

Abstract
p140 Ras-GRF1 and p130 Ras-GRF2 constitute a family of calcium/calmodulin-regulated guanine–nucleotide exchange factors that activate the Ras GTPases. Studies on mice lacking these exchange factors revealed that both p140 Ras-GRF1 and p130 Ras-GRF2 couple NMDA glutamate receptors (NMDARs) to the activation of the Ras/Erk signaling cascade and to the maintenance of CREB transcription factor activity in cortical neurons of adult mice. Consistent with this function for Ras-GRFs and the known neuroprotective effect of CREB activity, ischemia-induced CREB activation is reduced in the brains of adult Ras-GRF knockout mice and neuronal damage is enhanced. Interestingly, in cortical neurons of neonatal animals NMDARs signal through Sos rather than Ras-GRF exchange factors, implying that Ras-GRFs endow NMDARs with functions unique to mature neurons.
Keywords: calcium signaling, NMDA glutamate receptors, Ras, Ras-GRF, Sos
Introduction
Changes in intracellular calcium levels are used to regulate a wide variety of neuronal cell functions. Neuronal response to calcium is dependent not only on the magnitude and temporal properties of calcium changes but also on their spatial properties, including the specific channel through which calcium flows, presumably because this determines the specific signaling complexes with which calcium interacts, (West et al, 2001; Sheng and Kim, 2002). Thus, a major goal in understanding neuronal function is to define how calcium couples to specific signaling components under these different conditions.
Excitatory synapses of the brain primarily use glutamate as a transmitter, and elevation of intracellular calcium at the postsynaptic membrane is a common outcome of the engagement of the family of glutamate receptors (Ghosh and Greenberg, 1995). N-methyl-D-aspartate glutamate receptors (NMDARs) are a particularly important member of this class of receptors since they initiate signaling cascades that underlie synaptic plasticity, neuronal development, survival and excitoxicity (Hardingham and Bading, 2003).
Among the many signaling cascades activated by calcium entering cells through NMDARs are those mediated by the Ras-GTPases (H-Ras, N-Ras and K-Ras) (Sheng and Kim, 2002). Ras proteins can be activated by a wide array of upstream signals, including calcium. For the most part, Ras activation is mediated by one of a family of guanine–nucleotide exchange factors (GEFs) that promote the release of GDP bound to Ras, allowing activating GTP to take its place. Ras is inactivated by its intrinsic GTPase activity, which can be enhanced by GTPase-activating proteins (GAPs). In its active form, Ras can activate a variety of downstream target proteins and signaling pathways, the best characterized of which is the Raf/Mek/Erk kinase cascade (Takai et al, 2001). Although Ras proteins are known primarily for their role in promoting cell proliferation, a growing body of evidence argues that these proteins also play a key role in mediating the functions of both differentiating and fully differentiated neurons. Knockout mice deficient in the GEFs, p140 Ras-GRF1 (Brambilla et al, 1997; Giese et al, 2001) or p130 Ras-GRF2 (unpublished observations) or the GAP, NFI (Silva et al, 1998) impair specific aspects of learning, while H-Ras knockout mice display enhanced long-term potentiation (LTP) (Manabe et al, 2000). These effects of Ras are thought to be mediated in large part by its ability to regulate neuronal gene expression (Xia et al, 1996; Adams and Sweatt, 2002), at least partly through its contribution to the activation of the CREB transcription factor (Impey et al, 2002; Lonze and Ginty, 2002).
Despite the importance of NMDAR signaling to Ras in neurons, the mechanism by which this occurs is just now being revealed. Prime candidates for this function are the highly similar p140 Ras-GRF1 (Shou et al, 1992) and p130 Ras-GRF2 proteins (Fam et al, 1997), since they contain calmodulin-binding domains that promote activation of the Ras-GRFs in response to elevated calcium levels in cells (Farnsworth et al, 1995; Fam et al, 1997). Moreover, p140 Ras-GRF1 and p130 Ras-GRF2 are also expressed abundantly in the neurons, but not in surrounding glial cells of the adult central nervous system and only in a limited number of additional tissues (Guerrero et al, 1996; Gotoh et al, 1997) (ubiquitously expressed smaller versions of Ras-GRF2 have been detected at the mRNA level (Fam et al, 1997), but they have never been demonstrated to encode functional calcium-regulated exchange factors). However, the ubiquitous Sos exchange factors, Sos1 and Sos2, which are best known for their ability to couple tyrosine kinase receptors to Ras, can also mediate Ras activation by calcium. This occurs by calcium-induced activation of the Pyk2 and Src tyrosine kinases, which leads to tyrosine phosphorylation of the Shc adaptor protein. Shc then attracts a Grb2/Sos complex to the plasma membrane, where its target Ras is located (Takai et al, 2001).
In this paper, analysis of cortical tissue from Ras-GRF knockout mice shows that p140 GRF1 and p130 Ras-GRF2 are the predominant mediators of NMDAR activation of the Ras/Erk signaling pathway and maintain CREB activity in neurons from adult, but not neonatal animals, where Sos proteins function in this role instead.
Results
NMDA glutamate receptors signal through Ras-GRFs to activate the Ras/Erk/CREB signaling cascade in neurons from adult mice
Mice lacking Ras-GRF1 and Ras-GRF2 were generated to elucidate their role in calcium signaling in neurons. Behavioral analysis has already detected hippocampal learning defects in both strains of mice, and subtle distinctions were noticed (Giese et al, 2001; unpublished observations). Ras-GRF double knockout mice were then created by crossing these two single knockout mouse strains. Immunoblotting of brain lysates with antibodies that preferentially recognize either Ras-GRF1 or Ras-GRF2 confirmed that ras-grf1(−/−) mice expressed only Ras-GRF2 protein, ras-grf2(−/−) mice expressed only Ras-GRF1 protein and double knockout mice expressed neither Ras-GRF protein (Figure 1A). Similar to the findings of a previous report (Fernandez-Medarde et al, 2002), we detected no gross abnormalities in the brain histology of double Ras-GRF knockout mice (data not shown).
Figure 1.
