [82, 83]. EGR1 binds to two elements in the 0.3-kb heparanase-1 promoter region and has been shown to be responsible for the PMA-induced heparanase-1 gene transcription in T-lymphocytes [84], and also for the regulation of heparanase-1 transcription in several different tumour cells [85]. Blockage of the nuclear factor-kappa B (NF-κB) signal pathway results in the down regulation of heparanase-1 expression in a murine lung alveolar carcinoma cell line [86]. Human gastric carcinoma tissue shows a positive correlation between NF-κB activation and increased heparanase-1 gene expression [87]. Still, NF-κB was not found to be responsible for the TNFα-induced heparanase-1 expression in ECs, and a transcription factor search of the heparanase-1 promoter revealed no NF-κB elements [76]. Oestrogen can induce the expression of heparanase-1 mRNA in oestrogen receptor-positive, but not in receptor-negative breast cancer cells [88]. Four putative oestrogen response element sequences were found in the 3.5-kb heparanase-1 promoter, but whether the stimulation of heparanase-1 expression is attributable to the oestrogen receptor complex itself and/or to transcription factors (e.g. Ets or Sp1) induced by oestrogen is not known yet. High glucose conditions up-regulate heparanase-1 expression in renal epithelial cells [89] and a glucose- response element is located largely within the 0.3-kb promoter region, but the transcription factor(s) responsible for the glucose-dependent gene regulation remain elusive. Recently, it has been shown that the heparanase-1 gene is under a strict negative control by wild-type tumour suppressor p53, and that the loss of wild-type p53 activity or the expression of (cancer-associated) p53 mutants stimulates heparanase-1 expression [90]. Wild-type p53 binds to the 3.5-kb heparanase-1 promoter, which is suggested to alter the chromatin structure of the promoter by recruiting histone deacetylases.
4.
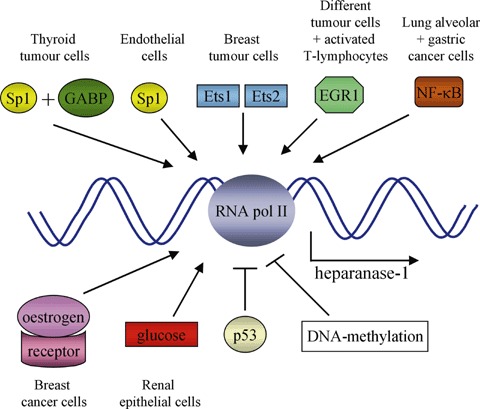
Regulation of heparanase-1 mRNA expression in different cell types. Heparanase-1 mRNA expression by RNA polymerase II (RNA pol II) is positively regulated by transcription factors such as Sp1, GA-binding protein (GABP), Ets1 and Ets2, early growth response-1 (EGR1), nuclear factor-kappa B (NF-κB), oestrogen and glucose (or transcription factors induced by the latter two). Expression is negatively regulated by the transcription factor p53 and by DNA-methylation.
Finally, heparanase-1 transcription may also be regulated by alterations in promoter methylation levels (cis-regulation). The heparanase-1 promoter appears to be hypo- and hypermethylated in high- and low-level heparanase-1 expressing cells, respectively [82, 83, 91]. Moreover, treating low-level expressing cells with demethylating agents or a DNA methylation inhibitor results in an increase of heparanase-1 mRNA expression [91, 92]. It must be noted, however, that pre-treating the cells with a protein synthesis inhibitor abolished this effect [92]. This result suggests that overexpression of heparanase-1 might not be (exclusively) caused by the aberrant demethylation of the heparanase-1 gene itself, but rather by other gene(s), encoding, e.g. transcriptional factors, that, in turn, regulate heparanase-1 expression.
Cellular activation and localization of human heparanase-1
Specific antibodies directed against human heparanase-1, as well as the expression of a heparanase-1-GFP fusion protein, have localized the enzyme predominantly within late endosomes/lysosomes, both in transfected normal cells (e.g. human fibroblasts) and in cells that express detectable levels of endogenous heparanase-1 (e.g. metastatic breast carcinoma cells) [67, 93]. Moreover, it has been shown that the addition of exogenous recombinant inactive pro-heparanase-1 to human fibroblasts results in the cell surface binding of this precursor, followed by its internalization and proteolytic activation [94]. The internalization occurs through endocytosis, involves the actin cytoskeleton, and is mediated by non-caveolar pathways. Also upon de novo synthesis, intracellular active heparanase-1 originates, at least in part, from the uptake of secreted inactive heparanase-1 precursor, suggesting that this possibly reflects an in vivo trafficking route [95]. Pro-heparanase-1 endocytosis was not blocked by the sole addition of mannose- 6-phosphate (Man-6-P) [94]. However, pre-incubation of the precursor with heparin partially inhibited binding and completely abolished endocytosis [94]. These results, together with more recent data from the same group [95], identified cell surface HSPGs, and in particular the syndecans, as mediators of proheparanase-1 binding and uptake. While confirming the importance of HSPGs, our laboratory identified low density lipoprotein receptor-related proteins (LRP) and Man-6-P receptors as key elements of the receptor system that mediates the internalization of the enzyme precursor [96]. The use of specific inhibitors and mutant cell lines, deficient in at least one type of receptor, showed that each of these receptors can work separately, and that LRP is a bona fide and major internalization receptor. Yet, we found that LRP also cooperates with HSPGs, to form a dual receptor-complex with an affinity for proheparanase-1 (Kd =∼0.05 nM) that is 22 times higher than that of LRP alone (Kd =∼1.06 nM) (Vreys et al., our own unpublished data). Man-6-P receptors appear to work as independent receptors, which have a lower affinity for pro-heparanase-1 (Kd =∼23.4 nM) but are ∼10 fold more abundant than the LRP-based receptors (Vreys et al., our own unpublished data).
Living cells that express heparanase-1-GFP in their lysosomes, show no signs of degranulation or trafficking of the enzyme, even over several hours of observation, suggesting that the enzyme is stored within these acidic compartments in a stable form [93]. Consistently, the half-life of the newly formed processed enzyme was estimated to be ∼30 h [95]. The acidic microenvironment within the late endosomes/lysosomes provides heparanase-1 with suitable conditions for storage and optimal enzymatic activity. Human heparanase-1 exhibits maximal endoglycosidase activity between pH 5.5 and 6.0, but shows no significant activity below pH 3.5 and above pH 7.0 [56, 71, 97]. The lysosomal compartment contains also a variety of proteolytic enzymes that may facilitate the conversion of latent heparanase-1 precursor to the active heterodimer form. Although it had originally been suggested that the proteolytic activation of heparanase-1 occurs at the cell surface [94], recent reports show that the processing occurs in acidic intracellular compartments [63, 67, 95, 96, 98].
Cell surface HSPGs are constitutively internalized and degraded [36, 99]. It is generally accepted that endosomal heparanase-1 is responsible for the initial decomposition of internalized HSPGs. However, the predominant (pathological) function of heparanase-1 is commonly attributed to degradation of HS-chains at the cell surface and in the ECM (see next section), a function that would require the presence of cell surface- bound or secreted active heparanase-1. Elevated levels of heparanase-activity have been detected in sera and urine samples of patients suffering from diabetic nephropathy [79, 89, 100] or aggressive metastatic disease [100–102], and also in the bone marrow plasma of myeloma patients [103]. Additionally, it has been shown that the active enzyme can be secreted from intracellular compartments in response to a proper and effective stimulus. In blood-borne cells, active heparanase-1 is stored in specific granules, but in response to chemoattractants or inflammatory stimuli it is redistributed to the cell surface and released by degranulation [104–107]. Inflammatory cytokines, such as TNFα and IL-1β, stimulate secretion of mature heparanase-1 by human ECs [76], and tumour-derived cells, stimulated with physiological concentrations of various nucleotides (ATP, ADP or adenosine) release active heparanase-1 from intracellular vesicles (most likely lysosomes) [108]. Yet, this secreted heparanase-1 appears also to be subject to uptake by cells. Recently, we found that this (re-)capture is mediated by the LRP HSPG receptor-complex and by LRP (Vreys et al., our own unpublished results). Thus, the presence of extracellular (active) heparanase-1 seems to be tightly regulated and restricted by at least two independent mechanisms, on one hand by regulated secretion and on the other hand by LRPmediated (re-)uptake.
Human gastric [109] and oesophagus [110, 111] cancer cells show also nuclear immunostaining for heparanase-1. The latent (∼65 kD) and active (∼50 kD) forms of human heparanase-1 were claimed to accumulate in the nucleus of non-transfected human breast carcinoma cells (expressing high levels of endogenous heparanase-1) and in the nucleus of glioma cells that were transfected or uploaded with exogenous human heparanase-1 [112]. The amount of nuclear heparanase-1 has been estimated to represent up to ∼7% of the total levels found in the cytosol [112]. It should be noted that the presence of HSPGs in the cell nucleus had been reported long before [113], and nuclear heparanase-1 is claimed to be able to degrade this nuclear HS [112, 114]. How heparanase-1 would translocate into the nucleus is not known, and different mechanisms have been suggested: (1) human heparanase-1 contains its own (yet unidentified) nuclear localization signal; (2) it uses HSPGs (its substrates) as vehicle (e.g. a functional nuclear localization signal has been detected in glypicans [115]); (3) it uses other proteins, e.g. β- and γ-catenin, as suggested by Schubert et al. [112], or heat shock protein 90, as suggested by Nobuhisa et al. [116]. The function for heparanase-1 in the nucleus has not been clarified yet, but recent data suggest that its nuclear localization is correlated with a role in cell differentiation (explained in Section ‘Normal development’).
