

Significance
Human IFN-inducible protein-16 (IFI16) is an essential intracellular foreign DNA receptor of innate immunity and also implicated in several autoimmune disorders. However, little is known about molecular mechanisms that underlie its function. We show here that IFI16 cooperatively assembles into filaments on dsDNA. IFI16 thus oligomerizes even in the presence of excess DNA, and it is the non–DNA-binding pyrin domain of IFI16 that drives the filament assembly. These results provide unifying mechanistic explanations for several previous in vivo observations regarding IFI16 and also suggest that assembling filaments on foreign nucleic acids is a broad host defense strategy.
Keywords: cooperative filament formation, inflammasome
Abstract
Whether host DNA receptors have any capacity to distinguish self from nonself at the molecular level is an outstanding question in the innate immunity of mammals. Here, by using quantitative assays and electron microscopy, we show that cooperatively assembling into filaments on dsDNA may serve as an integral mechanism by which human IFN-inducible protein-16 (IFI16) engages foreign DNA. IFI16 is essential for defense against a number of different pathogens, and its aberrant activity is also implicated in several autoimmune disorders, such as Sjögren syndrome. IFI16 cooperatively binds dsDNA in a length-dependent manner and clusters into distinct protein filaments even in the presence of excess dsDNA. Consequently, the assembled IFI16⋅dsDNA oligomers are clearly different from the conventional noninteracting entities resembling beads on a string. The isolated DNA-binding domains of IFI16 engage dsDNA without forming filaments and with weak affinity, and it is the non–DNA-binding pyrin domain of IFI16 that drives the cooperative filament assembly. The surface residues on the pyrin domain that mediate the cooperative DNA binding are conserved, suggesting that related receptors use a common mechanism. These results suggest that IFI16 clusters into signaling foci in a switch-like manner and that it is capable of using the size of naked dsDNA as a molecular ruler to distinguish self from nonself.
Recognition of foreign intracellular DNA is a widely conserved defense mechanism by which the innate immune system of mammals detects and responds to invading pathogens (1, 2). Using such a universal molecule as a major danger signal for detecting pathogens must be regulated in a stringent yet efficient manner. However, only a few deciding factors are known for the host innate immune system to selectively engage foreign DNA (i.e., nonself-DNA) while minimizing interactions with self-DNA, which include the compartmentalization of mammalian cells and the size of foreign DNA. These features, however, only raise more questions than provide answers. For example, the footprints of intracellular DNA receptors usually fall below 20 bp, and yet a long foreign DNA fragment [e.g., poly(dA:dT); ≥1,000 bp] is required to induce a robust innate immune response even in a normally DNA-free environment like the cytoplasm (1, 2). On the other hand, foreign DNA-sensing pathways also exist in the host nucleus in which DNA receptors must not respond to abundant self-DNA to prevent spurious activities (1, 2). Indeed, one of the major unresolved questions in understanding the DNA-sensing pathways of mammals is whether the host intracellular DNA receptors have any capacity to distinguish self- from nonself-DNA at the molecular level (2).
Human IFN inducible protein-16 (IFI16) is an intracellular DNA receptor of innate immunity that belongs to the family of absent-in-melanoma-2 (AIM2)-like receptors (ALRs) (1–4). IFI16 senses DNA from invading pathogens in both the nucleus and cytoplasm [e.g., vaccinia virus (VACV) and herpes simplex virus-1 (HSV-1)] and plays critical roles in production and maturation of major proinflammatory cytokines such as IFN-β and interleukin-1β. Notably, IFI16 is the only known host DNA sensor that operates in the nucleus, which is conventionally thought to be off limits to DNA receptors due to the abundant self-DNA. In addition to its role in defense against foreign DNA, the aberrant activity of IFI16 is also associated with several autoimmune disorders, such as systemic lupus erythematosus and Sjögren syndrome. IFI16 is involved in the onset and progression of these diseases not only by generating abnormally high levels of the proinflammatory cytokines but also by being recognized as an autoantigen by the host adaptive immune system (4–8).
Despite its implication to host defense and autoimmunity, little is known about molecular mechanisms that underlie IFI16. Indeed, there are three major unresolved questions regarding its behavior in vivo. First, as observed from several other DNA-sensing pathways in mammals, it is not known why long dsDNA (>60 bp) is required to induce IFI16 activity (3, 9, 10). Second, why IFI16 does not bind self-DNA in the nucleus is not known (11–14). Third, it is not known how IFI16 can selectively assemble into large signaling foci (e.g., inflammasome) with nonself-DNA (11–14).
Although mechanisms of many proteins can be inferred from their structures, the structure of IFI16 does not provide clear answers to the above questions. IFI16 is composed of two signature ALR domains, namely one N-terminal pyrin domain (PYD) (IFI16PYD) followed by two HIN200 domains (IFI16HinA and IFI16HinB). The HIN200 domains nonspecifically bind various ss- and dsDNA fragments on the phosphate backbone via electrostatic interactions with a footprint of eight to nine bases, thus confirming that DNA sequence (e.g., the CpG island) is not a recognition element (9, 15–17). On the other hand, the PYD is a homotypic protein-protein interaction domain whose function is thought to be limited to recruiting downstream effectors (3, 9, 10).
Based on crystallographic and binding studies of related AIM2 and the isolated HIN200 domains of IFI16, a general mechanistic model for ALR activation was recently proposed (9, 18). In this model, ALRs assume an autoinhibited conformation in which the PYD blocks the DNA-binding surface of the HIN200 domain. Upon encountering foreign dsDNA, the PYD is displaced and interacts with its downstream partners. A long dsDNA fragment is used as a pseudooligomerization platform, because the HIN200 domain independently binds to either strand of dsDNA with a footprint of about eight bases. The resulting ALR⋅DNA complex thus assumes a configuration similar to noninteracting beads on a string. Longer DNA fragments simply accommodate more ALRs, allowing the ALR pseudooligomers to recruit a greater number of downstream effectors.
However, these otherwise seminal studies (9, 18) do not explain any of the three questions regarding IFI16 behavior. First, it provides no rationale for the DNA length-dependent responses observed in vivo. By this model, IFI16 would bind equally well to either strand of any dsDNA exceeding its footprint. Thus, any dsDNA long enough to promote minimal IFI16 oligomer for downstream effector interaction (e.g., dimer for procaspase-1 activation; 10 bp) would produce similar responses at the same mass concentration. Second, this model provides no insight into how IFI16 suppresses its interaction with self-DNA in the nucleus. For instance, AIM2 has only one HIN200 domain and is exclusively localized in the cytoplasm (19). By contrast, IFI16 has two HIN200 domains that can bind either ss- or dsDNA and assumes an uninhibited open conformation, and its footprint falls within the range of the exposed dsDNA linker between host nucleosomes (10–20 bp) or transcription bubbles (∼17 bases) (12, 20–22). Indeed, the proposed autoinhibitory mechanism would be effective only in an intrinsically DNA-free environment like the cytoplasm but not in the nucleus, where abundant self-DNA would easily displace IFI16PYD from either HIN200 domain. Finally, it does not explain how IFI16 can selectively colocalize with foreign DNA and assemble into large signaling foci. The total amount of DNA under normal or infectious conditions easily exceeds that of IFI16. Thus, by this model, IFI16 would be scattered on foreign and self-DNA instead of selectively forming signaling foci on nonself-DNA (e.g., in principle, one genome of HSV-1 contains more than 30,000 binding sites).