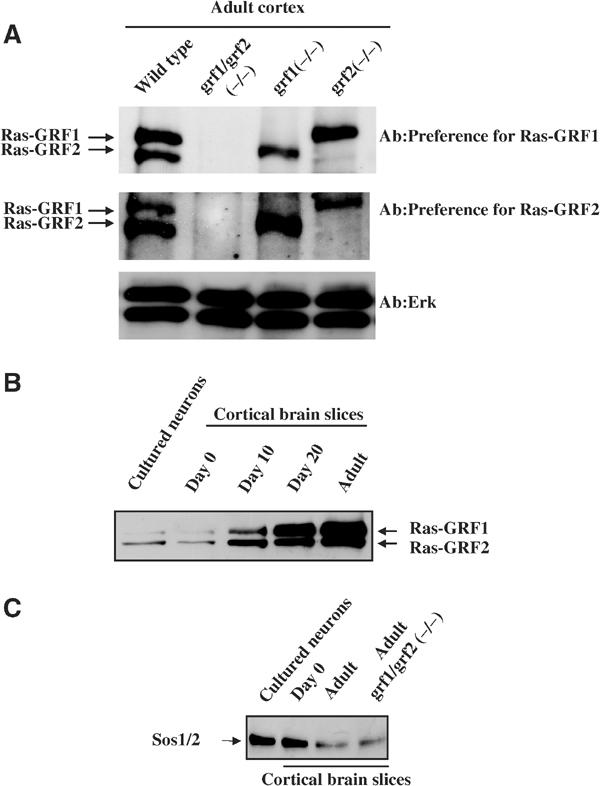
Ras-GRF and Sos expression in wild-type and knockout mice. (A) In all, 10 μg of cell lysates of brain from wild-type, double Ras-GRF knockout (DBKO), single Ras-GRF1 knockout (GRF1(−/−)) or single Ras-GRF2 knockout (GRF2(−/−)) adult mice were probed for Ras-GRF expression by immunoblotting with an antibody that preferentially recognizes Ras-GRF1(top panel) or Ras-GRF2 (middle panel). Double RasGRF knockout mice were generated by crossing Ras-GRF1(−/−) mice with Ras-GRF2 (−/−) mice until double knockout mice were obtained. (B) The expression levels of Ras-GRFs in primary cultures of cortical neurons isolated from newborn mice and cultured for 7 days in vitro, cortical brain slices from newborn (day 0), day 10, day 20 and day 30–40 (adult) mice are shown by immunoblotting 10 μg of cell lysates with antibodies that preferentially recognize Ras-GRF1. (C) The expression levels of Sos proteins in samples similar to those described in (B) using antibodies that recognize both Sos1 and Sos2 proteins, although lysates were from cortical brain slices from adult Ras-GRF double knockout mice, are also shown to indicate that loss of Ras-GRF proteins does not affect their expression.
To determine whether NMDARs function through Ras-GRFs, neurons from control and knockout mice were compared for their responses to NMDAR activation. The expressions of both Ras-GRFs are developmentally regulated. Their levels in the cortex are low in the neonate, with Ras-GRF2 expressed at levels higher than Ras-GRF1. Both GEFs increase during postnatal development, such that they reach peak and comparable levels in animals around day 20 when extensive dendrite elaboration and synaptogenesis occurs and are maintained at high levels through adulthood (7 weeks) (Ferrari et al, 1994; Figure 1B). Thus, studies were initiated using brain slices from mature mice to maximize the possible effects of the loss of Ras-GRF proteins.
Brain slices from the cortex of 30–40-day-old mice were treated with 100 μM NMDA for defined durations and then Erk activation was assessed by immunoblotting lysates with activation-specific Erk antibodies (Figure 2A). NMDA treatment led to an ∼3-fold increase in Erk activation within 5 min in samples from wild-type mice. Strikingly, this effect was completely blocked in brain slices from double Ras-GRF knockout mice. Brain slices from single Ras-GRF1 or Ras-GRF2 knockout mice displayed only small defects, arguing that both Ras-GRF1 and Ras-GRF2 contribute to Erk activation by NMDA. As Ras-GRFs are known to activate Erk through Ras, the activation state of Ras was also measured in these brain slices (Figure 2B). As expected, NMDA-induced Ras activation was observed in brain slices from wild-type mice but not in brain slices from double Ras-GRF knockout mice.
Figure 2.
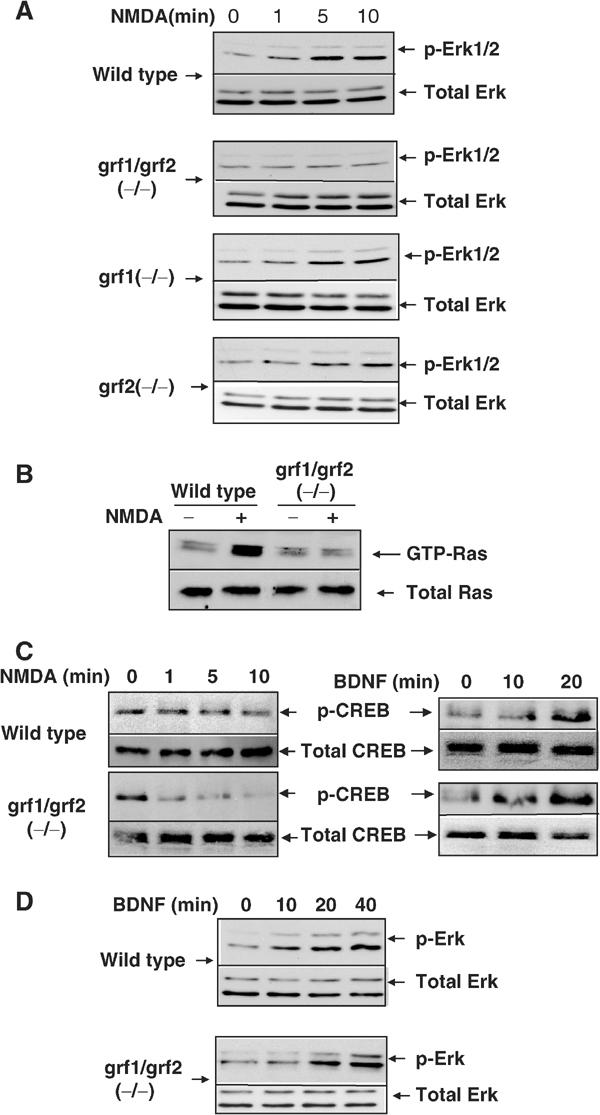
NMDA-induced Erk, Ras and CREB activation in cortical brain slices isolated from wild-type and Ras-GRF knockout mice. (A) Erk activation. Cortical brain slices from adult wild-type and single and double Ras-GRF knockout mice were treated with NMDA (100 μM) for various amounts of time. The slices were then lysed in detergent and immunoblots of lysates were performed using activation-specific phospho-Erk antibodies or antibodies against total Erk 1 and 2. (B) Ras activation. Lysates from 10 min time points described above in (A) were exposed to sepharose beads containing the Ras-binding domain of the Ras target Raf to affinity purify active Ras-GTP. The affinity-purified form of Ras and total Ras in the brain slice in lysates was assayed by immunoblotting with anti-Ras antibodies. The doublet band of Ras-GTP is routinely found in these preparations, presumably due to removal of the C-terminus, which is sensitive to proteolytic cleavage during incubation with beads (unpublished observations). (C) CREB activation. Cortical brain slices were stimulated with either NMDA as described above, or BDNF (100 ng/ml). Lysates immunoblotted with phospho-CREB (ser133) specific antibodies or with total CREB antibodies. (D) BDNF stimulation of Erk. Experiments were performed as in (A), except that BDNF (100 ng/ml) was used to stimulate the brain slices. The results for each section are representative of at least three independent experiments.