Involvement of human heparanase-1 in normal and pathological processes
Tumour metastasis
The degradation of various constituents of the ECM, including HSPGs, represents a critical step in the process of cancer invasion and metastasis. It has been shown for many years that the metastatic potential of tumour cells is related to their capacity to degrade HS [117, 118]. This observation was confirmed by the finding that increased expressions of heparanase-1 mRNA and protein correlate with increased heparanase activity and the metastatic capacity of rat and human breast cancer cells [53, 54]. Since then, this correlation has been observed in a variety of human tumour-derived cell lines and tissues, including those of the bladder [119], pancreas [120], cervix [121], colon [122], ovary [123], endometrium [124], prostate [83], thyroid [125], liver [126], oesophagus [110], stomach [109], oral cavity [127], galbladder [128], nasopharynx [129], brain [130], salivary gland [131] and in multiple myeloma [132] and acute myeloid leukaemia [133]. Consistently, as already mentioned above, elevated levels of heparanase-activity have been detected in sera and urine samples of patients suffering from an aggressive metastatic disease [100–102]. Moreover, several studies show that increased heparanase-1 expression correlates with a reduced post-operative survival time of cancer patients [109, 111, 119–121, 129, 131]. More convincing, direct evidence for a role of heparanase-1 in tumour metastasis was provided by the conversion of non-metastatic murine T-lymphoma and melanoma cells into highly metastatic cells, following transfection with human heparanase-1 cDNA [53]. Similar results were obtained upon over-expression of human heparanase-1 in pancreatic cancer [120], myeloma [132] and breast carcinoma [134] cells. Enhancement of metastasis was even more pronounced for a readily secreted and membrane-bound chimeric form of the enzyme [135]. It is interesting to note that breast cancer cells that express high levels of human heparanase-1, and that are grown in the mammary fat pads of SCID mice, stimulate bone resorption long before metastasis or even microscopic tumour foci can be detected in the bone marrow, suggesting that heparanase-1 conditions the bone marrow for metastasis by first stimulating osteoclastogenesis and bone resorption (e.g. by releasing HS-bound osteoclaststimulating factors) [136]. Consistent with the notion that heparanase-activity is correlated with cancer metastasis, in vivo administration of heparanaseinhibitors significantly reduces the incidence of experimental metastasis. Both JG3 (see above) and PI-88 (a mixture of highly sulphonated phosphomannan oligosaccharides derived from the yeast Pichia holstii that is currently undergoing Phase II clinical trials in patients with advanced malignancies) reduce metastasis by up to ∼90%[68, 137]. More rigorous and specific proof for this notion has been provided recently, using an adenovirus-mediated antisense gene-delivery strategy [138–140] or by applying siRNA and ribozyme technologies [141]. Tumours produced by cells infected or transfected with these gene-silencing vectors are less invasive in vitro and less metastatic in vivo than tumours produced by cells transfected with the control vectors.
Recently it has been found that heparanase-1 also contributes to tumour metastasis by mechanisms that are independent of its enzymatic activity. Catalytically inactive forms of heparanase-1 that are immobilized on a tissue culture plate facilitate in vitro cell adhesion and facilitate cell invasion when they are expressed as a chimera at the cell surface [142]. It should be noted, however, that (although the adhesive properties of the proteins are similar) cells that over-express the active membrane-bound chimera are significantly more invasive than cells that express the mutated, inactive chimeric form of the protein. The exact mechanism(s) by which (inactive) heparanase-1 promotes cell adhesion is still unclear. However, the effect cannot solely be explained by the enzyme binding to HS (potentially forming a bridge between the circulating cell and the adhesion surface) [142] and is suggested to be mediated, at least in part, by β1-integrin cell membrane localization and activation [143]. Addition of pro-heparanase-1 can induce Akt [144], p38 and Src signalling [145] in different mammalian cell types, independent of its uptake and activation. Activation of Akt does not require cell membrane HSPGs and can be augmented by heparin, mimicking heparin requirements for some growth factor receptors to exert maximal activities (e.g. FGF2- FGFR1 as explained above). Exogenous, added human heparanase-1 stimulates, independent of its enzymatic activity, in vitro EC migration and invasion (important for tumour angiogenesis, see next section) [144]. This effect is PI-3-kinase-dependent and is likely mediated by Akt activation. Such effects may not be restricted to ECs, as the enhanced in vitro migration of human breast carcinoma cells, resulting from the over-expression of heparanase-1, could be inhibited by treating the cells with inhibitors of p38 or Src [145].
Finally, it has also been suggested that heparanase-1 contributes to tumour growth by supporting cell survival under stress conditions. A marked stimulatory effect of heparanase-1 (overexpression) on cell proliferation and survival was noticed when breast carcinoma MCF-7 cells were cultured in vitro under low serum conditions [134]. However, this study did not address whether a function as survival factor is dedicated to the inactive proform (e.g. activating Akt, which has a well known function in cell survival [146]) and/or the active heterodimer form (e.g. releasing HS-bound ‘active’ FGF2 (see next topic), which is known to suppress apoptotic caspase 3 [147]).
Tumour angiogenesis
Angiogenesis is the process of generating new blood vessels, as extensions derived from the existing vasculature. A tumour must continuously recruit new blood vessels in order to sustain itself and grow. HSPGs are prominent components of blood vessels, and HSPG degrading enzymes have long been implicated in a number of angiogenesis-related cellular processes.
The principal cells involved in angiogenesis are ECs, which line all blood vessels and constitute virtually the entirety of capillaries. Immunohistochemical staining of several human carcinomas revealed preferential expression of heparanase-1 by ECs of sprouting capillaries in the vicinity of the tumour, but little or no staining of mature, quiescent vessels [75]. Moreover, a pronounced correlation between heparanase-1 expression and tumour microvessel density has been reported several times [103, 119, 148, 149]. Consistently, in vivo studies reveal that ‘on purpose’ elevation of the heparanase activity of tumour cells results in tumours that have a significantly higher microvessel density than tumours established from control cells with low heparanase activity [75, 103, 134]. Similar to effects on tumour metastasis, these effects were best demonstrated with a readily secreted and membrane-bound chimeric form of heparanase-1 [135]. Moreover, it has been shown that the anti-cancerous potential of JG3 or PI-88 is not restricted to suppression of the invasive metastatic phenotype, but is also due to suppression of tumour neovascularization [68, 137]. Microvessel density was significantly reduced in tumours developing from cells transfected with anti-heparanase-1 ribozyme or siRNA-vectors, in comparison with tumours produced by cells transfected with control vectors [141]. It remains to be clarified whether heparanase is also involved in normal processes of capillary sprouting, or whether heparanase-1 activity on sprouting is peculiar to tumours.
Similar to its effect on tumour metastasis, heparanase-1 is presumed to degrade the HS chains of the subendothelial capillary BM, disrupting a physical barrier and facilitating EC invasion and migration toward the growing tumour. Heparanase-1 may also promote angiogenesis by releasing HS-bound angiogenic factors (e.g. FGF2). As discussed in the first part of this review, heparanase-1 activity may also generate the precise HS-fragments that are required for the formation of, e.g. FGF2.FGFR1.HS ternary complexes, encrypted but present in ‘dormant’ state in native HS, with resulting biological consequences, e.g. EC proliferation [75, 150, 151]. Yet, heparanase-1 may also elicit angiogenic responses, like EC adhesion, migration, invasion and survival, independent of its enzymatic activity (e.g. by activating Akt signalling [144]). Upregulation of cyclooxygenase-2 (COX-2) [149, 152, 153] and vascular endothelial growth factor (VEGF) [145], two key regulators of vascular development, represent additional mechanisms by which heparanase-1 may have indirect pro-angiogenic functions. The up-regulation of COX-2 by heparanase-1 in human oesophageal cancer is strongly suppressed by the combined deletion of three transcription factor binding sites (cycling AMP response element, NF-κB and NF-interleukin-6) in the COX-2 promoter, but whether the (extra- or intracellular) precursor or active form of heparanase-1 is required for this process, is not known [149]. The marked elevation of VEGF expression and secretion in HEK 293-T, human breast carcinoma and rat glioma cells, by overexpression or exogenous addition of heparanase-1, is independent of the enzymatic activity of the enzyme and seems to be mediated by Src family members [145]. Similarly, (inactive) heparanase-1 was found to induce tissue factor expression in vascular endothelial and cancer cells, via the activation of the p38 signalling pathway [154]. Tissue factor not only possesses pro-coagulant activity, but also plays a role in cellular signalling, contributing to tumour growth, survival, metastasis and angiogenesis [155]. Recently, it has been shown that heparanase-1 enhances the synthesis and cell surface shedding of syndecan-1 [156, 157]. The shed ectodomain of syndecan-1 promotes the growth, metastasis and angiogenesis of tumours [158], possibly revealing a new mechanism whereby heparanase-1 and (proteolytically shed) syndecan-1 synergise to fine tune the tumour microenvironment. The effect of heparanase-1 on syndecan-1 shedding requires active enzyme, and shedding is also induced by exposing cells to bacterial heparitinase, suggesting shedding might be a (late, indirect) result from the enzymatic removal or reduction in size of the HS chains on syndecan-1 [156, 157]. Yet, it remains to be clarified whether syndecan-1 is acted upon at the cell surface, or encounters heparanase-1 along the stimulus-induced syntenin recycling pathway (see above).
Other pathologies
It has been known for many years that the extravasation of leucocytes, during inflammation, is linked to the degradation of HS from ECs and ECMs. Activated platelets can release heparanase-activity [159], and the ability of activated T cells, auto-sensitized to the myelin basic protein, to cause experimental autoimmune encephalomyelitis is closely correlated with their production of heparanase-activity [160]. Since then, heparanase-1 expression and activity have been reported in several other types of haematopoietic cells (e.g. neutrophils, monocytes, B-lymphocytes, macrophages and dendritic cells), with chemoattractants or inflammatory stimuli often dramatically enhancing heparanase-1 expression, and causing a redistribution of the active enzyme from specific intracellular granules to the cell surface and a release by degranulation [54, 104–107, 161–163]. Consistently, several inhibitors of heparanase-activity (e.g. PI-88) have been shown to exhibit in vivo antiinflammatory activity in rats [164] and more recently, it has been shown that local in vivo electroporation of anti-heparanase-1 siRNA into the ear skin of mice markedly inhibits the delayed-type hypersensitivity reactivity, which is mainly T-cell mediated [77].
Emerging evidence indicates that heparanase-1 may also be deregulated and play a critical role in the pathogenesis of different kidney disorders. Elevated levels of heparanase-1 protein and activity have been detected in urine of human patients with diabetic nephropathy [79, 89, 100] and in the glomerular epithelial cells and urine of rats with experimental induced nephropathy [78, 165, 166]. High glucose conditions up-regulate heparanase-1 expression in renal epithelial cells and, since urinary heparanase-1 antedates albuminuria, it's suggested that heparanase-1 disrupts the permselective properties of the glomerular BM, ultimately leading to urinary protein excretion [79, 89]. Consistently, in vivo administration of a polyclonal antiheparanase-1 antibody (raised against the Arg382- Phe398 peptide) [165] or of PI-88 [167] significantly prevents glomerular HS loss and reduces proteinuria.