Here, by using quantitative assays and electron microscopy, we find that cooperatively assembling into filamentous oligomers on dsDNA may underlie the observed behaviors of IFI16 in vivo. IFI16 binds dsDNA in a nonlinear length-dependent manner and forms oligomers that are clearly different from entities resembling beads on a string. The isolated HIN200 domains of IFI16 do not oligomerize and thus engage dsDNA with weak affinity. However, in contrast to the PYD of AIM2, IFI16PYD plays an unexpected positive role in DNA binding as it drives the filament assembly. The surface residues that are important for dsDNA binding in IFI16PYD are highly conserved, indicating that other ALRs use the same strategy. These results reveal that IFI16 is poised to rapidly engage even a single piece of long nonself-DNA with a switch-like mechanism (e.g., HSV-1 genome), while not responding to much higher concentrations of short self-DNA exposed under normal conditions (e.g., nucleosomal linker dsDNA).
Results
Assembling signal⋅receptor complex is a critical regulatory step in virtually all known signaling pathways, because this first step commits the rest of cascades in a switch-like mechanism (23, 24). We reasoned that it is unlikely that regulation of IFI16 pathways is only accomplished by yet unknown downstream events. The currently prevailing mechanistic model for IFI16 was established by studying its isolated HIN200 domains, which nonspecifically bind any DNA exceeding their footprints equally well (3, 9, 15). In contrast, we hypothesized that studying the full-length protein might better address the DNA-sensing strategy of IFI16. Thus, for this work, we first successfully generated full-length and the functional domains of IFI16 without solubility tags in high purity (Fig. S1 A–C) and also developed various in vitro assays to investigate how IFI16 engages DNA.
IFI16 Cooperatively Binds dsDNA in a Length-Dependent Manner.
We hypothesized that the DNA length-dependent responses of IFI16 in vivo arise directly from differences in binding affinity for different-length DNA ligands but not from an as yet unidentified regulatory mechanism mediated by downstream effectors. To test this idea, we developed fluorescence-anisotropy (FA) binding assays using various fluorescein-amidite (FAM)-labeled DNA fragments. Monitoring FA of FAM-dsVACV72 (a 72-bp fragment derived from VACV; Fig. S1D) (3) with increasing concentrations of IFI16 resulted in a sigmoidal binding isotherm best fit with a Hill equation [an apparent binding constant (KDapp) of 65 ± 19 nM and a Hill constant of 1.9 ± 0.2; Fig. 1A]. This result indicated that full-length IFI16, unlike its isolated HIN200 domains (3, 9, 15), binds dsDNA in a cooperative manner. We then tested the binding of shorter dsDNA variants of VACV and HSV fragments that were used in the previous in vivo study (3). In contrast to predictions from the previously proposed model (9), we found that the binding affinity decreased with decreasing length of the dsDNA fragments independent of sequence (Fig. 1B, Fig. S1D, and Table S1). Importantly, the calculated Hill constants also decreased with shorter dsDNA (e.g., Fig. 1 B and C), suggesting that an absence of cooperativity results in weaker affinity (see also Table S1).
Fig. 1.

IFI16 cooperatively binds dsDNA in a length dependent manner. (A) Binding of full-length IFI16 to FAM-dsVACV72 (2.5 nM) was monitored by changes in FA. The fraction bound was calculated as (A − A0)/(Amax − A0), where A0 is the anisotropy of the free DNA and Amax is the anisotropy of the saturating protein. The lines are fits to a Hill form of binding equation (number of independent experiments, n = 3; ±SD). (B) Binding of IFI16 to each FAM-dsDNA was determined by FA. The lines are fits to a Hill equation: fraction bound = 1/(1 + (KDapp/[IFI16])Hill constant). The determined values are listed in Table S1. (C) The data for dsHSV33 and dsHSV24 from B were replotted to demonstrate their noncooperative binding profiles. (D) The stoichiometry between IFI16 and dsVACV72 was determined using the concentration of FAM-dsVACV72 sixfold higher than its determined KDapp (Table S1). The lines are fits to a quadratic binding formula. Shown is representative of n =3. (E) Stoichiometry of IFI16 to dsDNA was determined by plotting the number of bound IFI16 molecules vs. dsDNA length (dsVACV72, dsHSV60, dsVACV52, and dsHSV33; see also Fig. S2A) (n = 3; ±SD). (F) A plot of binding-site normalized KDapp vs. dsDNA length for IFI16 (n = 3; ±SD).
A possible underlying mechanism for the cooperative length-dependent binding is that IFI16 assembles into a distinct oligomer on dsDNA, because such a protein cluster would result in a more stable complex than the previously proposed noninteracting beads on a string (9). To test this idea, we first determined the stoichiometry between IFI16 and dsDNA ligands by performing FA experiments using FAM-dsDNA concentrations at least six times higher than their respective KDapp values (e.g., Fig. 1D). The plot of dsDNA length vs. the number of bound molecules revealed that about 15 bp are required to accommodate one full-length IFI16 (Fig. 1E), which agrees with a previous report suggesting that one HIN200 domain takes up about eight to nine bases. Notably, because no significant binding was observed from FAM-dsHSV15, these results also suggest that oligomeric IFI16 is required for tight binding (see also below). To confirm the length-dependent binding, we normalized the KDapp of each fragment to the number of available binding sites on each dsDNA fragment (e.g., four for dsHSV60). Plotting normalized KDapp vs. dsDNA-length revealed that the binding affinity of IFI16 still increases with increasing DNA length (Fig. 1F), supporting the idea that IFI16 cooperatively oligomerizes on dsDNA in a length-dependent manner. Importantly, these results correlate with the previously reported dsDNA length-dependent responses in vivo (3) and thus suggest that cooperatively assembling stable IFI16⋅dsDNA complexes via oligomerization is critical for regulating its cellular activity.
