

Abstract
Accumulation of α-synuclein (α-syn) in the brain is a core feature of Parkinson disease (PD) and leads to microglial activation, production of inflammatory cytokines and chemokines, T-cell infiltration, and neurodegeneration. Here, we have used both an in vivo mouse model induced by viral overexpression of α-syn as well as in vitro systems to study the role of the MHCII complex in α-syn-induced neuroinflammation and neurodegeneration. We find that in vivo, expression of full-length human α-syn causes striking induction of MHCII expression by microglia, while knock-out of MHCII prevents α-syn-induced microglial activation, antigen presentation, IgG deposition, and the degeneration of dopaminergic neurons. In vitro, treatment of microglia with aggregated α-syn leads to activation of antigen processing and presentation of antigen sufficient to drive CD4 T-cell proliferation and to trigger cytokine release. These results indicate a central role for microglial MHCII in the activation of both the innate and adaptive immune responses to α-syn in PD and suggest that the MHCII signaling complex may be a target of neuroprotective therapies for the disease.
Introduction
Parkinson disease (PD) is the most common neurodegenerative movement disorder and is characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) along with the accumulation of aggregated forms of the protein α-synuclein (α-syn) in nigral neurons as well as in other regions of the brain. Genetic mutations or duplications, which cause increased expression or disruption of the native structure of α-syn, lead to familial forms of PD (Polymeropoulos et al., 1997; Ross et al., 2008), while genome-wide association studies (GWAS) show strong associations between the α-syn gene and sporadic PD (Maraganore et al., 2006). Collectively, these data point to a central role for α-syn in the etiology of PD, but the mechanisms by which α-syn induces neurodegeneration remain uncertain.
There is mounting evidence for a central role of the immune system in the pathophysiology of PD, with activation of both innate and adaptive immunity (Hirsch and Hunot, 2009; Appel, 2012). In human postmortem tissues and in animal models of PD, α-syn pathology is accompanied by inflammation and immune responses including reactive microgliosis (Gerhard et al., 2006), increased pro-inflammatory cytokine expression (Mogi et al., 1994a,b), infiltration of CD4 lymphocytes (Brochard et al., 2009), and IgG deposition surrounding degenerating neurons (Orr et al., 2005). Recent epidemiological studies have revealed an association between the long-term use of nonsteroidal anti-inflammatory medications and reduced risk of PD (Gao et al., 2011).
Expression of the MHCII complex is restricted to antigen-presenting cells and is a crucial regulator of the cellular immune response, responsible for the presentation of peptide antigens to CD4 T-cells. HLA-DR, a component of MHCII, is highly expressed on all antigen-presenting cells, and is particularly prominent on the reactive microglia found in postmortem PD brain tissue (McGeer et al., 1988). Genetic studies have linked polymorphisms in the HLA-DR locus with sporadic late-onset PD (Hamza et al., 2010). Antigen presentation is a critical step in the induction of adaptive immune responses, and there is mounting evidence for activation of T-cell-mediated processes both within the brain (Brochard et al., 2009; Hirsch and Hunot, 2009; Appel et al., 2010) and systemically (Reynolds et al., 2007; Reynolds et al., 2010). Here, we have used both an in vivo mouse model induced by viral overexpression of α-syn as well as in vitro systems to study the role of the MHCII complex in α-syn-induced neuroinflammation and neurodegeneration. We find that overexpression of full-length human α-syn causes striking induction of MHCII expression by microglia, activation of antigen processing, and presentation of antigen leading to CD4 T-cell proliferation and subsequent cytokine release. Knock-out (KO) of MHCII prevents α-syn-induced microglial activation, IgG deposition, and the degeneration of dopaminergic neurons. These results indicate a central role for microglial MHCII in the activation of both the innate and adaptive immune responses to α-syn in PD, and suggest that the MHCII signaling complex may be a target of neuroprotective therapies for the disease.
Materials and Methods
Animals and treatment.
C57BL/6 (catalog #000664) and MHCII KO mice (B6.129S-H2dlAb1−Ea, catalog #003584) (Madsen et al., 1999) maintained on a congenic background were used for these studies and were obtained from The Jackson Laboratory. The MHCII (KO) mice lack all four murine MHCII genes and exhibit immune system deficits, mainly a lack of CD4 T-lymphocytes in the thymus and spleen (Madsen et al., 1999). Construction and purification of the rAAV vectors, rAAV-CBA-IRES-EGFP-WPRE (CIGW) and rAAV-CBA-SYNUCLEIN-IRES-EGFP-WPRE (CSIGW), are described in previous publications (St Martin et al., 2007; Theodore et al., 2008; Cao et al., 2010). Male C57BL/6 and MHCII KO mice (8–12 weeks of age) were deeply anesthetized with isoflurane and unilaterally injected with 2 μl of AAV2-SYN or AAV2-GFP (4.0 × 1012 viral genome/ml diluted in sterile PBS) into the right SNpc. Coordinates were anterior–posterior −3.2 mm from bregma, mediolateral +1.2 mm from midline, and dorsoventral −4.6 mm from dura. All research conducted on animals was approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Immunohistochemistry.