It has also been reported that heparanase-1 contributes to restenosis. Following vascular injury (which results, e.g. in platelet degranulation), SMCs undergo proliferation, migration (from the medial to the innermost layer of the vessel wall) and deposit matrix proteins, contributing to the formation of an occlusive neointimal layer. It has been demonstrated that heparanase-1 releases FGF2 from the ECM deposited by vascular SMCs in vitro and from the arterial wall in vivo (when locally delivered to an uninjured rat artery), resulting in the induction of SMC proliferation and migration [150, 168]. Moreover, an antibody directed against the active site of heparanase-1 significantly inhibits in vivo FGF2 mobilization, SMC proliferation and neointima formation, when locally delivered to rats following a balloon injury [168].
Finally and interesting to note is that, in an experimental in vivo model for amyloid induction homozygous transgenic mice over-expressing human heparanase-1 (see further) revealed an inverse correlation between heparanase-1 over-expression and amyloid protein A amyloidosis [169]. Since formation of amyloid fibrils/plaques seems to proceed through intermediate stages of polymerization that are thought to depend on interactions with HS structures, it has been suggested that HS oligosaccharides generated by heparanase-1 might bind amyloid monomers, but are unable to polymerize and assemble these into larger aggregates
Normal development
Although most studies emphasize the involvement of heparanase-1 in pathophysiology, recent publications reveal some important normal physiological functions of the enzyme. Heparanase-1 protein is preferentially expressed in cells of the developing vascular and nervous systems of a 72 hrs chicken embryo [170]. Human heparanase-1 mRNA and protein are expressed in the liver and colon of 18- and 22-week human foetuses, but not in the corresponding mature tissues [171]. Moreover, a gain of heparanase-1 expression was observed in the adult rat liver shortly following partial hepatectomy [171]. All these data suggest a role for heparanase-1 in tissue morphogenesis, regeneration and repair during embryonic development and in the adult. Heparanase-1 may contribute to these processes by its known effects on ECM remodelling, cell migration, adhesion and proliferation. Recent publications reveal that it might also regulate cell differentiation. Neural cells of the olfactory epithelium express heparanase-1 in vitro depending on the degree of differentiation [172]. Similarly, heparanase-1 expression progressively increases in murine bone marrow-derived cells undergoing osteoblastic differentiation in vitro[173]. Moreover, it has been demonstrated that nuclear heparanase-1 expression is correlated with the expression of differentiation markers in normal oesophageal keratinocytes and in oesophageal cancer tissues [111, 114] and that overexpression of (active) heparanase-1 in the nucleus induces differentiation of leukemia and breast cancer cells in vitro[116, 174]. How nuclear heparanase-1 is involved during these differentiations has not been elucidated yet, but three hypotheses are: (1) affecting the transcriptional regulation associated with, e.g. nuclear HS-bound FGF2 [175]; (2) affecting the direct nuclear function of HSPGs itself (e.g. modulating the activity of DNA topoisomerase I [176]); (3) mediating the nuclear translocation of other proteins (e.g. β- and γ- catenins, as suggested by results of Schubert et al. [112]).
Recently, homozygous transgenic mice that overexpress human heparanase-1 in most of their tissues have been generated [177]. Biochemical analysis of HS isolated from newborn mice and adult tissues, where heparanase-1 is highly expressed, revealed a profound decrease in the size of the HS chains. Despite this change, the transgenic mice appeared normal, were fertile, and exhibited a normal life span. However, the mice had increased levels of urinary protein and creatinin, suggesting an effect on kidney function, which is consistent with the postulated role of heparanase-1 in the pathogenesis of different kidney disorders. In addition, the mice exhibited a decrease in food consumption and body weight. Although there was no effect on the litter size, overexpression of heparanase-1 was found to increase the number of implanted embryos during the first trimester of pregnancy. Consistently, incubation of mouse morulae with recombinant latent heparanase-1 significantly increased the embryo implantation rate in vivo[178] and high levels of heparanase-1 expression were detected in a variety of placental trophoblasts, as well as in the endothelium of angiogenic placental blood vessels [179, 180]. Another remarkable feature of the heparanase-1 overexpressing transgenic mice was an accelerated rate of wound healing [181] and hair growth [177, 182], both processes requiring an efficient migration and proliferation of keratinocytes and a proper angiogenic response. Consistently, in normal mice, heparanase-1 expression is restricted to keratinocytes in the stratum corneum of the epidermis (probably contributing to epidermal renewal [183]), as well as in the outer root sheath of a hair follicle [177]. However, during wound healing, heparanase-1 appears in most keratinocytes adjacent to the wound margins [181] and, after depilation, heparanase-1 is expressed in a highly coordinated temporospatial pattern in the hair follicle [182]. Moreover, addition of a heparanase-1 inhibitor (e.g. a N-acetylated, glycol-split derivative of heparin) or of recombinant active heparanase-1 to keratinocytes, respectively, decreases or increases the ability of these cells to migrate in vitro and, respectively, inhibits or promotes wound epithelialization and blood vessel maturation in vivo[181, 182]. Consistently, the skin of the transgenic mice shows an increase in vascular density [177]. Finally, haematoxylin/eosin staining of most tissues of the heparanase-1 over-expressing mice revealed no readily detectable histological differences, except for the mammary glands, which showed abundant side branches and alveolar structures, typical of normal pregnant mice [177, 184].
Heparanase-2
To potentially identify other heparanases, i.e. as members of a larger protein family, the published amino acid sequence of heparanase-1 was used to search EST databases [74]. This led to the molecular cloning of human heparanase-2, the product of a gene that localizes on chromosome 10q23-24. Alternative splicing of the heparanase-2 transcript gives rise to three different mRNAs, hep-2a, hep-2b and hep-2c, which encode putative proteins of 480, 534, 592 amino acids, respectively [74]. Our laboratory obtained similar results (patent WO 02/04645), yet also identified a fourth splice variant, hep2-B, of 528 amino acids (Fig. 5; where hep-2a from McKenzie et al. equals hep-2O, hep-2b equals hep-2A and hep-2c equals hep-2AB). The cDNA sequence features two possible ATG-start codons, separated from each other by 27 base pairs. Based on a context that corresponds to a Kozak-sequence, we surmize that translation is initiated from the second ATG, and results in heparanase-2 proteins starting with a signal peptide.
5.
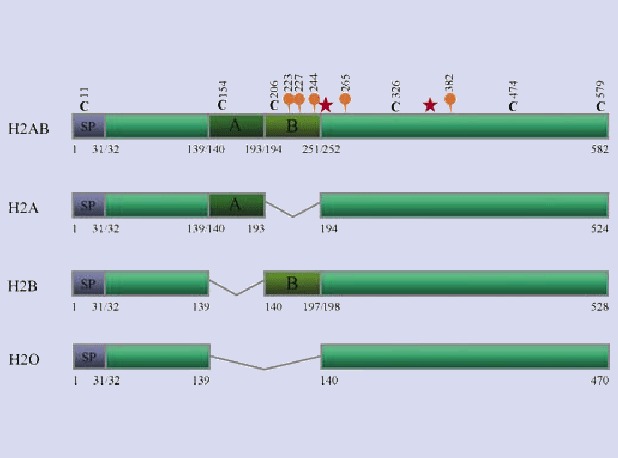
Four different splice-variants of human heparanase-2. Based on the predicted amino acid sequence, heparanase-2 contains two potential N-glycosylation sites (orange balloons) that are shared by all four isoforms, whereas the B-part contains three additional potential N-glycosylation sites. Moreover, six cysteine residues (black C's) occur in the pro-protein, one in the predicted signal peptide (SP), one in the A-part, one in the B-part and three in the C-terminal part that is present in all isoforms. The red asterisks indicate the two glutamates that correspond with the critical active sites of human heparanase-1 (according to Dürr and David; patent WO 02/04645).
Alignment of the predicted coding region of the four isoforms of human heparanase-2 with heparanase-1 reveals an overall identity of 40%, including conservation of the crucial active site residues of human heparanase-1 (Glu225 and Glu343). The highest identity (42%) is found with hep-2AB, for which the alignment is shown in Figure 6. The homology between the two proteins is striking over both subunits (∼8 and ∼50 kD) of heparanase-1, implying that models developed for that protein may also apply to heparanase-2. However, the segment corresponding to the ∼6 kD linker region and the residues corresponding to amino acids at, and surrounding the cleavage sites in heparanase-1 are not similar in heparanase-2. Moreover, western blot analyses of cell lysates of various mammalian cells, transiently transfected with the different human heparanase-2 isoforms (either alone or in combination with each other) provided no evidence for a post-translational processing that would mimic that of human heparanase-1 (Vreys et al., our own unpublished results). Analyses of the culture media of transiently transfected cell lines suggested that only hep-2B and hep-2AB can be released (Vreys et al., our own unpublished results). Binding and uptake experiments with wild type cell lines and with cell lines deficient in receptors involved in (re-)uptake of heparanase-1, and the use of various inhibitors of these receptors, showed that cells are able to bind the secreted variants of heparanase-2 and that HSPGs likely play a role in this binding, but no type of cell showed any evidence for (re-)uptake (Vreys et al., our own unpublished results).
6.
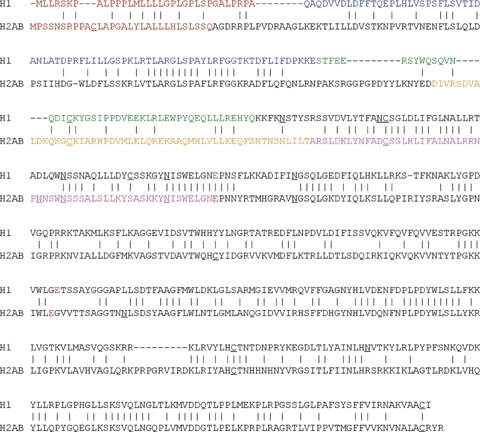
Amino acid alignment of human heparanase-1 with human heparanase-2AB. The predicted signal peptides are shown in brown, the ∼8 and the ∼6 kD fragment of heparanase-1 and the A- and B-parts of heparanase-2AB are shown in blue, green, orange and purple respectively. The potential N-glycosylation sites and cysteine residues are underlined, and the two critical active sites of human heparanase-1 and the corresponding glutamates of heparanase-2 are marked in red.
