

Abstract
The activation of trimeric HIV-1 envelope glycoprotein (Env) by its binding to the cell surface receptor CD4 and co-receptors (CCR5 or CXCR4) represents the first of a series of events that lead to fusion between viral and target cell membranes. Here, we present the cryo-electron microscopic structure, at ~ 6 Å resolution, of the closed, pre-fusion state of trimeric HIV-1 Env in complex with the broadly neutralizing antibody VRC03. We show that three gp41 helices at the core of the trimer serve as an anchor around which the rest of Env is reorganized upon activation to the open quaternary conformation. The architecture of trimeric HIV-1 Env in pre-fusion and activated intermediate states resembles the corresponding states of influenza hemagglutinin trimers, providing direct evidence for the similarity in entry mechanisms employed by HIV-1, influenza and related enveloped viruses.
Structural information on the trimeric envelope glycoprotein (Env), the only HIV-1 protein displayed on the surface of the viral membrane, is critical for rational vaccine design and for a better understanding of the detailed mechanisms of viral entry and its inhibition. Env is a heterodimer of a transmembrane glycoprotein (gp41) and a surface glycoprotein (gp120); these dimers are organized as trimers on the surface of the viral membrane1. Structural studies of Env have been carried out over the last two decades by application of a variety of complementary structural methodologies using preparations ranging from truncated variants of gp120 or gp41 to intact, native trimers. Starting with the first crystallographic structure2 of truncated monomeric gp120 in complex with soluble CD4 and Fab fragment of the monoclonal antibody 17b, numerous crystal structures of the core fragment of gp120 with and without bound ligands have been reported3–6. The conformation of gp120 in all of these structures is similar, irrespective of the presence or absence of bound ligands7. Numerous crystal structures of the six-helix bundle formed by gp41 in the post-fusion state are also available8,9. At the other end of the spectrum, cryo-electron tomographic methods, used in conjunction with newly developed tools for sub-volume averaging10,11, have enabled determination of several structures of the entire HIV-1 gp120-gp41 trimer, as displayed on intact viruses12–14. When trimeric Env is in the unliganded state, or when it is bound to CD4-binding-site directed broadly neutralizing antibodies VRC01, VRC02 or VRC03, it is in a “closed” quaternary conformation with the V1V2 loop located close to the apex of the spike12. When native trimeric HIV-1 Env is bound to CD4, or co-receptor mimics such as 17b or m36, it transitions to an open state. The transition requires a large movement of each gp120 protomer, which relocates the V1V2 loop to the periphery of the trimer12–14. These cryo-electron tomographic analyses of native HIV-1 Env thus delineate the “closed” and “open” quaternary conformations of trimeric HIV-1 Env and its connection to the activation of the trimer following its contact with cell surface receptors, thus defining key elements in the structural landscape of Env relevant to initial steps in viral entry.
While most of our analyses of trimeric HIV-1 Env structure have been carried out using native, membrane-bound trimeric HIV-1 Env12–14, we have also extended these studies to soluble variants of trimeric Env15. The ectodomain of HIV-1 Env is a heterodimer with a mass of ~ 140 kDa, composed of the entire gp120 component and ~ 20 kDa of gp41 which are displayed on the surface of the viral membrane. Many types of gp140 trimers have been studied over the years in efforts aimed at designing immunogens capable of eliciting protective humoral immune responses against HIV-1 infection16–18. Using SOSIP gp140 trimers16, which are soluble, proteolytically cleaved trimer variants stabilized by the presence of an engineered intermolecular disulfide bond between gp120 and gp41 (SOS) combined with a single residue change, I559P, within gp41, we established that they display the same closed and open quaternary conformations as that observed for native trimeric HIV-1 Env as assessed by cryo-electron tomography at ~ 20 Å resolution15. These studies with soluble trimers showed that as with native HIV-1 Env, similar open quaternary conformations are observed with the binding of either 17b alone, soluble CD4 alone or with both soluble CD4 and 17b bound. To further improve the resolution of the structures obtained, we later used single particle cryo-electron microscopy (cryo-EM) to determine the structure of the 17b-bound open conformation of soluble trimeric HIV-1 Env at a resolution of ~ 9 Å. These studies revealed the organization of three gp41 helices at the center of the trimer as a key structural signature of trimeric HIV-1 Env in this complex14. Despite these insights into Env conformational changes, no structural information is currently available on the structure and organization of the gp120-gp41 complex in mature, proteolytically cleaved Env trimers in the native conformation, nor is there an understanding of underlying structural changes in gp41 with Env activation. These are topics of fundamental interest relevant to the biology of HIV and to vaccine design. Here, we report the results from a structural analysis of trimeric HIV-1 Env in the closed, pre-fusion, state using cryo-electron microscopy. Our results provide new insights into the molecular architecture of trimeric Env and into the structural mechanisms involved in entry of HIV-1 into target cells.
