

Abstract
Autophagy is intimately associated with eukaryotic cell death and apoptosis. Indeed in some cases the same proteins control both autophagy and apoptosis. Apoptotic signaling can regulate autophagy and conversely autophagy can regulate apoptosis (and most likely other cell death mechanisms). However the molecular connections between autophagy and cell death are complicated and, in different contexts, autophagy may promote or inhibit cell death. Surprisingly, although we know that, at its core, autophagy involves degradation of sequestered cytoplasmic material, and therefore presumably must be mediating its effects on cell death by degrading something, in most cases we have little idea what is being degraded to promote autophagy’s pro- or anti-death activities. Because autophagy is known to play important roles in health and many diseases, it is critical to understand the mechanisms by which autophagy interacts with and affects the cell death machinery since this will perhaps allow new ways to prevent or treat disease. In this chapter we discuss the current state of understanding of these processes.
Keywords: Apoptosis, caspase, autophagic cell death, TRAIL, BCL proteins, autophagy-mediated protection, ATG protein function
Introduction
Autophagy delivers cytoplasmic components to lysosomes for degradation and recycling of the degradation products, such as amino acids, carbohydrates and lipids that are used to synthesize new proteins and organelles or metabolized to supply energy. Of the three main types of autophagy in mammalian cells, microautophagy, chaperone-mediated autophagy and macroautophagy, by far our best understanding of how autophagy regulates cell death relates to macroautophagy. In macroautophagy, which will simply be referred to as “autophagy” from now on in this chapter, a double-membrane structure called the autophagosome, sequesters proteins, organelles and other cytoplasmic components then fuses with lysosomes for degradation. The central problem in understanding how autophagy regulates and is regulated by cell death mechanisms is therefore to understand how the sequestration and degradation of cellular components in autophagosomes and autolysosomes affects apoptosis and other death pathways and, conversely how apoptosis may regulate autophagy. The term “autophagy” is often used to mean both the formation of autophagosomes and the full process whereby the autophagosome’s contents are subsequently degraded after fusion with the lysosome. It is important to remember that these two things are not necessarily the same. When we refer to “autophagic flux”, we mean that the whole process took place and something was degraded. However one can imagine situations where the effect on the cell could be due not to the flux but instead caused just by the formation of the autophagosomes themselves and, as will be discussed below, we now know that some mechanisms whereby cell death is regulated involve autophagosomes but not necessarily autophagic flux, while others involve autophagosome formation and flux.
A considerable amount of research has been carried out in recent years in this area (for recent reviews see [1, 2]). Much of this interest has been driven by the fact that manipulation of autophagy holds great promise for improving treatment of diverse diseases [3] with the central idea being that both diseases where we want to kill cells (e.g. cancer) and diseases where we want to improve cell survival (e.g. neurodegenerative disease) could benefit by manipulation of autophagy. However, as discussed below the connections between autophagy and cell death are complicated. Thus, before the practical application of autophagy manipulation can be undertaken to prevent or treat disease can be undertaken successfully, we need to understand how these complications will affect our plans.
An important concept that is often underappreciated is that even when autophagy modulates the amount of cell death in a particular context, this need not indicate a direct mechanistic connection between the process of autophagy and the process of cell death. For example, it is well established that autophagy can protect against neurodegenerative disease [4]; indeed, neural deficiency in autophagy is sufficient to cause neurodegenerative disease [5, 6]. Since neurodegenerative disease is ultimately caused by neurons dying, the obvious conclusion is that autophagy prevents neurons from dying. However, this doesn’t necessarily mean that in this context the process of autophagy interacts with the process of apoptosis and directly inhibits it. Instead the simpler (and the evidence suggests more likely) explanation for this protection is that neurodegenerative disease is often caused by toxic protein aggregates, and since autophagy is a process that degrades proteins, autophagy protects neurons by ensuring that the toxic stimulus (i.e. the protein aggregates) are disposed of and never have a chance to activate cell death mechanisms. This appears to be the case in autophagy-deficient mice, where aggregates of ubiquitin-containing proteins are found in the autophagy-deficient neurons and it is these aggregates that are normally removed by autophagy that activate cell death pathways [5, 6]. Similarly, autophagy can protect cells against nutrient and growth factor deprivation and under such circumstances autophagy inhibition leads to apoptosis [7, 8]. But, this doesn’t necessarily mean that autophagy was inhibiting apoptosis pathways in this case. It could simply mean that autophagic degradation of existing cellular macromolecules provides the nutrients and energy needed to maintain the essential functions required for cell survival during starvation, and that the cell therefore never activates the apoptosis pathway in the first place. In this chapter, we focus not on cases like this where autophagy regulates cell death indirectly by altering the presence or absence of an upstream signal that eventually activates death pathways. Instead we will focus on the questions– does the process of autophagy whereby certain substrates are degraded in the lysosome control the cell death machinery? And, how do the proteins that control autophagy interact with and regulate the cell death machinery?