Heparanase-1 and heparanase-2 also show very different patterns of mRNA distribution [74]. The highest expressions of heparanase-2 were detected in the human brain, mammary gland, prostate, small intestine, testis, uterus and bladder, with little or no expression in most other normal tissues examined, including the placenta and lymph nodes (in contrast to heparanase-1) [74]. As for heparanase-1, heparanase-2 mRNA was dramatically elevated in some pancreatic tumour samples, in comparison to normal pancreas. However, in tumour samples from other tissues (e.g. breast, colon, lung, ovarian, prostate) this seemed not to be the case [74].
In short, all data on processing, trafficking and tissue distribution suggest that heparanase-1 and heparanase-2 might have distinct biological functions. Indeed, up till now, no HS-degrading activity has been reported for the different isoforms of heparanase-2. Potential similarities in direct signalling functions, however, have not yet been explored. Intriguingly, also an alternatively spliced variant of human heparanase-1 has recently been described. Compared to the wild-type form, the variant cDNA skips exon 5, missing the 174bp that encode the residues Ser168-Glu225[185]. The protein encoded by the splice variant escapes proteolytic cleavage, does not possess HS-degrading activity, exhibits a diffuse intracellular staining and is not detectable in the incubation medium of transfected cells. All this is in clear contrast to wild-type heparanase-1, but is quite reminiscent of the findings, so far, for heparanase-2. Possibly, this further alludes to as yet to be discovered functions of this family of proteins.
Conclusions
Based on all information now available, it is clear that the identification of inhibitors of heparanase-1 is an attractive approach towards developing new therapeutics suitable for the treatment of cancer or inflammatory diseases. The unexpected identification of only a single predominant functional heparanase suggests that if its activity can be inhibited, other heparanases may not be available to cover for it. On the other hand, taking into account the normal roles of the enzyme, heparanase-inhibiting compounds might interfere with physiological processes such as wound healing and tissue-repair.
The existence of nearly ubiquitous and highly effective uptake mechanisms (LRP·HSPG and LRP), with very high affinities for both the secreted inactive precursor and the mature active enzyme, suggests that heparanase-1 is mostly an intracellular, lysosomal enzyme. Only in cases where the receptor-system is absent or defective, would the enzyme seem to have a chance to accumulate extracellularly. HS and LRP levels are known to be substantially decreased in several tumour cells [186, 187]. However, surrounding non-tumour cells (e.g. stromal and vascular endothelial cells) should still possess these high affine uptake mechanisms, quickly attracting and removing heparanase-1 from the extracellular space. One might therefore wonder whether an important, if not predominant patho-physiological function of heparanase-1 (e.g. during tumour angiogenesis) might not primarily involve the paracrine activation of intracellular signalling pathways, either by acting on the intracellular processing of (concomitantly) internalized HSPGs (e.g. syndecan-1) or by engaging receptors at the cell surface (activating e.g. Akt, p38 and/or Src) rather than by degrading the HS-chains in the extracellular matrix. If so, this raises the question to what extent the anti-metastatic, -angiogenic and -inflammatory activities of PI-88, JG3 or other sulphated oligosaccharides, of peptides that compete with the HS-binding domains of heparanase-1 or of antibodies directed against the enzyme, are due to their active-site inhibiting properties or to potential concomitant inhibitory effects on receptor-engagement and uptake or direct signalling of human heparanase-1. In that way, more studies are required to understand how the enzyme truly functions during pathological (and normal) processes. Crystallization and analysis of the 3D structure of heparanase-1 should aid the rational design of highly specific inhibitors directed, not only against the active site of the enzyme, but also against the domains that interact with the (different) cell-surface receptors, resulting in the development of novel strategies for inhibiting heparanase-1 activation and activity, inflammatory processes, and cancer progression. Conversely, future studies should also explore potential relationships of heparanase-1 deficiency to disease and the exploitation of heparanase-1 as a drug-in-aid.
References
- 1.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–77. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–73. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 4.Chen RL, Lander AD. Mechanisms underlying preferential assembly of heparan sulfate on glypican-1. J Biol Chem. 2001;276:7507–17. doi: 10.1074/jbc.M008283200. [DOI] [PubMed] [Google Scholar]
- 5.Esko JD, Zhang L. Influence of core protein sequence on glycosaminoglycan assembly. Curr Opin Struct Biol. 1996;6:663–70. doi: 10.1016/s0959-440x(96)80034-0. [DOI] [PubMed] [Google Scholar]
- 6.Duncan G, McCormick C, Tufaro F. The link between heparan sulfate and hereditary bone disease: finding a function for the EXT family of putative tumor suppressor proteins. J Clin Invest. 2001;108:511–6. doi: 10.1172/JCI13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grobe K, Ledin J, Ringvall M, Holmborn K, Forsberg E, Esko JD, Kjellen L. Heparan sulfate and development: differential roles of the N-acetylglucosamine N-deacetylase/N-sulfotransferase isozymes. Biochim Biophys Acta. 2002;1573:209–15. doi: 10.1016/s0304-4165(02)00386-0. [DOI] [PubMed] [Google Scholar]
- 8.Maccarana M, Sakura Y, Tawada A, Yoshida K, Lindahl U. Domain structure of heparan sulfates from bovine organs. J Biol Chem. 1996;271:17804–10. doi: 10.1074/jbc.271.30.17804. [DOI] [PubMed] [Google Scholar]
- 9.Gallagher JT. Multiprotein signalling complexes: regional assembly on heparan sulphate. Biochem Soc Trans. 2006;34:438–41. doi: 10.1042/BST0340438. [DOI] [PubMed] [Google Scholar]
- 10.Salmivirta M, Lidholt K, Lindahl U. Heparan sulfate: a piece of information. FASEB J. 1996;10:1270–9. doi: 10.1096/fasebj.10.11.8836040. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl B, Eriksson L, Lindahl U. Structure of heparan sulphate from human brain, with special regard to Alzheimer's disease. Biochem J. 1995;306:177–84. doi: 10.1042/bj3060177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledin J, Staatz W, Li JP, Gotte M, Selleck S, Kjellen L, Spillmann D. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem. 2004;279:42732–41. doi: 10.1074/jbc.M405382200. [DOI] [PubMed] [Google Scholar]
- 13.Kato M, Wang H, Bernfield M, Gallagher JT, Turnbull JE. Cell surface syndecan-1 on distinct cell types differs in fine structure and ligand binding of its heparan sulfate chains. J Biol Chem. 1994;269:18881–90. [PubMed] [Google Scholar]
- 14.Lindahl U, Kusche-Gullberg M, Kjellen L. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273:24979–82. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- 15.Jayson GC, Lyon M, Paraskeva C, Turnbull JE, Deakin JA, Gallagher JT. Heparan sulfate undergoes specific structural changes during the progression from human colon adenoma to carcinoma in vitro. J Biol Chem. 1998;273:51–7. doi: 10.1074/jbc.273.1.51. [DOI] [PubMed] [Google Scholar]
- 16.Safaiyan F, Lindahl U, Salmivirta M. Selective reduction of 6-O-sulfation in heparan sulfate from transformed mammary epithelial cells. Eur J. Biochem. 1998;252:576–82. doi: 10.1046/j.1432-1327.1998.2520576.x. [DOI] [PubMed] [Google Scholar]
- 17.Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–7. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nugent MA, Iozzo RV. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:115–20. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 19.Saksela O, Rifkin DB. Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. J Cell Biol. 1990;110:767–75. doi: 10.1083/jcb.110.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–8. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 21.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–8. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 22.Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem. 1992;267:10337–41. [PubMed] [Google Scholar]
- 23.Kreuger J, Salmivirta M, Sturiale L, Gimenez- Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–52. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 24.Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem. 1993;268:23906–14. [PubMed] [Google Scholar]
- 25.Rusnati M, Coltrini D, Caccia P, Dell'Era P, Zoppetti G, Oreste P, Valsasina B, Presta M. Distinct role of 2-O-, N-, and 6-O-sulfate groups of heparin in the formation of the ternary complex with basic fibroblast growth factor and soluble FGF receptor-1. Biochem Biophys Res Commun. 1994;203:450–8. doi: 10.1006/bbrc.1994.2203. [DOI] [PubMed] [Google Scholar]
- 26.Pye DA, Vives RR, Turnbull JE, Hyde P, Gallagher JT. Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J Biol Chem. 1998;273:22936–42. doi: 10.1074/jbc.273.36.22936. [DOI] [PubMed] [Google Scholar]
- 27.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGFFGFR- heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–50. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahimi OA, Zhang F, Hrstka SC, Mohammadi M, Linhardt RJ. Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry. 2004;43:4724–30. doi: 10.1021/bi0352320. [DOI] [PubMed] [Google Scholar]