RESULTS
Cryo-EM of gp140-VRC03 complex
To determine the structure of the pre-fusion state of trimeric HIV-1 Env, we carried out cryo-EM analysis of the complex of VRC03 Fab with cleaved, soluble SOSIP16 trimeric HIV-1 Env, taking advantage of our earlier findings that VRC03 binding to intact HIV-1 preserves Env in the closed quaternary conformation14. The presence of the bound Fab fragment increases the effective size and dimensions of the unliganded trimer and thus improves the accuracy with which orientations can be assigned to each molecular image in the 3D reconstruction procedure19. Cryo-electron micrographs recorded from vitrified suspensions of the Env-VRC03 complex show the expected dimensions for the complex (Supplementary Figure 1a, 1b). Individual molecular images and class averages (Supplementary Figure 1c) are readily recognized as representing projections of the Env-VRC03 complex previously determined by cryo-electron tomography of intact virus14. 3D reconstruction of the structure of the soluble Env-VRC03 Fab complex was carried out independently using two different 3D reconstruction programs (FREALIGN20 and RELION21) to provide greater confidence in structure determination (Supplementary Figures 2a, 2b). The final map resolution for both maps, measured using the 0.143 and 0.5 Fourier Shell Coefficient cutoff criteria, is ~ 6 Å and ~ 8 Å, respectively (Supplementary Figure 2c). We note that these density maps were derived solely from the information present in the experimentally obtained cryo-EM images, without recourse to atomic structural models of gp120 at any stage of processing which could result in apparent map improvement as a consequence of model bias. Features of the map that are more ordered and closer to the symmetry axis such as the central gp41 helices are consistent with the 6 Å resolution value, while features at the periphery such as the VRC03 Fab are more consistent with the lower 8 Å resolution value. There are several possible reasons for this radial gradient in resolution, which could include limited accuracy in the assignment of orientations given the small size of the complex, as illustrated by the point-spread in the tilt-pair parameter plot (Supplementary Figure 2d). These errors limit the resolution of the map and their effects are more pronounced at the periphery of the map because they are the furthest from the symmetry axis where small angular errors translate into larger displacements as compared to regions that are closer to the symmetry axis. It is also possible that there is greater flexibility in the peripheral Fab arms than at the central regions, which combined with some anisotropy in particle orientations can contribute to poorer resolution at the edges of the map.
Molecular architecture of HIV-1 Env trimer in “closed” state
The density map of the VRC03-Env complex, fitted (using automated procedures22) with three copies of the crystal structure of VRC03 Fab complexed to the truncated gp120 core (PDB 3SE823), reveals the overall structural organization of the complex, including the Fab arms (Figure 1). The crystal structure includes coordinates for residues 44–123, 198–301, 325–398, and 407–492 of gp120 which are accommodated in the density map. The map also includes density for the V1V2 loop region, the gp120 N- and C-termini and the gp41 base, which are absent in the 3SE8 crystal structure. A striking feature of the map is the appearance of a prominent set of three long central densities at the core of the trimer. Since all of the relevant portions of the gp120 polypeptide are accounted for within the rest of the map, these central densities can only arise from a gp41-derived α-helical segment. At the present resolution, this density cannot be definitively assigned to a specific region of the gp41 polypeptide, but likely includes a major contribution from the gp41 N-terminal helix region (NHR) as we suggested previously in the case of the open quaternary conformation14. With the exception of this additional feature, the overall structure of the trimeric complex at 6 Å resolution has all of the general features of the map obtained previously at 20 Å resolution by cryo-electron tomography14 (Supplementary Figure 3). We verified this by Fourier filtering of the 6 Å resolution density map to progressively lower resolutions (Supplementary Figure 4); this exercise shows that while the central helical features are still detectable at 10 Å resolution, they disappear into the background at 20 Å resolution, where individual α-helices are not expected to be resolved. The visual appearance of a cavity in isosurface representations of density maps of the pre-fusion state at 20 Å resolution is thus due to lower resolution, and not because of the presence of a large internal cavity in the trimer.
Figure 1.