To investigate the DNA length-dependent binding mechanism further, we then performed competition binding assays in which increasing concentrations of various unlabeled DNA fragments were added to the FAM-dsVACV72⋅IFI16 complex (Fig. 2A). dsDNA fragments shorter than ∼30 bp did not compete up to the highest mass concentrations used in these assays (Fig. 2A). For example, at the same mass concentration (140 µg/mL), unlabeled dsVACV72 completely competed off the labeled DNA while dsHSV33 did not show any competition (Fig. 2A). All lengths of ssDNA failed to show significant inhibition (15–72 bases), indicating that full-length IFI16 preferentially binds dsDNA despite the reported ssDNA-binding activity of IFI16HinA (15) [ssAG60 shown as an example (30 repeats of adenosine and guanosine); Fig. 2A]. Additionally, dsAG60 and dsHSV60 showed essentially the same IC50 values (the concentrations of competitor DNA at the half-maximal inhibition), confirming that IFI16 binds dsDNA independent of its sequence (Fig. 2A). dsDNA fragments longer than 120 bp competed well at similar mass concentrations (Fig. 2B); however, the FA of bound FAM-dsVACV72 increased with low concentrations of these longer dsDNA fragments, suggesting that the size of IFI16⋅FAM-dsDNA increased (Fig. 2B). Closer inspection of these results revealed that signals increased up to the binding-site normalized concentration of each competitor equivalent to the amount of IFI16 used in these assays (200 nM; indicated by the arrow in Fig. 2B). Because we have a large excess of IFI16 to FAM-dsVACV72 (5 nM; 0.2 µg/mL), we interpreted the initial increase to be caused by free IFI16 molecules preferentially binding to subsaturating concentrations of the longer competitor dsDNA and the resulting complexes interacting with IFI16⋅FAM-dsVACV72 (see also below). The apparent increase of bound fraction would result in significantly overestimated IC50 values for the longer fragments (thus true IC50 << 10µg/mL). Nonetheless, these results consistently suggest that IFI16 prefers to bind dsDNA fragments significantly exceeding its footprint with a switch-like selectivity.
Fig. 2.

Competition binding assays support the length-dependent binding mechanism. (A) Competition binding assays using IFI16 (600 nM) and FAM-dsVACV72 (5 nM; 0.2 µg/mL) against various DNA fragments; the lines are fits to a competition binding equation: 1/[1 + ([DNAcompetitor]/IC50)Hill constant]. The determined values 39-, 60-, and 72-bp fragments are 210 ± 30, 120 ± 20, and 38 ± 9 µg/ml, respectively. (B) Competition binding assays using various dsDNA fragments against IFI16⋅FAM-dsVACV72 complex (200 nM and 0.2 µg/mL, respectively). The arrow indicates the effective dsDNA concentration of 200 nM (n = 3; estimated SD, ≤15%).
IFI16 Oligomers Are Distinct Protein Clusters.
In contrast to the previous model in which IFI16⋅dsDNA complexes resemble noninteracting beads on a string (9), our binding experiments suggested that IFI16 assembles into distinct oligomeric clusters on dsDNA. These two models can be distinguished by monitoring formation of FAM-dsDNA⋅IFI16 complexes in the presence of increasing amounts of unlabeled DNA using native gel electrophoretic mobility-shift assays (EMSAs): the clustering mechanism would show a concerted transition without resulting in significant intermediates, whereas the noninteracting mechanism would clearly display intermediate species (25, 26). We first performed EMSAs with various FAM-dsDNA fragments. Despite extensive efforts to optimize conditions, IFI16⋅dsVACV72/HSV60 complexes did not fully enter into the gel matrix. This was likely caused by the unusually high pI of IFI16 (pI: 9.3), as similar results were observed for AIM2 (pI: 9.8) and other DNA-binding proteins with high pI values (10, 27); thus we limit analyses of EMSA for qualitative purposes only. Nevertheless, judging by the disappearance of free FAM-dsDNA and by the formation of only a few transient intermediates, these results are consistent with our FA assays in which IFI16 binds dsDNA cooperatively and eventually form an oligomer on each dsDNA in a length-dependent manner (Fig. 3A). We then performed a competition EMSA in which increasing concentrations of unlabeled dsAG60 were added to FAM-dsVACV72⋅IFI16 (Fig. 3B). As predicted from the clustering model, we found that IFI16 bound dsVACV72 in a concerted manner without forming distinct intermediates. Importantly, the lack of intermediates in this assay indicates that IFI16 preferentially clusters even if there are nonadjacent excess binding sites (i.e., excess dsDNA). Next, to further confirm these results, we cross-linked IFI16 using a cysteine-specific cross-linker [1,4 bismaleimidyl-2,3-dihydroxybutane (BMDB)] in the presence or absence of dsDNA and visualized resulting complexes using SDS/PAGE; IFI16 has eight cysteines spatially distributed on the opposite side of the DNA-binding patches of its two HIN200 domains. As expected, we found that IFI16 forms oligomers almost exclusively in the presence of dsDNA (Fig. 3C). Further supporting the clustering model, IFI16 was still efficiently cross-linked in the presence of excess DNA (i.e., excess nonadjacent binding sites) (Fig. 3D).
Fig. 3.

IFI16 cooperatively clusters on dsDNA. (A) EMSAs of various dsDNA (5 nM) against increasing concentrations of IFI16 (0, 1.5, 3, 6, 12, 24, 48, and 96 nM). (B) An EMSA in which increasing concentrations of dsAG60 (190, 95, 45, 23, 12, 6, 3, and 1,5 µg/mL) were added to IFI16⋅FAM-dsVACV72 (190 nM and 0.2 µg/mL respectively). (C) DNA-dependent cross-linking of IFI16. The ratio between available binding sites and IFI16 was one to one. The resulting complexes were separated by using a gradient SDS/PAGE and visualized by silver staining. (D) The cross-linking efficiency of IFI16 was monitored with increasing DNA. The indicated ratios indicate IFI16:binding sites. The resulting complexes were resolved as with C.
IFI16 Oligomerizes on dsDNA in a Switch-Like Manner.
To quantitatively determine the oligomerization efficiency of IFI16 in the presence of excess DNA, we developed a fluorescence resonance energy transfer (FRET) assay by labeling one batch of IFI16 with a FRET donor and another with an acceptor. Importantly, because we add increasing amounts of excess DNA to a fixed concentration of IFI16, FRET signals are expected to arise only if IFI16 preferentially binds next to one another even in the presence of excess nonadjacent binding sites. Indeed, adding increasing amounts of unlabeled dsHSV60 or dsVACV72 to an equimolar (20 nM each) mixture of FRET-labeled IFI16 produced changes of the emission ratios between the donor (decrease) and acceptor (increase) that could fit to a Hill equation (we denote the midpoints of these curves as KFapp values, apparent oligomerization constants; Fig. 4 A and C and Table S2). No significant FRET signals were observed from ssDNA (e.g., ssVACV72) or dsDNA shorter than 60 bp (e.g., Fig. 4B). The amplitudes of the FRET ratios from dsDNA fragments longer than 72 bp were greater but otherwise did not differ significantly from one another, indicating that all labeled IFI16 molecules used in the assay were bound to these fragments to form similarly sized oligomers (Fig. 4B). After determining the KFapp value for each dsDNA fragment, we plotted relative binding efficiency (normalized to the KFapp of dsDNA2000) vs. dsDNA length to analyze how the oligomerization (binding) efficiency of IFI16 changes with respect to the length of dsDNA. This plot showed a cooperative relationship best fit with a Hill constant of 5.2 ± 0.6 and the optimal oligomerization efficiency (infliction point) around 150 bp (indicated by the arrow in Fig. 4C). Consistent with the results from our FA binding experiments, the Hill constant of about 5 in this plot indicates that a small fixed amount of IFI16 can dramatically amplify its oligomerization efficiency with increasing length of dsDNA in a switch-like manner. Moreover, the infliction point at 150 bp suggests that about ten IFI16 molecules comprise an optimal binding cluster (15 bp per one IFI16).
Fig. 4.

