

Abstract
Fibroblast growth factor 2 (FGF-2) has multiple, pleiotropic effects on the nervous system that include neurogenesis, neuroprotection and neuroplasticity. Thus, alteration in FGF-2 expression patterns may have a profound impact in brain function, both in normal physiology and in pathology. Here, we used FGF-2 transgenic mice (TgFGF2) to study the effects of endogenous FGF-2 overexpression on susceptibility to seizures and to the pathological consequences of seizures. TgFGF2 mice display increased FGF-2 expression in hippocampal pyramidal neurons and dentate granule cells. Increased density of glutamatergic synaptic vesicles was observed in the hippocampus of TgFGF2 mice, and electrophysiological data (input/output curves and patch-clamp recordings in CA1) confirmed an increase in excitatory inputs in CA1, suggesting the presence of a latent hyperexcitability. Indeed, TgFGF2 mice displayed increased susceptibility to kainate-induced seizures compared with wild-type (WT) littermates, in that latency to generalized seizure onset was reduced, whereas behavioral seizure scores and lethality were increased. Finally, WT and TgFGF2 mice with similar seizure scores were used for examining seizure-induced cellular consequences. Neurogenesis and mossy fiber sprouting were not significantly different between the two groups. In contrast, cell damage (assessed with Fluoro-Jade B, silver impregnation and anti-caspase 3 immunohistochemistry) was significantly lower in TgFGF2 mice, especially in the areas of overexpression (CA1 and CA3), indicating reduction of seizure-induced necrosis and apoptosis. These data suggest that FGF-2 may be implicated in seizure susceptibility and in seizure-induced plasticity, exerting different, and apparently contrasting effects: favoring ictogenesis but reducing seizure-induced cell death.
Keywords: epilepsy, neurotrophic factors, transgenic, glutamate, neurodegeneration, neurogenesis
Introduction
Fibroblast growth factor-2 (FGF-2) is a neurotrophic factor belonging to a family of structurally related proteins, the FGFs, that encompasses 22 members in humans and mice (Itoh and Ornitz, 2004). Although the FGFs play key roles in a variety of physiological and pathological events in development and adult life (Bikfalvi et al., 1997), the specific function of each FGF family member in vivo has not been fully established.
FGF-2 is a single-chain polypeptide composed of 146 aa (Böhlen et al., 1984). It is found in at least 4 distinct isoforms: the high molecular weight (HMW) forms, ranging from 20 to 24 KDa, are predominantly localized in the nucleus, whereas the low molecular weight form (LMW, 18 KDa) resides in the cytoplasm (Florkiewicz et al., 1991; Quarto et al., 1991; Sørensen et al., 2006). FGF-2 can interact with four high-affinity tyrosine-kinase and one low-affinity cell surface receptors, termed FGFRs (Klint and Claesson-Welsh, 1999; Reuss and von Bohlen und Halbach, 2003).
Based on the existence of several isoforms with different subcellular localizations and of multiple receptor subtypes, FGF-2 can exert multiple, pleiotropic biological effects. It regulates proliferation, survival and differentiation of neural stem cells, and is also involved in the regulation of plasticity and in the maintenance of structural brain integrity (Grothe and Wewetzer, 1996; Lowenstein and Arsenault, 1996; Reuss and von Bohlen und Halbach, 2003). In particular, in vivo studies have shown that FGF-2 is a potent mitogenic factor for hippocampal neurogenesis (Gritti et al., 1999; Palmer et al., 1999; Wagner et al., 1999; Yoshimura et al., 2001). In vitro evidence supports the involvement of FGF-2 in neuronal morphogenesis, because it prompts bifurcation and axonal branching of hippocampal and cortical neurons (Aoyagi et al., 1994; Patel and McNamara, 1995; Szebenyi et al., 2001). Greater numbers of axon branches are expected to produce increased numbers of synaptic contacts and, indeed, FGF-2 has been found to increase the number of excitatory synapses between hippocampal neurons in vitro (Li et al., 2002).
