

Abstract
Background and Purpose: The orexin system regulates a multitude of key physiological processes, particularly involving maintenance of metabolic homeostasis. Consequently, there is considerable potential for pharmaceutical development for the treatment of disorders from narcolepsy to metabolic syndrome. It acts through the hormonal activity of two endogenous peptides, orexin A binding to orexin receptors 1 and 2 (OX1 and OX2) with similar affinity, and orexin B binding to OX2 with higher affinity than OX1 receptors. We have previously revealed data differentiating orexin receptor subtypes with respect to their relative stability in forming orexin receptor-arrestin-ubiquitin complexes measured by BRET. Recycling and cellular signalling distinctions were also observed. Here, we have investigated, using BRET, the molecular determinants involved in providing OX2 receptors with greater β-arrestin-ubiquitin complex stability.
Experimental Approach: The contribution of the C-terminal tail of the OX receptors was investigated by bulk substitution and site-specific mutagenesis using BRET and inositol phosphate assays.
Key Results: Replacement of the OX1 receptor C-terminus with that of the OX2 receptor did not result in the expected gain of function, indicating a role for intracellular domain configuration in addition to primary structure. Furthermore, two out of the three putative serine/threonine clusters in the C-terminus were found to be involved in OX2 receptor-β-arrestin-ubiquitin complex formation.
Conclusions and Implications: This study provides fundamental insights into the molecular elements that influence receptor-arrestin-ubiquitin complex formation. Understanding how and why the orexin receptors can be functionally differentiated brings us closer to exploiting these receptors as drug targets.
Linked Articles: This article is part of a themed section on Orexin Receptors. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-2
Keywords: BRET, arrestin, GPCR, orexin, hypocretin, ubiquitin
Introduction
The orexin system plays a critical role in maintaining and integrating primordial physiological functions including sleep-wake transitions and metabolic signals controlling energy homeostasis, as well as modulation of addictive behaviour processes and dependencies (Sakurai and Mieda, 2011; Kim et al., 2012).
Both orexin receptors (OX1 and OX2; receptor nomenclature follows Alexander et al., 2013) typically couple with the Gq subclass of G proteins upon stimulation, resulting in release of inositol phosphates and elevation of intracellular calcium levels (Sakurai et al., 1998). In addition, it is evident that both these receptors robustly recruit members of the multi-adaptor protein family, the arrestins (Milasta et al., 2005; Dalrymple et al., 2011).
The formation of arrestin-bound GPCR complexes can confer a wide range of regulatory and cellular signalling functions to the complex. Activated arrestin-bound complexes can desensitize certain signalling cascades, such as those mediated through Gα, and also propagate distinct signalling processes through scaffolding various precursors to pathways resulting in MAPK, c-Src and Akt activation (DeWire et al., 2007). Association with β-arrestin also influences compartmentalization of complexes in the cellular milieu including internalization, recycling and degradation through their ability to bind other structural and regulatory proteins (Moore et al., 2006). Within these complexes, ubiquitination of both receptors and β-arrestins introduces another level of regulation that can influence compartmentalization, trafficking and signalling properties of the complex (Becuwe et al., 2012).
Recruitment and stability of GPCR-β-arrestin interactions is dependent on the affinity of β-arrestin for the GPCR. This typically occurs through phosphorylation of serine and threonine residue clusters in the C-terminal tail of GPCRs by GPCR kinases (GRKs). Phosphorylation of these residues provides the necessary chemical energy to promote high affinity interaction between receptors and β-arrestins (Gurevich and Gurevich, 2006). The presence or absence of clusters of these residues can influence the temporal stability of GPCR-arrestin interactions. This has been characterized for a number of GPCRs including the β2-adrenoceptors, vasopressin V1A and V2 receptors, μ- and δ-opioid receptors, thyrotropin-releasing hormone TRH1 receptors, angiotensin II AT1A receptors and dopamine D2 receptors and may broadly promote separation of GPCRs towards different downstream trafficking and signalling outcomes based upon their relative degree of arrestin association (Oakley et al., 1999; 2000; 2001,,; Kafi et al., 2011).
Using BRET techniques, we have shown that both OX1 and OX2 receptors display relatively high stability in forming complexes with both β-arrestins (Dalrymple et al., 2011). However, in contrast to other receptors that display stable arrestin association, tangible differences between the BRET kinetics of the OX1 and OX2 receptor-β-arrestin complexes were only observed upon prolonged measurement. Extended BRET (eBRET) assays displayed kinetic profiles between β-arrestin, ubiquitin and OX1 receptors that were more transient over a period of 4 h of orexin A stimulation, compared with profiles between β-arrestin, OX2 receptors and ubiquitin which exhibited a more robust and stable kinetic profile (Dalrymple et al., 2011). In addition, temporal ERK1/2 phosphorylation could be similarly subtype specifically distinguished between the orexin receptors over extended periods of agonist stimulation. These long-term departures of orexin receptor-β-arrestin BRET kinetics suggest a mechanism for differential orexin receptor subtype function.
To gain an insight into such possible mechanisms involved in functional orexin receptor subtype distinctions, the contribution of the C-terminal tail of OX2 receptors was investigated through bulk substitution and site-specific mutagenesis. Previous studies that investigated molecular determinants involved in GPCR-β-arrestin interactions revealed the nature of serine/threonine cluster sites primarily phosphorylated by GRKs (Oakley et al., 1999; 2000; 2001,,) and specifically for OX1 (Milasta et al., 2005). Using these principles, the contributions of both the entire C-terminal tail as well as three putative GRK phosphorylation sites within the C-terminal tail of OX2 receptors were assessed to investigate the formation and stability of OX2 receptor-arrestin-ubiquitin complexes and to demonstrate key structural features that defined subtype-specific functions of orexin receptors.