IFI16 oligomerizes on dsDNA with a switch-like mechanism. (A) A sample fluorescence emission spectra of an equimolar mixture of FRET donor (Dylight 550)- and acceptor (Dylight 650)-labeled IFI16 with increasing concentrations of dsVACV72 (0, 1.5, 3, 6, 12 24, 48 µg/mL; excitation at 522 nm). A.U., arbitrary units; RFU, relative fluorescence units. (B) The emission spectra of FRET-labeled IFI16 with increasing concentrations of dsHSV24 (0, 10, 20, 50, 100 µg/mL). (C) A sample plot of changes in the ratio between the FRET donor emission (λmax: 578 nm) and the acceptor emission (λmax: 678 nm) at each indicated dsDNA concentration. The apparent oligomerization constants (KFapp) were obtained by fitting the data to a Hill equation (Table S2), and shown is a representative of n = 3. (D) A plot of binding efficiency vs. the length of dsDNA for IFI16. The data were fit to a Hill equation. The efficiency was determined by normalizing the mean KFapp of each fragment to that of dsDNA2000. The arrow indicates the infliction point (∼150 bp).
IFI16 Assembles into Filamentous Oligomers on dsDNA.
To define the structure of IFI16 oligomers on dsDNA, we examined IFI16 bound to various dsDNA fragments using negative-stain electron microscopy (EM). Electron micrographs of DNA-free IFI16 produced negatively stained particles of about 22 nm (Fig. 5A). This observation is consistent with the extended conformation (211 Å) seen from a previous small-angle X-ray scattering experiment (20) and confirms that apo-IFI16 does not assume an autoinhibited conformation (9) in which IFI16PYD docks on one of the HIN200 domains. With various dsDNA fragments (IFI16:binding sites, 1:1), we found that IFI16 forms filaments corresponding to the length of dsDNA used in each experiment (Fig. 5 B–D). For example, dsDNA600 is predicted to be 193 nm long, and the protein filaments we observed using this fragment ranged from 190 to 210 nm (Fig. 5D). The width of filaments, however, was consistently between 20–25 nm regardless of dsDNA length, indicating that IFI16 oligomerizes laterally along the length of dsDNA. Interestingly, when we generated IFI16 filaments with higher concentrations of protein and dsDNA (3 vs. 1 µM; still keeping IFI16:binding sites at 1:1), we also found a number of extended filaments that appeared to form by conjoining either end of two or more individual filaments (Fig. 5C, Left vs. Right). Notably, formation of such higher-order oligomers is consistent with our competition binding experiments in which IFI16⋅FAM-dsVACV72 was thought to interact with IFI16⋅competitor-dsDNA complexes. Finally, as expected from our solution assays, IFI16 still assembled into filaments in the presence of excess dsDNA (Fig. 5E). The incomplete coverage of dsDNA in this experiment is likely attributable to multiple nucleation events, as seen from other filament forming nucleic acid sensors (i.e., “optimal” decamer formation, presumably 50–60 nm) (26). Taken together with our biochemical data, we concluded that IFI16 cooperatively clusters into filamentous oligomers on dsDNA.
Fig. 5.

Negatively stained electron micrographs of IFI16⋅dsDNA complexes. (A) An electron micrograph of apo-IFI16. Electron micrographs of buffer A and buffer A plus dsDNA600 can be found in Fig. S2B. (B) IFI16 filaments were generated with dsDNA200 (∼66 nm). The observed lengths and widths of filaments were about 60–75 nm and 20–25 nm, respectively. (C) IFI16 filaments were generated with dsDNA300 (∼99 nm). (Left and Center) The observed lengths and widths of filaments were about 90–120 nm and 20–25 nm, respectively. (Right) Higher-order filaments formed by IFI16⋅dsDNA300 filaments. Arrows indicate the approximate size of one filament of IFI16⋅dsDNA300. (D) IFI16 filaments were generated with dsDNA600 (198 nm). The observed lengths and widths of filaments were about 190–210 nm and 20–25 nm, respectively. (E) IFI16 filaments were assembled in the presence of excess dsDNA300 (IFI16:binding sites, 1:3). The size of filaments ranged from 50 to 80 nm.
Noncooperative dsDNA Binding by the HIN200 Domains of IFI16 Results in Much Weaker Affinity.
To identify the functional domain of IFI16 that drives filament formation, we generated the individual and tandem HIN200 domains of IFI16 (IFI16HinA, IFI16HinB, and IFI16HinAB; Fig. S1) and assayed their binding to FAM-dsVACV72. Surprisingly, we did not observe significant dsDNA binding from these IFI16 variants (e.g., Fig. 6A, Upper; see also Fig. S2C and Fig. 7A). It was previously reported that dsDNA-binding affinity of IFI16HinB was significantly influenced by buffer salt concentrations (9). Indeed, we found that the near-physiological salt concentrations (160 mM KCl) in our reaction buffer interfered with binding (Fig. 6A, Lower). Unlike full-length, the disappearance of free FAM-dsVACV72 and dsHSV60 was apparently noncooperative even under 60 mM KCl, suggesting that the DNA-bound complexes formed by the tandem HIN200 domains of IFI16 must resemble noninteracting beads on a string (Fig. 6 A and B). To detect binding of the individual HIN200 domains, we decreased the KCl concentration even further to 20 mM. Here, we found that all IFI16 variants but IFI16PYD bound dsDNA in decreasing order of affinity: full-length, IF16HinAB, IFI16HinA, IFI16HinB. Importantly, as expected from the EMSA, the fits to the isolated HIN200 domains binding to dsVACV72 were noncooperative (Hill constants, ∼1, Fig. 6C and Fig. S2 D and E). These results suggest the HIN200 domains of IFI16, individually or in tandem, do not efficiently bind dsDNA because they fail to cooperatively oligomerize.
Fig. 6.