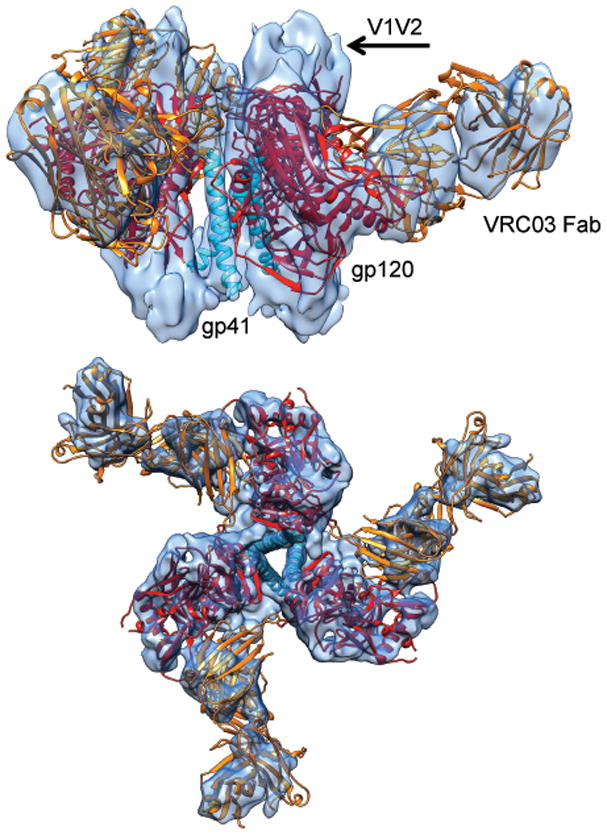
Structure of the pre-fusion state of trimeric HIV-1 Env bound to VRC03. Side (top) and top views (bottom) of the density map (from FREALIGN) of soluble HIV-1 Env trimer in complex with Fab fragment of the broadly neutralizing antibody VRC03. Three copies of the X-ray structure (PDB 3SE823) of the complex of the truncated gp120 core (red) with VRC03 Fab (orange), shown as a ribbon representation are fitted into the density map. The three central densities arise from gp41 and are fitted with the long central helix from influenza HA2 (PDB 3HMG39)(cyan). Densities that are not interpreted by the ribbon diagrams include the V1V2 loop region, N- and C-terminal ends of gp120 and the portion of gp41 outside of the central rods of density.
Inspection of selected sub-regions of the interior of the density map reveals the general structural features of gp120-gp41 architecture in the pre-fusion state. Each of the three central α-helical features is associated with one of the three gp120 protomers (Figure 2a), and spans almost the entire length of the trimer along the 3-fold axis. Front and top views of a single gp120 protomer (Figures 2b) show that many of the secondary structural elements present in the crystal structure (Figure 2c) can be identified. The molecular architecture of trimeric HIV-1 Env we present here is not in agreement with the structure recently reported by Mao et al24 for tail-truncated, uncleaved trimeric HIV-1 Env, where gp41 was indicated as being located on the outer edge of the ectodomain of the trimeric spike with the presence of a large cavity at the center of the spike (comparison is presented in Supplementary Figure 5). Further, the structural elements in gp120 (α0, α1 and α5) reported by Mao et al24 to require flexible fitting in order to be accommodated within their map are present in our map at the locations expected based on the crystal structure of the gp120-VRC03 complex (Figure 2d). Although it is likely that there will be important differences in conformation between the structure of gp120 in the trimeric state as compared to that seen in 3D crystals that contain monomeric gp12025, our density map suggests that at the present resolution of our map, the general features of the structure of the gp120 component of the trimer, at least in the core region, are well described by the 3SE8 crystal structure.
Figure 2.
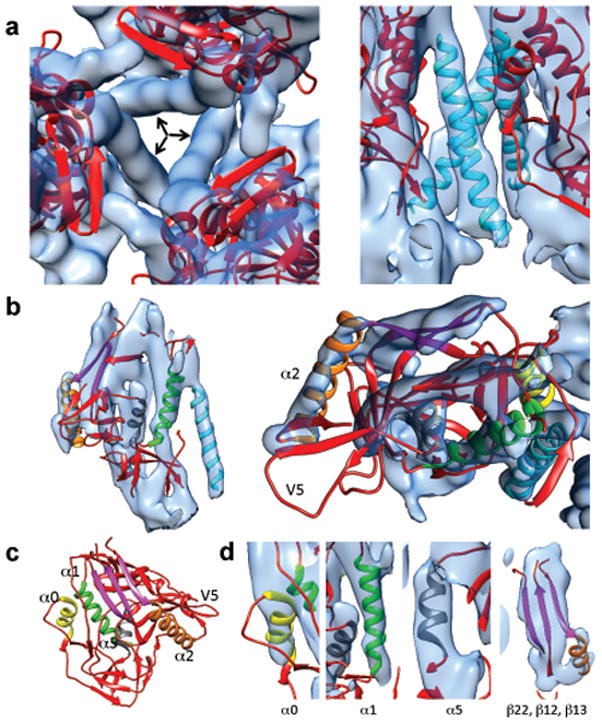
Detailed view of gp120 and gp41 structural elements. (a) Close-up of the arrangement of the three gp120 protomers around the three long rods of central density (indicated by arrows) as viewed from the apex of the trimer (left) or the side (right). (b) Close-up of the fit of the gp120 portion of the 3SE8 coordinates into the map shown in side (left) and top views (right). (c) Ribbon diagram of the crystal structure of the gp120 portion of the 3SE823 coordinates highlighting some of the major secondary structural elements α0 (yellow), α1 (green), α2 (orange) and α5 (gray) and a sheet formed from strands β 12, β 13, and β 22 (magenta) in the crystal structure of gp1202. (d) Zoomed-in views of selected regions of the density map illustrating quality of fit of secondary structural elements color-coded as in (c).