- 29.Pellegrini L, Burke DF, Von Delft F, Mulloy B, Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature. 2000;407:1029–34. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Coomans C, David G. Membrane heparan sulfate proteoglycan-supported FGF2- FGFR1 signaling: evidence in support of the “cooperative end structures” model. J Biol Chem. 2001;276:41921–9. doi: 10.1074/jbc.M106608200. [DOI] [PubMed] [Google Scholar]
- 31.Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646–56. doi: 10.1038/nrm1702. [DOI] [PubMed] [Google Scholar]
- 32.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 33.Zimmermann P, David G. The syndecans, tuners of transmembrane signaling. FASEB J. 1999;13:91–100. doi: 10.1096/fasebj.13.9001.s91. [DOI] [PubMed] [Google Scholar]
- 34.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 35.De Cat B, David G. Developmental roles of the glypicans. Semin Cell Dev Biol. 2001;12:117–25. doi: 10.1006/scdb.2000.0240. [DOI] [PubMed] [Google Scholar]
- 36.Yanagishita M, Hascall VC. Cell surface heparan sulfate proteoglycans. J Biol Chem. 1992;267:9451–4. [PubMed] [Google Scholar]
- 37.Rapraeger AC. Molecular interactions of syndecans during development. Semin Cell Dev Biol. 2001;12:107–16. doi: 10.1006/scdb.2000.0239. [DOI] [PubMed] [Google Scholar]
- 38.Asundi VK, Carey DJ. Self-association of N-syndecan (syndecan-3) core protein is mediated by a novel structural motif in the transmembrane domain and ectodomain flanking region. J Biol Chem. 1995;270:26404–10. doi: 10.1074/jbc.270.44.26404. [DOI] [PubMed] [Google Scholar]
- 39.Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, Swenson TL, Fisher EA, Williams KJ. The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro. J Clin Invest. 1997;100:1611–22. doi: 10.1172/JCI119685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grootjans JJ, Reekmans G, Ceulemans H, David G. Syntenin-syndecan binding requires syndecan-synteny and the co-operation of both PDZ domains of syntenin. J Biol Chem. 2000;275:19933–41. doi: 10.1074/jbc.M002459200. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann P, Meerschaert K, Reekmans G, Leenaerts I, Small JV, Vandekerckhove J, David G, Gettemans J. PIP(2)-PDZ domain binding controls the association of syntenin with the plasma membrane. Mol Cell. 2002;9:1215–25. doi: 10.1016/s1097-2765(02)00549-x. [DOI] [PubMed] [Google Scholar]
- 42.Zimmermann P, Zhang Z, Degeest G, Mortier E, Leenaerts I, Coomans C, Schulz J, N'Kuli F, Courtoy PJ, David G. Syndecan recycling [corrected] is controlled by syntenin-PIP2 interaction and Arf6. Dev Cell. 2005;9:377–88. doi: 10.1016/j.devcel.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R, Neri G, Cao A, Forabosco A, Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi- Behmel overgrowth syndrome. Nat Genet. 1996;12:241–7. doi: 10.1038/ng0396-241. [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Papageorgiou A, Polychronakos C. Developmental regulation of the soluble form of insulin-like growth factor-II/mannose 6-phosphate receptor in human serum and amniotic fluid. J Clin Endocrinol Metab. 1998;83:437–42. doi: 10.1210/jcem.83.2.4537. [DOI] [PubMed] [Google Scholar]
- 45.Song HH, Shi W, Filmus J. OCI-5/rat glypican-3 binds to fibroblast growth factor-2 but not to insulinlike growth factor-2. J Biol Chem. 1997;272:7574–7. doi: 10.1074/jbc.272.12.7574. [DOI] [PubMed] [Google Scholar]
- 46.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–21. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 47.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–41. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 48.Han C, Yan D, Belenkaya TY, Lin X. Drosophila glypicans Dally and Dally-like shape the extracellular Wingless morphogen gradient in the wing disc. Development. 2005;132:667–79. doi: 10.1242/dev.01636. [DOI] [PubMed] [Google Scholar]
- 49.De Cat B, Muyldermans SY, Coomans C, Degeest G, Vanderschueren B, Creemers J, Biemar F, Peers B, David G. Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J Cell Biol. 2003;163:625–35. doi: 10.1083/jcb.200302152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogren S, Lindahl U. Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. J Biol Chem. 1975;250:2690–7. [PubMed] [Google Scholar]
- 51.Hook M, Wasteson A, Oldberg A. A heparan sulfate- degrading endoglycosidase from rat liver tissue. Biochem Biophys Res Commun. 1975;67:1422–8. doi: 10.1016/0006-291x(75)90185-0. [DOI] [PubMed] [Google Scholar]
- 52.Bame KJ. Heparanases: endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology. 2001;11:91R–98R. doi: 10.1093/glycob/11.6.91r. [DOI] [PubMed] [Google Scholar]
- 53.Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 54.Hulett MD, Freeman C, Hamdorf BJ, Baker RT, Harris MJ, Parish CR. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med. 1999;5:803–9. doi: 10.1038/10525. [DOI] [PubMed] [Google Scholar]
- 55.Kussie PH, Hulmes JD, Ludwig DL, Patel S, Navarro EC, Seddon AP, Giorgio NA, Bohlen P. Cloning and functional expression of a human heparanase gene. Biochem Biophys Res Commun. 1999;261:183–7. doi: 10.1006/bbrc.1999.0962. [DOI] [PubMed] [Google Scholar]
- 56.Toyoshima M, Nakajima M. Human heparanase. Purification, characterization, cloning, and expression. J Biol Chem. 1999;274:24153–60. doi: 10.1074/jbc.274.34.24153. [DOI] [PubMed] [Google Scholar]
- 57.Dong J, Kukula AK, Toyoshima M, Nakajima M. Genomic organization and chromosome localization of the newly identified human heparanase gene. Gene. 2000;253:171–8. doi: 10.1016/s0378-1119(00)00251-1. [DOI] [PubMed] [Google Scholar]
- 58.Simizu S, Ishida K, Wierzba MK, Osada H. Secretion of heparanase protein is regulated by glycosylation in human tumor cell lines. J Biol Chem. 2004;279:2697–703. doi: 10.1074/jbc.M300541200. [DOI] [PubMed] [Google Scholar]
- 59.Fairbanks MB, Mildner AM, Leone JW, Cavey GS, Mathews WR, Drong RF, Slightom JL, Bienkowski MJ, Smith CW, Bannow CA, Heinrikson RL. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. J Biol Chem. 1999;274:29587–90. doi: 10.1074/jbc.274.42.29587. [DOI] [PubMed] [Google Scholar]
- 60.McKenzie E, Young K, Hircock M, Bennett J, Bhaman M, Felix R, Turner P, Stamps A, McMillan D, Saville G, Ng S, Mason S, Snell D, Schofield D, Gong H, Townsend R, Gallagher J, Page M, Parekh R, Stubberfield C. Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochem J. 2003;373:423–35. doi: 10.1042/BJ20030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levy-Adam F, Miao HQ, Heinrikson RL, Vlodavsky I, Ilan N. Heterodimer formation is essential for heparanase enzymatic activity. Biochem Biophys Res Commun. 2003;308:885–91. doi: 10.1016/s0006-291x(03)01478-5. [DOI] [PubMed] [Google Scholar]
- 62.Levy-Adam F, Abboud-Jarrous G, Guerrini M, Beccati D, Vlodavsky I, Ilan N. Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J Biol Chem. 2005;280:20457–66. doi: 10.1074/jbc.M414546200. [DOI] [PubMed] [Google Scholar]
- 63.Abboud-Jarrous G, Rangini-Guetta Z, Aingorn H, Atzmon R, Elgavish S, Peretz T, Vlodavsky I. Sitedirected mutagenesis, proteolytic cleavage, and activation of human proheparanase. J Biol Chem. 2005;280:13568–75. doi: 10.1074/jbc.M413370200. [DOI] [PubMed] [Google Scholar]
- 64.Nardella C, Lahm A, Pallaoro M, Brunetti M, Vannini A, Steinkuhler C. Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry. 2004;43:1862–73. doi: 10.1021/bi030203a. [DOI] [PubMed] [Google Scholar]
- 65.Hulett MD, Hornby JR, Ohms SJ, Zuegg J, Freeman C, Gready JE, Parish CR. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry. 2000;39:15659–67. doi: 10.1021/bi002080p. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Z, Bates M, Madura JD. Structure modeling, ligand binding, and binding affinity calculation (LRMM- PBSA) of human heparanase for inhibition and drug design. Proteins. 2006;65:580–92. doi: 10.1002/prot.21065. [DOI] [PubMed] [Google Scholar]
- 67.Zetser A, Levy-Adam F, Kaplan V, Gingis-Velitski S, Bashenko Y, Schubert S, Flugelman MY, Vlodavsky I, Ilan N. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–58. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 68.Zhao H, Liu H, Chen Y, Xin X, Li J, Hou Y, Zhang Z, Zhang X, Xie C, Geng M, Ding J. Oligomannurarate sulfate, a novel heparanase inhibitor simultaneously targeting basic fibroblast growth factor, combats tumor angiogenesis and metastasis. Cancer Res. 2006;66:8779–87. doi: 10.1158/0008-5472.CAN-06-1382. [DOI] [PubMed] [Google Scholar]
- 69.Gong F, Jemth P, Escobar Galvis ML, Vlodavsky I, Horner A, Lindahl U, Li JP. Processing of macromolecular heparin by heparanase. J Biol Chem. 278:35152–8. doi: 10.1074/jbc.M300925200. [DOI] [PubMed] [Google Scholar]
- 70.Pikas DS, Li JP, Vlodavsky I, Lindahl U. Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem. 1998;273:18770–7. doi: 10.1074/jbc.273.30.18770. [DOI] [PubMed] [Google Scholar]
- 71.Freeman C, Parish CR. Human platelet heparanase: purification, characterization and catalytic activity. Biochem J. 1998;330:1341–50. doi: 10.1042/bj3301341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Podyma-Inoue KA, Yokote H, Sakaguchi K, Ikuta M, Yanagishita M. Characterization of heparanase from a rat parathyroid cell line. J Biol Chem. 2002;277:32459–65. doi: 10.1074/jbc.M203282200. [DOI] [PubMed] [Google Scholar]