NMDA-induced CREB phosphorylation depends at least in part on Erk activation (Shaywitz and Greenberg, 1999; Hardingham et al, 2001). It is also developmentally regulated, such that NMDA produces robust CREB phosphorylation at position Ser133 in neonatal animals. However, in more mature neurons NMDARs also activate a CREB phosphatase that limits the steady-state levels of activated CREB induced by cellular kinases (Sala et al, 2000; Hardingham and Bading, 2002). In fact, we could not reproducibly detect NMDA stimulation of CREB phosphorylation at Ser133 in adult brain slices, presumably because in this mixture of cell types the small enhancement that may have occurred was masked by cells not affected by NMDA treatment (Figure 2C, left). Nevertheless, a role for Ras-GRFs in CREB regulation was supported by the observation that NMDA induced a clear decrease in CREB phosphorylation at Ser133 in brain slices from adult Ras-GRF double knockout mice. Again, brain slices from single p140 Ras-GRF1 or p130 Ras-GRF2 showed only small and barely detectable changes in NMDA-induced phosphorylation of CREB (data not shown). In contrast, BDNF, which functions through the Sos exchange factor, activated CREB similarly in both control and Ras-GRF double knockout mice (Figure 2C, right). Thus, both Ras-GRF family members contribute to the maintenance of CREB phosphorylation after NMDA stimulation, presumably through positive regulation of Erk activity. Ras-GRFs could also maintain CREB activity through negative regulation of a CREB phosphatase.
Not all NMDA-induced changes were altered in knockout mice, since NMDA-induced Jnk kinase dephosphorylation was similar in brain slices from wild-type and mutant mice (data not shown). Moreover, not all Erk activation pathways were blocked because BDNF-mediated activation was normal in brain slices from double knockout mice (Figure 2D).
Ras-GRFs play a protective role in stroke-induced neurotoxicity
To test whether physiological changes associated with the loss of Ras-GRF expression are consistent with defects in NMDA signaling observed in isolated brain slices, we examined the response of knockout mice to an established model for stroke (Asahi et al, 2001), since excessive glutamate signaling through NMDARs is known to occur during ischemia-induced neuronal damage (Lo et al, 2003). In this paradigm, the right middle cerebral artery of mice was occluded for 2 h and then re-perfused to induce transient focal cerebral ischemia. After 24 h, mice were killed, and the volumes of ischemic lesions were measured on both the untreated and treated sides of the brain. Lesions in the brains on the treated side of double knockout mice were ∼2-fold larger than those from wild-type mice (Figure 3A and B). Consistent with previous results on CREB and ERK regulation, single p140 Ras-GRF1 or p130 Ras-GRF2 knockout mice displayed lesions that were only slighter, if at all larger, than those observed on the treated side of control mice (data not shown). These data demonstrated that both Ras-GRF family members normally play a protective role in stroke-induced neuronal damage. This finding is consistent with our conclusion from in vitro experiments using brain slices that Ras-GRFs are involved in the regulation of the activity of CREB, an established survival-promoting transcription factor (Lonze and Ginty, 2002). To gain further support for this model, CREB phosphorylation was measured in brain cell lysates from both treated and untreated sides of the brain of wild-type and double knockout mice 30 min after occlusion. While CREB phosphorylation at Ser133 increased ∼3-fold on the occluded side of the brain of wild-type mice, this effect was reduced dramatically on the occluded side of Ras-GRF double knockout mice (Figure 3C).
Figure 3.
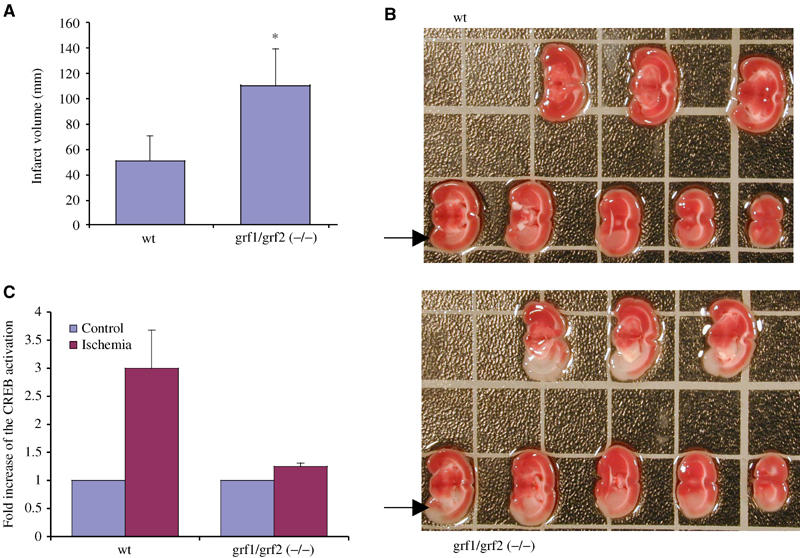
Comparison of neuronal damage and CREB activation in wild-type and Ras-GRF double knockout mice after stroke-induced ischemia. (A) Ischemic lesion volumes were measured 24 h after transient focal cerebral ischemia was produced, by occluding the right internal carotid artery of wild-type (wt) and Ras-GRF double knockout mice (grf1/grf2(−/−)). Data represent the average of eight mice for each group±s.d., P<0.01. (B) Cross sections of a representative experiment used to generate data in (A), with an area of ischemia marked with an arrow. (C) CREB activation in the control half or occluded half of the brain was measured 30 min after the initiation of ischemia in either wild-type or double Ras-GRF knockout mice. The data are the average of three experiments±s.d.
Sos rather than Ras-GRF mediates NMDA induction of Ras/Erk/CREB signaling in neonatal neurons
The vast majority of experiments performed to date to analyze calcium signaling through the Ras/Erk signaling cascade have been carried out on primary neurons dissociated from neonatal mice. However, Ras-GRF levels are relatively low in the neonatal brain (see Figure 1B). To determine whether Ras-GRFs contribute to NMDA-induced Erk activation in neonatal cells, cerebral cortical neurons were isolated from newborn mice and cultured for 7 days in vitro (Xia et al, 1996). Neurons from wild-type and double knockout mice showed no significant differences in the amount of NMDA-stimulated Erk activation (Figure 4A). Similar results were obtained from primary cultures of cerebellar granule neurons isolated from 6–day-old mice and cultured for 7 days (data not shown). Finally, CREB phosphorylation by NMDA treatment was also not affected by the loss of Ras-GRF proteins in these neuronal cultures from double knockout mice (Figure 4B). These results show that Ras-GRF proteins do not play a major role in coupling NMDARs to Erk Map kinase and CREB activation in isolated cortical neurons derived from neonatal mice and cultured for a brief time in vitro.