Structural mechanism of Env activation
A view of only the gp120 and gp41 components of the Env-VRC03 complex provides a clearer interpretation of the gp41 densities and the arrangement of the three gp120 protomers around the central gp41 stalk (Figure 3). Most of the residues in the V1V2 loop are not included in the construct used to obtain the crystal structure of the monomeric gp120-VRC03 complex, but density for this region is evident at the top of the trimer density map. From the location of the stump of the V3 loop near the apex, it appears likely that the V3 loop is partially buried by the V1V2 loop in the closed conformation of the trimer (Figure 3). The unassigned densities at the bottom of the map include contributions from gp41 and from the N- and C-terminal residues of gp120 (1–43 and 493–510) that are absent in the crystal structure of the gp120-VRC03 complex. The three central gp41 helices emerge from this cradle of density at the bottom, and are also joined to the rest of Env near the apex at what appears as a sharp bend. The central gp41 helices are thus supported by interactions at both ends, with the rest of gp41 ectodomain closely nestled against gp120.
Figure 3.
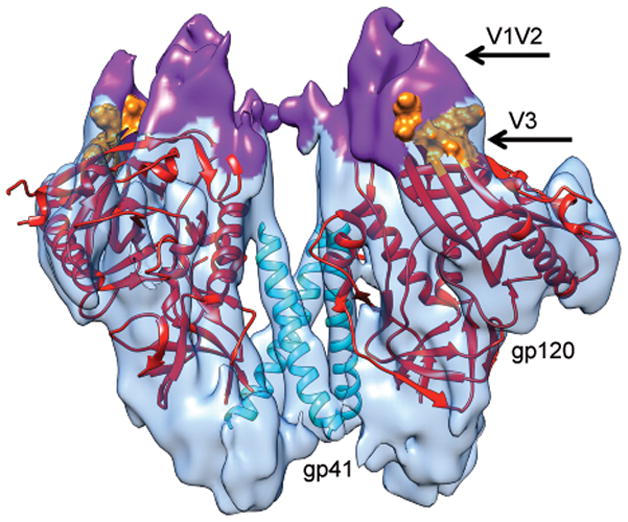
Molecular structure of soluble trimeric HIV-1 Env in the closed state. The density for the three VRC03 Fab moieties from the map shown in Figure 1 was computationally removed to provide a view of only the Env component of the complex illustrating the locations of gp120, gp41 and the V1V2 and V3 loops. The density map was fit with three copies of the gp120 core (Chain G of 3SE823), shown in red. Residues 120–204 are not shown and residues at the base of the V3 loop are shown in surface representation and colored orange. The density corresponding to the V1V2 loop is highlighted in purple and the three central gp41 helices (derived from PDB ID 3HMG39) are shown in cyan.
Comparison of the structures of the closed, pre-fusion and open, activated conformations (Supplementary Figure 6) reveals that in each case, the central densities are at the same location, despite the substantial change in location and orientation of gp120 (Figure 4a). This remarkable conservation of the structure at the core, while allowing changes at the periphery, shows that the central gp41 helices in trimeric HIV-1 Env must serve as an anchor around which the three gp120 protomers pivot outwards upon ligand activation (Figure 4b). This large conformational change, which is seen here to involve the same rearrangements as those that occur with native trimeric Env on intact virions involves relocation of the V1V2 loop to the periphery of the trimer upon activation12,14. The base of the V3 loop is in roughly the same position in both pre-fusion and activated states, but the visualization of these structures explains clearly how the outward movement of the V1V2 loop uncloaks the partially buried V3 loop in the open quaternary conformation. The preservation of similar gp41 architecture in the central region thus suggests an elegant structural mechanism for how the overall trimeric assembly remains intact despite the large quaternary structural rearrangements of gp120 that occur with transition to the open state.
Figure 4.