Autophagy as a promoter of cell death
Mammalian cell death occurs by multiple mechanisms some of which are deliberate “suicide” that are clearly programmed and some of which seem to be accidental or the result of damage [9]. Apoptosis is by far the best-described programmed death mechanism and it is now also clear that necrosis can be programmed in many instances. It has been proposed that “autophagic cell death” is another type of “active” programmed death [10]. The suggestion that autophagic cell death occurs came about was based on the frequent observation that cell death is often accompanied by high levels of autophagosomes and features of active autophagy. However, just because cells that will die induce autophagy before their demise, this doesn’t necessarily implicate a causal relationship between the autophagy and cell death. Indeed, high levels of autophagy could be an indication that the cell is attempting to survive by inducing autophagy and that it is only when this effort fails, that death occurs. Thus the question is: does the process of autophagy actually lead to the death of the cell? There are many publications in the literature concluding that death stimuli in mammalian cells and non-mammalian systems are caused by autophagy [11], however whether these situations are a case of autophagy being upstream of apoptosis or autophagy killing the cell by a mechanism that has nothing to do with the other established death mechanisms such as apoptosis is often unclear. So, does autophagy kill cells and, if so, how?
One situation where autophagy may cause cell death is in situations where the cell has no ability to activate canonical apoptosis, which is usually the preferred mechanism of death. One of the first such examples was described in cells from BAX and BAK double knockout mice, which lack the two main proteins that regulate the release of mitochondrial proteins during apoptosis. These cells are severely compromised in apoptosis and do not display any activation of pro-apoptotic caspases but still die in response to stimuli such as DNA damaging agents [12] (in fact most stressful stimuli that kill cells by inducing apoptosis generally still kill cells when apoptosis is prevented). This non-apoptotic death was dependent on autophagy regulators ATG5 and BECLIN1 and knockdown of these genes was sufficient to provide long term protection to the cells after treatment with death stimuli. This implies that in the absence of apoptosis, the DNA damage activates autophagy that kills the cells. In another example, pharmacological inhibition of caspases was found to induce death of L929 fibroblasts and this too was reported to rely on autophagy regulators but not apoptosis [13] and to involve autophagic degradation of catalase that ultimately led to increased reactive oxygen species that killed the cells [14]. It is unclear whether the explanation of catalase degradation being the ultimate cause of the death applies more broadly to other situations of autophagic cell death or just to the caspase-inhibitor treated L929 cells. However, the major concern with these examples is that they represent a very artificial situation. Normal cells are probably never as defective in apoptosis as BAX/BAK double knockout murine embryonic fibroblasts. Thus, even if autophagy is inducing cell death in these contexts, this may be merely a reflection that the cells use this pathway to die because they have no other option rather than an indication that autophagy is a bona fide death mechanism that is able to promote death in cells under more normal circumstances.
Forced expression of oncogenic Ras was recently reported to induce very high levels of autophagy that led to autophagy-dependent death and loss of clonogenic capacity in an ovarian cancer cell line [15]. Cell death occurred after proliferative arrest, was inhibited by pharmacological and genetic inhibition of autophagy and was not associated with any detectable caspase activation or other markers of apoptosis. However, unlike the examples in the previous paragraph where apoptosis was artificially blocked, the dying cells in Elgendy et. al.’s study [15] were still able to activate canonical caspase-dependent apoptosis in response to other stimuli– i.e. they are not unable to activate caspase-dependent apoptosis, they just don’t do so in this instance. Interestingly, death in this case required Ras-induced expression of NOXA, a Bcl family protein that is a well known as a regulator of p53-induced apoptosis [16]. Examples like this one suggest that autophagy can induce cell death even in cells where apoptosis has not been artificially blocked. However, this example is also rather artificial– the death stimulus was a massive forced overexpression of a mutated signaling protein and one might therefore reasonably wonder if it is physiologically significant.