Figure 4.
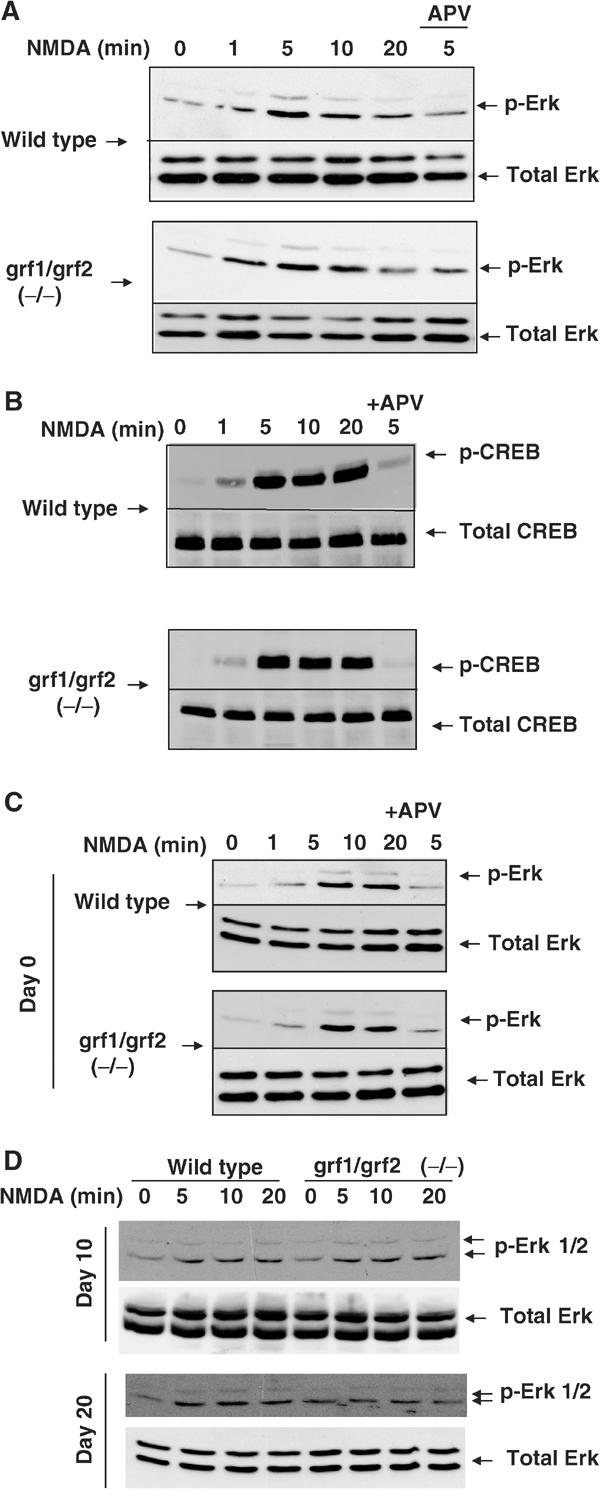
NMDA-induced Erk and CREB activation in neurons from neonatal animals. (A) Primary cultures of cortical neurons isolated from newborn mice of either wild-type or Ras-GRF double knockout mice were cultured in vitro for 7 days and then stimulated with NMDA (100 μM) for various amounts of time. Cell lysates were then assayed for Erk activation as described previously. Some neurons were exposed to the NMDAR inhibitor APV, to show that the effect of NMDA was specific. (B) Wild-type and Ras-GRF double knockout neurons were stimulated as described in (A) and then assayed for CREB activation as described previously. (C) Cortical brain slices were prepared from newborn mice as described previously for brain slices from adult animals. The samples were then stimulated and assayed as described in Figure 2. (D) Cortical brain slices from day 10 or day 20 wild-type or Ras-GRF double knockout mice were prepared and stimulated and assayed as described in Figure 2.
To confirm that the difference in the way NMDARs activate the Ras/Erk signaling was a function of age of the mice and not a function of the neuron preparation, cortical brain slices from newborn, day 10 and day 20 wild-type and Ras-GRF double knockout mice were compared with previous results from adult (30–40 days) mice. Consistent with the hypothesis that the role for Ras-GRFs in coupling NMDARs to Erk is age dependent, brain slices from newborn (Figure 4C) and day 10 double Ras-GRF knockout mice (Figure 4D) showed no impairment of NMDA-induced Erk activation, while brain slices from day 20 (Figure 4D) were similar to adult brain slices (Figure 2A) in showing complete abrogation (Figure 4C) of NMDA-induced Erk activation. A similar age-dependent regulation of CREB phosphorylation by Ras-GRFs was also obtained (data not shown).
In addition to responding directly to tyrosine kinase receptors, the Sos1 and Sos2 Ras exchange factors can also be activated by calcium influx. This occurs through calcium-induced activation of the Pyk (Lev et al, 1995) and Src kinases (Rusanescu et al, 1995), which leads to tyrosine phosphorylation of the Shc adaptor protein and then complex formation with a Grb2/Sos complex. To determine whether Sos proteins could be involved in NMDA-induced activation of the Ras/Erk/CREB signaling pathway in neonatal neurons, complex formation between Shc and Grb2 was assessed as an indicator of potential Sos involvement. Enhanced Shc/Grb2 complex formation was observed after NMDA stimulation of brain slices from newborn mice (Figure 5A, left). In contrast, little, if any, enhancement of Shc/Grb2 complex formation was detected after NMDA stimulation of brain slices from adult mice, consistent with our observation that Ras-GRF is the major mediator of NMDA signaling to Ras in mature neurons (Figure 5A, right). Moreover, preincubation of neurons from newborn animals with the Src inhibitor PP1 suppressed both Erk and CREB activation induced by NMDA (Figure 5B). In contrast, treatment of adult brain slices with the Src inhibitor PP1 had no effect on either Erk or CREB activity (Figure 5C), consistent with the fact that Ras-GRFs are activated directly by calcium/calmodulin binding. Finally, the developmental pattern of Sos protein expression is opposite to that of Ras-GRF, such that Sos levels are higher in the cortex of neonatal animals than in those from adult mice (Figure 1C).
Figure 5.