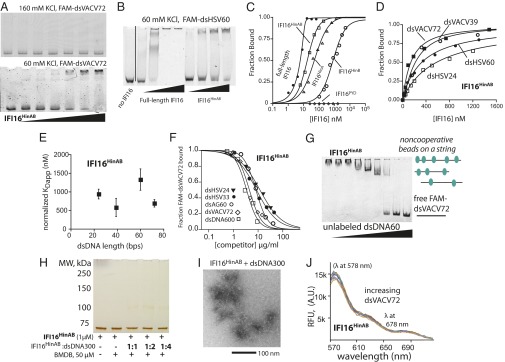
The HIN200 domains of IFI16 do not oligomerize on dsDNA. (A) An EMSA of IFI16HinAB to FAM-dsVACV72 (2 nM) at 160 mM KCl (Upper) and 60 mM KCl (Lower) (IFI16HinAB: 0, 1.5, 3, 6, 12, 24, 48, and 96 nM). (B) An EMSA of full-length IFI16 (50, 100, 200, 400 nM) and IFI16HinAB (100, 200, 400, 800 nM) to FAM-dsHSV60 (20 nM). (C) Binding of IFI16 variants to FAM-dsVACV72 were determined by FA at 20 mM KCl. The determined values are listed in Table S3. The data for full-length IFI16 were fit with a quadratic form of binding equation, and the others were fit with a Hill equation. Shown is a representative of n = 4. (D) Binding of IFI16HinAB toward indicated FAM-dsDNA fragments (5 nM each) was determined by FA at 60 mM KCl. The lines are fits to a Hill equation. Shown is a representative of four independent experiments. (E) Binding-site normalized KDapp values of IFI16HinAB from D (n = 4; ±SD). (F) Competition binding assays were performed at 60 mM KCl using various dsDNA fragments against IFI16HinAB⋅FAM-dsVACV72 complex (300 nM and 0.2 µg/mL, respectively). Shown is the average of n = 2. (G) An EMSA in which increasing concentrations of dsAG60 (190, 95, 45, 23, 12, 6, 3, and 1,5 µg/mL) were added to IFI16HinAB⋅FAM-dsVACV72 (250 nM and 0.2 µg/mL, respectively) using 60 mM KCl. (H) The cross-linking efficiency of IFI16HinAB was monitored with increasing DNA using 60 mM KCl. The ratios indicate IFI16HinAB:binding sites. The resulting complexes were resolved as with D. (I) An electron micrograph of IFI16HinAB (1 µM) with dsDNA300. (IFI16HinAB:binding sites, 1:1.) (J) The lack of FRET signals for donor- and acceptor-labeled IFI16HinAB (100 nM each) with increasing dsVACV72 (0, 0.5, 1, 2, 4, 8, 17, 35, 70, 140 µg/mL) at 60 mM KCl.
Fig. 7.
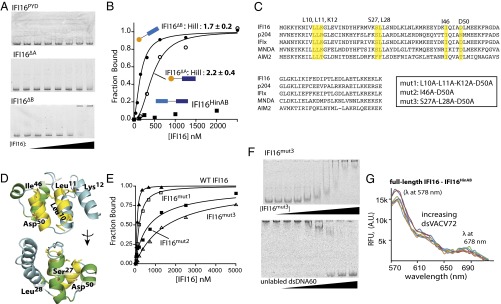
IFI16PYD plays a positive role in cooperative oligomerization. (A) EMSAs of IFI16PYD (Top), IFI16∆A (Middle), and IFI16∆B (Bottom) to FAM-dsVACV72 at 160 mM KCl (0, 1.5, 3, 6, 12, 24, 48, and 96 nM; for IFI16PYD: 15, 30, 62, 125, 250, 500, and 1,000 nM). (B) Binding of IFI16HinAB, IFI16∆A, and IFI16∆B to FAM-dsVACV72 was determined by FA at 160 mM KCl. The data were fit with a Hill equation (Table S3). Shown is a representative of n = 3. (C) A sequence alignment of the PYDs of various ALRs. The mutated residues are highlighted, and the alignment was generated using ClustalOmega (www.clustal.org). P204 is a mouse homolog of IFI16, and all of the other proteins are human ALRs. (D) A homology model of IFI16PYD was generated using the PYD of MNDA as a template and the SWISS-MODEL server. Mutated side chains are shown with a stick representation; the figure was generated using PyMOL (DeLano Scientific). (E) Binding of each mutant IFI16 to FAM-dsVACV72 was determined by FA at 160 mM KCl. The wild-type data from Fig. 1A are shown for comparison. The determined values are listed in Table S3, and shown is a representative of n = 3. (F, Upper) an EMSA-binding assay of IFI16mut3 (0, 3, 6, 12, 24, 48, 96, 192, 384, 768 nM) to FAM-dsVACV72 (0.4 µg/mL). (F, Lower) An EMSA in which increasing concentrations of dsAG60 (0, 1.5, 3, 6, 12, 23, 45, and 190 µg/mL) were added to IFI16mut3⋅FAM-dsVACV72 (768 nM and 0.2 µg/mL, respectively). (G) The lack of FRET signals from an equal mixture of donor-labeled IFI16HinAB and acceptor-labeled full-length IFI16 (50 nM each) with increasing dsVACV72 (0, 1.2, 6, 28, 140 µg/mL) at 60 mM KCl.
Several additional experiments performed at 60 mM KCl confirmed that the tandem HIN200 domains of IFI16 do not oligomerize. First, in FA binding assays, we found that unlike full-length, IFI16HinAB bound all FAM-dsDNA fragments noncooperatively and without apparent length-dependency (Fig. 6 D and E vs. Fig. 1). Second, in FA competition assays, all dsDNA fragments competed almost equally well without showing any apparent increase in bound fractions (Fig. 6F vs. Fig. 2). Third, in an EMSA competition experiment similar to Fig. 3B, IFI16HinAB clearly displayed intermediates (Fig. 6G). Fourth, the cross-linking of IFI16HinAB was minimal with various concentrations of dsDNA300 (Fig. 6H vs. Fig. 3 C and D). Fifth, no filament was observed in EM (Fig. 6I vs. Fig. 5B). Importantly, the lack of cross-linking even at the 1:1 complex suggests that IFI16HinAB⋅dsDNA is organized differently than the one assembled by full-length. Finally, no significant FRET signals were observed from donor- and acceptor-labeled IFI16HinAB using increasing concentrations of dsVACV72 (Fig. 6I vs. Fig. 4A). All of these results consistently support the idea that the HIN200 domains of IFI16 do not cooperatively cluster on dsDNA but independently bind dsDNA analogous to beads on a string.
The PYD of IFI16 Is Necessary for Cooperative DNA Binding.
Conventionally, the role of PYDs in ALRs is thought to be limited to recruiting downstream effectors (1, 19, 28). However, several independent experiments we have performed thus far suggested that IFI16PYD is important for oligomerization on dsDNA. To test this possibility, we first confirmed that IFI16PYD is a monomer and does not directly bind dsDNA (Figs. 6A and 7A; see also Fig. S1). Next, we constructed IFI16 variants consisting of either IFI16PYD and IFI16HinA (IFI16∆B; residues 1–393) or IFI16PYD and IFI16HinB (IFI16∆A; residues 1–191 plus 459–729) (see also Fig. S1). At 160 mM KCl, we found that IFI16∆B efficiently binds dsVACV72 and IFI16∆A binds dsVACV72 more weakly than IFI16∆B, likely reflecting the weaker binding affinity of the IFI16HinB in our hands compared with the previous report (3) (Fig. 7 A and B and Table S3). Importantly, unlike the IFI16 constructs without the PYD, the binding profiles were positively cooperative (Fig. 7B and Table S3). Additionally, IFI16∆B did not show any intermediates in an EMSA competition assay (Fig. S2F).