However, under certain circumstances, the process of autophagy does indeed seem to promote cell death under normal physiological situations. The best understood examples occur during the cell death events that drive tissue remodeling occur during Drosophila development [17–19]. However it is important to note that even in these cases, it does not necessarily follow that autophagy alone kills the cells. Instead these examples of autophagy-dependent cell death under physiological conditions are largely due to autophagy making other death pathways (especially caspase-dependent apoptosis) more likely. For example during Drosophila oogenesis, autophagy controls developmental cell death by selectively degrading the protein dBruce, which itself functions to inhibit caspase activation [19] (Figure 1). Thus, in this case autophagy promotes cell death by degrading a particular target that itself acts as a negative regulator of caspase activity thus stimulating higher levels of caspase activity, i.e. something that we would normally classify as being a form of apoptosis [10]. Additionally, at least some developmental cell death in flies occurs in an autophagy-dependent but caspase-independent manner [20] although in this case the underlying target that is degraded by autophagy and which causes the caspase-independent death is unclear.
Figure 1. The promotion of cell death by autophagy.
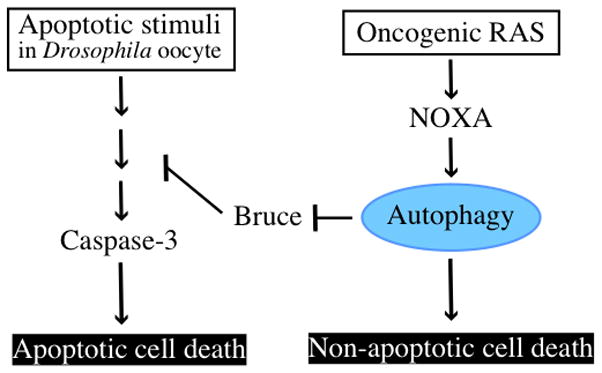
In Drosophila oocyte, apoptotic cell death occurs via caspase-3. Autophagy targets and degrades dBruce, inhibitor of apoptosis, to promote apoptosis. Therefore, inhibition of autophagy prevents nurse cells in late oocyte from causing apoptotic cell death. In some mammalian cells, overexpression of oncogenic mutated RAS increases autophagy level and leads to cell death that is dependent on autophagy but not apoptosis.
The dBruce example is a case where autophagic flux is necessary for the effect on cell death– that is, the dBruce protein was degraded and this is why apoptosis was stimulated. Sometimes however autophagosome formation is important but flux is not. An example comes from recent work where it was shown that autophagosomes induce apoptosis by serving as a platform upon which caspases (specifically caspase-8) can be activated [21]. This mechanism does not necessarily require any actual degradation of autophagosome cargo and so may not indicate a need for autophagic flux in order to get induction of apoptosis and cell death. However it does still require the core autophagy machinery that is required for autophagosome formation. This distinction has important practical implications. Suppose we wanted to block disease-associated apoptosis where autophagy is involved by a flux-dependent or a flux-independent mechanism. How would this affect the intervention in the autophagy process that is needed to achieve the goal of inhibiting apoptosis? In a case like that in fly oocytes where autophagic flux and degradation of a substrate is required to alter sensitivity to apoptosis, it may be sufficient just to block the degradation step without affecting autophagosome formation in order to get the result you want. This can be done by inactivating lysosomes and we actually have drugs like chloroquine that can do this. However, if the way that autophagy is promoting apoptosis is because autophagosomes serve as a platform upon which caspase activation is achieved then just blocking the lysosome wouldn’t have any effect. Instead in this case, an intervention would need to prevent formation of the autophagosomes if you want to prevent apoptosis.
Thus although it seems clear that autophagy can promote cell death and the literature is filled with examples where claims of “autophagic” death can be found, we still lack a detailed understanding of whether this can really occur under normal situations in a manner that is truly independent of other better established death mechanisms such as caspase-dependent apoptosis. And, in most cases, it is quite unclear how autophagy—i.e. a process that degrades things– is mediating its effects. The example of dBruce degradation [19] suggests that there may be other pro-survival signaling proteins that if degraded would lead to autophagy-dependent death. However while it makes sense that they should exist, the full spectrum of such molecules is unknown. Without knowing what autophagy has to degrade in order to cause cell death, one cannot claim to understand mechanisms of autophagy dependent death.