At 4 weeks, 3 months, and 6 months post-transduction, animals were deeply anesthetized and transcardially perfused with heparinized 0.01 m PBS, pH 7.4, followed by 4% paraformaldehyde (PFA) in 0.01 m PBS. Brains were postfixed for 24 h in 4% PFA and then cryoprotected in a 30% sucrose solution in PBS. Brains were frozen on dry ice and cryosectioned coronally on a sliding microtome (cut thickness: 40 μm); sections were collected serially throughout the striatum and SNpc, placed into tissue collection solution (50% 0.01 m PBS, 50% glycerol), and stored at −20 for immunohistochemical analysis.
For fluorescent analysis, free-floating sections were labeled with anti-MHCII (M5/114.15.2; eBioscience, 1:100), anti-CD11b (Serotec, 1:500), anti-GFP (Rockland, 1:1000), or anti-tyrosine hydroxylase (TH) (Millipore, 1:2000) antibodies overnight at 4°C. Appropriate Alexa-conjugated secondary antibodies diluted 1:1000 (Invitrogen) were used at room temperature for 2.5 h. For IgG staining, Cy3-conjugated goat anti-mouse IgG was used (Jackson ImmunoResearch, 1:500). Sections were mounted onto plus-coated glass slides, and coverslips were added using Vectashield Hard Set mounting medium.
For TH neuron quantification using unbiased stereological analysis, free-floating sections were stained as previously described (Cao et al., 2010), coded, and analyzed with an Olympus BX51 microscope and MicroBrightfield software (MicroBrightfield). A total of five sections covering the rostrocaudal extent of the SNpc, both ipsilateral and contralateral to the injection site, were quantified using the optical fractionator method and StereoInvestigator software. TH-positive neurons were counted and weighted section thickness was used to correct for variations in tissue thickness at varying sites.
Imaging and quantification.
Confocal images were captured using a Leica TCS-SP5 laser-scanning confocal microscope. Images were processed using the Leica LASAF software and exported and processed using Adobe Photoshop. For quantification of MHCII and CD11b staining, slides were observed using a Nikon Eclipse E800 m fluorescent microscope. Coded slides were scored by using a numerical scale 0 (no staining) to 4 (most intense) by a single observer blind to the treatment paradigm. Staining within the vicinity of viral transduction (GFP) was considered for scoring while staining immediately surrounding the needle tract was ignored. Scores obtained from 6 to 8 mice per group were plotted and statistically analyzed using the Kruskal–Wallis and Dunn's multiple-comparisons test. For antigen processing and presentation, immunofluorescence from four representative confocal images was quantified using ImageJ software. The corrected total cell fluorescence was quantified by the following: Integrated density (area of selected cell × mean fluorescence of background readings).
Midbrain fractionation protocol.
Ventral midbrains were homogenized on ice in 200 μl of lysis buffer containing 50 mm Tris/HCl, pH 7.4, 175 mm NaCl, 5 mm EDTA and protease and phosphatase inhibitors (Sigma-Aldrich). Triton X-100 (Sigma-Aldrich) was next added to homogenates at a final concentration of 1%. Following 30 min incubation on ice, 50 μl aliquots were saved from each sample and designated as “whole” fractions. Remaining homogenate from each sample was spun at 15,000 × g for 1 h; supernatants from each sample were transferred to a fresh tube and designated as “detergent (Triton X-100)-soluble” fractions. Pellets were resuspended in lysis buffer containing 2% SDS and following sonication on ice were designated as “detergent (Triton X-100)-insoluble” fractions. Protein concentrations were determined by the BCA protein assay.
Western blot analysis of brain homogenates.
Twenty micrograms of each sample were run on 12% gels using SDS-PAGE and transferred to PVDF membranes. Following the blocking of membranes for 30 min at room temperature in wash buffer (1× TBS-T containing 5% milk) to minimize nonspecific binding, primary antibody against human selective ASYN (clone 211; S5566) was added at a 1:500 dilution and incubated overnight at 4°C. Following three 5 min room temperature washes with wash buffer, secondary antibody (horseradish peroxidase-conjugated anti-mouse IgG; Sigma-Aldrich) was added at a 1:500 dilution in wash buffer containing 5% milk and incubated for 1 h at room temperature. Following four 5 min room temperature washes, signal on membranes was detected using enhanced chemiluminescence (ThermoFisher Scientific). Blots were then stripped using stripping buffer (ThermoFisher Scientific) and reprobed for the actin loading control (Sigma-Aldrich).
Primary cultures, α-syn treatment, and flow cytometry.
Primary murine microglia were isolated from postnatal day 0–2 pups according to previously published protocols (Harms et al., 2012) with the following modifications. Briefly, brains were isolated, meninges removed and dissociated for 10 min at 37°C with agitation every few minutes. Mixed glial populations were plated in T75 flasks in DMEM/F12 supplemented with 20% heat inactivated fetal bovine serum (FBS; Sigma-Aldrich), 1% penicillin/streptomycin, 1% l-glutamine (Sigma-Aldrich), and 10 ng/ml granulocyte monocyte colony stimulating factor (GM-CSF; PeproTech) for 14 d. Microglia were isolated from the astrocyte bed by mechanical shaking 195 rpm for 1 h at 37°C. Before assays, microglia were plated and allowed to adhere overnight in serum-free media at 37°C. Purified recombinant human α-syn (r-Peptide) was resuspended and incubated at 37°C with constant agitation for 7 d as previously described (Cao et al., 2012). Before use, aggregated α-syn was sonicated and added to the primary microglia at 500 nm at various time points.