FGF-2 may be involved in activity-dependent plasticity associated with brain pathologies. For example, FGF-2 could be a leading regulator for physiopathological events induced by prolonged seizures, including neuronal loss, neurogenesis, axonal and dendritic sprouting, gliosis, and network changes (Pitkänen and Sutula, 2002). Indeed, several lines of evidence suggest a link between FGF-2 and epilepsy: (1) seizures increase FGF-2 mRNA and protein levels (Jankowsky and Patterson, 2001) and upregulate FGFR1 receptors (Gómez-Pinilla et al., 1995); (2) chronic i.c.v. infusion of low dose FGF-2 does not affect kainate seizures, but improves behavioral recovery and reduces hippocampal damage (Liu et al., 1993; Liu and Holmes, 1997); (3) acute intrahippocampal injection of FGF-2 induces seizures in a dose-dependent manner (Liu and Holmes, 1997); (4) kainate seizure severity is not significantly altered in FGF-2 knock-out mice, whereas seizure-induced neurogenesis is almost abolished (Yoshimura et al., 2001).
To further elucidate the effect of FGF-2 in the regulation of brain function in vivo and, specifically, in susceptibility to seizures and to seizure-associated pathological consequences, we used transgenic mice expressing the human FGF-2 (TgFGF2 mice). In contrast to the studies cited above, in which FGF-2 was exogenously applied, these mice provide the opportunity to investigate the role of the FGF-2 generated endogenously during development and in adulthood.
Materials and Methods
Animals.
TgFGF2 mice were used for all experiments. These mice harbor a transgene that includes the full-length human FGF-2 cDNA under the control of phosphoglycerokinase (pgk) 5′ regulatory elements (Coffin et al., 1995). These animals were created in the FVB/N strain, because of its suitability for transgenic manipulation (Taketo et al., 1991). The TgFGF2 mice express all four isoforms of human FGF-2 that arise from alternative initiation sites for translation (Florkiewicz and Sommer, 1989). TgFGF2 mice have been previously characterized from both molecular and phenotypic perspectives. Western blot analysis has shown that the murine and human FGF-2 isoforms are tissue-specifically expressed and translationally regulated (Coffin et al., 1995). By gross examination, the majority of mice seem to be indistinguishable from their WT littermates, but some of them are affected by skeletal malformations, such as shortening and flattening of long bones and moderate macrocephaly (Coffin et al., 1995).
Mating of TgFGF2 males with nontransgenic females generated 1:1 nontransgenic (or wildtype, WT) to TgFGF2 littermates that were used to generate experimental groups at 6–8 weeks of age. Presence of the FGF-2 transgene was confirmed by genomic PCR as described previously (Coffin et al., 1995). The genotype of each animal was assessed twice using PCR of genomic DNA isolated from tail (before experiment and after kill). The mice that were used for all experiments were 6–8 months old WT and TgFGF2 littermates. All animals were housed under standard conditions: constant temperature (22−24°C) and humidity (55–65%), 12 h dark-light cycle, free access to food and water. Procedures involving animals were conducted in accordance with European Community and national (Ministry of Health) laws and policies (authorizations: D.M. 82/2001-B and 93/2004-B).
In situ hybridization.
Mice were killed by decapitation under light diethyl-ether anesthesia, and their brains rapidly removed, frozen in isopentane cooled in a dry ice/methanol bath, and then stored at −70°C until use. Twenty μm coronal sections at the level of the dorsal hippocampus (plate 45; Paxinos and Franklin, 2001) were thaw-mounted onto polylisine coated slides, fixed in paraformaldehyde 4%, soaked in phosphate saline buffer (PBS 1×: PO42− 0.01 m; NaCl 0.15 m) for 10 min at room temperature, then rinsed in a graded ethanol series and dried. Sections were stored at −70°C until use. Immediately before in situ hybridization, they were pretreated with proteinase K (10 μg/ml in Tris-EDTA buffer) for 10 min at 37°C and with acetic anhydride 0.25% v/v for 10 min at room temperature.