Autophagy as a protector against cell death
Although autophagy has been described as a death mechanism, the current consensus is that autophagy’s role as regards cell death is primarily protective [22, 23]. However, as noted above, we need to be careful not to be confused by protective effects that, while real, are not necessarily indicative of a direct functional role of autophagy in blocking the death machinery. As explained above in the case of aggregate prone proteins killing neurons, autophagy might protect without having any direct effect on the death pathways that actually carry out programmed cell deaths. With this in mind, how good is the evidence that autophagy and, in particular, autophagic flux to degrade cargo sequestered in autophagosomes directly inhibits apoptosis (or other death pathways)?
The first thing to consider is that autophagy has been reported to protect against a very diverse set of death stimuli. For example, a myriad of different chemotherapeutic agents used in cancer treatment have been reported to be sensitized by autophagy inhibition [24] implying that in each case autophagy is acting to reduce death in response to the chemotherapy in question. Since these agents work by very diverse means (e.g. DNA damage, interference with metabolic pathways, inhibition of steroid receptors, disruption of cytoskeleton, interference with growth promoting kinase pathways, activation of cell surface death receptor signaling, inhibition of the proteasome etc.), it is hard to imagine that autophagy could always be merely working to remove a toxic stimulus as we discussed in the case of aggregated proteins in neurodegenerative disease. Instead, since the vast majority of these instances of autophagy protecting against cell death are in fact protection against apoptotic death, a simpler explanation would be that autophagy directly inhibits the apoptotic machinery.
Apoptosis is usually considered to occur by two distinct but connected pathways. The extrinsic pathway occurs after activation of death receptors such as Fas/CD95 or the Tumor Necrosis Related Apoptosis Inducing Ligand (TRAIL) receptors [25] and these pathways are well understood and have even been mathematically modeled [26]. Upon ligand binding to the receptor, an adaptor protein called FADD is recruited to the active receptor. FADD recruits the inactive pro-form of caspase-8 and this receptor/adaptor/caspase complex, which has been called the Death Inducing Signaling Complex, DISC, allows the dimerization and catalytic activation of caspase-8. Proteolytically active caspase-8 then cleaves itself and either directly cleaves the effector caspase, caspase-3 or more commonly cleaves a protein called Bid, which translocates to the mitochondria to cause activation of the intrinsic apoptosis pathway. Other apoptotic stimuli also use the intrinsic pathway, which is activated by pro-apoptotic BH3-only proteins from the Bcl family of apoptosis regulators (Bid is one of this family) that induce mitochondrial permeabilization causing release of cytochrome c and other mitochondrial proteins. Cytochrome c binds to a scaffolding protein called Apaf1 to form a framework called the apoptosome for caspase-9 activation (i.e. the apoptosome functions rather similarly to the DISC in that it creates a scaffold on which dimerization of the caspase that will start the ball rolling, caspase-8 for the DISC and caspase-9 for the apoptosome). In both pathways, small amounts of active caspase-3 (and caspase-7) are rapidly amplified through self cleavage and cleavage of other caspases such as caspase-6, and the resulting burst of high intensity caspase protease activity cleaves hundreds of proteins to result in the common morphology (cellular fragmentation, contraction, breakup of the nucleus etc.) that is seen as apoptosis.
Given the very large number of examples where autophagy protects against apoptosis, it is somewhat surprising that we have few mechanistic examples that can explain how autophagy inhibits the apoptotic machinery. Thus, while hundreds of papers convincingly demonstrate that autophagy protects against apoptosis in response to very different stimuli, the key mechanistic question– what is it that autophagy is degrading to do this – is unknown for the vast majority of cases. One exception comes from examination of the effects of autophagy on TRAIL receptor signaling. As mentioned above death receptors work in a quite straightforward way, the activated receptor recruits caspase-8 to the DISC and it is the catalytic activation of this caspase that initiates the cascade of proteolysis that leads to apoptosis. It has been reported that autophagy can inhibit TRAIL-induced apoptosis and at least in part and in certain cell lines, this appears to be due to autophagic degradation of cleaved caspase-8, but not the uncleaved (and inactive precursor) [27] (Figure 2). However, other mechanisms that would more generally apply to apoptosis whereby autophagy could inhibit the core apoptosis machinery have not yet been identified. One possibility is that since autophagy degrades mitochondria (a process known as mitophagy), that this reduces the sensitivity of cells to apoptotic stimuli. However, since mitophagy occurs preferentially for impaired mitochondria with low membrane potential [28, 29] and apoptosis involves the release of mitochondrial proteins from all or almost all the mitochondria in the cell, it’s not immediately obvious why this on its own would lead to apoptosis inhibition by autophagy unless the mitochondrial mass was greatly reduced, which in most cases is not the case.