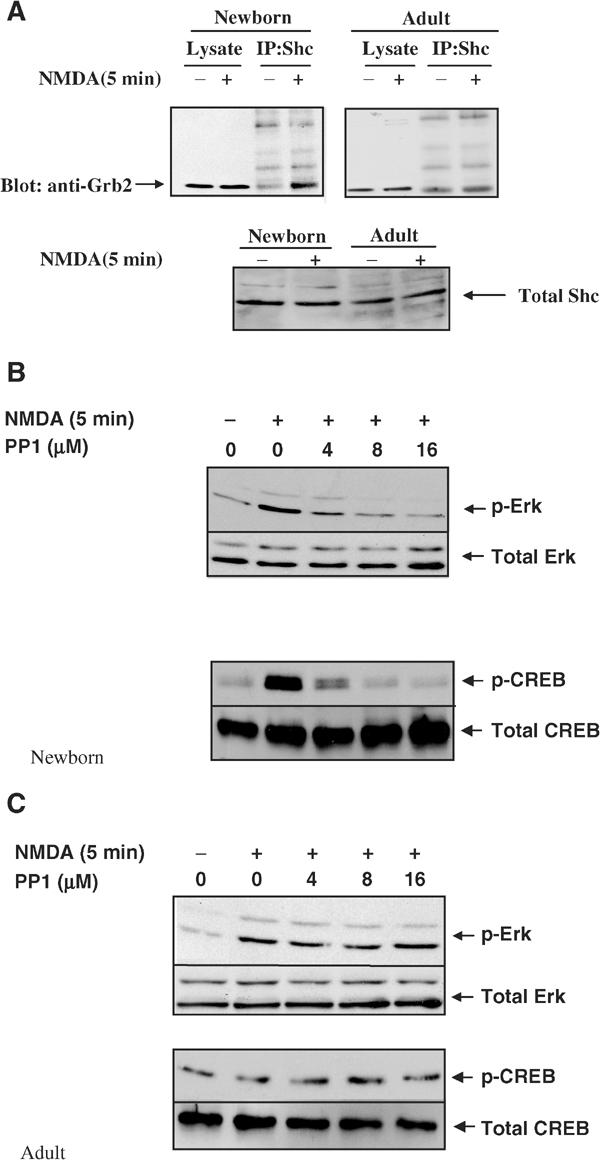
Sos protein involvement in NMDA-induced Erk activation in neurons from neonatal mice. (A) Shc/Grb2 complex formation. Cortical brain slices from neonatal or adult mice were treated with buffer (−) or NMDA (100 μm) (+) for 5 min and then lysates were immunoprecipitated with anti-Shc antibodies and then immunoblotted with anti-Grb2 antibodies. Total Shc in cell lysates in samples was also assessed. The data are representative of two independent experiments. (B, C) PP1 inhibition. Buffer or various concentrations of the Src family inhibitor PP1 were preincubated for 30 min with brain slices from either neonatal mice (B) or adult mice (C). The samples were then stimulated with NMDA and processed as described previously for Erk (top panels) or CREB (bottom panels) activation. The data are representative of at least two independent experiments.
These data argue that NMDARs switch from using Sos exchange factors to Ras-GRF exchange factors to activate Ras and Erk and to maintain CREB signaling upon postnatal development, concomitant with an increase in the expression of Ras-GRF proteins and a decrease in Sos expression.
Discussion
The experiments described in this paper reveal that both Ras-GRF proteins, Ras-GRF1 and Ras-GRF2, couple NMDA glutamate receptors to the Ras/Erk/CREB signaling cascade in neurons from adult mice. This conclusion is based on studies on tissue from knockout mice lacking either Ras-GRF1, Ras-GRF2 or both Ras-GRF family members. Knockout of only one Ras-GRF family member generated only small, difficult to reproducibly detect, defects in NMDAR regulation of Ras and Erk in brain slices from adult. The lack of the expected 50% signaling defect in single knockout mice was not due to a significant increase in the expression of the remaining family member, which suggests instead that, under normal conditions, excess Ras-GRF exchange factor activity exists. Thus, a clear defect in the regulation of Ras and Erk by NMDARs was only observed when both Ras-GRF family members were missing. CREB regulation by NMDARs was also disrupted in brain tissue from these mice. CREB activity in adult tissues is enhanced by kinases including Erk that phosphorylate it at Ser 133. However, CREB phosphatases are highly active in mature neurons and blunt NMDA activation of the transcription factor (Sala et al, 2000; Hardingham and Bading, 2002). Consistent with these findings, we failed to reproducibly detect an increase in CREB phosphorylation at Ser 133 in brain slices from adult mice after NMDA stimulation. However, in Ras-GRF double (but not single) knockout mice, we observed a clear NMDA-induced decrease in CREB phosphorylation. Thus, both Ras-GRFs likely promote the maintenance of CREB activity in the cortex of adult mice upon NMDA signaling, since Erk activity has been shown to contribute to the late phase of CREB phosphorylation after NMDA stimulation (Impey et al, 2002). Ras-GRFs could also potentially accomplish this task by suppressing CREB phosphatases, although this possibility is less likely since we found that the lack of both Ras-GRFs in Ras-GRF double knockout mice did not suppress BDNF-induced CREB phosphorylation at Ser 133.
These findings imply that Ras-GRF1 and Ras-GRF2 play redundant roles in coupling NMDARs to Ras/Erk and CREB signaling, which is consistent with the fact that most CNS neurons contain both proteins (unpublished observation). However, subtle differences in the functions of an individual Ras-GRF family member may be revealed when neurons are activated by glutamate under conditions that are more natural than those used in this study. Moreover, neurons that predominantly express one Ras-GRF family member likely exist in the CNS, and thus in these cells one Ras-GRF may play the major role in calcium signaling. These possibilities are consistent with the fact that Ras-GRF1 (Itier et al, 1998), but not Ras-GRF2 (Fernandez-Medarde et al, 2002), knockout mice have been found to be smaller in size than normal mice, due at least in part to the impaired growth hormone secretion in Ras-GRF1 knockout mice (Itier et al, 1998).
An equally important conclusion from these studies is that the participation of Ras-GRFs in coupling NMDARs to Ras/Erk/CREB signaling is developmentally regulated. No detectable defect in NMDAR activation of the Ras/Erk/CREB signaling cascade was observed in brain slices derived from newborn or postnatal day 10 double Ras-GRF knockout mice. However, at postnatal day 20 and beyond, contributions from both Ras-GRFs to this signaling cascade became apparent.
We found evidence to support the idea that in neonatal neurons, NMDARs signal through the Sos exchange factors instead of the Ras-GRFs. Sos proteins are best known for their ability to be activated in response to ligands that activate tyrosine kinase receptors, and they play such a role by mediating neurotrophin function through Trk receptors in neurons. However, Sos proteins also become stimulated in response to elevated calcium levels in neurons through activation of intracellular tyrosine kinases, such as Src and Pyk2. These kinases phosphorylate the Shc adaptor protein, which then recruits a Grb2/Sos complex to its target Ras in the membrane (Takai et al, 2001). We demonstrated the engagement of Shc with Grb2 in neonatal brain slices stimulated with NMDA. In addition, NMDA receptor activation of the Ras/Erk/CREB signaling cascade in this tissue was blocked when an Src inhibitor was introduced.