The FGF-2 riboprobe was obtained using the plasmid pBluescript-mFGF-2 (kindly provided by Dr. G.R. Martin, Department of Anatomy, University of California, San Francisco, CA, USA) containing a 475 bp portion of mouse FGF-2 cDNA. The human and mouse FGF-2 mRNAs in this region are 89.3% homologous (Huang and Miller, 1991). The plasmid was linearized with XbaI and transcribed with T3 RNA polymerase to obtain antisense riboprobes, or linearized with EcoRI and transcribed with T7 RNA polymerase to obtain sense riboprobes. Riboprobes were obtained by running the respective transcription assays in the presence of α[33P]rUTP (PerkinElmer Life Sciences).
In situ hybridization was performed as previously described (Simonato et al., 1996, 1998). The in situ hybridization mixture contained 50% deionized formamide, 0.6 m NaCl, 2 mm EDTA, 20 mm Tris (2× NTE), 5× Denhart's solution, 100 μg/ml ssDNA, 100 μg/ml tRNA, 0.05% sodium pyrophosphate and 60 ng/ml 33P-riboprobe. Sections were incubated overnight at 52°C with 40 μl of hybridization mixture. After hybridization, they were rinsed twice for 10 min in 4× SSC (0.6 m NaCl, 0.06 m citric acid) and treated with RNase A (20 μg/ml) for 30 min at 37°C. Sections were then rinsed in 1× SSC for 10 min, 0.1× SSC at 52°C for 30 min, 0.1× SSC at room temperature for 10 min, and dehydrated through an ethanol series. Autoradiograms were generated by apposing these dried sections alongside 33P-riboprobe standards to Hyperfilm β-max (GE Healthcare Bio-Sciences) at −70°C for 4 weeks. Films were developed in Kodak D-19 (5 min) and fixed in Kodak fixer (5 min).
After development of the film, a digital analysis system (RBR Altair, Firenze, Italy) was used to obtain optical density measurements over structures of interest in the autoradiogram. The mean total optical density within a particular area was calculated by multiple sampling of that area in 4 sections taken from each animal. Background (nonspecific) hybridization was estimated using other sections incubated with the sense probe, and subtracted from the calculated values of total optical density. These specific optical density measurements were then compared with those obtained from the 33P-riboprobe standards. By using this method of standardization, small differences in OD resulting from differences in film development were corrected, allowing values obtained from homologous regions to be compared across different films.
FGF-2 immunohistochemistry.
Mice were killed and their brains were rapidly removed, immersed in 10% formaline and then paraffin-embedded. Coronal sections (6 μm thick) were cut at the level of the dorsal hippocampus and mounted onto poly-lysine-coated slides.
The immunohistochemistry protocol was based on a previously described one (Marconi et al., 1999, 2005). Briefly, sections were deparaffinized (2 washes in xylol 10 min, 5 min in ethanol 100%, 5 min in ethanol 95%) and then rehydratated in distilled water for 5 min and in PBS 1× for 10 min. The FGF-2 antigen was unmasked using a commercially available kit (Unmasker Diapath), according to the manufacturer's instructions. After incubation in H2O2 3V for 15 min at room temperature, sections were rapidly rinsed in distilled water and washed again in PBS. They were then incubated with Ultra V Block (Ultra Vision Detection System; Lab Vision Corporation) for 5 min at room temperature, to block nonspecific background staining. After washing in PBS 1× for 5 min, sections were incubated overnight at 4°C in humid atmosphere with the primary antibody for FGF-2 (mouse monoclonal; 5 μg/ml; BD Transduction Laboratories). After rinsing in PBS 1×, they were incubated with biotynilated goat anti-polyvalent serum (Ultra Vision Detection System; Lab Vision Corporation) at room temperature for 10 min, washed in PBS 1× for 5 min and then incubated in Streptavidine Peroxidase (Ultra Vision Detection System; Lab Vision Corporation) at room temperature for 10 min. The reaction product was detected as a brown substrate using a 10 mg tablet of 3,3-diaminobenzidinetetrahydrochloride (DAB; Sigma) in a solution containing 16.6 ml of PBS 1× and 130 μl of H2O2 24V. Finally, sections were washed three times in PBS 1× (5 min each), counterstained with hematoxylin for 2 min and washed again in PBS 1× (5 min). Coverslips were mounted using Gel/mount (Biomeda). The specificity of immunolabeling was verified in all experiments by controls in which the primary antibody was omitted.