Figure 2. Example of the evasion from apoptotic cell death by autophagy.
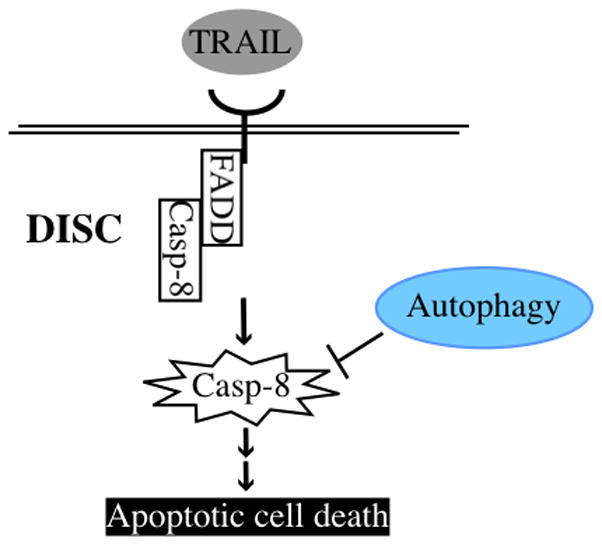
TRAIL induces DISC formation and caspase-8 is converted to active caspase-8 by self-processing in the DISC, which transduces a downstream signal cascade resulting in apoptosis. Autophagy can inhibit TRAIL-induced apoptosis by selective degradation of active caspase-8.
Thus a major open question in the field is to determine how autophagy has generally inhibitory effects on many apoptotic stimuli. One more radical possibility is that autophagy does not generally regulate the core apoptosis machinery per se, but rather that autophagy regulators do so but in an autophagy-independent fashion. And it is these activities that are responsible for the observed inhibitory effect of autophagy on apoptosis stimuli. The conclusion that autophagy protects against apoptosis is based on the following type of experiment. An apoptotic stimulus (e.g. a chemotherapy drug) is shown to have increased effectiveness (i.e. it kills more cells at an intermediate dose) when autophagy is blocked either pharmacologically or genetically. By far the most common pharmacological inhibitors that have been used in the studies reported to date are agents that affect the lysosome– most commonly chloroquine. For genetic inhibition, the most common genes that are targeted are those encoding ATG proteins that are required for autophagosome formation (most often ATG5, ATG7, ATG12 or BECLIN1, although sometimes other core ATGs such as LC3 are also targeted), and the inhibition is most commonly done by RNA interference. The rationale behind these experiments is that the inhibition of autophagy is specific– i.e. autophagy and only autophagy is inhibited by the inhibitor and if sensitivity to apoptosis is increased, this means that autophagy inhibited apoptosis. The problem is that these conclusions may be mistaken because the rationale is not correct– the manipulation may not truly be specific for autophagy. This is not too surprising with pharmacological inhibitors of autophagy, which are well known to be non-specific e.g. chloroquine inhibits lysosomes and lysosomes do other things (including regulation of apoptosis) that may be unrelated to autophagy. Indeed a recent paper demonstrated that chloroquine can sensitize cancer cells to anti-cancer drugs in an autophagy-independent manner [30].