The mechanism by which NMDARs switch from Sos to Ras-GRF signaling during postnatal mouse development is not yet clear, although developmentally regulated changes in the expression of both Ras-GRFs and Sos proteins may be involved. In particular, we have shown that Sos protein expression is at its highest in neonatal neurons, when we have shown that it is functionally engaged with NMDARs, while the levels of Ras-GRF1 and Ras-GRF2 are at their lowest. In the adult, the pattern of expression and functional significance of exchange factors in NMDAR signaling is reversed. Sos levels drop dramatically during postnatal development and are at their lowest levels in the adult. Moreover, we found no evidence for NMDA signaling to Sos proteins in adult tissue, since we no longer detected an NMDA-induced increase in Shc/Grb2 complex formation, and we no longer observed suppression of NMDA induction of the Ras/Erk/CREB signaling cascade by an Src inhibitor. The reason for the loss of Sos coupling to NMDA signaling in the adult is clearly more complex than just a decrease in Sos expression, because Sos proteins are still capable of coupling neurotrophins to Ras in adult neurons and the defect we found was upstream of Sos. Presumably, signaling components needed to activate Sos through NMDARs, such as Shc or Grb2, are unable to respond to a calcium influx mediated by NMDARs in adult tissue, possibly because of their distinct subcellular localization in adult neurons.
In contrast to Sos, Ras-GRF1 and Ras-GRF2 accumulate during postnatal development and are at their highest levels in the adult. It is at this time, not in the neonate, that they play a dominant role in connecting NMDARs to Ras/ERK/CREB activity. The subunit structure of NMDARs changes during postnatal development, such that the receptors contain predominantly NR1/NR2B subunits in neonatal animals and NR1/NR2A subunits in more mature animals. This change is associated with an age-dependent increase in the concentration of NR2A, while the level of the NR2B remains constant (Cull-Candy et al, 2001). This raised the possibility that the age-dependent coupling of NMDARs to Ras-GRFs derives at least in part by the co-appearance of NR2A subunits in NMDARs along with Ras-GRFs during postnatal development. However, while the present paper was under review, Krapivinsky et al (2003) reported that Ras-GRF1 couples NMDARs to Erk activation by binding to the NR2B, not NR2A subunit of the NMDA receptor. The predominance of NR2B-containing NMDARs in young animals led to the hypothesis that Ras-GRF1 is mainly involved in regulating neonatal neurons, presumably to contribute to neuronal differentiation (Krapivinsky et al, 2003). Despite this correlation, our data show that neither Ras-GRF1 nor Ras-GRF2 play a significant role in regulating NMDA activation of Ras and Erk in neonatal animals, but instead are the predominant mediators of this function in fully differentiated neurons. Consistent with this view is the fact that complexes between endogenous Ras-GRF1 and NR2B can be detected in brain lysates from adult animals (Krapivinsky et al, 2003). Also, we did not detect any gross defects in brain morphology in Ras-GRF1, Ras-GRF2 or double Ras-GRF knockout mice (unpublished observations). Moreover, we have shown that Ras-GRF2 plays an equally important role in coupling NMDARs to Erk signaling in mature cortical neurons, yet it is different from Ras-GRF1 in the region critical for binding to the NR2B subunit of the NMDAR (Krapivinsky et al, 2003). These results imply that either Ras-GRF2 responds specifically to calcium influx from NR2B-containing NMDARs through a different localizing mechanism, that Ras-GRF2 couples Erk activation to a different NMDAR subunit or that Ras-GRF2 couples Erk indiscriminately to all forms of NMDARs. These possibilities are presently under investigation.
To complement our biochemical studies assessing the loss of Ras-GRF function in neurons, we searched for physiological defects in Ras-GRF knockout mice. We began by examining the response of Ras-GRF knockout mice to an established model for stroke (Asahi et al, 2001), since excess signaling through NMDARs is known to play a central role in promoting ischemia-induced neuronal damage (Lo et al, 2003), and CREB is a known survival-promoting transcription factor. Consistent with our biochemical studies in brain slices that showed a positive role for both Ras-GRF1 and Ras-GRF2 in the maintenance of CREB phosphorylation, we found that both Ras-GRF family members normally play protective roles in this process, since ischemia-induced lesions were over two-fold larger in double Ras-GRF knockout mice than control mice, while single knockout mice showed only small, if any, effects.
Furthermore, we compared CREB phosphorylation in the brain lysates of wild-type and Ras-GRF double knockout mice 30 min after the initiation of ischemia, since cerebral artery occlusion has been shown to activate CREB phosphorylation through NMDARs (Mabuchi et al, 2001). The signaling events that occur after ischemia are clearly more complex than in our in vitro experiments, where we did not see enhanced CREB phosphorylation, presumably because of concurrent activation of CREB phosphatases (Sala et al, 2000). Nevertheless, we observed that ischemia-induced CREB phosphorylation on Ser133 observed on the treated side of the brain of control mice was suppressed in double Ras-GRF knockout mice. Thus, these in vivo experiments are consistent with our in vitro experiments on brain slices, both of which concluded that Ras-GRFs contribute to the maintenance of CREB phosphorylation after NMDAR stimulation.
The finding of a neuroprotective effect and a positive role in CREB regulation for Ras-GRF1 and Ras-GRF2 are striking in light of recent reports showing that NR2B subunits are enriched in extra-synaptic NMDARs (Hardingham and Bading, 2002; Brickley et al, 2003), which promote both the suppression of CREB activity through CREB phosphatase activation and the stimulation of cell death (Hardingham et al, 2002). As Ras-GRF1 binds to NMDARs containing NRB2 subunits (Krapivinsky et al, 2003), the neuroprotective effect of Ras-GRF1 presumably limits the negative effects that other downstream effectors of extra-synaptic NMDARs have on cell survival. Through which class of NMDARs Ras-GRF2 exerts its neuroprotective effects remains to be determined.
In contrast to the present study, Krapivinsky et al (2003) reported no effect of Ras-GRF1 on the phosphorylation state of CREB at Ser 133. The reason for this difference is not yet clear, but it may be the consequence of differences in the types of neurons studied and/or differences in the method used to inhibit Ras-GRF function. Krapivinsky et al used hippocampal neurons isolated from day 18 rat embyros and then cultured in vitro for 14 days, compared to cortical brain slices from day 20 and older mice used in the present study. Another important difference may be that Krapivinsky et al suppressed Ras-GRF function in cells by transfecting a cDNA encoding a peptide thought to block the interaction of NMDARs with Ras-GRF1, but not Ras-GRF2 in primary hippocampal neurons. Our results with knockout mice show that we can only clearly detect a defect in NMDAR signaling when the functions of both Ras-GRFs are missing.