FGFR1 immunohistochemistry.
FGFR1 immunohistochemistry was performed using the method described for FGF-2. The primary antibody directed against FGFR1 (rabbit polyclonal; Santa Cruz Biotecnology) was diluted 1:100 in PBS and applied for 1 h at 37°C.
FGF-2 and GFAP double immunofluorescence.
WT and TgFGF2 mice were killed and their brains were rapidly removed, immersed in 10% formalin and then paraffin-embedded. Coronal sections (6 μm thick) were cut at the level of the dorsal hippocampus (plate 45, Paxinos and Franklin, 2001) and mounted onto polarized slides (Superfrost slides, Diapath).
Sections were deparaffinized, rehydratated, and the FGF-2 and GFAP antigens were unmasked as described for FGF-2 immunohistochemistry. Then, after a 1× PBS wash, they were preincubated with PBS containing 0.3% Triton X-100 for 10 min. After two rinses in PBS, they were blocked for 30 min in a solution of PBS containing 5% bovine serum albumin (BSA) and 5% normal goat serum. Finally, sections were incubated overnight at 4°C with the primary antibodies, anti-FGF-2 (mouse monoclonal; 10 μg/ml; BD Transduction Laboratories) and anti-GFAP (rabbit polyclonal; 1:100; Sigma).
The next day, sections were rinsed twice with PBS and preincubated with PBS containing 0.3% Triton X-100 for 30 min. Next, they were rinsed with PBS again and then incubated for 3 h at room temperature with a secondary antibody mixture containing goat anti-mouse, fluorescein isothiocyanate (FITC)-conjugate (1:100 dilution; Jackson ImmunoResearch) and a goat anti-rabbit, TX red-conjugate (1:100; Jackson ImmunoResearch). Finally, samples were washed three times with PBS, the last wash containing 100 ng/ml 4′, 6-diamidino-2-phenylindole (DAPI, Sigma). DAPI was used as a fluorescent counterstain for cell nuclei. Coverslips were mounted using anti-fading Gel/Mount water based media (Biomeda). The specificity of immunolabeling was verified in all experiments by controls in which the primary antibody was omitted.
VGLUT1 and VGAT immunohistochemistry.
On coronal, adjacent sections to those used for FGF-2 immunostaining, we performed VGLUT1 and VGAT immunohistochemistry, similar to the procedures described above for FGF-2. After pretreatment, tissue sections were incubated overnight in a humidity chamber at room temperature in PBS containing a primary antibody against either VGLUT1 (rabbit polyclonal; 1:500 dilution; Synaptic Systems) or VGAT (mouse monoclonal; 1:200 dilution; Synaptic Systems). DAB staining was performed as described above and coverslips were mounted using Gel/Mount (Biomeda). The specificity of immunolabeling was verified in all experiments by controls in which the primary antibody was omitted.
Extracellular recordings.
WT and TgFGF2 mice were anesthetized with ether and decapitated. Brains were quickly removed and transferred in cold oxygenated artificial CSF (aCSF; composition in mm: NaCl 124, KCl 2, KH2PO4 1.25, NaHCO3 26, MgSO4 2, CaCl2 2, glucose 10; pH 7.4). Transverse dorsal hippocampal slices of 450 μm were cut with a vibroslicer (Campden Instruments Ltd.). After at least 1 h of incubation at room temperature for recovery, slices were placed in a recording chamber and continuously perfused with oxygenated aCSF at a flow rate of 2.5 ml/min. Experiments were performed at 30°C.
To probe for differential excitability in the two experimental groups, input-output (I/O) curves were generated. CA1 Schaffer collateral fibers were stimulated electrically (trains of 0.05–0.1 ms rectangular pulses at 0.033 Hz) through a twisted tungsten bipolar electrode, using a computer-driven stimulator unit (Digitimer DS2A, from AutoMate Scientific Inc.), and field excitatory postsynaptic potentials (fEPSPs) were recorded from CA1 stratum radiatum by means of an extracellular microelectrode filled with aCSF. The stimulating electrode was positioned 0.5–1 mm away from the recording electrode. Acquisition was done using an AC amplifier (A-M System 3000, A-M Systems) controlled by the LTP 2.30D software (Anderson and Collingridge, 2001). After several minutes of settling, evoked responses for a given stimulus were stable throughout the recording.