In fact, the same thing is true for the ATG genes that regulate autophagy– the proteins they encode don’t just regulate autophagy, they also regulate other processes, especially apoptosis (Figure 3). Thus, for example, ATG7 interacts with and controls p53 [31]. ATG12 can regulate apoptosis by two quite separate autophagy-independent mechanisms [32, 33], and, ATG5 can also regulate apoptosis independently of autophagy [34]. UV radiation resistance-associated gene (UVRAG) induces autophagosome formation as part of the BECLIN 1 complex [35] but also regulates BAX activation at the mitochondrial membrane to control intrinsic apoptosis pathway [36]. Conversely, well known apoptosis regulators also control autophagy. This is particularly evident for members of the BCL family of proteins. Anti-apoptotic BCL-2 and BCL-xL proteins do not just regulate apoptosis; they also inhibit autophagy [37]. And the core autophagy regulator BECLIN1 itself possesses one of the domains, a BH3 domain, that defines the BCL family [38]. Moreover, many BH3-only proteins that work in concert with BCL-2, BCL-xL and BAX and BAK to regulate mitochondrial permeability [39] also regulate autophagy. As noted above, NOXA can induce autophagy [15]. However, other BH3 proteins, BAD and BNIP3 as well as drugs that serve as BH3 mimetics can disrupt the interaction between BECLIN1 and BCL-xL and to promote autophagy [40, 41]. Conversely, another pro-apoptotic BH3 protein, BIM, can inhibit autophagy [42] and this is critical in the regulation of starvation-induced autophagy. Another well known apoptosis regulator, FLIP, also controls autophagy [43]. The problem for our experiments that conclude that autophagy regulates apoptosis is clear. The experiment may have showed that ATG knockdown altered apoptosis but this might have been due to an effect on an autophagy-independent function of that ATG protein. Additionally since many of these effects occur in different directions (a protein promotes death on one instance but inhibits in another), a problem might also arise where a true effect could be missed because of these competing effects. Imagine a case where an apoptotic stimulus kills 50% of the cells on a dish when ATG12 is knocked down. If autophagy protects against apoptosis, perhaps the amount of death that would have been obtained in the absence of knockdown would have been much less. However since the ATG12 protein may also have been required for efficient induction of mitochondrial protein release through a completely different mechanism, e.g. inhibition of anti-apoptotic proteins like MCL-1 [33], the knockdown would at the same time inhibit this effect. Thus one could imagine a situation where the balance of pro-and anti- death effects of knocking down ATG12 may add up to little overall difference in the number of dead cells whether or not it was knocked down.
Figure 3. Crosstalk between autophagy and apoptosis regulators.
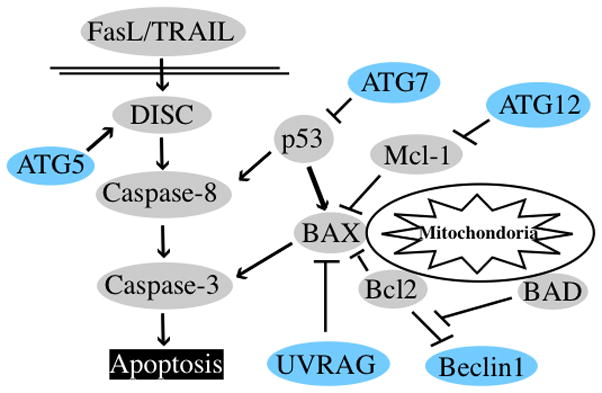
Examples of autophagy regulator (blue) interaction with apoptosis regulators (gray). ATG5 helps activation of DISC via interaction with FADD. ATG7 and UVRAG can bind and inhibit apoptosis regulators, p53 and BAX respectively; at least theoretically this could explain a mechanism by which some autophagy proteins directly suppress apoptosis. ATG12 binds to anti-apoptotic members of the BCL-2 family such as Mcl-1 and inhibits their antiapoptotic activity. Conversely, BCL-2 (and BCL-xL) binds and inhibits an autophagy positive regulator, Beclin1.
It is difficult to design experiments that discriminate between different effects by the same protein. One way is to identify mutant versions of the protein that affect one function but not the other. Jay Debnath and colleagues recently used this approach of making such separation of function mutants to show that while ATG12 can be conjugated to ATG3, this does not affect autophagy as ATG12-ATG5 does, but rather affects mitochondrial homeostasis and apoptosis [32]. The practical problem with this strategy is that mutants that really separate out different functions may be hard to identify, however this strategy does allow one to at least begin to determine what function of a given autophagy regulator is important for the effects that are being measured.