NMDARs switch from signaling through Sos proteins to signaling through Ras-GRFs in the cortex between postnatal day 10 and day 20, and continue using the latter GEF throughout adulthood. Thus, for the most part, NMDARs use Ras-GRFs to signal to Ras and Erk after gross morphological development in the cortex has taken place and active synaptogenesis has been completed. Presumably, Ras-GRFs endow NMDARs with new functions required of differentiated neurons that cannot be accomplished by Sos proteins in neonatal neurons. Although Ras-GRFs and Sos1 and Sos2 are similar in that they both activate Ras and the Raf/Erk kinase cascade in response to calcium, the Raf/Erk signaling cascade is only one of the many known downstream effector pathways emanating from Ras. Others are mediated by additional Ras targets, including PI3 kinase, Ral-GEFs, Tiam1, Nore1 and Rin proteins (Takai et al, 2001). Moreover, both Ras-GRFs and Sos have a second exchange factor domain that activates Rac GTPases, which modulate the activity of at least 10 different effector proteins (Takai et al, 2001). Recent experiments have shown that GEFs not only activate GTPases but also participate in the selection of specific GTPase targets for activation (Zhou et al, 1998; Buchsbaum et al, 2003) by binding specific GTPase effectors directly or by binding scaffold proteins that recruit specific effectors to GEF target GTPases. Also, the Ras-activating CDC25 GEF domain of Ras-GRF1, but not that of Sos proteins, activates the Ras-related R-Ras protein (Gotoh et al, 1997; Tian and Feig, 2001), which has been implicated in inside/out integrin signaling. Thus, NMDARs may switch from Sos to Ras-GRF exchange factors upon postnatal development to alter the pattern of effector pathways activated by Ras and Rac family GTPases.
Regulation of gene expression is thought to be a key mechanism by which NMDARs mediate synaptic plasticity, learning and memory. The fact that Ras-GRFs contribute to NMDA receptor signaling to the Ras/Erk/CREB signaling pathway in adults implies that this family of exchange factors plays a key role in mediating some aspects of this physiological process in adults. In fact, previous studies on single Ras-GRF1 and Ras-GRF2 knockout mice have detected changes in amygdala- and hippocampal-dependent learning, although some significant inconsistencies in conclusions from these studies exist (Brambilla et al, 1997; Giese et al, 2001).
Finally, developmental changes in NMDAR signaling have been proposed to contribute to age-dependent neuronal functions, such as increased sensitivity to excitoxicity and the loss of plasticity associated with brain maturation. These changes include alterations in gating properties of the NMDAR (Sheng et al, 1994; Quinlan et al, 1999). NMDARs also activate a CREB phosphatase (Sala et al, 2000; Hardingham and Bading, 2002) and increase their association with specific scaffold proteins in an age-dependent manner (Rao et al, 1998). In this study, we have detected a new developmentally regulated change in NMDAR function, where the receptor changes the mechanism by which it couples to Ras family GTPases. How this ‘exchange factor switching' contributes to the unique properties displayed by mature neurons is presently under investigation.
Materials and methods
Generation of ras-grf1/ras-grf2 double knockout mice
ras-grf1−/− mice have been described previously (Giese et al, 2001). The generation and full characterization of ras-grf2−/−mice will be described in detail in a manuscript in preparation. Briefly, a genomic clone of ras-grf2 was isolated from a 129/Sv mouse genome library. A neomycin gene was inserted into a 10 kb Sal/EcoRI fragment of the first exon to generate a targeting vector (see supplementary material). ES cells were selected for homologous recombination by PCR and chimeras were generated in 129/Sv mice. Male chimeras were mated and heterozygous mutants derived from one targeted clone were intercrossed to obtain homozygous mutants. For the generation of ras-grf1−/−/ras-grf2−/− double knockout mice, homozygous mice of ras-grf1−/− and ras-grf2−/− were crossbred as follows. The first-generation heterozygous for both ras-grf1 and ras-grf2 (ras-grf1+/−/ras-grf2+/−) were crossbred with the ras-grf1−/− mice for the second generation. The expected 1/4 of the offspring from the second generation were ras-grf1−/−/ras-grf2+/− mice. After crossbreeding the ras-grf1−/−/ras-grf2+/− mice for the third generation, the ras-grf1−/−/ras-grf2−/− double knockout mice were obtained in the expected ratio of 1/4. Genotyping of the offspring was performed by PCR of tail DNA extracts and by Western blot for both Ras-GRF1 and Ras-GRF2 proteins. As Ras-GRF1 knockout mice were previously bred into the C57BL/6J background, hybrid mice of the C57BL/6J × 129S1/SvImJ background of the same generation were obtained from The Jackson Laboratory (B6129SF2/J, Bar Harbor, ME, USA) for control experiments. For experiments on single knockout mice, C57Bl/6J or 129/Sv were used. ras/grf1(−/−) mice and double knockout mice were smaller than controls, while ras/grf2(−/−) mice were of normal size.
Primary cultures of cortical neurons
Cortical neurons were prepared from the brains of newborn mice as previously described (Xia et al, 1996). For the NMDA (RBI/Sigma, USA) stimulation, the cultures were incubated with 100 μM NMDA/10 mM KCl for the times indicated in the figures. Inhibitors, 100 μM DL-2-amino-5-phosphonovaleric acid (APV, RBI/Sigma, USA) or PP1 (Sigma), were added to the cultures 30 min before stimulation. Cells were lysed in buffer A containing 10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM NaVO3. Nucleus-free supernatants were subjected to SDS–PAGE and immunoblotting with different antibodies as indicated.
Cortical brain slices preparation
Cortical brain slices (300 μm thick) were prepared as previously described (Vanhoutte et al, 1999), from day 0 pups or 3-month adult mice. The slices were placed into six-well plates (two to four slices per well) with Krebs-Ringer solution (11.1 mM glucose, 1.1 mM MgCl2, 1 mM Na2HPO4, 1.3 mM CaCl2, 25 mM NaHCO3, 120 mM NaCl, 4.7 mM KCl) saturated with 95% O2–5% CO2 at 25°C. The brain slices were incubated 60 min before pharmacological treatment to prevent initial neuronal firing due to the slicing procedure. For the protein activation experiments, 100 μM NMDA or 100 ng/ml brain-derived neurotrophic factor (BDNF, Chemicon, CA, USA) was applied, respectively, at different times as indicated in the figures. For the inhibition experiments, inhibitors were added as described above 30 min before treatment of the brain slices culture. At the end of the experiment, cortical slices were rapidly removed from the wells and immediately lysed in buffer A. Insoluble material was removed by centrifugation (13 000 g for 20 min at 4°C). The prepared samples were subject to immunoblotting for different proteins.
Antibodies
Anti-phospho-Erk1/2 antibody, anti-phospho-Ser133-CREB antibody, anti-phospho-JNK antibody, anti-CREB antibody and anti-JNK antibody were all obtained from Cell SignallingTechnology (MA, USA). Anti-GRF1(C-20), anti-GRF2, anti-Sos and anti-Erk1(K-23) antibodies were obtained from Santa Cruz (CA, USA). Anti-Ras antibodies were obtained from Transduction Laboratories (USA).