Methods
cDNA constructs and mutagenesis
Haemagglutinin (HA)-tagged human OX1 receptor cDNA was from Missouri S&T Resource Center (Rolla, MO, USA; Cat. No. HCR010TN00). Wild-type human OX2 receptor cDNA was kindly provided by M. Yanagisawa (Howard Hughes Medical Institute, Dallas, TX, USA; Accession No. NM_001526). β-arrestin1 and β-arrestin2 cDNAs were from RZPD GenomeCube, Berlin, Germany. OX2 multiple point mutants were generated in the C-terminus using site-directed mutagenesis to replace serine and threonine residues with alanine. cDNA mutations in OX2 receptors are as follows: ‘Δ399’ (a1195g, a1198g, g1199c, a1201g, a1207g, g1208c) resulting in amino acid mutations T399A, S400A, T401A and S403A; ‘Δ406’ (t1216g, a1222g, a1225g) resulting in amino acid mutations S406A, T408A and T409A; ‘Δ427’ (a1279g, a1282g, g1283c, a1288g, g1289c, a1291g) resulting in amino acid mutations T427A, S428A, S430A and T431A, and a single point mutation (g1204c) ‘E402Q’. The chimeric receptor, OX1 receptor with the C-terminal tail of OX2 receptor (OX1ctOX2), was generated by cleaving OX1 receptors with the PflM1 restriction enzyme, and introducing a PflM1 restriction site in OX2 receptors at the equivalent site by PCR mutagenesis. The C-terminal fragment of OX2 receptor (bases 1054–1335) was ligated with the N-terminal fragment of OX1 receptor (bases 1–1035). To generate cDNA encoding for BRET fusion proteins, sequences were PCR-amplified and subcloned into pcDNA3.1(+) backbone vectors containing Venus yellow fluorescent protein kindly provided by Atsushi Miyawaki (RIKEN Brain Science Institute, Wako City, Japan) or Renilla luciferase 8 (Rluc8) cDNA kindly provided by Andreas Loening and Sanjiv Gambhir (Stanford University, Stanford, CA, USA) as described previously for other GPCR constructs (Kocan et al., 2008). The stop codon between the sequences was removed to generate constructs capable of being translated into fusion proteins upon transfection, as described previously (Pfleger and Eidne, 2003; Jaeger et al., 2010), and all receptors were HA-tagged. BRET-tagged Kras constructs were generously provided by Nevin Lambert, Georgia Regents University, Augusta, GA, USA, and their use has been described previously (Lan et al., 2011; 2012,; Jensen et al., 2013). cDNA encoding Venus-ubiquitin fusion proteins has been described previously (Dalrymple et al., 2011). Fusion cDNA constructs were verified by BDT labelling and capillary separation on an AB3730xl sequencer (Australian Genome Research Facility, Brisbane, Australia) and compared with published sequence data.
Test systems
Cell culture and transfection
HEK293FT cells (Life Technologies, Mulgrave, Vic., Australia) were maintained at 37°C in 5% CO2 and complete media (DMEM) containing 0.3 mg mL−1 glutamine, 100 IU mL−1 penicillin, and 100 μg mL−1 streptomycin (Life Technologies) supplemented with 10% fetal calf serum (FCS, Life Technologies). HEK293FT media also contained geneticin (G418; 400 μg mL−1; Life Technologies). Transfections were carried out 24 h after cell seeding using GeneJuice (Novagen, Merck KGaA, Darmstadt, Germany) according to manufacturer's instructions.
Measurements
Inositol phosphate assays
Inositol phosphate was measured through determination of inositol-1-phosphate accumulation and performed in 96-well microplates using the IP-One HTRF® assay (CisBio Bioassays, Bagnol sur Ceze, France) according to manufacturer's instructions, as described previously (Mustafa et al., 2012). Cells were stimulated with orexin A ligand for 30 min at 37°C before addition of measurement reagents. The assay was incubated for 2 h at room temperature and terbium cryptate fluorescence and time-resolved fluorescence resonance energy transfer signals were measured at 620 and 665 nm, respectively, 60 μs after excitation at 340 nm using the EnVision 2102 multilabel plate reader (PerkinElmer Life Sciences, Melbourne, Vic., Australia).
BRET assays
HEK293FT cells transfected 48 h earlier were harvested and prepared as described previously in 96-well plates (Nunc, Thermo Scientific, Waltham, MA, USA) (Dalrymple et al., 2011). Cells for eBRET assays were resuspended in HEPES-buffered (25 mM) phenol-red free DMEM with 5% FCS to maintain viability. EnduRen™ substrate (Promega, Madison, WI, USA) was added to each well at a final concentration of 60 μM. Cells were maintained for 2 h at 37°C, 5% CO2 for the cell permeable substrate to reach equilibrium. Samples were read sequentially using a VICTOR Light™ 1420 luminescence counter (PerkinElmer Life Sciences) with 400–475 nm (‘donor emission’) and 520–540 nm (‘acceptor emission’) filters, except for Figures 2B, 6A and 6B. For these figures, data were generated using a POLARstar Omega (BMG Labtech, Mornington, Vic., Australia) with 460–490 nm (‘donor emission’) and 520–550 nm (‘acceptor emission’) filters. eBRET kinetics were measured for approximately 30 min to obtain a basal signal. Cells were then treated with vehicle or ligand and read continuously for several hours. Ligand-induced BRET signals were calculated by subtracting the ratio of ‘acceptor emission’ over the ‘donor emission’ for a vehicle-treated cell sample containing both the Rluc8 and Venus fusion proteins from the same ratio for a second aliquot of the same cells that was treated with ligand as described previously (Kocan et al., 2008). The final pretreatment measurement is presented at the zero time point (time of ligand or vehicle addition). BRET signals for assays in the presence of BRET-tagged Kras were calculated by subtracting the ratio of ‘acceptor emission’ over the ‘donor emission’ for a cell sample containing only the Rluc8 fusion protein from the same ratio of a second aliquot of cells containing both the Rluc8 and Venus fusion proteins. For these assays, 15 measurements were taken over the course of 25 min and averaged for each construct.
Data analysis
Data were presented and analysed using Prism 6 graphing software (GraphPad, San Diego, CA, USA). Sigmoidal dose-response curves were fitted using non-linear regression. Statistical significance for dose-response and eBRET kinetic data was determined using one-way anova and Tukey's multiple comparison post hoc tests.
Materials
Orexin A was sourced from the American Peptide Company (Sunnyvale, CA, USA).
Results
Investigation of an OX1 receptor chimera with C-terminal tail of OX2 receptor
Based on the findings of our previous study (Dalrymple et al., 2011), we hypothesized that a chimeric receptor involving the replacement of the C-terminal region of OX1 receptor with that of OX2 receptor (OX1ctOX2; Figure 1) would result in a gain-of-function with respect to arrestin binding stability. BRET data indicating proximity between the cell surface marker, Kras, and OX1, OX2 receptors and the OX1ctOX2 mutant (Figure 2A, B) revealed that a decreased level of the mutant OX1ctOX2 appears to be present at the cell surface, despite maintaining potency and maximal efficacy for orexin A-stimulated inositol phosphate production that was not different from that of either OX1 or OX2 receptors (Figure 2C). These data indicate that although G protein-mediated functions with regard to Gαq-coupling were not sensitive to this bulk alteration, this mutant was impaired in being suitably targeted to the cell surface compared to OX1 and OX2 receptors (although presumably not enough to deplete the receptor reserve given the lack of effect on inositol phosphate signalling). Ligand-induced BRET assays for β-arrestin proximity were carried out in both BRET-tag orientations for comparison (Figure 3). Importantly, for both BRET-tag orientations, OX2 receptors provided a more stable BRET signal due to β-arrestin proximity, than observed with OX1 receptors (Figure 3), consistent with our previous findings using EGFP-tagged OX receptors and Rluc-tagged β-arrestins (Dalrymple et al., 2011). The Venus-tagged receptor orientation resulted in a very low ligand-induced BRET signal for the mutant compared to either wild-type OX receptor subtype (Figure 3A, B). In contrast, the Rluc8-tagged receptor orientation resulted in substantially greater BRET signals between the OX1ctOX2 mutant and both β-arrestin1 and 2 (Figure 3C, D). Interestingly, this OX1ctOX2 mutant displays similar kinetics to OX1 receptor, its profile being less stable over the 4 h measurement period compared to that of OX2 receptor (Figure 3C, D).