A series of test stimuli of increasing intensity were initially applied to identify the response threshold. The threshold stimulus intensity was always in the range of 3–10 V, and was not different in slices taken from WT or TgFGF2 mice (average threshold intensity in WT: 4.5 ± 0.9 V; in TgFGF2 5.0 ± 0.9 V). Intensity of input stimuli was increased by steps, starting at threshold (the maximal stimulus intensity that did not produce any detectable response) up to a maximal saturating value.
The slope of dendritic fEPSPs was calculated using the LTP 2.30D software and plotted versus increasing stimulus intensity. For each animal, the fold-increase in input stimulus intensity necessary for evoking a half-maximal response was calculated (based on the individual input-output curve) using the GraphPad Prism 4.0 software. These individual values were used for statistical analysis of the data that was conducted using the Student's t test for unpaired data.
Patch-clamp procedures.
Adult mice were deeply anesthetized (i.p. injection of 60 mg/kg sodium pentobarbital) and decapitated. The brain was exposed, chilled with aCSF, and the hippocampus was dissected. Transverse hippocampal slices (300–350 μm) were cut with a vibratome and stored at room temperature in a holding bath containing aCSF. After a recovery period of at least 1 h, slices were transferred to the recording chamber (1 cm3 volume) and mounted on an Olympus BX50WI microscope. Slices were constantly superfused using a gravity flow system (2 ml/min), with a aCSF having the following composition (mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 15 glucose, continuously bubbled with 95% O2/5% CO2; the osmolarity was adjusted at 305 mOsm with glucose.
The pipette-filling solution contained the following (mm): 120 KCl, 10 NaCl, 2 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, 2 Na-ATP, 10 glucose. The osmolarity was adjusted to 295 mOsm with glucose and the pH was set to 7.2 with NaOH. Membrane currents were recorded and acquired with an Axopatch 200A amplifier (Molecular Devices), and a 12 bit A/D-D/A converter (Digidata 1200B; Molecular Devices); off-line analysis was performed using version 10 of pClamp (Molecular Devices). A 70–80% compensation of the series resistance was routinely used.
Excitatory synaptic transmission was examined in CA1 pyramidal cells in both WT and TgFGF2 mice using the whole-cell patch-clamp technique in thin slices continuously perfused with bicuculline (10 μm). Schaffer collateral fibers were cut between CA3 and CA1 to avoid the onset of status epilepticus after perfusion with bicuculline. Bipolar twisted NiCr-insulated electrodes localized into the Schaffer collaterals bundle were used to evoke synaptic responses. We used minimal stimulation at the frequency of 0.05 Hz to activate only one presynaptic fiber connected to the neuron under recording. According to the technique described by Jonas et al. (1993) and Allen and Stevens (1994), the stimulation intensity was decreased until only a single axon connected to the recorded CA1 neuron was activated. This was achieved when the mean amplitude of the postsynaptic currents and the failure probability remained constant over a range of stimulus intensities near threshold for detecting a response (in the range of 2.5–6.5 mV). The latency and the shape of individual synaptic responses remained constant for repeated stimuli; in all recordings paired stimuli were applied at 50 ms intervals. Failures of transmission were judged subjectively for each trace. Data were statistically analyzed using the Origin 7.5 software (OriginLab), with the Student's t test.
Ca2+-dependent inactivation of NMDA currents.