PYDs are homotypic protein-protein interaction domains in which multiple binding surfaces are used in their interactions (29). For example, helices one and four dock on helices two and three to create a high-affinity oligomeric interaction (30) (see also Fig. 7D). To identify surface residues in IFI16PYD important for the cooperative filament formation, we generated a set of mutations on the conserved surfaces residues using a homology model of IFI16PYD based on the PYD of myeloid cell nuclear differentiation antigen (MNDA) (Protein Data Bank ID code 2DBG; 88% sequence similarity; Fig. 7 C and D). We found that mutating these surface side-chains on IFI16PYD resulted in up to 25-fold increase in KDapp values for FAM-dsVACV72 compared with wild-type (Fig. 7E and Table S3). In addition, the binding profiles of IFI16mut2 and IFI16mut3 were essentially noncooperative (Hill constants: ∼1 for both; Fig. 7E), indicating that the cooperative dsDNA-binding mechanism requires intact IFI16PYD. Also supporting our FA results, IFI16mut3 bound FAM-dsVACV72 significantly weakly compared with wild-type with an altered migration pattern in an EMSA (Fig. 7F, Upper). Moreover, unlike wild-type, IFI16mut3 showed intermediates in a competition ESMA (Fig. 7F, Lower). Finally, no FRET signal was observed between an equal mixture of donor-labeled IFI16HinAB and acceptor-labeled full-length IFI16 with increasing dsVACV72 at 60 mM KCl (Fig. 7G). The lack of FRET signal in this experiment suggests that IFI16PYD interacts with one another instead of interacting with adjacent HIN200 domains on the dsDNA scaffold. Taken together, we concluded that the conditional proximity induced upon encountering dsDNA by the HIN200 domains triggers IFI16PYD-driven cooperative filament formation of IFI16.
Discussion
We show here that IFI16 binds dsDNA in a length-dependent manner and cooperatively assembles into filamentous oligomers. Our findings depart from the conventional noncooperative model for how ALRs engage foreign DNA, which precludes any possibility for assembling into distinct oligomers that are capable of triggering the entire pathway in a switch-like manner (9, 28). We begin our discussion by describing how the results from this report provide mechanistic explanations for three major questions regarding the in vivo behavior of IFI16 in a consistent manner (Fig. 8).
Fig. 8.

Two models for IFI16. (A) The conventional model lacks any regulatory mechanism that can account for the observed behavior of IFI16 in vivo. (B) A competing model established based on the results presented in this report.
What Could Be the Molecular Basis for DNA Length-Dependent Responses?
Conventionally, it was thought that IFI16 and its related ALRs prefer long dsDNA as it can accommodate a greater number of ALRs, which would be required to promote multimeric protein⋅protein interactions with their downstream partners (3, 9, 18). However, an important point to consider here is that if IFI16 binds dsDNA resembling beads on a string as previously thought, longer DNA fragments can be as counterproductive for multimeric interactions as short fragments. For example, even one femtogram of transfected 100-bp DNA fragments (≥9,000 copy number) provide more than 100,000 equally plausible nonadjacent binding sites for the HIN200 domains of IFI16. Furthermore, in principle, one HSV-1 genome contains more than 30,000 binding sites. Thus, because IFI16 lacks any DNA sequence specificity, without an intrinsic clustering mechanism mediated by protein⋅protein interactions, IFI16 would be randomly scattered on any self- or nonself-DNA (Fig. 8A) instead of selectively forming signaling foci on foreign DNA as observed in vivo (11–14). Our results suggest that the PYD-driven filament formation by IFI16 can provide an effective mechanism that allows multiple IFI16 (or its related ALRs) to bind adjacent to one another to form signaling foci even in the presence of excess DNA (Fig. 8B).
What Is a Potential Regulatory Mechanism of IFI16 in the Nucleus?
A striking behavior of IFI16 is that even though it is localized in the nucleus, it is randomly diffused without forming distinct foci with self-DNA (11–14). In fact, one of the longest-standing questions regarding IFI16 is how it does not engage self-DNA in the nucleus (11–14, 31, 32). We show here that the HIN200 domains of IFI16 possess negligible DNA-binding capacities under physiologically relevant salt concentrations [i.e., KD >> 10 µM for the HIN domains in the ∼200 mM effective salt concentrations of the host cell nucleus (33)]. Importantly, the interaction between the HIN200 domains and dsDNA are exclusively mediated by the phosphate backbone [i.e., nonspecific electrostatics (9)]. Although useful in engaging a wide variety of foreign DNA, this intrinsically weak intermolecular force does not generate enough binding energy beyond formation of an encounter complex (34). Thus, despite exceeding the footprint of the HIN200 domains, the length of the exposed linker-dsDNA between nucleosomes [10–20 bp (21)] or even that of the transcription bubble [∼17 bases (22)] is too short to promote robust filament assembly of IFI16. Additionally, we envision that requiring oligomerization to achieve tight binding also plays a negative role in competing against replication/transcription machinery. Thus, our data suggest that the weak binding capacity of its HIN200 domains coupled with filament formation could sufficiently suppress its interaction with self-DNA.
How Can IFI16 Selectively Engage Foreign DNA and Assemble into Large Signaling Foci?
IFI16 is known to selectively colocalize with viral genomic DNA to form signaling foci within the nucleus (e.g., HSV-1, KSHV) (11–14). Although the total amount of viral DNA would never exceed that of self-DNA, there are two features of foreign DNA that are much more conducive to filament formation by IFI16. First, the entire naked DNA genome is exposed immediately after the invasion (35–37). Second, although the viral genome packages into chromatin with the host histones, it is less dense and much more loosely packed than the nuclear self-DNA (35–37). Importantly, we find that the relative binding affinity of IFI16 to various dsDNA fragments changes cooperatively not only with increasing IFI16 concentrations (Fig. 1) but also with increasing number of binding sites (dsDNA lengths; Figs. 3 and 4). These highly cooperative relationships suggest that IFI16 is capable of amplifying its clustering behavior in a switch-like manner. For example, the Hill constant of about 5 from Fig. 4C suggests that even the low basal amount of IFI16 (38) prefers by more than 2,000-fold to oligomerize on a 150-bp fragment rather than simply binding to a 15-bp fragment. Additionally, by these cooperative mechanisms, the binding efficiency diminishes in the same manner it is amplified; thus, these results suggest that IFI16 can clearly define an “on” or “off” state with respect to its concentration and the length of dsDNA. Collectively, the filament formation by IFI16 provides a compelling mechanism by which it could selectively engage foreign DNA while minimizing its interaction with self-DNA.
The Role of PYD.
In contrast to the autoinhibitory role of the PYD of AIM2 (9, 18), we find that IFI16PYD plays an unexpected positive role in dsDNA binding. Several surface residues in IFI16PYD that mediate the cooperative dsDNA binding of IFI16 are highly conserved (Fig. 7C), thus suggesting that other related ALRs use a common oligomerization mechanism. Interestingly, we find that the PYD-driven filament assembly could provide two important tactical advantages that might not be attainable by the previously proposed noninteracting model (9). First, it allows formation of an ordered array of IFI16PYD oligomers on foreign DNA even with the low basal concentration of IFI16 in vivo (38). The IFI16PYD cluster would then be instrumental for subsequent steps in IFI16 signaling pathways, because assembling PYD/CARD oligomers by receptor molecules is required for recruiting downstream partners (23, 24, 39). Second, the PYD-driven filament assembly ensures formation of a high-affinity IFI16⋅foreign DNA complex for signaling, which would also directly interfere with replication of pathogens. Indeed, IFI16 can directly suppress replication of human cytomegalovirus (HCMV) without downstream activation of IFN-β (40). The authors of this report (40) also showed that this intrinsic antiviral activity of IFI16 required its PYD, supporting the positive role in engaging foreign DNA in vivo. Finally, further consistent with the positive role of IFI16PYD, very recent results published while this paper was under review showed that IFI16 oligomerizes on HCMV genome via its PYD in human fibroblasts and that a viral protein (pUL83) sequesters IFI16PYD to evade the host immune response (41). Future studies could reveal how PYDs regulate the function of IFI16 and its related ALRs in more detail.