Figure 1.
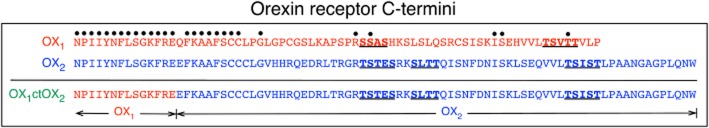
Diagrammatic representation of the primary amino acid structures of the C-termini of OX1, OX2 and the OX1ctOX2 mutant receptors. Dots above residues indicate identical amino acids in OX1 and OX2 receptors when aligned from the NPIIY motif at the end of transmembrane domain 7. The OX1ctOX2 mutant contains amino acids 1–367 of the OX1 receptor and amino acids 374–444 of the OX2 receptor, as indicated. Underlined, bold residues are putative GRK phosphorylation cluster sites in OX1 and OX2 receptors.
Figure 2.
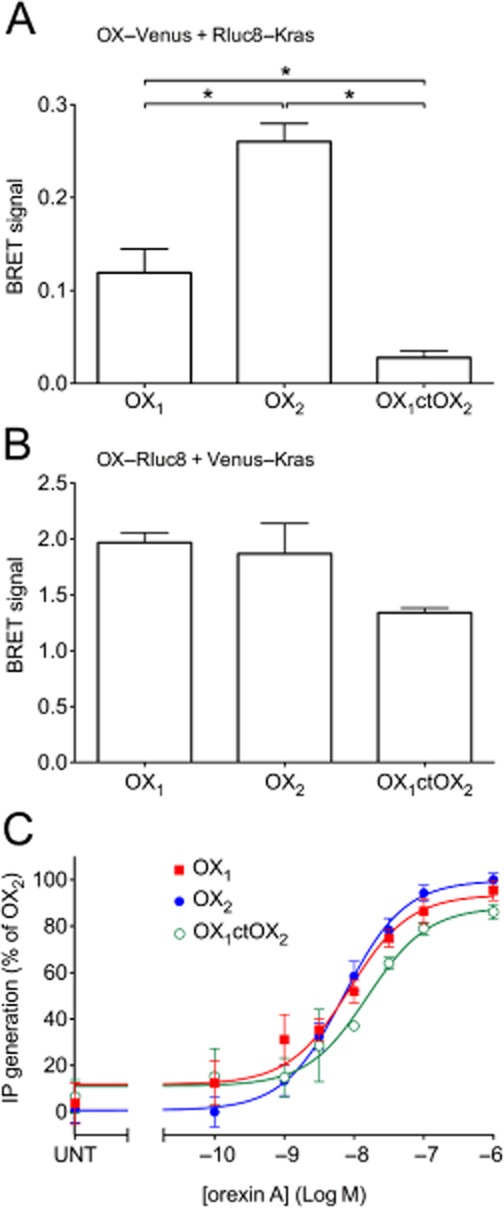
BRET proximity data between Rluc8-Kras and Venus-tagged OX1, OX2, OX1ctOX2 receptors (A), or Venus-Kras and Rluc8-tagged OX1, OX2 and OX1ctOX2 receptors (B). Concentration-response data of inositol phosphate production for OX1, OX2 and OX1ctOX2 receptors. HEK293FT cells were transiently transfected with C-terminally Venus-tagged OX1, OX2 or OX1ctOX2 receptors and treated with orexin A at concentrations shown (C). pEC50 values were as follows: 8.07 ± 0.15 (OX1), 8.15 ± 0.09 (OX2) and 7.81 ± 0.17 (OX1ctOX2). These values were not significantly different from each other (anova; P = 0.28). Significant differences in maximal efficacy were also not observed (anova; P = 0.052). Values for maximal efficacy of OX1 and OX1ctOX2 receptors were 95.4 ± 4.4% and 86.1 ± 3.3% of OX2 receptor respectively. ‘UNT’ refers to untreated cells transfected with each OX receptor construct (C). Data are expressed as mean ± SEM of at least three independent experiments. * P < 0.05, significantly different, as indicated.
Figure 3.
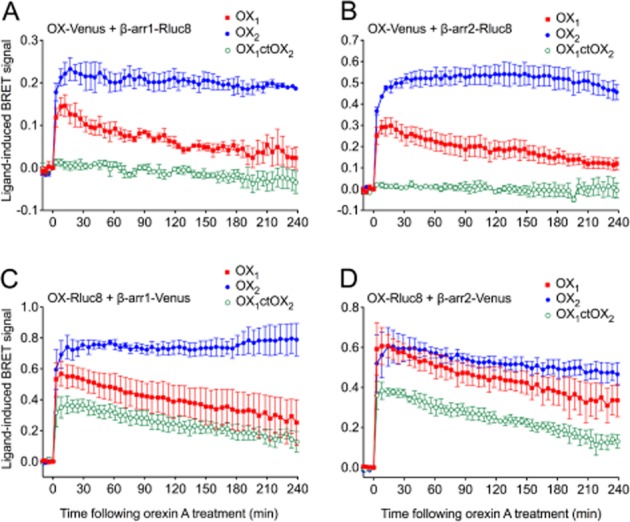
eBRET kinetic data for OX1, OX2 and OX1ctOX2 receptors. HEK293FT cells transiently transfected with C-terminally Venus-tagged receptors and Rluc8-tagged β-arrestin1 (A) or β-arrestin2 (B), or C-terminally Rluc8-tagged receptors and Venus-tagged β-arrestin1 (C) or β-arrestin2 (D) were treated with 0.6 μM orexin A. Data are presented as mean ± SEM of three independent experiments.
Effect of serine/threonine clusters on OX2 receptor-arrestin proximity
To gain more specific insights into the mechanism of orexin receptor-arrestin interaction, serine and threonine residues in defined clusters in the C-terminal tail were mutated to alanine, generating a series of OX2 receptor mutants (Figure 4). BRET proximity time course assays were subsequently carried out between these mutants and β-arrestin1 or 2 in both BRET-tag orientations (Figure 5). Mutation of a single cluster in isolation, except for the Δ406, did not notably reduce the strength of the ligand-induced BRET signal compared to wild-type OX2 receptors (Figure 5A, C, E, G). Interestingly with β-arrestin2, the BRET signal for the Δ406 mutant displays a dramatic change in BRET kinetics when Venus-tagged (Figure 5C). Rluc8-tagged OX2 receptors and each of the single cluster mutants displayed greater BRET signal stability. Nevertheless, the Δ406 mutant appears to display a marginally suppressed BRET signal compared to the other single mutants (Figure 5E, G). The data from both the Venus-tagged and Rluc8-tagged double and triple mutants indicate that the Δ406-Δ427 and Δ399-Δ406-Δ427 mutants display a substantially lowered BRET signal compared to wild-type OX2 receptors (Figure 5B, D, F, H). Interestingly, the Rluc8-tagged receptors display a hierarchy in BRET signal magnitude. A ‘step-wise’ reduction in BRET signal was observed with both β-arrestin subtypes, dependent upon the presence of either the Δ406 or Δ427 cluster, and appears to be additive independently of the Δ399 mutation (Figure 5F, H). These results should also be considered in the context of relative receptor expression levels at the plasma membrane (see below).