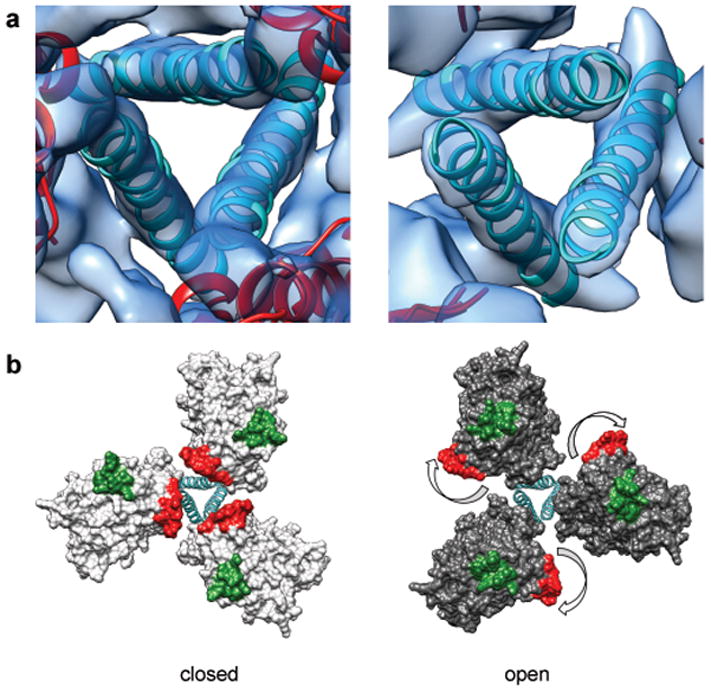
Comparison of structures of trimeric Env in the closed, pre-fusion and open, activated conformations. (a) Zoomed-in top views of the closed (left) and open (right) quaternary states derived from structures of trimeric HIV-1 Env in complex with VRC03 and 17b antibodies, respectively. The ribbons representing the central helices are in an identical position in both panels indicating that the location of the central density is approximately the same in closed and open quaternary conformations. (b) Molecular models for the two conformations in (a) show how Env activation results in major rearrangements of gp120 location relative to the central gp41 stalk. The outward rotation of each gp120 protomer repositions the V1V2 loop (base of loop shown in red) from the center to the periphery and alters its position relative to the location of the V3 loop (base of loop shown in green).
DISCUSSION
The overall organization of trimeric HIV-1 Env, which we have now derived at sub-nanometer resolution in both pre-fusion and activated intermediate states, bears a striking resemblance to the structures of corresponding states previously observed for influenza hemagglutinin trimers26,27. The possible similarity in spike architecture and activation mechanism between HIV, influenza and other viruses such as Ebola has long been hypothesized8,9,28,29. In influenza hemagglutinin, three copies of HA1 are arranged around the central stalk formed by HA2 trimers30 in the pre-fusion state (Figure 5a). Movement of HA1 protomers away from the central axis during the activation process ultimately leads to formation of the post-fusion six-bundle state formed from HA2 trimers31. The role of co-receptor binding in the case of the HIV-1 spike is most likely analogous to the role of low pH in induction of conformational changes in the influenza spike. How these changes lead to the formation of the pre-hairpin intermediate state, and to the slow conversions involved32 in the steps leading to membrane fusion in both viruses, is still not understood in mechanistic detail. Nevertheless, our findings with HIV-1 Env showing a closed pre-fusion state with a central stalk made up of three α-helices and an activation process that leads to an outward displacement of the three gp120 protomers (Figures 5b) provides further and strong evidence for a fundamental similarity between HIV and influenza, extending previously noted similarities in the metastability of the pre-fusion state25,28,33 and the architecture of the final post fusion state8,9. Considering all of these structural and functional similarities in the fusion machinery, it is not surprising that the broadest human monoclonal antibodies against HIV and influenza target gp41 and HA2, the respective central anchors of these trimeric glycoproteins34,35.
Figure 5.
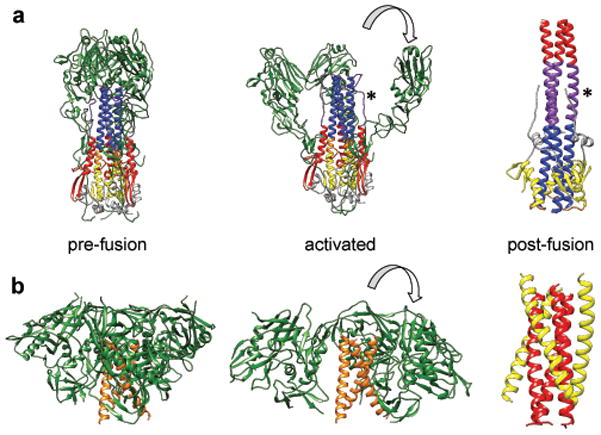
Comparison of the structures of influenza hemagglutinin and HIV-1 Env trimers in different states. (a) Structure of hemagglutinin trimer in the pre-fusion state30 (left panel) displaying the organization of the HA1 trimers (green) around a central stalk formed by HA2 trimers (mixed colors). A model for the initial stages of how the HA1 chains may become repositioned once conformational changes are triggered in hemagglutinin by low pH26 (middle panel). Structure of post-fusion six-helix bundle state31 in which the HA2 chain is rearranged (left panel). The HA2 chains are shown in yellow, purple, blue, gray, red and orange to highlight how these regions are rearranged between pre-fusion and post-fusion states. An asterisk is used to help locate the segment of the polypeptide shown in purple that is extended in the middle panel and is helical in the left panel. (b) Structures of trimeric HIV-1 Env in the closed, pre-fusion state described here (left panel), the open, activated state14 (middle panel) and the post-fusion six-helix bundle state8,9 (right panel). At the present resolution of the maps for the pre-fusion and activated states of HIV-1 Env, we cannot assign different regions of the gp41 polypeptide that correspond to the N-terminal helix region (red) and the C-terminal helix region (yellow).