For primary microglia and T-cell cocultures, primary microglia were isolated by mechanical shaking and plated in serum-free RPMI with 1% penicillin/streptomycin, 1% sodium pyruvate, 1% nonessential amino acids, 1% l-glutamine, and 0.1% β-mercaptoethanol and allowed to adhere overnight. Primary microglia were treated for 6 h with 500 nm α-syn in the presence or absence of OVA323–339 peptide (1 μg/ml, 10 μg/ml). Primary CD4 T-cells were isolated from OTII-11.1 TCR transgenic mice (Barnden et al., 1998) on a B6 congenic background with the following modifications. Briefly, male mice were deeply anesthetized and their spleens were removed, dissociated through a 70 μm cell strainer, and red blood cells were hypotonically lysed. CD4 T-cells were positively selected using the FlowComp CD4 isolation kit according to manufacturer's protocols (Life Technologies, Invitrogen). Isolated CD4 T-cells were cocultured with primary microglia for 60 h in RPMI supplemented with 10% heat inactive FBS, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% nonessential amino acids, 1% l-glutamine, 0.1% β-mercaptoethanol, and OVA323–339 peptide during the 60 h coculture. Immediately following coculture, conditioned medium was removed and frozen at −80°C for multiplex ELISA analysis. Remaining cells were pulsed with 13 μm EdU for 1 h at 37°C. Cocultured cells were then processed for flow cytometry by washing twice with fluorescence-activated cell sorting buffer (0.01 m PBS, pH 7.4, with 1% bovine serum albumin and 2 mm EDTA), Fcγ receptors were blocked with 2.4G2 (1:500), and cells were surface stained with anti-CD4-APC-eFluor 780 (eBioscience, 1:400) and anti-CD5-Alexa 488 (1:1600) for 15 min in the dark. A Fixable Viability Dye eFluor 450 (eBioscience, 1:1000) was used per manufacturer's instruction. Cells were then stained with Click-iT EdU Alexa Fluor 647 Imaging Kit (Life Technologies, Invitrogen) according to manufacturer's protocols with the following modifications: reactions were scaled to 200 μl and Alexa Fluor 647 azide was used at 1:400. Cells were immediately analyzed on the LSR-II flow cytometer (BD Biosciences) and analyzed using FlowJo software.
For antigen processing, primary microglia cells were isolated and plated into serum-free DMEM/F12 with 1% penicillin/streptomycin and 1% l-glutamine and allowed to adhere overnight. Following a complete media change, microglial cells were pulsed with 500 nm α-syn for either 4 h or overnight. Cells were then pulsed with 5 μl of DQ-Ovalbumin (Invitrogen, Life Technologies) for 1 h. Cells were then fixed and imaged on a Leica TCS-SP5 laser-scanning confocal microscope. Images were processed using the Leica LASAF software and quantified using ImageJ.
Multiplex ELISA.
Conditioned media were collected from the primary microglia T-cell cocultures treated with aggregated human α-syn immediately before EdU pulse and from primary microglia cultures alone 66 h after α-syn treatment for each of three independent experiments, and analyzed for mouse cytokine and chemokine production on an assay panel with 25 analytes (G-CSF, GM-CSF, IFNγ, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-15, IL-17, IL-1α, IL-1b, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IP-10, KC, MCP-1, MIP-1α, MIP-1β, MIP-2, RANTES, TNF) per manufacturer's instructions (Millipore).
Results
α-Syn overexpression increases MHCII expression in vivo
We have previously shown that overexpression of human full-length α-syn via adeno-associated virus (AAV) in the SNpc of mice results in reactive microgliosis, elevated pro-inflammatory cytokine expression, infiltration of T-lymphocytes, and ipsilateral IgG deposition as early as 2 weeks post-transduction (Theodore et al., 2008), demonstrating activation of both innate and adaptive immunity and replicating many of the features of inflammation observed in human PD. MHCII proteins are a critical link between innate and adaptive responses; they are expressed on activated microglia, and are responsible for the presentation of foreign proteins to CD4 T-lymphocytes. To examine the effects of α-syn overexpression on MHCII expression in vivo, 8-week-old male C57BL/6 mice received a unilateral stereotactic injection of recombinant AAV encoding human full-length α-syn (AAV2-SYN) or AAV2-GFP control virus (4 × 1012 viral genome/ml) into the right SNpc. Four weeks and 12 weeks post-transduction, there was strong expression of α-syn and GFP proteins in nigral neurons (Fig. 1A,C). This α-syn expression was accompanied by a striking increase in staining for MHCII in cells with the morphological characteristics of activated microglia (Fig. 1A–C). Although the expression of α-syn in neurons was intense, MHCII staining was not observed in virally transduced neurons. The enhancement of MHCII staining was persistent; a similar increase in microglial MHCII expression was also observed at 3 months post-transduction (Fig. 1C).
Figure 1.