Figure 4.

Diagrammatic representation of OX2 and each of the OX2 C-terminal mutant receptors used in this study in terms of primary amino acid structure. Amino acids 360–444 corresponding to the C-terminal tail region of the OX2 receptor are shown. Residues indicated in bold are in the serine/threonine (S/T) clusters that were assessed as putative GRK phosphorylation sites (Oakley et al., 2001). Underlined bold residues (in red) indicate amino acids within each of the clusters that were mutated to alanine. Additionally, glutamate 402 was mutated to glutamine as indicated (in green).
Figure 5.
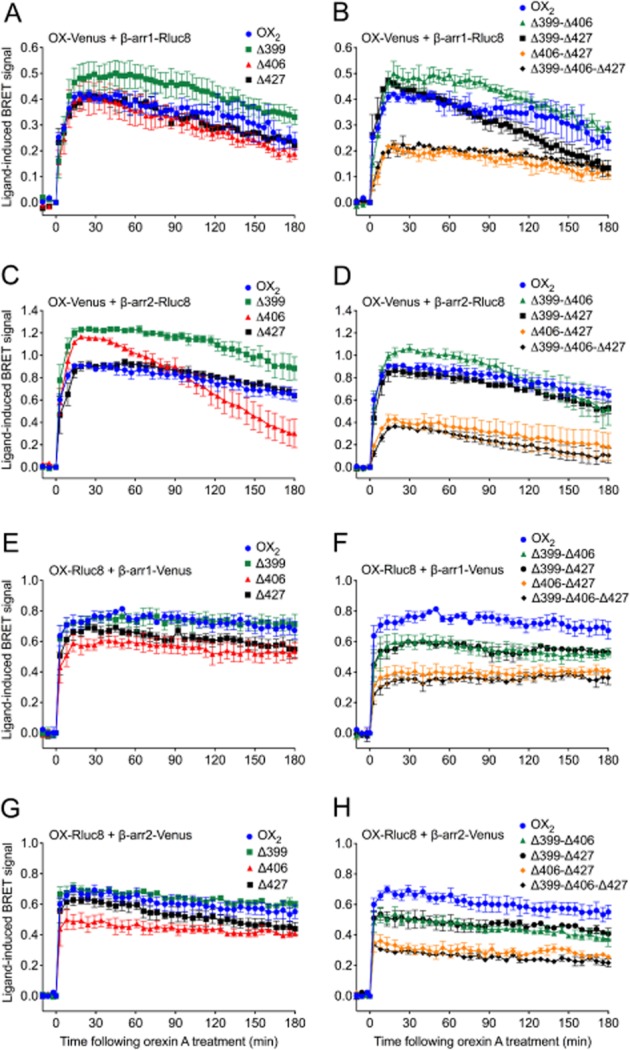
eBRET data indicating proximity between OX2 or OX2 C-terminal tail mutant receptors with β-arrestin1 or 2. HEK293FT cells were transiently transfected with either C-terminally Venus-tagged (A-D), or Rluc8-tagged (E-H) OX2 or each of the single (A, C, E, G) or double/triple (B, D, F, H) C-terminal OX2 mutant receptors in the presence of either Rluc8-tagged β-arrestin1 (A, B) or β-arrestin2 (C, D), or Venus-tagged β-arrestin1 (E, F) or β-arrestin2 (G, H). The zero time point indicates the point at which 0.6 μM orexin A was added. Data are presented as mean ± SEM of three independent experiments.
Effect of serine/threonine cluster mutations on cell surface expression and inositol phosphate production
BRET proximity of OX2 receptors and each of the mutants with the cell surface marker Kras provides insights into their relative plasma membrane expression levels. Notably, there were no significant reductions in receptor-Kras BRET signal with either BRET-tag orientation (Figure 6A, B), indicating that reductions in receptor-arrestin BRET signals relative to wild-type were not as a consequence of reduced receptor plasma membrane expression. Interestingly, with the Venus-tagged receptors, some mutants appear to be expressed at higher levels. More specifically, it is notable that the receptor-arrestin BRET signals for Δ399 are higher than wild-type in Figure 5A and C, and the signal for Δ406 is initially higher in Figure 5C, which correlates with these mutants appearing to be expressed at substantially higher levels on the plasma membrane from the receptor-Kras BRET data. Furthermore, the Δ399-Δ406 double mutant gave the highest receptor-Kras BRET signal of the Venus-tagged double mutants, which may account for the receptor-arrestin BRET signals for this mutant being higher than observed for wild type OX2 receptor (Figure 5B, D). In contrast, all of the Rluc8-tagged mutants displayed similar BRET signals to wild-type OX2 receptor with Kras (Figure 6B). The Venus-tagged Δ406, Δ406-Δ427 and Δ399-Δ406-Δ427 mutants were also analysed and compared to wild-type Venus-tagged OX2 receptor to assess their ability to stimulate the turnover of inositol phosphates (Figure 6C). The potency of all of these mutants was significantly increased compared to wild-type OX2 receptor (P < 0.05). These data indicate that no loss of G protein coupling results from these mutations. Indeed, it is hypothesized that decreased desensitization of G protein-mediated signalling as a consequence of the reduced recruitment of β-arrestins contributes to this increase in G protein coupling potency.
Figure 6.