Mice were decapitated under deep anesthesia and their brains were quickly removed and placed in a cold carboxygenated solution with the following composition (in mm): KCl 3, NaH2PO4 1.25, NaHCO3 21, MgCl2 2, CaCl2 1.6, glucose10; sucrose 200, α-tocopherol 0.1; pH 7.4, osmolarity 300–305 mOsm. The Na+ concentration was lowered to prevent hypoxia-induced Na+ influx into neurons and α-tocopherol was added as a scavenger of free radicals (Gabriel et al., 2004). Transverse slices (300–350 μm) were cut by using a tissue vibratome and then transferred to an interface tissue chamber and maintained in a bicarbonate buffer solution (BBS) bubbled with 95% O2 and 5% CO2 at room temperature at least 1 h before their use. The BBS composition was (in mm): NaCl 125, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, MgCl2 1, CaCl2 2, glucose 15.
In conventional whole-cell patch-clamp recordings, some of the endogenous Ca2+ buffer systems and of other intracellular components that regulate NMDA receptor function may be washed out. To avoid this problem, we used perforated patch recordings to study the calcium-dependent inactivation of NMDA receptors. This configuration, maintaining a relatively intact intracellular environment, has been shown to ensure optimal results in the study of calcium-dependent inactivation (Kyrozis et al., 1996). The antibiotic amphotericin B was used as perforating agent (Horn and Marty, 1988). Before each experiment, 2 mg of amphotericin B were dissolved in 50 μl of dimethylsulphoxide (DMSO). This solution was sonicated for 2 min and then added to the pipette solution.
NMDA (50 μm) was focally applied to the cell body of neurons via a glass tube with an internal diameter of 0.8 mm, positioned at ∼2 mm distance from the cell (Biologic RSC-160, Claix, France). Membrane currents were recorded and acquired with an Axopatch 200A amplifier (Axon Instruments) and a 12 bit A/D-D/A converter (Digidata 1440A; Axon Instruments). Off line analysis was performed using pClamp version 10.1 (Axon Instruments).
Kainate.
Kainate was administered i.p. (20 mg/kg), and the mouse behavior was observed for 2 h thereafter. The occurrence of seizures and their severity were blind-scored by two investigators, using the following classification (Janumpalli et al., 1998; Bregola et al., 2002): 1, chewing and drooling; 2, head nodding; 3, unilateral forelimb clonus; 4, bilateral forelimb clonus; 5, bilateral forelimb and/or hindlimb clonus with falling; 6, running or jumping seizure; 7, tonic hindlimb extension; 8, death.
Bromodeoxyuridine in vivo labeling and immunohistochemistry.
For bromodeoxyuridine (BrdU) immunohistochemistry, WT and TgFGF2 mice were divided in two groups after kainate administration. The first group was used to study proliferation of neural stem cells and progenitors. Three days after kainate administration, mice were given a series of four BrdU injections (50 mg/kg i.p., dissolved in propilenic glycol and water, Boehringer Mannheim), every 2 h over a period of 6 h. These animals were killed 24 h after the last BrdU injection. The second group was used to study neurogenesis. For 5 d, beginning 3 d after kainate, WT and TgFGF2 mice in this group received a single BrdU injection per day (50 mg/kg i.p.). These animals were killed 9 d after kainate.
BrdU immunostaining was performed as previously described (Marconi et al., 2005). Briefly, DNA was denatured by incubating tissue sections in 2 N HCl for 30 min at 37°C. Sections were rinsed in PBS 1× and treated with 3% H2O2 3V to block endogenous peroxidase. Slices were then washed in PBS 1× and incubated with Ultra V Block. Incubation with a primary antibody for BrdU (mouse monoclonal; 1:100 dilution in PBS 1×; Boehringer Mannheim) was conducted overnight at room temperature in a humidity chamber. After 3× 5 min rinses in PBS, they were incubated with biotinylated secondary antibody and then in Streptavidine Peroxidase. The reaction product was detected using DAB, as described above. Finally, sections were counterstained with hematoxylin and the coverslips mounted using Gel/mount. The specificity of immunolabeling was verified in all experiments by controls in which the primary antibody was omitted.