AAV2-SYN overexpression induces MHCII expression in vivo. A, α-syn (α-GFP, green) results in enhanced MHCII expression (α-MHCII, red) in the SNpc (α-TH, blue) 4 weeks post-transduction. Scale bar, 125 μm in the Merge part. AAV2-GFP and AAV2-SYN Zoom insets from white boxes shown. Scale bar, 75 μm. B, Quantification of MHCII staining in the SNpc of AAV2-GFP (control) and AAV2-SYN mice at 4 weeks. C, α-Syn (α-GFP, green) results in enhanced MHCII expression (α-MHCII, red) in the SNpc 12 weeks post-transduction in AAV2-SYN-injected animals. Confocal images were captured using a Leica TCS-SP5 laser-scanning confocal microscope. Images were processed using the Leica LASAF software, exported, and processed using Adobe Photoshop. For quantification of MHCII slides were observed using a Nikon Eclipse E800M fluorescent microscope. Coded slides were scored by using a numerical scale 0 (no staining) to 4 (most intense) by a single observer blind to the treatment paradigm. Data represent the median (n = 6/group) **p < 0.0043, Mann–Whitney test.
MHCII is critical for α-syn-induced microglial activation and IgG deposition in vivo
We have previously shown that deletion of the gamma chain of Fc receptors, which are found on microglia and form high-affinity binding sites for immunoglobulins, attenuates inflammation and neurodegeneration induced by α-syn overexpression (Cao et al., 2010, 2012). MHCII proteins are responsible for presenting foreign proteins to CD4 T-lymphocytes, and subsequently provide help to B-cells required for triggering Ig production. We hypothesized that an upstream step in α-syn-induced inflammation might be MHCII-mediated antigen presentation to CD4 T-cells. To examine this question, male C57BL/6 and age-matched MHCII KO males on a congenic background were stereotactically injected with AAV2-SYN or AAV2-GFP. Four weeks post-transduction, a marked and statistically significant increase in CD11b-positive reactive microglia in the SNpc in WT AAV2-SYN-injected animals was noted, but in the MHCII KO animals this response was greatly attenuated (Fig. 2A–C) and persisted for at least 12 weeks (Fig. 2D). In the same animals, we also observed striking IgG deposition in WT AAV2-SYN animals in the ipsilateral hemisphere but a complete lack of IgG deposition in the AAV2-SYN MHCII KO animals at both 4 weeks (Fig. 3A) and 12 weeks (Fig. 3B).
Figure 2.
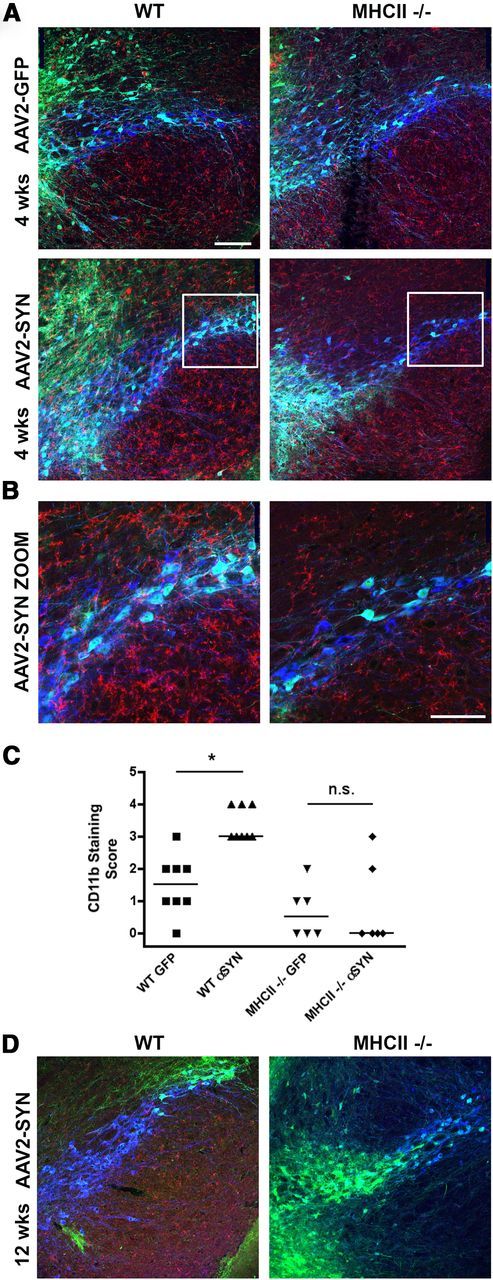
Genetic KO of MHCII attenuates AAV2-SYN-induced reactive microgliosis in vivo. A, α-Syn (α-GFP, green) results in enhanced CD11b+ microgliosis (α-CD11b, red) in the SNpc (α-TH, blue) 4 weeks post-transduction of WT mice but not MHCII KO mice. Scale bar, 125 μm. B, AAV2-GFP and AAV2-SYN Zoom insets from white boxes shown in A. Scale bar, 75 μm. C, Quantification of CD11b microgliosis in the SNpc of WT and MHCII KO mice 4 weeks post-transduction. D, α-Syn (α-GFP, green) results in enhanced CD11b+microgliosis (α-CD11b, red) in the SNpc (α-TH, blue) 12 weeks post-transduction of WT mice but not MHCII KO mice. Data represent the median (n = 6–8/group) *p < 0.05, **p < 0.01, Kruskal–Wallis test with Dunn's multiple-comparison post hoc test.