Potential Role of Extended Filaments.
A growing body of evidence suggests that cooperative assembly of supermolecular structures plays a critical role in regulating signal transduction pathways (23). Indeed, formation of large aggregates by other PYD/CARD-containing inflammatory signaling molecules has been observed under overexpression conditions (28, 30). However, we find that extending the size of filaments by conjoining two or more individual oligomers in an end-to-end manner is unique to IFI16 (Figs. 2B and 5C). In fact, this behavior of IFI16 also provides a tangible explanation for why IFI16 and relatively short DNA (60–70 bp) also cluster into a few large foci as observed from viral DNA (3). We envision that formation of such supermolecular structures would increase the cooperative nature of IFI16 signaling pathways, thus further enhancing its fidelity. Moreover, such oligomers may represent its aberrant state that could promote autoimmunity. First, it might provide means to amplify the signaling efficiency (and stability) of IFI16⋅dsDNA complexes formed by relatively short dsDNA fragments (e.g., apoptotic self-DNA). Second, such an abnormal structure could be recognized as a danger signal by other host immune receptors, leading to activation of inflammatory responses. Future studies could reveal the mechanism of the higher-order oligomer assembly and its function in more detail.
Filament Formation as a Broad Host Defense Strategy.
Conventionally, assembling filaments on foreign nucleic acids has been considered as a defense strategy reserved for sensing intracellular RNA (28, 42). For example, a principal mechanism by which melanoma differentiation associated protein (MDA)5 distinguishes self- from nonself-RNA is via cooperatively assembling into filaments along the length of long dsRNA with its two CARDs forming clusters tracing the center of filaments in a helical trajectory (25, 26, 42–45). IFI16 and MDA5 are unrelated proteins, and, not surprisingly, mechanisms allowing these proteins to assemble into filaments are significantly different. For example, the RecA-like RNA-sensing domain of MDA5 wraps around dsRNA and promotes filament formation in a head-to-tail manner; the CARD clusters are not required for filament assembly. In contrast, the DNA-binding domains of IFI16 do not directly promote filament formation, and yet its PYD is necessary to drive this process. Interestingly, our data suggest that an optimal binding unit consists of approximately ten IFI16 protomers (Fig. 4C). It is tempting to speculate that the filament assembly is then accomplished by propagating these decameric units along the length of dsDNA. Another difference between IFI16 and MDA5 is that the helicase domain regulates the lifetime of MDA5 filaments via ATP hydrolysis, although it is not clear what triggers the disassembly of IFI16 filaments. Further structural and kinetic studies could reveal the architecture and dynamics of IFI16 filaments in more detail. Overall, regardless of these mechanistic differences, our results suggest that assembling filaments of intracellular dsDNA/dsRNA receptors is not only an effective mechanism but also a broadly conserved host defense strategy against foreign nucleic acids.
Experimental Procedures
Protein Expression and Purification.
Human IFI16 full-length (residues 1–729), IFI16PYD (residues 1–93), IFI16HinA (residues 192–393), IFI16HinB (residues 518–729), IFI16HinAB (residues 192–729), IFI16∆A (residues 1–191 + 459–729), and IFI16∆B (residues 1–393) were cloned into a pET21b vector (Novagen) using standard PCR methods. The identity of each IFI16 construct and mutant was confirmed by DNA sequencing. IFI16 constructs were expressed using T7 express cells (NEB). The cells were grown until OD600: 0.5 at 37 °C and protein expression was induced by 0.2 mM isopropyl β-d-1-thiogalactopyranoside overnight at 18 °C. Each protein construct was purified using Ni2+-nitrilotriacetic acid (NTA) affinity chromatography (Qiagen), followed by cation exchange chromatography using a Resource S column (GE Healthcare) with a NaCl gradient (50 mM to 1 M) and Superdex-200 or Superdex-75 size-exclusion chromatography (GE Healthcare). Micrococcal nuclease (NEB; 100 units per 1 L of cells) was added during cell lysis to eliminate bacterial DNA contamination. All proteins were eluted as monomers in size-exclusion chromatography (Fig. S1A), and UV-visible spectroscopy confirmed that they were free from nucleic acid contamination (Fig. S1B). Each protein was greater than 95% pure (Fig. S1C) and concentrated and stored at −80 °C in 20 mM Hepes⋅NaOH at pH 7.4, 200 mM NaCl, 1 mM EDTA, 5 mM DTT, and 10% (vol/vol) glycerol.
DNA.
Each FAM-labeled and unlabeled DNA fragment shorter than 120 bp was commercially purchased from Integrated DNA Technologies (IDT; Fig. S1D). Duplex formation was achieved by mixing the sense and the complementary strands in 1:1 molar ratio in buffer A (see below) and heating in 95 °C for 10 min. The duplex samples were subsequently cooled to room temperature (25 ± 2 °C) on the bench top. The longer dsDNA fragments were generated by PCR using a plasmid. Under our reaction conditions, all duplex DNAs are at least 15 °C below their predicted melting temperatures (SciTools; IDT).
Biochemical Assays.
Unless noted otherwise, all assays were performed at room temperature (25 ± 2 °C) with 40 mM Hepes⋅NaOH at pH 7.4, 160 mM KCl, 1 mM EDTA, 2 mM DTT, 10% glycerol, and 0.1% Triton-X100 (referred to as buffer A). The FA of FAM-labeled DNA (at least 15- to 20-fold lower concentrations than the KDapp for each experiment) was monitored with increasing concentrations of IFI16 variants by using a Tecan M1000 plate reader (excitation at 470 nm and emission at 528 nm) and 384-well nonbinding plates (Corning), and FA values were determined by iControl data analysis software (Tecan). The concentration of each FAM-dsDNA was kept at least 20-fold less than its respective KDapp in each of our binding assay to prevent any length bias (typically 2.5–5 nM per assay). For EMSA, each DNA was incubated with indicated concentrations of IF16 variants for 10 min and the bound complexes were separated from unbound dsDNA using 6% Tris⋅boric acid-EDTA (TBE) gels with 0.25× TBE as a running buffer. The results were then visualized by scanning on a Typhoon imager (GE Healthcare; excitation at 488 nm; emission at 532 nm). FRET assays were also performed in a 384-well plate format using the Tecan M1000. All experiments were performed at least three times independent of one another, and errors were calculated by using the SDs (number of independent experiments, n ≥ 3). Data were fit to a Hill, quadratic, or competition form of binding equation using Kaleidagraph (Synergy Soft).
Protein Cross-Linking and Labeling.