The general similarity between the molecular architectures of the gp120-gp41 trimer in HIV-1 and HA1-HA2 trimer in influenza provides an insight into the likely origin of the long central helices in the pre-fusion spike structure we report here. In both HIV and influenza, the final post-fusion state of the spike involves a closely-packed, six-helix bundle arrangement (derived from HA2 or gp41), in which three central helices are surrounded by three peripheral helices8,31. In the pre-fusion state and activated intermediate states of the influenza spike, the central stalk is a trimer of three long helices where each helix is made up of a composite of three regions from HA2 (Figure 5a, left and middle panels). The segment at the bottom (yellow) becomes the outer helix of the post-fusion six-helix bundle, while the segment at the top (blue) becomes a portion of the inner helix of the six-helix bundle. The segment in the middle (orange) transforms into the loop connecting the inner and outer helices (Figure 5a, right panel). If the central helix in the HIV-1 Env pre-fusion state also undergoes similar rearrangements to reach the final six-helix bundle state, it seems plausible that the central helix in the closed and open quaternary states of the HIV-1 Env spike may be composed of regions that include the corresponding segments from the N-terminal and C-terminal helices in the post-fusion six helix bundle state (Figure 5b). This mode of packing also implies that in both the closed and open quaternary conformations, the gp41 fusion peptide is likely buried towards the base of the HIV-1 spike as observed for hemagglutinin and surface spikes in Ebola29 and RSV36, which represent “class I” fusion proteins37. This model is consistent with our previously articulated expectation14 that the central helix likely includes much of the gp41 NHR sequence, with the bend at the top end most likely representing the point where the polypeptide is bent downwards to enable the fusion peptide to be buried.
Structural analyses at the resolution we report here provide a starting point to explore the conformational changes that occur in trimeric Env with binding of ligands such as CD4, co-receptor and various antibodies and fusion inhibitors that target gp120 and gp41. As we have shown previously, some broadly neutralizing antibodies such as VRC01 or VRC03 can access the CD4 binding site with no change in quaternary conformation relative to the unliganded state, while others such as b12 bind almost the same region of gp120, but require a partial opening to be accommodated on the surface of the trimer12,14. The structural plasticity of the trimer is thus fundamentally related to neutralization potency of antibodies that require quaternary structural change for efficient binding. To advance beyond the visualization of individual α-helices we report here, and to obtain an unambiguous chain trace of the gp41 and gp120 polypeptides in the structure of trimeric Env it will be necessary to obtain structures at resolutions of 4 Å or better. At these higher resolutions, it should be possible to unambiguously identify all of the ordered portions of the gp120 variable loops and N-linked carbohydrate moieties on the surface of Env. Recent advances in direct electron detectors for cryo-EM have enabled structure determination at 3.3 Å resolution for a highly symmetric 800 kDa complex derived from a thermophilic organism38. Application of these and related technological advances to achieve similar resolutions for small <500 kDa-sized, dynamic protein complexes such as soluble trimers is thus an exciting and challenging frontier in structural biology. Such advances could provide a window into the detailed landscape of spike structural changes in enveloped viruses such as HIV and influenza, allowing characterization of intermediate states that may not always be accessible to analysis by X-ray crystallography. Finally, while structural investigations of soluble Env trimers can be highly informative as we have shown, it will not be possible to define the biologically relevant structure of Env trimers unless high-resolution structures can also be obtained from native, membrane-associated trimeric HIV-1 Env, laying the structural foundation for the design of effective immunogens.
ONLINE METHODS
Proteins and preparation of specimens for microscopy
Purified samples of soluble Clade A strain KNH1144 SOSIP, cleaved HIV-1 Env gp140 trimers containing the entire Env ectodomain including the membrane-proximal external region (MPER, residues 661–681) were kindly provided by K. Kang and W. Olson (Progenics Inc.), and were the same as those previously described and used for the structural analysis of Env structure at 20 Å resolution15. Purified VRC03 IgG was kindly provided by J. Macsola (Vaccine Research Center, NIAID, NIH). VRC03 Fab fragments were prepared by papain digestion, concentrated to 3 mg/ml, and incubated on ice for 30–60 minutes with soluble Env trimers (650 μg/ml) with five times higher estimated molar excess of Fab. 2.5 μl of the mixture was deposited on 400 mesh C-flat grids from Protochips Inc. (Raleigh, NC) with 2 μm-wide holes spaced by 4 μm, and vitrified specimens for cryo-electron microscopy were prepared using a Mark IV Vitrobot from FEI Company (Hillsboro, OR).