Figure 3.

Genetic KO of MHCII attenuates AAV2-SYN-induced IgG deposition in vivo. A, Representative images of IgG deposition (α-IgG, red) on the ipsilateral side of α-syn overexpression (α-GFP, green) in WT versus MHCII KO mice. IgG deposition is markedly attenuated in virally transduced MHCII KO animals at 4 weeks (A) and 12 weeks (B).
MHCII is critical for α-syn-induced neurodegeneration
We and others have previously observed that viral-mediated α-syn overexpression in the SNpc results in progressive neurodegeneration of dopaminergic neurons with a 25–30% loss of TH-immunopositive cells at 6 months post-transduction in mice (St Martin et al., 2007; Theodore et al., 2008; Cao et al., 2010). To determine whether deletion of MHCII proteins modifies the neurodegenerative process, 8-week-old male C57BL/6 wild-type (WT) and MHCII KO mice on a congenic background received unilateral stereotactic injections of AAV2-SYN or AAV2-GFP control virus (4 × 1012 viral genome/ml) into the right SNpc. Six months post-transduction, nigral cell number was evaluated by immunohistochemistry for TH and cell counting using an unbiased optical dissector method. We found that genetic KO of MHCII completely prevents α-syn-induced dopaminergic cell loss in this model system (Fig. 4A,B).
Figure 4.

Genetic KO of MHCII attenuates AAV2-SYN-induced neurodegeneration in vivo. Six months post-transduction TH-immunopositive neurons were quantified to determine observed neuroprotection. A, Representative images of the ipsilateral SNpc stained for TH. B, Genetic KO of MHCII attenuates AAV2-SYN-induced neuron loss. Neuron loss is reported as a percentage of contralateral side. n = 6–8/group one way ANOVA with Bonferroni selected comparison post hoc test. *p < 0.05.
α-Syn overexpression results in accumulation of high molecular weight α-syn species
Recent studies have suggested that the secondary structure of α-syn may be important for the observed toxicity in animal models (Lashuel et al., 2013). To determine the predominant α-syn species present in our in vivo model, Western blot analysis of α-syn was performed on ventral midbrain homogenates from AAV2-SYN transduced mice. We used solubility in 1% Triton X-100 to separate soluble from insoluble forms. Four weeks post-transduction, we found viral transduction led to enhancement of high molecular weight forms of α-syn, along with appearance of α-syn monomers that were not detectable in the untreated SNpc homogenate (Fig. 5). Most striking, however, was the appearance of Triton-insoluble high molecular weight aggregates in the virally transduced SNpc; these were not observed in untreated mouse SNpc, and are similar to the Triton-insoluble high molecular weight forms that we and others have described in human synucleinopathies (Cantuti-Castelvetri et al., 2005).
Figure 5.

Overexpression of AAV2-SYN in mouse SNpc results in the accumulation of high molecular weight α-syn species. Western blot analysis of α-syn of midbrain homogenates obtained from mice 4 weeks post-transduction into the right substantia nigra, using an antibody that is selective for human α-syn. There is an increase in high molecular weight α-syn species (≥50 kDa) is both the “whole” and Triton-soluble (“T-X-Sol”) fractions in homogenate fractions derived from right (R), infected ventral midbrain samples compared with that of noninfected left (L) control samples. There is also appearance of detectable soluble α-syn monomers (17 kDa). Insoluble high molecular weight forms of α-syn (“T-X-100-Insol” fraction) were observed only after AAV2-SYN treatment. Actin (42 kDa) was used to normalize for gel loading.
α-Syn increases antigen processing and presentation
Within the cell, MHCII proteins traffic to endosomal or lysosomal compartments where they are stripped of the MHCII invariant chain, loaded with antigen that has been processed by lysosomal enzymes and shipped to the plasma membrane for presentation. Once on the plasma membrane, MHCII can present antigen to CD4 T-lymphocytes via the T-cell receptor (TCR), subsequently activating the adaptive immune response (Dani et al., 2004). We have previously shown that α-syn is taken up into autophagosomal compartments in microglia (Cao et al., 2012). To examine the effects of α-syn on antigen processing and presentation, primary WT microglia were isolated from postnatal pups and pretreated with 500 nm α-syn for 4 h and overnight before addition of DQ-Ovalbumin. DQ-Ovalbumin is a self-quenched conjugate of ovalbumin that exhibits fluorescence upon proteolytic degradation in the lysosome, allowing visualization of antigen processing and presentation. We observed that 4 h after α-syn treatment of microglia there was a marked induction of antigen processing and presentation (Fig. 6B,D). This effect was time limited, and 24 h after the addition of α-syn the rate of antigen processing was reduced below baseline (Fig. 6C,D). These data indicate a potent ability of aggregated α-syn to stimulate the lysosomal processing required for microglial antigen processing and presentation. To determine whether the effect is specific to aggregated α-syn, we tested monomeric α-syn on primary microglia and found no significant induction of MHCII or antigen processing (Fig. 6E) suggesting the effect is specific to aggregated α-syn.
Figure 6.