In summary, we already have numerous examples where the same proteins, whether identified initially as an apoptosis regulator or as an autophagy regulator, also regulate the other process. This suggests a general model that regulation of both autophagy and apoptosis occurs again and again by the same proteins, but also raises a different potential explanation for the data suggesting that autophagy generally inhibits apoptosis– could some of these cases actually be due to the knockdown of autophagy regulators affecting steps in the apoptosis pathway without implicating any requirement for altered autophagic flux itself being necessary for the effects? Could the sequestration or depletion of a protein like UVRAG that otherwise might be involved in regulating the BECLIN1 complex to control autophagosome formation result in less available UVRAG to inhibit BAX induced apoptosis? While such mechanisms still indicate that connections between autophagy and apoptosis are important, they lead to a very different view of how autophagy controls apoptosis– i.e. they suggest it may sometimes have nothing to do with the actual process of autophagosome formation, fusion with lysosomes and degradation of the contents.
Concluding remarks
We now have a large amount of evidence showing interactions between autophagy and cell death pathways, particularly the canonical apoptosis pathway. However, their larger significance for normal biology and disease is still unclear and we lack a full understanding of how mechanistically these connections take place. The first examples whereby autophagic degradation of specific targets such as dBRUCE or caspase-8 can promote or inhibit apoptosis have been found. However, in the vast majority of cases where autophagy appears to regulate apoptosis whether positively or negatively are still unclear in any mechanistic sense. Moreover the big question, of how autophagy generally regulates the core apoptotic machinery, or even whether it does so, is unresolved. Given the intense focus on this area of research, better understanding of these mechanisms will certainly be obtained in the next few years. And, this knowledge may help guide the manipulation of these processes to treat diseases where we might want to see more or less cell death.
Summary.
There are some reports that demonstrate autophagy promotes cell death. Apoptosis-null cell indicates autophagy-dependent cell death in response to DNA damage agents and overexpression of oncogenic RAS causes apoptosis-independent cell death. Moreover, in Drosophila oocyte, autophagy is necessary for apoptotic cell death of nurse cells. Therefore, autophagy can be needed for apoptotic and non-apoptotic cell death.
Autophagy also inhibits apoptosis by degrading apoptosis regulators. For example, autophagy selectively degrades active caspase-8 in some TRAIL-induced apoptotic cells. Although we have many examples that show autophagy protects cells against apoptosis, few reports explain those mechanisms.
Recent studies have demonstrated that there are many physical and functional interactions between autophagy and apoptosis regulators. Some studies have shown that autophagy proteins regulate apoptosis proteins, whereas other studies have demonstrated that apoptosis proteins regulate autophagy proteins. Additionally, positive regulators of autophagy can positively or negatively (or differentially) regulate apoptosis proteins in different situations (or cases or contexts), and it is not clear whether this regulation is conditional or comprehensive. Further analysis is needed to solve these questions.
Acknowledgments
Work in our laboratory is supported by RO1 grants from the National Institutes of Health (CA111421 and CA150925) and Shared Resources that are supported by P30 CA04934.
References
- 1.Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21:387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125:5259–5268. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- 3.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews Drug discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 7.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 8.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DR, Victor B. The pantheon of the fallen: why are there so many forms of cell death? Trends in cell biology. 2012;22:555–556. doi: 10.1016/j.tcb.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon H-U, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 13.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 Program of Autophagic Cell Death by Caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 14.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic ras-induced expression of noxa and beclin-1 promotes autophagic cell death and limits clonogenic survival. Molecular Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CY, Cooksey BA, Baehrecke EH. Steroid regulation of midgut cell death during Drosophila development. Dev Biol. 2002;250:101–111. doi: 10.1006/dbio.2002.0784. [DOI] [PubMed] [Google Scholar]
- 19.Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–531. doi: 10.1083/jcb.201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu C-D, Zhang J, Kester M, Wang H-G. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130–141. doi: 10.1016/j.pharmthera.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorburn A. Death Receptor-induced cell killing. Cellular Signalling. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJR, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–228. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Molecular Cell. 2011;44:698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 35.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–698. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 36.Yin X, Cao L, Kang R, Yang M, Wang Z, Peng Y, Tan Y, Liu L, Xie M, Zhao Y, Livesey KM, Tang D. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 2011;12:727–734. doi: 10.1038/embor.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 39.Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Molecular Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PPC, Sadiq O, Rubinsztein DC. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Molecular Cell. 2012;47:359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]