Imaging conditions
Plunge-frozen specimens of the Env-VRC03 Fab complex were imaged using a Titan Krios electron microscope aligned for parallel illumination at an operating voltage of 80 kV. Electron micrographs were collected on a Gatan 4k x 4K CCD optimized to maximize detective quantum efficiency (DQE) at lower voltages. The use of lower voltages comes at the expense of sacrificing the higher microscope performance at 300 kV, but the excellent DQE performance provides increased image contrast at 80 kV which, in turn, improves image alignment accuracy. Data collection utilized EPU automated data acquisition software, a total electron dose of 10 electrons/Å2/image and a nominal microscope magnification of 75,000x corresponding to a pixel size at the specimen plane of 1.08 Å. A total of 4713 micrographs were collected, with roughly equal numbers of images recorded at defocus values of −1.5, −1.8, −2, −2.35 and −2.5 μm. From these, the subset of micrographs displaying the highest resolution Thon ring profiles, least astigmatism, and clearly visible, well-separated particles were then selected for further analysis.
Image processing
Hot-pixel removal to correct defects in electron microscope images was done using IMOD’s ccderaser using options “-scan 10 -xyscan 128 -edge 64 -radius 2.1” 40. CTFFIND341 was used to estimate the defocus of each micrograph using an FFT box size of 512 pixels with microscope parameters for Cs and WGH of 2.7 and 0.1 respectively. The resolution range used for defocus estimation was set from 50 Å to 7 Å, with a defocus step size of 250 Å and astigmatism step size of 2500 Å.
Particles were picked manually from 8x-binned versions of the original micrographs using EMAN’s boxer program42 and subsequently extracted with the batchboxer command using a box size of 256 pixels, ignoring particles that extended outside the micrographs. Particle stacks were normalized using EMAN’s proc2d with option “edgenormalize”. All entities that had densities consistent with sizes of ~200 Å in diameter were initially selected. Characteristic top-views showing the three-fold symmetry were easily recognizable and more abundant, but side and edge-on views orthogonal to the 3-fold symmetry axis were also selected. Particles that were too close to each other were not included. 114,713 particles from 3755 micrographs were selected, giving an average of ~30 particles per image.
In order to obtain a clean subset of particles, 2D classification of the 114,713 particles into 500 classes was done in IMAGIC43 with the MSA command using the MODULATION distance for 25 iterations using 25 eigenimages. This analysis was done without pre-centering or aligning the particles in order to rule out any type of reference bias in the analysis. Classification was done with the HAC option using all 25 eigenimages and a 0.15 fraction of worst class members were removed from the analysis. 48 classes were visually identified as being of poor quality or heterogeneous and were de-selected giving a cleaned dataset of 88,125 particles that were subsequently used for 3D refinement. The particle selection and classification was independently carried out numerous times to verify that consistent class averages were obtained each time. The class averages (Supplementary Figure 1c) invariably showed agreement with the projection images expected from the tomographically reconstructed density map of the native Env-VRC03 complex at 20 Å resolution (Supplementary Figure 3a), which was used as a starting model for 3D refinement.
Single particle refinement using the tomographic map as an initial model was done with programs FREALIGN v8.1020 and RELION v1.221 imposing 3-fold (C3) symmetry. FREALIGN refinement was carried out using frequency components between 100 Å and 8 Å as follows: 4 initial rounds of orientation assignment using randomized global search (MODE 4) were done using parameters DANG=200 and ITMAX=50 followed by 4 additional rounds of local refinement (MODE 1). At each iteration round, the top 75% of particles according to phase residual were included in the reconstruction and value of PBC=100 was used in all iterations. RELION refinement using the “3D auto-refine” option was carried out for 19 iterations. Refinement parameters were as follows: CTFs until first peak were ignored and an initial low-pass filter of 20 Å was used. Particle diameter was set to 220 Å and no reference mask was used. Initial angular sampling interval was set to the default value of 7.5 degrees and the offset search range and offset search step also set to default values of 5 and 1 pixels respectively. Local searches from auto-sampling were set to the default value of 1.8 degrees as well.