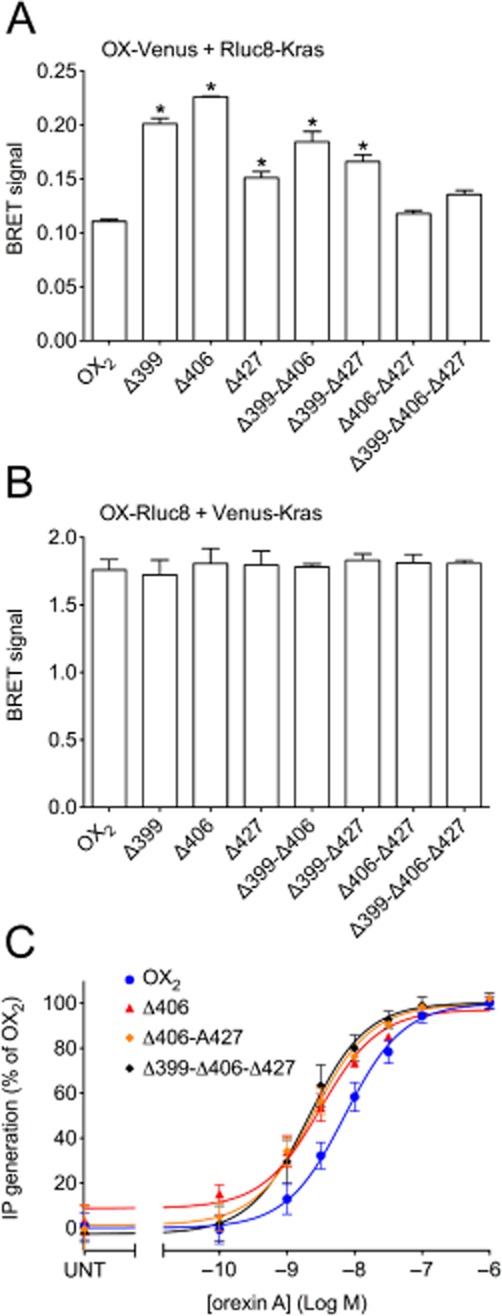
eBRET data indicating proximity between Rluc8-Kras and Venus-tagged OX2 wild-type and mutant receptors (A), or Venus-Kras and Rluc8-tagged OX2 wild-type and mutant receptors (B). Inositol phosphate concentration-response data for OX2 wild-type and mutant receptors. Transiently transfected HEK293FT cells with C-terminally Venus-tagged wild-type OX2 or OX2 mutant receptors (Δ406, Δ406-Δ427 or Δ399-Δ406-Δ427) were treated with doses of orexin A as shown (C). pEC50 values were: 8.15 ± 0.09 (OX2); 8.51 ± 0.08 (Δ406); 8.62 ± 0.09 (Δ406-Δ427) and 8.69 ± 0.10 (Δ399-Δ406-Δ427). Values for maximal efficacy as a percentage of OX2 receptors are as follows: 101.2 ± 0.2 (Δ406), 102.1 ± 1.7 (Δ406-Δ427), 101.1 ± 3.3 (Δ399-Δ406-Δ427). ‘UNT’ refers to untreated cells transfected with each OX receptor construct (C). Data are presented as mean ± SEM of at least three independent experiments. * P < 0.05, significantly different from wild-type OX2 receptor.
Comparison of receptor-arrestin interaction potency
Dose-response data of orexin A-stimulated BRET between Venus-tagged OX2, OX2 Δ406 or OX2 Δ406-Δ427 receptors and β-arrestin2 are shown for an early time point (20 min; Figure 7A) and a later time point (120 min; Figure 7B). The potency of β-arrestin2 recruitment to the Δ406 mutant was not reduced sufficiently to reach statistical significance compared to wild-type OX2 receptor (Figure 7). In contrast, the potency observed with the Δ406-Δ427 mutant was fivefold lower compared to OX2 receptor, and this difference was statistically significant at the earlier time point (Figure 7A; P < 0.05). Maximal efficacy of the Δ406-Δ427 mutant was also substantially reduced compared to both OX2 and the Δ406 mutant receptors (Figure 7). These findings are consistent with the eBRET kinetic data suggesting that the Δ406-Δ427 mutant is substantially impaired in its ability to recruit β-arrestins (Figure 5B, D, F, H). Interestingly, maximal efficacy of the Δ406 mutant was significantly reduced relative to the wild-type OX2 receptor after 120 min of orexin A stimulation (Figure 7B), but not after 20 min of stimulation (Figure 7A), consistent with the kinetic profile shown in Figure 5C.
Figure 7.
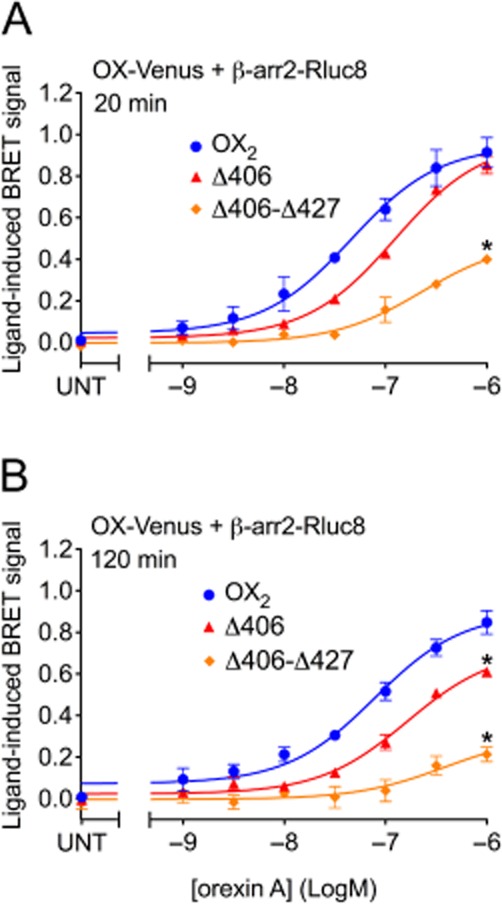
eBRET dose-response data indicating proximity between β-arrestin2 and OX2, or OX2 mutants Δ406 or Δ406-Δ427, at 20 and 120 min post-agonist stimulation. pEC50 values were as follows: 7.34 ± 0.11 (OX2), 6.91 ± 0.05 (Δ406), 6.64 ± 0.14 (Δ406-Δ427) at 20 min; 7.11 ± 0.10 (OX2), 6.79 ± 0.08 (Δ406), 6.42 ± 0.43 (Δ406-Δ427) at 120 min. Maximal BRET efficacy values are as follows: 0.91 ± 0.07 (OX2), 0.85 ± 0.04 (Δ406), 0.40 ± 0.02 (Δ406-Δ427) at 20 min; 0.85 ± 0.06 (OX2), 0.61 ± 0.01 (Δ406), 0.21 ± 0.03 (Δ406-Δ427) at 120 min. Data are presented as mean ± SEM of three independent experiments. * P < 0.05, significantly different from OX2 receptors.
Investigation of glutamate as a potential phosphate mimic in the proximal serine/threonine cluster
To investigate the possibility that the negatively charged glutamate residue in the Δ399 cluster (E402) may have a contributing effect on the stability of the interaction with β-arrestin, a comparison was made between the mutants Δ399 and Δ399-E402Q, and between Δ399-Δ406-Δ427 and Δ399-Δ406-Δ427-E402Q (Figure 4). However, no notable deviations in BRET signal kinetics or magnitude were observed as a consequence of this additional mutation (Figure 8).
Figure 8.