α-Syn regulates antigen processing and presentation in vitro. Primary microglia were treated with 500 nm aggregated α-syn or vehicle control (A) for 4 h (B) or overnight (C) before a 1 h pulse with DQ-Ovalbumin. Immunofluorescence was quantified from confocal images using ImageJ software (D). Four hour aggregated α-syn pretreatment resulted in an increase in antigen processing and presentation while overnight α-syn pretreatment decreased antigen processing and presentation. These effects were not observed with monomeric α-syn (E). One way ANOVA with Bonferroni multiple-comparisons post hoc test. ***p < 0.001, **p < 0.01 compared with control. Scale bar, 25 μm.
α-Syn increases CD4 T-cell proliferation and promotes expression of cytokines and chemokines
The MHCII complex presents antigen exclusively to CD4 T-lymphocytes, and this interaction is likely to be critical for PD-associated inflammation. To determine whether the α-syn-mediated increase in microglial antigen processing and MHCII expression can drive downstream proliferation of CD4 T-cells, primary WT microglia were isolated and pretreated with 500 nm recombinant full-length aggregated α-syn for 6 h before coculture with primary CD4 OTII-transgenic TCR (OTII-TCR) T-cells. OTII-TCR T-cells express the ova peptide-specific transgenic TCR and are capable of an MHCII-dependent proliferative response (Barnden et al., 1998). Sixty hours after coculture with primary microglial cells, we found that α-syn pretreatment led to marked stimulation of T-cell proliferation in the presence of OVA323–339 peptide (Fig. 7A–C) indicating that the enhanced expression of MHCII induced by α-syn in these cells can mediate MHCII-dependent antigen presentation that is sufficient to drive a subsequent TCR- and antigen-specific CD4 T-cell proliferative response.
Figure 7.

α-Syn-induces CD4 T-cell activation and proliferation in vitro. Primary microglia (previously treated with OVA323–339 and 500 nm α-syn for 6 h) and CD4 T-cell cocultures were pulsed with EdU for 1 h before flow cytometry analysis. A, No peptide control. Vehicle represented with solid line and shading, α-syn treatment represented with a dotted line. Primary microglia and CD4 T-cell cocultures revealed little or no EdU incorporation events. OVA peptide 1 μg/ml (B) and 10 μg/ml (C) treatment results in an increase in EdU+ events 60 h after coculture. Positive events were gated by live/dead, CD4, CD5, and EdU. D–I, α-syn pretreatment (500 nm) of primary microglia 6 h before T-cell coculture resulted in a statistically significant increase in pro-inflammatory cytokines and chemokines. All cytokine and chemokine expression levels were normalized to the level of vehicle-treated primary microglia and T-cell coculture. Student's t test (n = 3–4/group, representative of 2 independent experiments). D, 10 μg/ml OVA323–339 IL-1α *p < 0.0193. E, 10 μg/ml OVA323–339 IFNγ **p < 0.0087. F, 10 μg/ml OVA323–339 IL-1β ***p < 0.0009. G, 1 μg/ml OVA323–339 TNF **p < 0.0022. H, 10 μg/ml OVA323–339 TNF *p < 0.011. I, 10 μg/ml OVA323–339 IL-10 **p < 0.0021.
We also examined the role of MHCII in mediating interactions between microglia and T-cells. We have previously found that α-syn can interact directly with microglia and can be internalized and trafficked to autophagosomes, but uptake of α-syn alone is not sufficient to activate microglia or to initiate a pro-inflammatory response (Cao et al., 2012). Other studies using nitrated α-syn have shown that CD4 T-cells are capable of activating the pro-inflammatory transcription factor NF-κB, thereby triggering pro-inflammatory gene expression in microglia (Reynolds et al., 2008, 2009). We analyzed the conditioned media from primary microglia alone and microglia cocultured with CD4 OTII T-cells and ova-peptide by multiplex ELISA. Conditioned media from microglia or CD4 OTII T-cells alone revealed that α-syn did not induce any significant inflammatory response as measured by the 25 cytokines and chemokines present on the array (Cao et al., 2012) (data not shown). In contrast, treatment of microglia followed by addition of CD4 OTII T-cells led to a robust inflammatory response, with statistically significant enhancement in the production of IL-1α, IFNγ, IL-1β, TNF, and IL-10 (Fig. 7D–I). Significant TNF induction was even seen with ova peptide as low as 1 μg/ml (Fig. 7G). These observations demonstrate the critical role for interactions between microglia and CD4 T-cells in establishing the inflammatory response to α-syn.
Discussion
In these studies we have identified a key upstream mediator of α-syn-induced inflammation and neurodegeneration, the MHCII complex. We have observed that overexpression of α-syn causes induction of MHCII expression on microglia, while genetic KO of MHCII prevents α-syn-induced microglial activation, IgG deposition, and neurodegeneration in vivo. In vitro, overabundance of α-syn enhances processing, presentation of antigen, and CD4 T-cell proliferation. Interaction of microglia and CD4 T-cells leads to a potent cytokine and response to aggregated α-syn. These results indicate a central role for microglial MHCII in the activation of both the innate (microglial) and adaptive immune responses to α-syn in our PD model.