Maps obtained using either FREALIGN or RELION were verified to display the same salient structural features, and displayed similar resolution values in Fourier Shell Correlation (FSC) plots (Supplementary Figure 2a-2c). FSC resolution plots were obtained with EMAN’s proc3d command using the unfiltered half-maps with soft spherical masks applied to eliminate the influence of the background in the resolution estimates. The curves show the “gold-standard” curve obtained using RELION and the curve obtained from the correlation of two halves of the data set obtained using FREALIGN and indicate resolution values of ~ 6 Å at 0.143 FSC and ~ 8 Å at 0.5 FSC cutoff values. For the purpose of visualization, maps were corrected by a B-factor of −600 and low-pass filtered to the 0.143 FSC-cutoff resolution of 6 Å. The final map was also validated using the tilt-pair parameter plot 44,45 (Supplementary Figure 2d). 383 particles were selected manually from 19 sets of tilt-pair images using the program e2RCTboxer.py in the EMAN2 suite of programs46. Using the 6 Å density map as the reference 3D model, particle orientations were assigned using FREALIGN and plotted using the TILTMULTIDIFF program44. The spread of angular orientations for most of the tilt pairs is within ~ 12.5°, consistent with expectations of a density map at the resolution obtained for a complex with a polypeptide mass of ~ 400 kD (not counting the disordered glycan shell). Fits of coordinates to the density map and rendering of maps and coordinates were carried out using UCSF Chimera22.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by funds to S.S. and J.L.S.M. from the Center for Cancer Research at the National Cancer Institute, NIH and to S.S. from the NIH IATAP program. We thank J. Mascola (Vaccine Research Center, NIH) for providing VRC03 antibodies, K. Kang and W. Olson (Progenics Inc.) for providing soluble KNH1144 gp140 trimers, S. Fellini, S. Chacko and their colleagues for continued support with use of the Biowulf cluster for computing at NIH, D. Schauder and H. He for assistance with data collection, P. Rao for assistance with collection of the tilt pair images, and L. Earl for helpful discussions and comments.
Footnotes
Author Contributions
A.B, A.M., M.J.B., J.L.S.M. and S.S. analyzed and interpreted data; S.S. collected the cryo-EM data; all authors helped compose the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
ACCESSION CODES:
The density map for the gp140-VRC03 complex and the fitted coordinates have been deposited with the Electron Microscopy Data Bank with accession codes of EMD-xxxx and PDB-xxxx, respectively.
References
- 1.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 2.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang CC, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou T, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pancera M, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci U S A. 2010;107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merk A, Subramaniam S. HIV-1 envelope glycoprotein structure. Curr Opin Struct Biol. 2013;23:268–276. doi: 10.1016/j.sbi.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 9.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 10.Bartesaghi A, et al. Classification and 3D averaging with missing wedge correction in biological electron tomography. J Struct Biol. 2008;162:436–450. doi: 10.1016/j.jsb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank GA, et al. Computational separation of conformational heterogeneity using cryo-electron tomography and 3D sub-volume averaging. J Struct Biol. 2012;178:165–176. doi: 10.1016/j.jsb.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyerson JR, et al. Molecular structures of trimeric HIV-1 Env in complex with small antibody derivatives. Proc Natl Acad Sci U S A. 2013;110:513–518. doi: 10.1073/pnas.1214810110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran EE, et al. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 2012;8:e1002797. doi: 10.1371/journal.ppat.1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris A, et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci U S A. 2011;108:11440–11445. doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders RW, et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol. 2002;76:8875–8889. doi: 10.1128/JVI.76.17.8875-8889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moscoso CG, et al. Quaternary structures of HIV Env immunogen exhibit conformational vicissitudes and interface diminution elicited by ligand binding. Proc Natl Acad Sci U S A. 2011;108:6091–6096. doi: 10.1073/pnas.1016113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu SR, et al. Single-particle cryoelectron microscopy analysis reveals the HIV-1 spike as a tripod structure. Proc Natl Acad Sci U S A. 2010;107:18844–18849. doi: 10.1073/pnas.1007227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, et al. Fabs enable single particle cryoEM studies of small proteins. Structure. 2012;20:582–592. doi: 10.1016/j.str.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Wu X, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao Y, et al. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc Natl Acad Sci U S A. 2013;110:12438–12443. doi: 10.1073/pnas.1307382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon YD, et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci U S A. 2012;109:5663–5668. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J Virol. 2012;86:2919–2929. doi: 10.1128/JVI.06698-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu R, Wilson IA. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol. 2011;85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrison SC. Viral membrane fusion. Nature structural & molecular biology. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Molecular cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 30.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 31.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 32.Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife. 2013;2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annual review of biochemistry. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 34.Huang J, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annual review of immunology. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- 36.McLellan JS, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. 2013;340:1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baquero E, et al. Intermediate conformations during viral fusion glycoprotein structural transition. Current opinion in virology. 2013;3:143–150. doi: 10.1016/j.coviro.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weis WI, Brunger AT, Skehel JJ, Wiley DC. Refinement of the influenza virus hemagglutinin by simulated annealing. Journal of molecular biology. 1990;212:737–761. doi: 10.1016/0022-2836(90)90234-D. [DOI] [PubMed] [Google Scholar]
- 40.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 41.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 42.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 43.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 44.Henderson R, et al. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. Journal of molecular biology. 2011;413:1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. Journal of molecular biology. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 46.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.