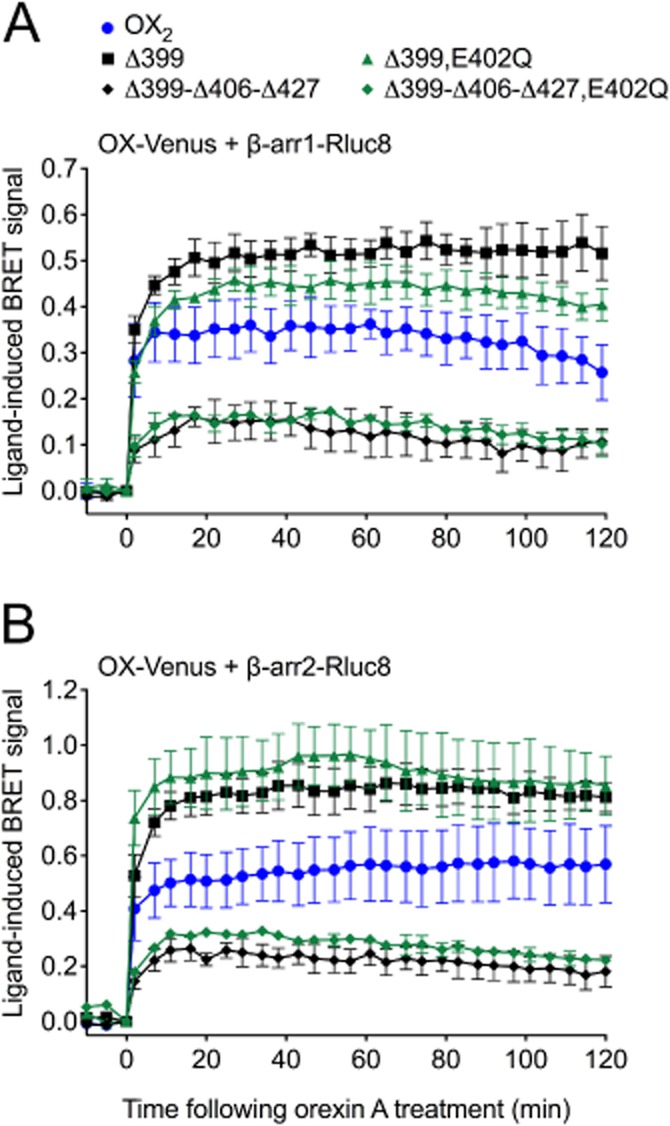
eBRET data comparing proximity between β-arrestin1 or 2 and OX2, OX2 Δ399 or OX2 Δ399-Δ406-Δ427 receptors, with or without the E402Q mutation. HEK293FT cells were transiently transfected with Venus-tagged OX2 or mutant receptors and either Rluc8-tagged β-arrestin1 (A) or β-arrestin2 (B). The zero time point indicates when 0.6 μM orexin A was added. Data are presented as mean ± SEM of three independent experiments.
Ubiquitin-arrestin proximity in the presence of OX2 receptor and serine/threonine cluster mutants
In an alternate BRET configuration, the orexin receptor complex was observed through the measurement of proximity between β-arrestin2 and ubiquitin in the presence of non-BRET-tagged receptors to reveal further properties of these mutant OX2 receptor complexes. BRET proximity assays revealed a robust and stable signal for OX2 and a less sustained kinetic signal for OX1 receptors (Figure 9), as observed previously (Dalrymple et al., 2011). In contrast, and consistent with the receptor-arrestin proximity (Figure 5) and dose-response data (Figure 7), diminished arrestin-ubiquitin proximity was observed in the presence of the stimulated OX2 Δ406-Δ427 mutant, with the BRET signal returning to baseline levels sooner than with OX1 or OX2 receptors. Interestingly, the kinetic BRET profile observed in the presence of OX2 Δ406 mutant receptors was essentially identical to that observed with OX1 receptors (Figure 9).
Figure 9.
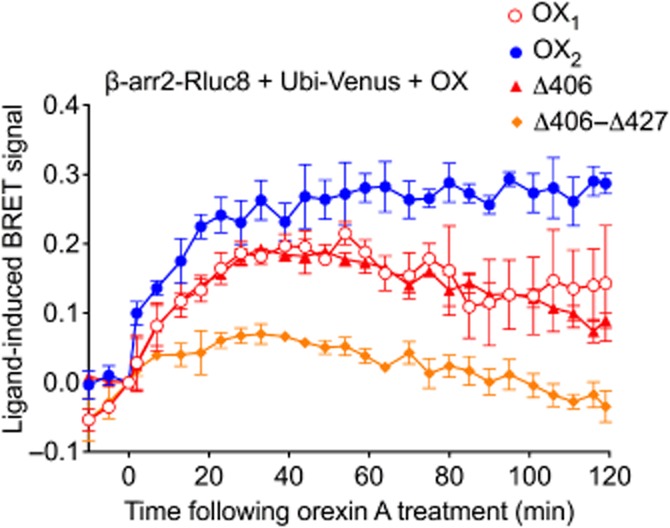
eBRET data indicating proximity between ubiquitin and β-arrestin2 in the presence of wild-type OX1, OX2 or OX2 mutant receptors. HEK293FT cells were transiently transfected with N-terminally Venus-tagged ubiquitin, C-terminally Rluc8-tagged β-arrestin2 and non-BRET-tagged OX1, OX2, OX2 Δ406 or OX2 Δ406-Δ427 receptors. The zero time point indicates when 0.6 μM orexin A was added. Data are presented as mean ± SEM of three independent experiments.
Discussion and conclusions
We have previously shown that the two orexin receptor subtypes display diverging BRET kinetic profiles when forming β-arrestin and ubiquitin complexes, with differences in cellular localization and signalling also being observed (Dalrymple et al., 2011). OX2 receptors displayed greater stability over time when forming β-arrestin complexes compared with OX1 receptors, along with a sustained ability to maintain phosphorylated ERK1/2 while being unable to rapidly recycle upon internalization. Prior to that study, OX1 receptors had been found to colocalize with β-arrestin1 upon orexin A stimulation using confocal microscopy (Evans et al., 2001) and specific sites in these receptors were shown to be involved in β-arrestin interaction (Milasta et al., 2005). Our aim was therefore to establish the molecular determinants responsible for OX2 receptor interactions with β-arrestin and to understand the molecular basis for the differences in OX1 and OX2 receptor function that we had observed, in terms of receptor-arrestin-ubiquitin complex stability.
The OX1ctOX2 chimera (Figure 1) was generated to investigate whether the C-terminal tail of OX2 receptors could bestow greater stability to the OX1 receptor-β-arrestin complex. However, ligand-induced BRET assays indicated that BRET-tag orientation made a substantial difference to our ability to detect a β-arrestin proximity BRET signal specifically for this chimera, in contrast to the wild-type receptors (Figure 3). The cell surface expression of the Venus-tagged OX1ctOX2 chimera was significantly reduced, compared with that of OX1 and OX2 receptors (Figure 2A), which may contribute to the reduction in β-arrestin proximity BRET signal observed. However, as the inositol phosphate generation was not affected (Figure 2C), it is unlikely that cell surface expression alone was responsible for the almost complete abolition of this BRET signal. Indeed, this finding implies that the conformation of the OX2 C-terminal tail is different (and therefore orients the BRET-tag differently) when attached to the rest of the OX1 receptor, compared with the conformation when attached to the rest of the OX2 receptor.