- 73.Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M, Sugahara K. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis. Hierarchical sulfate groups with different effects and the essential target disulfated trisaccharide sequence. J Biol Chem. 2002;277:42488–95. doi: 10.1074/jbc.M206510200. [DOI] [PubMed] [Google Scholar]
- 74.McKenzie E, Tyson K, Stamps A, Smith P, Turner P, Barry R, Hircock M, Patel S, Barry E, Stubberfield C, Terrett J, Page M. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem Biophys Res Commun. 2000;276:1170–7. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 75.Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. FASEB J. 2001;15:1661–3. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 76.Chen G, Wang D, Vikramadithyan R, Yagyu H, Saxena U, Pillarisetti S, Goldberg IJ. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry. 2004;43:4971–7. doi: 10.1021/bi0356552. [DOI] [PubMed] [Google Scholar]
- 77.Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107:3609–16. doi: 10.1182/blood-2005-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Levidiotis V, Kanellis J, Ierino FL, Power DA. Increased expression of heparanase in puromycin aminonucleoside nephrosis. Kidney Int. 2001;60:1287–96. doi: 10.1046/j.1523-1755.2001.00934.x. [DOI] [PubMed] [Google Scholar]
- 79.Katz A, Van-Dijk DJ, Aingorn H, Erman A, Davies M, Darmon D, Hurvitz H, Vlodavsky I. Involvement of human heparanase in the pathogenesis of diabetic nephropathy. Isr Med Assoc J. 2002;4:996–1002. [PubMed] [Google Scholar]
- 80.Jiang P, Kumar A, Parrillo JE, Dempsey LA, Platt JL, Prinz RA, Xu X. Cloning and characterization of the human heparanase-1 (HPR1) gene promoter: role of GA-binding protein and Sp1 in regulating HPR1 basal promoter activity. J Biol Chem. 2002;277:8989–98. doi: 10.1074/jbc.M105682200. [DOI] [PubMed] [Google Scholar]
- 81.Lu WC, Liu YN, Kang BB, Chen JH. Trans-activation of heparanase promoter by ETS transcription factors. Oncogene. 2003;22:919–23. doi: 10.1038/sj.onc.1206201. [DOI] [PubMed] [Google Scholar]
- 82.Ogishima T, Shiina H, Breault JE, Terashima M, Honda S, Enokida H, Urakami S, Tokizane T, Kawakami T, Ribeiro-Filho LA, Fujime M, Kane CJ, Carroll PR, Igawa M, Dahiya R. Promoter CpG hypomethylation and transcription factor EGR1 hyperactivate heparanase expression in bladder cancer. Oncogene. 2005;24:6765–72. doi: 10.1038/sj.onc.1208811. [DOI] [PubMed] [Google Scholar]
- 83.Ogishima T, Shiina H, Breault JE, Tabatabai L, Bassett WW, Enokida H, Li LC, Kawakami T, Urakami S, Ribeiro-Filho LA, Terashima M, Fujime M, Igawa M, Dahiya R. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11:1028–36. [PubMed] [Google Scholar]
- 84.De Mestre AM, Khachigian LM, Santiago FS, Staykova MA, Hulett MD. Regulation of inducible heparanase gene transcription in activated T cells by early growth response 1. J Biol Chem. 2003;278:50377–85. doi: 10.1074/jbc.M310154200. [DOI] [PubMed] [Google Scholar]
- 85.De Mestre AM, Rao S, Hornby JR, Soe-Htwe T, Khachigian LM, Hulett MD. Early growth response gene 1 (EGR1) regulates heparanase gene transcription in tumor cells. J Biol Chem. 2005;280:35136–47. doi: 10.1074/jbc.M503414200. [DOI] [PubMed] [Google Scholar]
- 86.Andela VB, Schwarz EM, Puzas JE, O'Keefe RJ, Rosier RN. Tumor metastasis and the reciprocal regulation of prometastatic and antimetastatic factors by nuclear factor kappaB. Cancer Res. 2000;60:6557–62. [PubMed] [Google Scholar]
- 87.Cao HJ, Fang Y, Zhang X, Chen WJ, Zhou WP, Wang H, Wang LB, Wu JM. Tumor metastasis and the reciprocal regulation of heparanase gene expression by nuclear factor kappa B in human gastric carcinoma tissue. World J Gastroenterol. 2005;11:903–7. doi: 10.3748/wjg.v11.i6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elkin M, Cohen I, Zcharia E, Orgel A, Guatta- Rangini Z, Peretz T, Vlodavsky I, Kleinman HK. Regulation of heparanase gene expression by estrogen in breast cancer. Cancer Res. 2003;63:8821–6. [PubMed] [Google Scholar]
- 89.Maxhimer JB, Somenek M, Rao G, Pesce CE, Baldwin D, Gattuso P, Schwartz MM, Lewis EJ, Prinz RA, Xu X. Heparanase-1 gene expression and regulation by high glucose in renal epithelial cells: a potential role in the pathogenesis of proteinuria in diabetic patients. Diabetes. 2005;54:2172–8. doi: 10.2337/diabetes.54.7.2172. [DOI] [PubMed] [Google Scholar]
- 90.Baraz L, Haupt Y, Elkin M, Peretz T, Vlodavsky I. Tumor suppressor p53 regulates heparanase gene expression. Oncogene. 2006;25:3939–47. doi: 10.1038/sj.onc.1209425. [DOI] [PubMed] [Google Scholar]
- 91.Shteper PJ, Zcharia E, Ashhab Y, Peretz T, Vlodavsky I, Ben-Yehuda D. Role of promoter methylation in regulation of the mammalian heparanase gene. Oncogene. 2003;22:7737–49. doi: 10.1038/sj.onc.1207056. [DOI] [PubMed] [Google Scholar]
- 92.Simizu S, Ishida K, Wierzba MK, Sato TA, Osada H. Expression of heparanase in human tumor cell lines and human head and neck tumors. Cancer Lett. 2003;193:83–9. doi: 10.1016/s0304-3835(02)00719-x. [DOI] [PubMed] [Google Scholar]
- 93.Goldshmidt O, Nadav L, Aingorn H, Irit C, Feinstein N, Ilan N, Zamir E, Geiger B, Vlodavsky I, Katz BZ. Human heparanase is localized within lysosomes in a stable form. Exp Cell Res. 2002;281:50–62. doi: 10.1006/excr.2002.5651. [DOI] [PubMed] [Google Scholar]
- 94.Nadav L, Eldor A, Yacoby-Zeevi O, Zamir E, Pecker I, Ilan N, Geiger B, Vlodavsky I, Katz BZ. Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. J Cell Sci. 2002;115:2179–87. doi: 10.1242/jcs.115.10.2179. [DOI] [PubMed] [Google Scholar]
- 95.Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279:44084–92. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 96.Vreys V, Delande N, Zhang Z, Coomans C, Roebroek A, Durr J, David G. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6- phosphate receptors, and heparan sulfate proteoglycans. J Biol Chem. 2005;280:33141–8. doi: 10.1074/jbc.M503007200. [DOI] [PubMed] [Google Scholar]
- 97.Gilat D, Hershkoviz R, Goldkorn I, Cahalon L, Korner G, Vlodavsky I, Lider O. Molecular behavior adapts to context: heparanase functions as an extracellular matrix-degrading enzyme or as a T cell adhesion molecule, depending on the local pH. J Exp Med. 1995;181:1929–34. doi: 10.1084/jem.181.5.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cohen E, Atzmon R, Vlodavsky I, Ilan N. Heparanase processing by lysosomal/endosomal protein preparation. FEBS Lett. 2005;579:2334–8. doi: 10.1016/j.febslet.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 99.Egeberg M, Kjeken R, Kolset SO, Berg T, Prydz K. Internalization and stepwise degradation of heparan sulfate proteoglycans in rat hepatocytes. Biochim Biophys Acta. 2001;1541:135–49. doi: 10.1016/s0167-4889(01)00132-x. [DOI] [PubMed] [Google Scholar]
- 100.Shafat I, Zcharia E, Nisman B, Nadir Y, Nakhoul F, Vlodavsky I, Ilan N. An ELISA method for the detection and quantification of human heparanase. Biochem Biophys Res Commun. 2006;341:958–63. doi: 10.1016/j.bbrc.2006.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Quiros RM, Rao G, Plate J, Harris JE, Brunn GJ, Platt JL, Gattuso P, Prinz RA, Xu X. Elevated serum heparanase-1 levels in patients with pancreatic carcinoma are associated with poor survival. Cancer. 2006;106:532–40. doi: 10.1002/cncr.21648. [DOI] [PubMed] [Google Scholar]
- 103.Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–56. [PubMed] [Google Scholar]
- 104.Sasaki N, Higashi N, Taka T, Nakajima M, Irimura T. Cell surface localization of heparanase on macrophages regulates degradation of extracellular matrix heparan sulfate. J Immunol. 2004;172:3830–5. doi: 10.4049/jimmunol.172.6.3830. [DOI] [PubMed] [Google Scholar]
- 105.Mollinedo F, Nakajima M, Llorens A, Barbosa E, Callejo S, Gajate C, Fabra A. Major co-localization of the extracellular-matrix degradative enzymes heparanase and gelatinase in tertiary granules of human neutrophils. Biochem J. 1997;327:917–23. doi: 10.1042/bj3270917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ihrcke NS, Parker W, Reissner KJ, Platt JL. Regulation of platelet heparanase during inflammation: role of pH and proteinases. J Cell Physiol. 1998;175:255–67. doi: 10.1002/(SICI)1097-4652(199806)175:3<255::AID-JCP3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 107.Benhamron S, Nechushtan H, Verbovetski I, Krispin A, Abboud-Jarrous G, Zcharia E, Edovitsky E, Nahari E, Peretz T, Vlodavsky I, Mevorach D. Translocation of active heparanase to cell surface regulates degradation of extracellular matrix heparan sulfate upon transmigration of mature monocyte-derived dendritic cells. J Immunol. 2006;176:6417–24. doi: 10.4049/jimmunol.176.11.6417. [DOI] [PubMed] [Google Scholar]
- 108.Shafat I, Vlodavsky I, Ilan N. Characterization of mechanisms involved in secretion of active heparanase. J Biol Chem. 2006;281:23804–11. doi: 10.1074/jbc.M602762200. [DOI] [PubMed] [Google Scholar]
- 109.Takaoka M, Naomoto Y, Ohkawa T, Uetsuka H, Shirakawa Y, Uno F, Fujiwara T, Gunduz M, Nagatsuka H, Nakajima M, Tanaka N, Haisa M. Heparanase expression correlates with invasion and poor prognosis in gastric cancers. Lab Invest. 2003;83:613–22. doi: 10.1097/01.lab.0000067482.84946.bd. [DOI] [PubMed] [Google Scholar]