Measurement of GTP-bound state of Ras
Ras activation was measured as described previously (de Rooij and Bos, 1997). Briefly, the slices were lysated in buffer 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 200 mM NaCl, 2% NP40, 10% glycerol, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM NaVO3 and 1 mM DTT. Nucleus-free supernatants containing 150 μg total protein were affinity purified using a GST fusion with Ras-GTP-binding domain of c-Raf (GST-Raf-BD) immobilized on S-hexylglutathione-agarose beads (Sigma) by incubating for 1 h at 4°C, after washing 3 × with 25 mM Tris–HCl (pH 7.5), 30 mM MgCl2, 40 mM NaCl, 1% NP40 and 1 mM DTT. Immunoblots were visualized with anti-Ras antibodies (Transduction Laboratories) by ECL.
Focal cerebral ischemia model and evaluation of brain infarction
Forebrain ischemia was induced by bilateral carotid artery occlusion (BCAO) in male WT mice or ras-grf1−/− oras-grf2−/− double knockout mice as described (Asahi et al, 2001). Focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO) using a silicon-coated 8-0 nylon filament. Regional cerebral blood flow was monitored by laser-Doppler flowmetry (FLO-C1, Omegawave, Tokyo, Japan). Animal protocols followed the National Cardiovascular Center's guidelines for animal care and experiments. Brains were cut into coronal slices, and incubated with 2% 2,3,5-triphenyltetrazolium chloride, as described (Asahi et al, 2001). The infarcted areas were measured on each section by an image analysis system (Olympus, Tokyo, Japan), and infarction volume was calculated by summing the infarct areas. Evaluation of brain atrophy volume was carried out using the following formula: (contralateral volume−ipsilateral volume) × 100/contralateral volume.
Supplementary Material
Suppl. Fig 1.
Acknowledgments
We thank Dr Yiping Shen for his help in analyzing brain slices, and Dr Timothy Turner for helpful discussions on the manuscript. This research was supported by grants to LAF from the NCI and the GRASP Digestive Disease Center P30-DK34928, to EHL from NINDS and to XT from the Medical Foundation.
References
- Adams JP, Sweatt JD (2002) Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42: 135–163 [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH (2001) Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci 21: 7724–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R (1997) A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390: 281–286 [DOI] [PubMed] [Google Scholar]
- Brickley SG, Misra C, Mok MH, Mishina M, Cull-Candy SG (2003) NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci 23: 4958–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum RJ, Connolly BA, Feig LA (2003) Regulation of p70 S6 kinase by complex formation between the Rac guanine nucleotide exchange factor (Rac-GEF) Tiam1 and the scaffold spinophilin. J Biol Chem 278: 18833–18841 [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327–335 [DOI] [PubMed] [Google Scholar]
- de Rooij J, Bos JL (1997) Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 14: 623–625 [DOI] [PubMed] [Google Scholar]
- Fam NP, Fan W-T, Wang Z, Zhang L-J, Chen Z, Moran MF (1997) Cloning and characterization of Ras-GRF2, a novel guanine nucleotide exchange factor for Ras. Mol Cell Biol 17: 1396–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995) Calcium activation of Ras mediated by the neuronal exchange factor Ras-GRF. Nature 376: 524–527 [DOI] [PubMed] [Google Scholar]
- Fernandez-Medarde A, Esteban LM, Nunez A, Porteros A, Tessarollo L, Santos E (2002) Targeted disruption of Ras-Grf2 shows its dispensability for mouse growth and development. Mol Cell Biol 22: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari C, Zippel R, Martegani E, Gnesutta N, Carrera V, Sturani E (1994) Expression of two different produces of CDC25Mm, a mammalian Ras activator, during development of mouse brain. Exp Cell Res 210: 353–357 [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268: 239–247 [DOI] [PubMed] [Google Scholar]
- Giese KP, Friedman E, Telliez JB, Fedorov NB, Wines M, Feig LA, Silva AJ (2001) Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1). Neuropharmacology 41: 791–800 [DOI] [PubMed] [Google Scholar]
- Gotoh T, Niino Y, Tokuda M, Hatase O, Nakamura S, Matsuda M, Hattori S (1997) Activation of R-Ras by Ras-guanine nucleotide-releasing factor. J Biol Chem 272: 18602–18607 [DOI] [PubMed] [Google Scholar]
- Guerrero C, Rojas JM, Chedid M, Esteban LM, Zimonjic DB, Popescu NC, Font de Mora J, Santos E (1996) Expression of alternative forms of Ras exchange factors GRF and SOS1 in different human tissues and cell lines. Oncogene 12: 1097–1107 [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001) A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4: 565–566 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2002) Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta 1600: 148–153 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2003) The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26: 81–89 [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414 [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH (2002) Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34: 235–244 [DOI] [PubMed] [Google Scholar]
- Itier JM, Tremp GL, Leonard JF, Multon MC, Ret G, Schweighoffer F, Tocque B, Bluet-Pajot MT, Cormier V, Dautry F (1998) Imprinted gene in postnatal growth role. Nature 393: 125–126 [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40: 775–784 [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J (1995) Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376: 737–745 [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA (2003) Neurological diseases: mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4: 399–414 [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605–623 [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M (2001) Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci 21: 9204–9213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Aiba A, Yamada A, Ichise T, Sakagami H, Kondo H, Katsuki M (2000) Regulation of long-term potentiation by H-Ras through NMDA receptor phosphorylation. J Neurosci 20: 2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999) Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2: 352–357 [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM (1998) Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 18: 1217–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusanescu G, Qi H, Thomas SM, Brugge JS, Halegoua S (1995) Calcium influx induces neurite growth through a Src-Ras signaling cassette. Neuron 15: 1415–1425 [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M (2000) Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci 20: 3529–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861 [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147 [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776–780 [DOI] [PubMed] [Google Scholar]
- Shou C, Farnsworth CL, Neel BG, Feig LA (1992) Molecular cloning of cDNAs encoding a guanine-nucleotide releasing factor for Ras p21. Nature 358: 351–354 [DOI] [PubMed] [Google Scholar]
- Silva AJ, Elgersma Y, Friedman E, Stern J, Kogan J (1998) A mouse model for learning and memory defects associated with neurofibromatosis type I. Pathol Biol (Paris) 46: 697–698 [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T (2001) Small GTP-binding proteins. Physiol Rev 81: 153–208 [DOI] [PubMed] [Google Scholar]
- Tian X, Feig LA (2001) Basis for signaling specificity difference between Sos and Ras-GRF guanine nucleotide exchange factors. J Biol Chem 276: 47248–47256 [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J (1999) Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol 19: 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024–11031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 16: 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Wang Y, Gorski JL, Nomura N, Collard J, Bokoch GM (1998) Guanine nucleotide exchange factors regulate specificity of downstream signaling from Rac and Cdc42. J Biol Chem 273: 16782–16786 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig 1.