In contrast, although the cell surface expression may have been slightly reduced with the Rluc8-tagged OX1ctOX2 chimera (Figure 2B), a BRET signal was observed for proximity to the Venus-tagged β-arrestins (Figure 3C, D). Notably, the resultant kinetic profile of Rluc8-tagged OX1ctOX2 was similar to that of OX1 and not OX2 receptors. These findings indicate that the primary structure of the OX C-terminal tail is not wholly responsible for determining the stability of the interaction with β-arrestins.
OX1 and OX2 receptors share significant homology in primary structure except for the N-terminus, distal region of the C-terminal tail and the third intracellular loop (ICL3) (Sakurai et al., 1998; Voisin et al., 2003). There are very few differences in ICL1 and 2, with those present not likely to have a major effect on β-arrestin recruitment. Regarding ICL3, the only putative high affinity GRK phosphorylation site is conserved (T250, T251, S252 in OX1 and T258, S259, S260 in OX2) and is in the proximal region of the loop that is itself highly conserved. Beyond this, OX1 ICL3 has two serines and a threonine spread through the loop, whereas OX2 ICL3 has three serines and two threonines, again not clustered. Therefore, from the perspective of primary structure, there are no obvious differences between the ICL3 of OX1 and OX2 receptors in terms of high affinity GRK phosphorylation sites. However, it is likely that the conformation of ICL3 differs between the receptor subtypes and how configuration of this with the C-terminus influences high affinity β-arrestin interaction is certainly worthy of future investigation.
It is our hypothesis that the lack of gain of function observed with the OX1ctOX2 mutant with respect to β-arrestin binding is due to disruption of secondary or tertiary structure, within or between the intracellular domains. This indicates that appropriate GRK phosphorylation sites need to be not only present, but correctly positioned and orientated for both phosphorylation and β-arrestin interaction. Therefore, although there are notable examples of C-terminal tail chimeras adopting the characteristics of the substituted C-terminal tail, such as β2-adrenoceptor-V2-tail or V2-β2-adrenoceptor tail chimeras (Oakley et al., 1999; Shenoy and Lefkowitz, 2003; Tohgo et al., 2003) as well as chimeras of β2-adrenoceptor-AT1A (Anborgh et al., 2000) and NK1-PAR2 receptors (Pal et al., 2012), our data show that this is not always the case. Indeed, we have previously published work investigating the effect of extracellular loop substitution on ligand binding and signalling properties of the gonadotrophin-releasing hormone receptor (Pfleger et al., 2008), where interactions between loops appeared to play a role. It is likely that similar interactions between the intracellular loops and C-terminal tail are involved in configuring intracellular binding sites for β-arrestin. Therefore, although the C-terminal tail of OX2 receptors may function well in the spatial context of the rest of the OX2 receptor, it may not when set amongst the intracellular loops of the OX1 receptor. It is also possible that homo- or heteromerization could play a role.
To further elucidate the molecular determinants of orexin receptor-arrestin interactions, three serine/threonine clusters in the C-terminal tail of OX2 receptors were analysed for their ability to affect OX2 receptor-β-arrestin binding strength and stability (Figure 4). The BRET-tag orientation of the OX receptor constructs appears to have some influence on their relative cell surface expression levels (Figure 6A, B) and general stability of the kinetic profiles (Figure 5), however, taking this into account, the overall effects of the mutations relative to wild-type are largely consistent, regardless of BRET-tag orientation (Figure 5). Mutation of any of the cluster sites in isolation had little detrimental impact on the initial strength of the receptor-arrestin interaction, with the possible exception of the Δ406 mutant with Rluc8-tagged receptors. This contrasts with receptors such as AT1A and oxytocin receptors that have a similar complement of phosphorylation clusters, but only require mutation of a single cluster to significantly disturb arrestin translocation (Oakley et al., 2001).
As GRK phosphorylation increases receptor affinity for β-arrestins as a consequence of introducing negative charge, it was postulated that the negatively charged glutamate within the 399 cluster could act as a phosphate mimic, as described previously (Gurevich and Gurevich, 2006). However, our data from the Δ399-E402Q mutant compared to Δ399 (and indeed the triple cluster mutant with and without E402Q) provide evidence against any such role for this residue (Figure 8).
A previous study revealed that mutation of the single serine/threonine cluster in the distal end of the C-terminal tail of OX1 receptors severely impaired arrestin translocation, but surprisingly mutation of the proximal cluster had little effect (Milasta et al., 2005). Upon comparison of the primary structures of OX1 and OX2 receptors, the serine/threonine clusters at 399 and 427 in OX2 receptor are similarly present at corresponding sites in OX1 receptor (Figure 10). However, an additional cluster present in OX2 receptors at position 406 is absent in the same corresponding region of OX1 receptors (Figure 10). Our findings indicate that mutation of the 406 serine/threonine cluster has the greatest effect on destabilizing the OX2 receptor-β-arrestin interaction, this being particularly clear with Venus-tagged receptor and Rluc8-tagged β-arrestin 2 (Figures 5C and 7). Indeed, this profile exhibits similarities to that observed in our previous study with OX1 receptors (Dalrymple et al., 2011). This alteration in kinetics is also demonstrated through arrestin-ubiquitin kinetics where the profiles of OX2 Δ406 and OX1 receptors overlap (Figure 9), in contrast to the separation in β-arrestin-ubiquitin kinetics observed for the wild-type receptors (Dalrymple et al., 2011). These data indicate that, in addition to the potential role of C-terminus/ICL3 configuration discussed above, the 406 cluster may contribute to the differentiation of the orexin receptor subtypes, by conferring upon OX2 receptors the ability to form more stable complexes with β-arrestin and ubiquitin.
Figure 10.
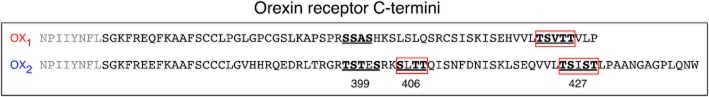
Diagram summarizing the apparently critical putative GRK phosphorylation sites in the C-terminal tail of OX1 and OX2 receptors. Underlined residues indicate the clusters that were examined for OX1 receptors previously (Milasta et al., 2005) and for OX2 receptors in this study. The boxed residues indicate clusters that had a notable effect on β-arrestin-mediated recruitment/colocalization with OX1 in the work by Milasta et al. (2005), and with OX2 receptors in the current study. Note that from our data, mutation of the 406 cluster in OX2 receptors had the most influence on the receptor-arrestin-ubiquitin complex over time, but mutation of both 406 and 427 clusters was required to substantially reduce the initial strength of complex formation.