IFI16 constructs were incubated with or without indicated dsDNA fragments for 20 min in buffer A minus glycerol and with DTT replaced by tris(2-carboxyethyl)phosphine (TCEP). BMDB (10.2-Å spacer arm) was then added to each sample and cross-linking reactions proceeded at room temperature for 20 min. After quenching the reactions with 50 mM DTT, the final mixtures were applied to 4–15% TGX SDS/PAGE (Bio-Rad) and visualized by silver-stain.
IFI16 and IFI16HinAB were labeled using twofold molar excess of Dylight 550 maleimide or Dylight 650 maleimide (Pierce) in buffer A with DTT replaced by TCEP. Labeling reactions were quenched by adding excess β-mercaptoethanol and the labeled-proteins were purified using Ni2+-NTA chromatography followed by size-exclusion chromatography. The labeling efficiency of IFI16 to each dye was approximately one to one, and there was no conceivable difference in FRET data from two different batches of labeled proteins.
Electron Microscopy.
To preserve the stability of IFI16 oligomers during EM experiments, we first cross-linked IFI16⋅dsDNA complexes using BMDB. The resulting complexes were adsorbed to glow discharged carbon grids for 2 min, then blotted and transferred through two consecutive drops of 1% uranyl formate or 1% uranyl acetate for a total of 1–2 min. The carbon film was then quickly dried by aspiration. Images were collected with a Philips BioTwin CM120 (FEI) transmission electron microscope, operating at 80 kV, and digitally captured with an XR-80 CCD. The images from control experiments show no protein contamination in our buffer or DNA samples (Fig. S2B).
Supplementary Material
Acknowledgments
We thank Drs. L. Mario Amzel, Scott Bailey, Brent Cezairliyan, Daniel Leahy, Emily Lin, Antony Rosen, and Cynthia Wolberger for critical reading of the manuscript. This work is supported by Johns Hopkins School of Medicine startup funds (to J.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313577111/-/DCSupplemental.
References
- 1.Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. 2011;243(1):109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- 2.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38(5):870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veeranki S, Choubey D. Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: Regulation of subcellular localization. Mol Immunol. 2012;49(4):567–571. doi: 10.1016/j.molimm.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mondini M, et al. The interferon-inducible HIN-200 gene family in apoptosis and inflammation: Implication for autoimmunity. Autoimmunity. 2010;43(3):226–231. doi: 10.3109/08916930903510922. [DOI] [PubMed] [Google Scholar]
- 6.Uchida K, et al. Identification of specific autoantigens in Sjogren’s syndrome by SEREX. Immunology. 2005;116(1):53–63. doi: 10.1111/j.1365-2567.2005.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondini M, et al. Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci. 2007;1110:47–56. doi: 10.1196/annals.1423.006. [DOI] [PubMed] [Google Scholar]
- 8.Gugliesi F, et al. Nuclear DNA sensor IFI16 as circulating protein in autoimmune diseases is a signal of damage that impairs endothelial cells through high-affinity membrane binding. PLoS ONE. 2013;8(5):e63045. doi: 10.1371/journal.pone.0063045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin T, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36(4):561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kerur N, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011;9(5):363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci USA. 2012;109(26):10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA. 2012;109(44):E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh VV, et al. Kaposi's sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J Virol. 2013;87(8):4417–4431. doi: 10.1128/JVI.03282-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan H, et al. RPA nucleic acid-binding properties of IFI16-HIN200. Biochim Biophys Acta. 2008;1784(7-8):1087–1097. doi: 10.1016/j.bbapap.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Brazda V, Coufal J, Liao JC, Arrowsmith CH. Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem Biophys Res Commun. 2012;422(4):716–720. doi: 10.1016/j.bbrc.2012.05.065. [DOI] [PubMed] [Google Scholar]
- 17.Jones JW, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA. 2010;107(21):9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem. 2013;288(19):13225–13235. doi: 10.1074/jbc.M113.468033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alnemri ES. Sensing cytoplasmic danger signals by the inflammasome. J Clin Immunol. 2010;30(4):512–519. doi: 10.1007/s10875-010-9419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao JC, et al. Interferon-inducible protein 16: Insight into the interaction with tumor suppressor p53. Structure. 2011;19(3):418–429. doi: 10.1016/j.str.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato M, et al. Dinucleosome DNA of human K562 cells: Experimental and computational characterizations. J Mol Biol. 2003;332(1):111–125. doi: 10.1016/s0022-2836(03)00838-6. [DOI] [PubMed] [Google Scholar]
- 22.Pal M, Ponticelli AS, Luse DS. The role of the transcription bubble and TFIIB in promoter clearance by RNA polymerase II. Mol Cell. 2005;19(1):101–110. doi: 10.1016/j.molcel.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Wu H. Higher-order assemblies in a new paradigm of signal transduction. Cell. 2013;153(2):287–292. doi: 10.1016/j.cell.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 25.Peisley A, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. 2011;108(52):21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peisley A, et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci USA. 2012;109(49):E3340–E3349. doi: 10.1073/pnas.1208618109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiros I, Timmins J, Hall DR, McSweeney S. Crystal structure and DNA-binding analysis of RecO from Deinococcus radiodurans. EMBO J. 2005;24(5):906–918. doi: 10.1038/sj.emboj.7600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park HH, et al. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J Biol Chem. 2012;287(50):41732–41743. doi: 10.1074/jbc.M112.381228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unterholzner L. The interferon response to intracellular DNA: Why so many receptors? Immunobiology. 2013;218(11):1312–1321. doi: 10.1016/j.imbio.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Unterholzner L, Bowie AG. Innate DNA sensing moves to the nucleus. Cell Host Microbe. 2011;9(5):351–353. doi: 10.1016/j.chom.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Arbely E, et al. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci USA. 2011;108(20):8251–8256. doi: 10.1073/pnas.1105028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3(5):427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 35.Cliffe AR, Knipe DM. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol. 2008;82(24):12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacasse JJ, Schang LM. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J Virol. 2010;84(4):1920–1933. doi: 10.1128/JVI.01934-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oh J, Fraser NW. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol. 2008;82(7):3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veeranki S, Duan X, Panchanathan R, Liu H, Choubey D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE. 2011;6(10):e27040. doi: 10.1371/journal.pone.0027040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pagliusi S, et al. Developing Countries Vaccine Manufacturers Network (DCVMN): Engaging to step up for vaccine discovery and access. Meeting report 2012. Vaccine. 2013;31(31):3111–3115. doi: 10.1016/j.vaccine.2013.04.082. [DOI] [PubMed] [Google Scholar]
- 40.Gariano GR, et al. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012;8(1):e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li T, Chen J, Cristea IM. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013;14(5):591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berke IC, Li Y, Modis Y. Structural basis of innate immune recognition of viral RNA. Cell Microbiol. 2013;15(3):386–394. doi: 10.1111/cmi.12061. [DOI] [PubMed] [Google Scholar]
- 43.Wu B, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152(1-2):276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 44.Berke IC, Yu X, Modis Y, Egelman EH. MDA5 assembles into a polar helical filament on dsRNA. Proc Natl Acad Sci USA. 2012;109(45):18437–18441. doi: 10.1073/pnas.1212186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31(7):1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.