In human PD, there is substantial evidence that inflammation and immune system activation are key mediators in the PD neurodegenerative process (Whitton, 2007; Hirsch and Hunot, 2009; Tansey and Goldberg, 2010). In human disease, α-syn pathology is accompanied by reactive microgliosis (McGeer et al., 1988; Langston et al., 1999; Gerhard et al., 2006), increased pro-inflammatory cytokine expression (Mogi et al., 1994a,b; Blum-Degen et al., 1995; Mount et al., 2007; Reale et al., 2009), infiltration of CD4 T lymphocytes (Brochard et al., 2009), and IgG deposition surrounding degenerating neurons (Orr et al., 2005). Epidemiological studies have demonstrated a consistent association between use of the anti-inflammatory drug ibuprofen and reduced risk of PD (Gao et al., 2011). HLA-DR, a component of MHCII, is highly expressed on all antigen-presenting cells, and is particularly prominent on the reactive microglia found in postmortem PD brain tissue (McGeer et al., 1988). A GWAS study found that genetic polymorphisms in the HLA-DR locus are associated with late-onset PD, implicating its importance in disease pathogenesis (Hamza et al., 2010). More recently, several studies have provided additional confirmation of the infiltration of CD4 (helper) and CD8 (cytotoxic) T-cell populations in human brain and animal models of PD, and have shown that these are concentrated in areas of degeneration, particularly the SNpc (Brochard et al., 2009).
Inflammatory responses have been replicated in a variety of different animal models of PD. In the current studies we used the AAV2-SYN model of PD, a model in which inflammation is a key mediator before the neurodegenerative process. We have previously shown that in this model system, overexpression of human full-length α-syn results in NF-κB activation, localized inflammation, microglial activation, lymphocyte infiltration, IgG deposition, and gradual degeneration of dopaminergic neurons (Theodore et al., 2008; Cao et al., 2010). Other types of model systems also demonstrated the importance of inflammatory mechanisms. In neurotoxin models, such as 6-OHDA and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), it has been shown that pro-inflammatory cytokines TNF, IL-1β, and IFNγ are key regulators of dopaminergic cell death, and neutralizing or blocking expression attenuates dopaminergic cell loss and reactive microgliosis associated with these models (Mount et al., 2007; Pott Godoy et al., 2008; Harms et al., 2011). Furthermore, in an MPTP neurotoxin mouse model of PD, CD4 T-cell infiltration into the SNpc is required for the observed dopaminergic neurotoxicity, and this process involves a Fas/FasL-dependent mechanism (Brochard et al., 2009) indicating the importance of MHCII antigen presentation in the immunological synapse and the subsequent activation of the adaptive immune response via CD4 T-cell activation. While MHCII exclusively presents antigen to CD4 T-cells, CD8-positive T-cells are also found in areas of neurodegeneration in human PD and models systems (Brochard et al., 2009; Appel, 2012). CD8 T-cells are found in the brain in the MPTP model, but at least in this system, CD8 cells do not seem to play a significant role in the neurodegenerative process (Brochard et al., 2009). MHCII KO mice have been reported to have increased numbers of CD8 T-cells (Madsen et al., 1999). We have not directly evaluated the role of CD8 T-cells in our α-syn degenerative model, but the complete attenuation of IgG deposition and CD11b microgliosis with deletion of MHCII alone suggest CD8 T-cells are likely to have a very limited role at most in this model.
In these studies we have shown that KO of MHCII markedly attenuates the inflammatory response associated with α-syn, with decreased CD11b+ reactive microgliosis (Fig. 2) at 4 weeks and 3 months post-transduction. In addition, we found that genetic KO of MHCII attenuates ipsilateral IgG deposition (Fig. 3) at 4 weeks and 3 months post-transduction indicating that the inflammation observed in our model is not simply delayed by the KO of MHCII proteins. We also found that genetic KO of MHCII is neuroprotective, and completely blocks the observed nigral neuron loss in the AAV2-SYN model at 6 months post-transduction (Fig. 4), indicating a critical role for MHCII expression in α-syn-induced neurodegeneration.
We have also found that one of the early steps in the inflammation associated with overabundance of α-syn is likely to involve antigen-presenting cells such as microglia and the MHCII signaling complex. Our data demonstrate that aggregated α-syn is a potent stimulus that induces microglia to enhance antigen processing (Fig. 6) and express MHCII (Fig. 1). Studies using an EAE model of multiple sclerosis (MS), a neuro-inflammatory disorder associated with cell loss, have shown that antigen processing of myelin auto-antigens is an essential part of MHCII antigen presentation and subsequent CD4 T-cell activation, and we believe that there may be a similar requirement for processing of α-syn (Slavin et al., 2001). The data presented here clearly implicates MHCII in α-syn-mediated immune responses, but the exact nature of the antigen presented by MHCII is at present unknown. It is possible that the antigen in these studies is either a form of α-syn itself, or a protein that is induced by overabundance of α-syn. Identification of the specific antigen is a difficult task that has been accomplished only in a few specialized settings, such as the extensively studied EAE model, and even here the full spectrum of antigens involved in the inflammatory response remains uncertain (Hohlfeld et al., 2008). Regardless of the antigen presented, the downstream consequences of α-syn-driven MHCII-dependent CD4 T proliferation are likely to be several, and include the induction of effector Th1 and or Th17 cells, and interactions with B-cell populations to promote adaptive immunity.