Two of the three hypothesized GRK phosphorylation cluster sites, 406 and 427, appear to be involved in achieving stable OX2 receptor-β-arrestin complexes. Mutation of these clusters in combination appears to render substantial loss-of-function with regard to β-arrestin interaction strength, despite no apparent reduction in cell surface expression as determined by assessment of proximity to the Kras cell surface marker. These findings imply a degree of redundancy, and hint at another potential molecular mechanism to explain the previously observed difference in receptor-arrestin-ubiquitin complex stability with OX1 compared to OX2 receptors (Dalrymple et al., 2011). As a result of our findings, we propose a simplistic model that may help to explain some of the complex stability differences, particularly as observed in Figures 5C, 7 and 9. This model would suggest that OX1 receptors are phosphorylated on the distal serine/threonine cluster, as reported by Milasta et al. (2005), but OX2 receptors are phosphorylated on the two clusters that we have designated 406 and 427 (Figure 10). Perhaps only one of these sites in the OX2 receptor requires phosphorylation in order for the receptor to adopt a high-affinity state for arrestin binding. As the receptors are internalized, it is hypothesized that dephosphorylation of the distal serine/threonine cluster of OX1 receptor switches the receptor to a lower-affinity state for arrestin binding, whereas OX2 receptor requires both sites to be dephosphorylated for this to occur. This may then extend the time during which individual receptors remain in the high-affinity state for arrestin binding, which is consistent with the OX2 receptor-arrestin-ubiquitin complex being more stable over time and OX2 receptors recycling more slowly to the plasma membrane as a consequence (Dalrymple et al., 2011). This is of course not the only possible explanation, and our findings with the C-terminal chimera indicate a role for other parts of the intracellular domain as well, as discussed above.
This study therefore provides fundamental insights into the molecular determinants that govern orexin receptor subtype-specific arrestin-ubiquitin complex formation and stability. Our findings, and the conceptual models they have elicited, provide further potential molecular explanations for our previous observations (Dalrymple et al., 2011) and enable us to begin making key correlations between structure and function.
Acknowledgments
This research was funded by an Australian Postgraduate Award to W. C. J. and an Australian Research Council Future Fellowship (FT100100271) to K. D. G. P.
Glossary
- eBRET
extended BRET
- FCS
fetal calf serum
- GRK
GPCR kinase
- HA
hemagglutinin
- ICL
intracellular loop
- OX1ctOX2
chimera of OX1 receptor with OX2 receptor C-terminal tail
- Rluc8
Renilla luciferase 8
Conflict of interest
None.
References
- 1.Anborgh PH, Seachrist JL, Dale LB, Ferguson SS. Receptor/beta-arrestin complex formation and the differential trafficking and resensitization of beta2-adrenergic and angiotensin II type 1A receptors. Mol Endocrinol. 2000;14:2040–2053. doi: 10.1210/mend.14.12.0565. [DOI] [PubMed] [Google Scholar]
- 2.Becuwe M, Herrador A, Haguenauer-Tsapis R, Vincent O, Leon S. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem Res Int. 2012;2012:1–12. doi: 10.1155/2012/242764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalrymple MB, Jaeger WC, Eidne KA, Pfleger KD. Temporal profiling of orexin receptor-arrestin-ubiquitin complexes reveals differences between receptor subtypes. J Biol Chem. 2011;286:16726–16733. doi: 10.1074/jbc.M111.223537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeWire S, Ahn S, Lefkowitz R, Shenoy S. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 5.Evans NA, Groarke DA, Warrack J, Greenwood CJ, Dodgson K, Milligan G, et al. Visualizing differences in ligand-induced beta-arrestin-GFP interactions and trafficking between three recently characterized G protein-coupled receptors. J Neurochem. 2001;77:476–485. doi: 10.1046/j.1471-4159.2001.00269.x. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaeger WC, Pfleger KDG, Eidne KA. Monitoring GPCR–protein complexes using bioluminescence resonance energy transfer. In: Poyner DR, Wheatley M, editors. G Protein-Coupled Receptors. West Sussex, UK: Wiley-Blackwell; 2010. pp. 111–132. [Google Scholar]
- 8.Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, et al. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem. 2013;288:22942–22960. doi: 10.1074/jbc.M113.455774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kafi AKM, Hattori M, Misawa N, Ozawa T. Dual-color bioluminescence analysis for quantitatively monitoring G-protein-coupled receptor and β-arrestin interactions. Pharmaceuticals. 2011;4:457–469. [Google Scholar]
- 10.Kim AK, Brown RM, Lawrence AJ. The role of orexins/hypocretins in alcohol use and abuse: an appetitive-reward relationship. Front Behav Neurosci. 2012;6:78. doi: 10.3389/fnbeh.2012.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kocan M, See HB, Seeber RM, Eidne KA, Pfleger KD. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J Biomol Screen. 2008;13:888–898. doi: 10.1177/1087057108324032. [DOI] [PubMed] [Google Scholar]
- 12.Lan TH, Kuravi S, Lambert NA. Internalization dissociates beta2-adrenergic receptors. PLoS One. 2011;6:e17361. doi: 10.1371/journal.pone.0017361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan TH, Liu Q, Li C, Wu G, Lambert NA. Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic. 2012;13:1450–1456. doi: 10.1111/j.1600-0854.2012.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milasta S, Evans NA, Ormiston L, Wilson S, Lefkowitz RJ, Milligan G. The sustainability of interactions between the orexin-1 receptor and beta-arrestin-2 is defined by a single C-terminal cluster of hydroxy amino acids and modulates the kinetics of ERK MAPK regulation. Biochem J. 2005;387:573–584. doi: 10.1042/BJ20041745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore C, Milano S, Benovic J. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2006;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 16.Mustafa S, See HB, Seeber RM, Armstrong SP, White CW, Ventura S, et al. Identification and profiling of novel alpha1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J Biol Chem. 2012;287:12952–12965. doi: 10.1074/jbc.M111.322834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- 18.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 19.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- 20.Pal K, Mathur M, Kumar P, Defea K. Divergent beta-arrestin-dependent signaling events are dependent upon sequences within G-protein-coupled-receptor C-termini. J Biol Chem. 2012;288:3265–3274. doi: 10.1074/jbc.M112.400234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfleger KD, Eidne KA. New technologies: bioluminescence resonance energy transfer (BRET) for the detection of real time interactions involving G-protein coupled receptors. Pituitary. 2003;6:141–151. doi: 10.1023/b:pitu.0000011175.41760.5d. [DOI] [PubMed] [Google Scholar]
- 22.Pfleger KD, Pawson AJ, Millar RP. Changes to gonadotropin-releasing hormone (GnRH) receptor extracellular loops differentially affect GnRH analog binding and activation: evidence for distinct ligand-stabilized receptor conformations. Endocrinology. 2008;149:3118–3129. doi: 10.1210/en.2008-0002. [DOI] [PubMed] [Google Scholar]
- 23.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 24.Sakurai T, Mieda M. Connectomics of orexin-producing neurons: interface of systems of emotion, energy homeostasis and arousal. Trends Pharmacol Sci. 2011;32:451–462. doi: 10.1016/j.tips.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 26.Tohgo A, Choy E, Gesty-Palmer D, Pierce K, Laporte S, Oakley R, et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 27.Voisin T, Rouet-Benzineb P, Reuter N, Laburthe M. Orexins and their receptors: structural aspects and role in peripheral tissues. Cell Mol Life Sci. 2003;60:72–87. doi: 10.1007/s000180300005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander SPH, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]