Quantification of BrdU immunohistochemistry experiments was performed in a blinded manner by two investigators, counting the number of BrdU-positive cells in the granular layer and in the hilus of the dorsal hippocampus dentate gyrus. Paraffin-embedded brains were cut in successive 6 μm sections across the entire dorsal hippocampus (i.e., 141 sections, corresponding to 846 μm; plates 42–49, Paxinos and Franklin, 2001). One section every 28 (that is, every 168 μm) was used for BrdU immunohistochemistry. Five sections have been examined per each animal (progressive section numbers: 1, 29, 57, 85, 113). An estimate of the total BrdU-positive cells in the area under examination was obtained by multiplying the number of BrdU-positive cells in each section by 28 and summing the five resulting counts. Statistical analysis has been performed using ANOVA and post hoc the Newman–Keuls test.
Activin A immunohistochemistry.
Activin A (ActA) immunohistochemistry was performed using the method described for FGF-2. The primary antibody directed against ActA (rabbit polyclonal; dilution 1:50; R&D Systems) was applied overnight in a humidity chamber at 4°C. The specificity of immunolabeling was verified by controls in which the primary antibody was omitted.
BrdU and doublecortin double immunofluorescence.
BrdU and doublecortin (DCX) double immunofluorescence was performed as described above for FGF-2 and GFAP immunofluorescence. Sections were incubated overnight at 4°C with the primary antibodies, anti-BrdU (rat polyclonal; dilution 1:50; AbD Serotec) and anti-DCX (goat polyclonal; dilution 1:50; Santa Cruz Biotechnology).
The next day, they were incubated for 3 h at room temperature with a secondary antibody mixture containing donkey anti-goat, FITC-conjugate (dilution 1:25; Jackson ImmunoResearch) and a donkey anti-rat, TX red-conjugate (1:50; Jackson ImmunoResearch). DAPI was used as a fluorescent counterstain for nuclei. The specificity of immunolabeling was verified by controls in which the primary antibody was omitted. The quantification of the number of DCX-positive cells in the dentate gyrus was performed as described above for BrdU. It must be noted that the use of very thin (6 μm), paraffin-embedded sections offers the advantages of very high resolution, of clear identification of stained cells (in that no overlap of multiple cell layers can occur), and of precise anatomical localization (thanks to the possibility of counterstaining the sections with standard procedures, such as DAPI).
Analysis of cell damage.
Analysis of seizure-induced cell damage was performed in WT and TgFGF2 mice killed 3 d after kainate administration, by using Fluoro-Jade B (FJB) staining and silver impregnation. As described above for BrdU, paraffin-embedded brains were cut in successive 6 μm sections across the entire dorsal hippocampus (142 sections, corresponding to 852 μm; plates 42–49, Paxinos and Franklin, 2001). Two adjacent sections every 35 were used for FJB staining and silver impregnation. FJB staining was performed as described (Schmued and Hopkins, 2000; Zucchini et al., 2002). Briefly, sections were mounted onto poly-lysine coated slides, deparaffinized (as described above) and rehydrated in a solution containing 1% NaOH in 80% ethanol (5 min), then 70% ethanol (2 min), and then distilled water (2 min). Subsequently, they were incubated for 10 min in 0.06% potassium permanganate, washed for 2 min in distilled water and transferred to a 0.001% FJB staining solution. After staining, sections were washed 3× 1 min in distilled water, dried, immersed for 1 min in xylene and then coverslipped.
Silver staining impregnation has been performed according to the method of Gallyas et al. (1980). Sections were deparaffinized and immersed in a pretreating solution (0.5% ammonium nitrate in 4.5% NaOH) to prepare the tissue for reaction with silver nitrate solution (0.75% silver nitrate in 8% ammonium nitrate and 4.5% NaOH). Neurodegenerated cells were identified by inclusion of silver in the nuclei. To avoid aspecific precipitation of silver nuclei, washes of 3× 5 min were performed for eliminating the excess of silver nitrate accumulated on sections. Slices were then dipped in developing solution (0.6% formaldehyde, 0.05% citric acid, 10% ethanol and 0.001% ammonium nitrate) for 1 min, to increase the size of the silver nuclei, which become visible at light microscopy. Finally, sections were soaked in a 0.5% acetic acid solution 3× 10 min.