- 110.Mikami S, Ohashi K, Usui Y, Nemoto T, Katsube K, Yanagishita M, Nakajima M, Nakamura K, Koike M. Loss of syndecan-1 and increased expression of heparanase in invasive esophageal carcinomas. Jpn J Cancer Res. 2001;92:1062–73. doi: 10.1111/j.1349-7006.2001.tb01061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ohkawa T, Naomoto Y, Takaoka M, Nobuhisa T, Noma K, Motoki T, Murata T, Uetsuka H, Kobayashi M, Shirakawa Y, Yamatsuji T, Matsubara N, Matsuoka J, Haisa M, Gunduz M, Tsujigiwa H, Nagatsuka H, Hosokawa M, Nakajima M, Tanaka N. Localization of heparanase in esophageal cancer cells: respective roles in prognosis and differentiation. Lab Invest. 2004;84:1289–304. doi: 10.1038/labinvest.3700159. [DOI] [PubMed] [Google Scholar]
- 112.Schubert SY, Ilan N, Shushy M, Ben-Izhak O, Vlodavsky I, Goldshmidt O. Human heparanase nuclear localization and enzymatic activity. Lab Invest. 2004;84:535–44. doi: 10.1038/labinvest.3700084. [DOI] [PubMed] [Google Scholar]
- 113.Ishihara M, Fedarko NS, Conrad HE. Transport of heparan sulfate into the nuclei of hepatocytes. J Biol Chem. 1986;261:13575–80. [PubMed] [Google Scholar]
- 114.Kobayashi M, Naomoto Y, Nobuhisa T, Okawa T, Takaoka M, Shirakawa Y, Yamatsuji T, Matsuoka J, Mizushima T, Matsuura H, Nakajima M, Nakagawa H, Rustgi A, Tanaka N. Heparanase regulates esophageal keratinocyte differentiation through nuclear translocation and heparan sulfate cleavage. Differentiation. 2006;74:235–43. doi: 10.1111/j.1432-0436.2006.00072.x. [DOI] [PubMed] [Google Scholar]
- 115.Liang Y, Haring M, Roughley PJ, Margolis RK, Margolis RU. Glypican and biglycan in the nuclei of neurons and glioma cells: presence of functional nuclear localization signals and dynamic changes in glypican during the cell cycle. J Cell Biol. 1997;139:851–64. doi: 10.1083/jcb.139.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nobuhisa T, Naomoto Y, Okawa T, Takaoka M, Gunduz M, Motoki T, Nagatsuka H, Tsujigiwa H, Shirakawa Y, Yamatsuji T, Haisa M, Matsuoka J, Kurebayashi J, Nakajima M, Taniguchi S, Sagara J, Dong J, Tanaka N. Translocation of heparanase into nucleus results in cell differentiation. Cancer Sci. 2007;98:535–40. doi: 10.1111/j.1349-7006.2007.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nakajima M, Irimura T, Di Ferrante D, Di Ferrante N, Nicolson GL. Heparan sulfate degradation: relation to tumor invasive and metastatic properties of mouse B16 melanoma sublines. Science. 1983;220:611–3. doi: 10.1126/science.6220468. [DOI] [PubMed] [Google Scholar]
- 118.Ricoveri W, Cappelletti R. Heparan sulfate endoglycosidase and metastatic potential in murine fibrosarcoma and melanoma. Cancer Res. 1986;46:3855–61. [PubMed] [Google Scholar]
- 119.Gohji K, Hirano H, Okamoto M, Kitazawa S, Toyoshima M, Dong J, Katsuoka Y, Nakajima M. Expression of three extracellular matrix degradative enzymes in bladder cancer. Int J Cancer. 2001;95:295–301. doi: 10.1002/1097-0215(20010920)95:5<295::aid-ijc1051>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 120.Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Buchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001;61:4655–9. [PubMed] [Google Scholar]
- 121.Shinyo Y, Kodama J, Hongo A, Yoshinouchi M, Hiramatsu Y. Heparanase expression is an independent prognostic factor in patients with invasive cervical cancer. Ann Oncol. 2003;14:1505–10. doi: 10.1093/annonc/mdg407. [DOI] [PubMed] [Google Scholar]
- 122.Friedmann Y, Vlodavsky I, Aingorn H, Aviv A, Peretz T, Pecker I, Pappo O. Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and stroma. Evidence for its role in colonic tumorigenesis. Am J Pathol. 2000;157:1167–75. doi: 10.1016/S0002-9440(10)64632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Davidson B, Shafat I, Risberg B, Ilan N, Trope CG, Vlodavsky I, Reich R. Heparanase expression correlates with poor survival in metastatic ovarian carcinoma. Gynecol Oncol. 2006;104:311–9. doi: 10.1016/j.ygyno.2006.08.045. [DOI] [PubMed] [Google Scholar]
- 124.Hasengaowa, Kodama J, Kusumoto T, Seki N, Matsuo T, Ojima Y, Nakamura K, Hongo A, Hiramatsu Y. Heparanase expression in both normal endometrium and endometrial cancer. Int J Gynecol Cancer. 2006;16:1401–6. doi: 10.1111/j.1525-1438.2006.00614.x. [DOI] [PubMed] [Google Scholar]
- 125.Xu X, Quiros RM, Maxhimer JB, Jiang P, Marcinek R, Ain KB, Platt JL, Shen J, Gattuso P, Prinz RA. Inverse correlation between heparan sulfate composition and heparanase-1 gene expression in thyroid papillary carcinomas: a potential role in tumor metastasis. Clin Cancer Res. 2003;9:5968–79. [PubMed] [Google Scholar]
- 126.El-Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res. 2001;7:1299–305. [PubMed] [Google Scholar]
- 127.Ikuta M, Podyma KA, Maruyama K, Enomoto S, Yanagishita M. Expression of heparanase in oral cancer cell lines and oral cancer tissues. Oral Oncol. 2001;37:177–84. doi: 10.1016/s1368-8375(00)00077-4. [DOI] [PubMed] [Google Scholar]
- 128.Chang XZ, Wang ZM, Yu JM, Tian FG, Jin W, Zhang Y, Yu J, Li LF, Liu XF, Li ZW, Shao ZM. Isolation of a human gallbladder cancer cell clone with high invasive phenotype in vitro and metastatic potential in orthotopic model and inhibition of its invasiveness by heparanase antisense oligodeoxynucleotides. Clin Exp Metastasis. 2007;24:25–38. doi: 10.1007/s10585-006-9053-7. [DOI] [PubMed] [Google Scholar]
- 129.Bar-Sela G, Kaplan-Cohen V, Ilan N, Vlodavsky I, Ben-Izhak O. Heparanase expression in nasopharyngeal carcinoma inversely correlates with patient survival. Histopathology. 2006;49:188–93. doi: 10.1111/j.1365-2559.2006.02469.x. [DOI] [PubMed] [Google Scholar]
- 130.Marchetti D, Nicolson GL. Human heparanase: a molecular determinant of brain metastasis. Adv Enzyme Regul. 2001;41:343–59. doi: 10.1016/s0065-2571(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 131.Ben-Izhak O, Kaplan-Cohen V, Ilan N, Gan S, Vlodavsky I, Nagler R. Heparanase expression in malignant salivary gland tumors inversely correlates with long-term survival. Neoplasia. 2006;8:879–84. doi: 10.1593/neo.06382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, Sanderson RD. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood. 2005;105:1303–9. doi: 10.1182/blood-2004-06-2141. [DOI] [PubMed] [Google Scholar]
- 133.Bitan M, Polliack A, Zecchina G, Nagler A, Friedmann Y, Nadav L, Deutsch V, Pecker I, Eldor A, Vlodavsky I, Katz BZ. Heparanase expression in human leukemias is restricted to acute myeloid leukemias. Exp Hematol. 2002;30:34–41. doi: 10.1016/s0301-472x(01)00766-4. [DOI] [PubMed] [Google Scholar]
- 134.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–17. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 135.Goldshmidt O, Zcharia E, Abramovitch R, Metzger S, Aingorn H, Friedmann Y, Schirrmacher V, Mitrani E, Vlodavsky I. Cell surface expression and secretion of heparanase markedly promote tumor angiogenesis and metastasis. Proc Natl Acad Sci USA. 2002;99:10031–6. doi: 10.1073/pnas.152070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kelly T, Suva LJ, Huang Y, Macleod V, Miao HQ, Walker RC, Sanderson RD. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005;65:5778–84. doi: 10.1158/0008-5472.CAN-05-0749. [DOI] [PubMed] [Google Scholar]
- 137.Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB. Identification of sulfated oligosaccharide- based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999;59:3433–41. [PubMed] [Google Scholar]
- 138.Uno F, Fujiwara T, Takata Y, Ohtani S, Katsuda K, Takaoka M, Ohkawa T, Naomoto Y, Nakajima M, Tanaka N. Antisense-mediated suppression of human heparanase gene expression inhibits pleural dissemination of human cancer cells. Cancer Res. 2001;61:7855–60. [PubMed] [Google Scholar]
- 139.Roy M, Reiland J, Murry BP, Chouljenko V, Kousoulas KG, Marchetti D. Antisense-mediated suppression of Heparanase gene inhibits melanoma cell invasion. Neoplasia. 2005;7:253–62. doi: 10.1593/neo.04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gao J, Su L, Qin R, Chang Q, Huang T, Feng Y. Transfection of antisense oligodeoxynucleotide inhibits heparanase gene expression and invasive ability of human pancreatic cancer cell in vitro. J Huazhong Univ Sci Technolog Med Sci. 2006;26:72–4. doi: 10.1007/BF02828042. [DOI] [PubMed] [Google Scholar]
- 141.Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J Natl Cancer Inst. 2004;96:1219–30. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- 142.Goldshmidt O, Zcharia E, Cohen M, Aingorn H, Cohen I, Nadav L, Katz BZ, Geiger B, Vlodavsky I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003;17:1015–25. doi: 10.1096/fj.02-0773com. [DOI] [PubMed] [Google Scholar]
- 143.Zetser A, Bashenko Y, Miao HQ, Vlodavsky I, Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–41. [PubMed] [Google Scholar]
- 144.Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–41. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- 145.Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–63. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 146.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 147.Miho Y, Kouroku Y, Fujita E, Mukasa T, Urase K, Kasahara T, Isoai A, Momoi MY, Momoi T. bFGF inhibits the activation of caspase-3 and apoptosis of P19 embryonal carcinoma cells during neuronal differentiation. Cell Death Differ. 1999;6:463–70. doi: 10.1038/sj.cdd.4400506. [DOI] [PubMed] [Google Scholar]
- 148.Watanabe M, Aoki Y, Kase H, Tanaka K. Heparanase expression and angiogenesis in endometrial cancer. Gynecol Obstet Invest. 2003;56:77–82. doi: 10.1159/000072821. [DOI] [PubMed] [Google Scholar]
- 149.Okawa T, Naomoto Y, Nobuhisa T, Takaoka M, Motoki T, Shirakawa Y, Yamatsuji T, Inoue H, Ouchida M, Gunduz M, Nakajima M, Tanaka N. Heparanase is involved in angiogenesis in esophageal cancer through induction of cyclooxygenase- 2. Clin Cancer Res. 2005;11:7995–8005. doi: 10.1158/1078-0432.CCR-05-1103. [DOI] [PubMed] [Google Scholar]