It is important to note that MHCII expression is critical not only for antigen presentation, but also for the maturation and selection of CD4 T-cells in the thymus. Therefore, the MHCII KO animals used here are expected to have a deficiency of CD4 T-cells (Madsen et al., 1999) making it difficult to conclude whether it is the deficiency of MHCII in microglia or the lack of CD4 T-cells that is responsible for the neuroprotective effects of MHCII depletion in vivo. On the other hand, our in vitro studies unequivocally indicate an important role of microglial MHCII in both T-cell proliferation and cytokine and chemokine induction. This communication between microglia and CD4 T-cells may also involve interferon gamma and other signaling pathways (Aloisi et al., 2000).
The development of neuroprotective or immunomodulatory strategies for PD is a vital unmet need. Most currently available treatments are based on restoration and/or replacement of dopaminergic function that has been depleted by the loss of neurons in the SNpc. It is increasingly clear, however, that PD encompasses far more than simply dopaminergic degeneration and that the non-motor aspects of the disease, which include cognitive and autonomic impairment, are important contributors to the disability. The complexity of the disease process points to the vital need for the development of a strategy that can prevent the onset of PD, slow the progression, or reverse the disease process once it is established. Our data point to a critical role of MHCII signaling early in the neuroinflammatory process associated with overabundance of α-syn. The MHCII signaling appears to be essential both for induction of microglial inflammatory responses before observed neurodegeneration, as well as the engagement of systemic adaptive immunity. This signaling may be a key target for the development of potentially neuroprotective immunotherapies.
Footnotes
The work from these studies is generously supported by the RJG Foundation, the University of Alabama (UAB) Comprehensive Arthritis, Musculoskeletal, and Autoimmunity Center (CAMAC) Comprehensive Flow Cytometry Core (NIH Grant P30 AR48311), the UAB Animal Resources Program (NIH Grants G20 RR025858 and G20 RR022807-01), and NIH Grant T32 AR007450-30. We thank Dr. Chad Steele for his help with the multiplex ELISA assay at University of Alabama at Birmingham.
The authors declare no competing financial interests.
References
- Aloisi F, Serafini B, Adorini L. Glia-T-cell dialogue. J Neuroimmunol. 2000;107:111–117. doi: 10.1016/S0165-5728(00)00231-9. [DOI] [PubMed] [Google Scholar]
- Appel SH. Inflammation in Parkinson's disease: cause or consequence? Mov Disord. 2012;27:1075–1077. doi: 10.1002/mds.25111. [DOI] [PubMed] [Google Scholar]
- Appel SH, Beers DR, Henkel JS. T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol. 2010;31:7–17. doi: 10.1016/j.it.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the CSF of Alzheimer's and de novo Parkinson's disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, Hyman BT, Standaert DG. Alpha-synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2005;64:1058–1066. doi: 10.1097/01.jnen.0000190063.90440.69. [DOI] [PubMed] [Google Scholar]
- Cao S, Theodore S, Standaert DG. Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson's disease. Mol Neurodegener. 2010;5:42. doi: 10.1186/1750-1326-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Standaert DG, Harms AS. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J Neuroinflammation. 2012;9:259. doi: 10.1186/1742-2094-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A, Chaudhry A, Mukherjee P, Rajagopal D, Bhatia S, George A, Bal V, Rath S, Mayor S. The pathway for MHCII-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J Cell Sci. 2004;117:4219–4230. doi: 10.1242/jcs.01288. [DOI] [PubMed] [Google Scholar]
- Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology. 2011;76:863–869. doi: 10.1212/WNL.0b013e31820f2d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Barnum CJ, Ruhn KA, Varghese S, Treviño I, Blesch A, Tansey MG. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson's disease. Mol Ther. 2011;19:46–52. doi: 10.1038/mt.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Lee JK, Nguyen TA, Chang J, Ruhn KM, Treviño I, Tansey MG. Regulation of microglia effector functions by tumor necrosis factor signaling. Glia. 2012;60:189–202. doi: 10.1002/glia.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Meinl E, Dornmair K. B- and T-cell responses in multiple sclerosis: novel approaches offer new insights. J Neurol Sci. 2008;274:5–8. doi: 10.1016/j.jns.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::AID-ANA7>3.0.CO%3B2-F. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, Fugger L. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96:10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/WNL.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994a;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994b;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S, Park DS. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27:3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain. 2005;128:2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, Onofrj M. Peripheral cytokines profile in Parkinson's disease. Brain Behav Immun. 2009;23:55–63. doi: 10.1016/j.bbi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82:1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol. 2009;182:4137–4149. doi: 10.4049/jimmunol.0803982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, Nishioka K, Fuchs J, Gasser T, Maraganore DM, Adler CH, Larvor L, Chartier-Harlin MC, Nilsson C, Langston JW, Gwinn K, Hattori N, Farrer MJ. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63:743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin AJ, Soos JM, Stuve O, Patarroyo JC, Weiner HL, Fontana A, Bikoff EK, Zamvil SS. Requirement for endocytic antigen processing and influence of invariant chain and H-2M deficiencies in CNS autoimmunity. J Clin Invest. 2001;108:1133–1139. doi: 10.1172/JCI200113360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted overexpression of alpha-synuclein in mouse substantia nigra. J Neurochem. 2007;100:1449–1457. doi: 10.1111/j.1471-4159.2006.04310.x. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]