- 150.Myler HA, West JL. Heparanase and platelet factor- 4 induce smooth muscle cell proliferation and migration via bFGF release from the ECM. J Biochem. 2002;131:913–22. doi: 10.1093/oxfordjournals.jbchem.a003182. [DOI] [PubMed] [Google Scholar]
- 151.Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006;8:596–606. doi: 10.1593/neo.06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Imada T, Matsuoka J, Nobuhisa T, Okawa T, Murata T, Tabuchi Y, Shirakawa Y, Ohara N, Gunduz M, Nagatsuka H, Umeoka T, Yamamoto Y, Nakajima M, Tanaka N, Naomoto Y. COX-2 induction by heparanase in the progression of breast cancer. Int J Mol Med. 2006;17:221–8. [PubMed] [Google Scholar]
- 153.Ohtawa Y, Naomoto Y, Shirakawa Y, Takaoka M, Murata T, Sonoda R, Sakurama K, Yamatsuji T, Gunduz M, Tsujigiwa H, Nagatsuka H, Terada N, Itano S, Horiki S, Yanagihara K, Nakajima M, Tanaka N. The close relationship between heparanase and cyclooxygenase-2 expressions in signet-ring cell carcinoma of the stomach. Hum Pathol. 2006;37:1145–52. doi: 10.1016/j.humpath.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 154.Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, Goldshmidt O, Vlodavsky I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006;4:2443–51. doi: 10.1111/j.1538-7836.2006.02212.x. [DOI] [PubMed] [Google Scholar]
- 155.Versteeg HH, Ruf W. Emerging insights in tissue factor- dependent signaling events. Semin Thromb Hemost. 2006;32:24–32. doi: 10.1055/s-2006-933337. [DOI] [PubMed] [Google Scholar]
- 156.Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–33. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 157.Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, Klein B. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–23. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sanderson RD, Yang Y, Suva LJ, Kelly T. Heparan sulfate proteoglycans and heparanase-partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–52. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 159.Oldberg A, Heldin CH, Wasteson A, Busch C, Hook M. Characterization of a platelet endoglycosidase degrading heparin-like polysaccharides. Biochemistry. 1980;19:5755–62. doi: 10.1021/bi00566a014. [DOI] [PubMed] [Google Scholar]
- 160.Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature. 1984;310:241–4. doi: 10.1038/310241a0. [DOI] [PubMed] [Google Scholar]
- 161.Vlodavsky I, Eldor A, Haimovitz-Friedman A, Matzner Y, Ishai-Michaeli R, Lider O, Naparstek Y, Cohen IR, Fuks Z. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis. 1992;12:112–27. [PubMed] [Google Scholar]
- 162.Laskov R, Michaeli RI, Sharir H, Yefenof E, Vlodavsky I. Production of heparanase by normal and neoplastic murine B-lymphocytes. Int J Cancer. 1991;47:92–8. doi: 10.1002/ijc.2910470117. [DOI] [PubMed] [Google Scholar]
- 163.Bartlett MR, Underwood PA, Parish CR. Comparative analysis of the ability of leucocytes, endothelial cells and platelets to degrade the subendothelial basement membrane: evidence for cytokine dependence and detection of a novel sulfatase. Immunol Cell Biol. 1995;73:113–24. doi: 10.1038/icb.1995.19. [DOI] [PubMed] [Google Scholar]
- 164.Parish CR, Hindmarsh EJ, Bartlett MR, Staykova MA, Cowden WB, Willenborg DO. Treatment of central nervous system inflammation with inhibitors of basement membrane degradation. Immunol Cell Biol. 1998;76:104–13. doi: 10.1046/j.1440-1711.1998.00722.x. [DOI] [PubMed] [Google Scholar]
- 165.Levidiotis V, Freeman C, Tikellis C, Cooper ME, Power DA. Heparanase is involved in the pathogenesis of proteinuria as a result of glomerulonephritis. J Am Soc Nephrol. 2004;15:68–78. doi: 10.1097/01.asn.0000103229.25389.40. [DOI] [PubMed] [Google Scholar]
- 166.Kramer A, Van Den Hoven M, Rops A, Wijnhoven T, Van Den Heuvel L, Lensen J, Van Kuppevelt T, Van Goor H, Van Der Vlag J, Navis G, Berden JH. Induction of glomerular heparanase expression in rats with adriamycin nephropathy is regulated by reactive oxygen species and the Renin-Angiotensin system. J Am Soc Nephrol. 2006;17:2513–20. doi: 10.1681/ASN.2006020184. [DOI] [PubMed] [Google Scholar]
- 167.Levidiotis V, Freeman C, Punler M, Martinello P, Creese B, Ferro V, Van Der Vlag J, Berden JH, Parish CR, Power DA. A synthetic heparanase inhibitor reduces proteinuria in passive Heymann nephritis. J Am Soc Nephrol. 2004;15:2882–92. doi: 10.1097/01.ASN.0000142426.55612.6D. [DOI] [PubMed] [Google Scholar]
- 168.Myler HA, Lipke EA, Rice EE, West JL. Novel heparanase-inhibiting antibody reduces neointima formation. J Biochem. 2006;139:339–45. doi: 10.1093/jb/mvj061. [DOI] [PubMed] [Google Scholar]
- 169.Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, Vlodavsky I, Kisilevsky R, Lindahl U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci USA. 2005;102:6473–7. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Goldshmidt O, Zcharia E, Aingorn H, Guatta- Rangini Z, Atzmon R, Michal I, Pecker I, Mitrani E, Vlodavsky I. Expression pattern and secretion of human and chicken heparanase are determined by their signal peptide sequence. J Biol Chem. 2001;276:29178–87. doi: 10.1074/jbc.M102462200. [DOI] [PubMed] [Google Scholar]
- 171.Goldshmidt O, Yeikilis R, Mawasi N, Paizi M, Gan N, Ilan N, Pappo O, Vlodavsky I, Spira G. Heparanase expression during normal liver development and following partial hepatectomy. J Pathol. 2004;203:594–602. doi: 10.1002/path.1554. [DOI] [PubMed] [Google Scholar]
- 172.Moretti M, Sinnappah-Kang ND, Toller M, Curcio F, Marchetti D. HPSE-1 expression and functionality in differentiating neural cells. J Neurosci Res. 2006;83:694–701. doi: 10.1002/jnr.20753. [DOI] [PubMed] [Google Scholar]
- 173.Kram V, Zcharia E, Yacoby-Zeevi O, Metzger S, Chajek-Shaul T, Gabet Y, Muller R, Vlodavsky I, Bab I. Heparanase is expressed in osteoblastic cells and stimulates bone formation and bone mass. J Cell Physiol. 2006;207:784–92. doi: 10.1002/jcp.20625. [DOI] [PubMed] [Google Scholar]
- 174.Nobuhisa T, Naomoto Y, Takaoka M, Tabuchi Y, Ookawa K, Kitamoto D, Gunduz E, Gunduz M, Nagatsuka H, Haisa M, Matsuoka J, Nakajima M, Tanaka N. Emergence of nuclear heparanase induces differentiation of human mammary cancer cells. Biochem Biophys Res Commun. 2005;331:175–80. doi: 10.1016/j.bbrc.2005.03.129. [DOI] [PubMed] [Google Scholar]
- 175.Sheng Z, Liang Y, Lin CY, Comai L, Chirico WJ. Direct regulation of rRNA transcription by fibroblast growth factor 2. Mol Cell Biol. 2005;25:9419–26. doi: 10.1128/MCB.25.21.9419-9426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kovalszky I, Dudas J, Olah-Nagy J, Pogany G, Tovary J, Timar J, Kopper L, Jeney A, Iozzo RV. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol Cell Biochem. 1998;183:11–23. doi: 10.1023/a:1006898920637. [DOI] [PubMed] [Google Scholar]
- 177.Zcharia E, Metzger S, Chajek-Shaul T, Aingorn H, Elkin M, Friedmann Y, Weinstein T, Li JP, Lindahl U, Vlodavsky I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004;18:252–63. doi: 10.1096/fj.03-0572com. [DOI] [PubMed] [Google Scholar]
- 178.Revel A, Helman A, Koler M, Shushan A, Goldshmidt O, Zcharia E, Aingorn H, Vlodavsky I. Heparanase improves mouse embryo implantation. Fertil Steril. 2005;83:580–6. doi: 10.1016/j.fertnstert.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 179.Haimov-Kochman R, Friedmann Y, Prus D, Goldman-Wohl DS, Greenfield C, Anteby EY, Aviv A, Vlodavsky I, Yagel S. Localization of heparanase in normal and pathological human placenta. Mol Hum Reprod. 2002;8:566–73. doi: 10.1093/molehr/8.6.566. [DOI] [PubMed] [Google Scholar]
- 180.Dempsey LA, Plummer TB, Coombes SL, Platt JL. Heparanase expression in invasive trophoblasts and acute vascular damage. Glycobiology. 2000;10:467–75. doi: 10.1093/glycob/10.5.467. [DOI] [PubMed] [Google Scholar]
- 181.Zcharia E, Zilka R, Yaar A, Yacoby-Zeevi O, Zetser A, Metzger S, Sarid R, Naggi A, Casu B, Ilan N, Vlodavsky I, Abramovitch R. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005;19:211–21. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- 182.Zcharia E, Philp D, Edovitsky E, Aingorn H, Metzger S, Kleinman HK, Vlodavsky I, Elkin M. Heparanase regulates murine hair growth. Am J Pathol. 2005;166:999–1008. doi: 10.1016/S0002-9440(10)62321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bernard D, Mehul B, Delattre C, Simonetti L, Thomas-Collignon A, Schmidt R. Purification and characterization of the endoglycosidase heparanase 1 from human plantar stratum corneum: a key enzyme in epidermal physiology? J Invest Dermatol. 2001;117:1266–73. doi: 10.1046/j.1523-1747.2001.15401.x. [DOI] [PubMed] [Google Scholar]
- 184.Zcharia E, Metzger S, Chajek-Shaul T, Friedmann Y, Pappo O, Aviv A, Elkin M, Pecker I, Peretz T, Vlodavsky I. Molecular properties and involvement of heparanase in cancer progression and mammary gland morphogenesis. J Mammary Gland Biol Neoplasia. 2001;6:311–22. doi: 10.1023/a:1011375624902. [DOI] [PubMed] [Google Scholar]
- 185.Nasser NJ, Avivi A, Shushy M, Vlodavsky I, Nevo E. Cloning, expression, and characterization of an alternatively spliced variant of human heparanase. Biochem Biophys Res Commun. 2007;354:33–8. doi: 10.1016/j.bbrc.2006.12.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–8. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 187.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–84. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]