Analysis was conducted using a Leica microscope (DMRA2, Leica). The degree of cell damage was quantified blinded by two investigators, based on two distinct approaches. The first was a modification of a previously described scoring system named “neurodegeneration score” (Zucchini et al., 2002; Franceschetti et al., 2007). The score range spanned from 0 (absence of FJB- or silver impregnation-positive neurons) to 5 (maximal neurodegeneration pattern in the hippocampus). A score of 1 was given for observation of positive cells either in the hilus, in the CA3 pyramidal layer, or in the CA1 pyramidal layer. Whenever a very intense neurodegeneration was observed in the CA3 or CA1 pyramidal layer (many positive cells with confluent fluorescence), the score related to that subarea was increased by 0.5; another 0.5 was added when fluorescent cells were detectable in the strata oriens and/or radiatum (therefore, the score related to CA3 and to CA1 could reach a maximal value of 2). The second quantification method was based on the thresholding of FJB digital images. Images of the hippocampus were captured using a Leica DFC300FX camera and transformed into gray levels. Using Adobe Systems Photoshop CS2 (version 9.0.2), the hippocampus was cut out and the mean ± SD gray level was calculated. FJB-positive pixels were identified by thresholding at the gray level corresponding to the mean plus three SDs. Using this approach, only those pixels that were significantly above background (that is, positive to FJB) were selected. Data were then expressed as percentage of FJB-positive pixels over total hippocampal pixels.
As stated above, two every 35 of the 142 6 μm sections cut across the dorsal hippocampus were stained (one with FJB, the other with silver impregnation). Thus, five FJB (progressive section numbers: 1, 36, 71, 106, 141) and five silver impregnation sections (progressive numbers: 2, 37, 72, 107, 142) have been examined for each animal. The mean neurodegeneration scores in the five sections examined were obtained for each staining approach (FJB and silver impregnation). Finally, these two neurodegeneration scores were averaged, obtaining a single value per animal that was used for statistical analysis. Alternatively, the mean percentage of FJB-positive pixels in the five sections examined was calculated for each animal and used for statistical analysis. Statistical analysis was conducted using the Mann–Whitney two-tailed U test.
Cleaved caspase-3 immunohistochemistry.
Cleaved caspase-3 immunohistochemistry was performed using a method very similar to the one described above, for FGF-2 immunostaining. The essential difference was that the primary antibody was directed against active caspase-3 (rabbit polyclonal; Cell Signaling Inc.) instead of FGF-2. Anti-capsase-3 was diluted 1:100 in PBS and applied overnight at 4°C. Subsequent application of secondary antibody, processing for microscopy and analysis of the samples was identical to procedures described above.
Timm staining.
Thirty days after kainate treatment, a group of WT and TgFGF2 mice was trans-cardially perfused with 4% paraformaldehyde and 1.25% glutaraldehyde in phosphate buffer. After postfixation (0.4% Na2S for 30 min, 1% paraformaldehyde and 1.25% glutaraldehyde overnight), 20 μm coronal brain sections were cut at the level of the dorsal hippocampus and Timm staining was performed as described by Wenzel et al. (1997), to reveal axonal sprouting of the mossy fibers. Briefly, free-floating sections were put in a developer [30 ml of 25% arabic gum, 5 ml of 2 m citrate buffer, pH 3.7, 15 ml of 5.67% hydroquinone and 0.25 ml of 17% AgNO3] for at least 60 min in the dark, under constant agitation. Sections were then washed in distilled water for 5 min and dehydrated with alcohol and xylene.
Analysis was performed using a digital analysis system (RBR Altair, Firenze, Italy). The areas of interest were divided in circular areas according to procedures described by Adams et al. (2002) and a score (from 0 to 5, according to Cavazos et al., 1991) was given to every circle. The “Timm index” was the sum of these scores.
Results
Expression of the FGF-2 transgene in the brain
Based on Western blot analysis of tissue extracts, the human FGF-2 transgene is highly expressed in the brain (Coffin et al., 1995). However, its distribution in the adult brain was not previously investigated and, therefore, we examined it first. Unfortunately, the possibility of specifically identifying transgene-derived FGF-2 is precluded by the high degree of homology between human and mouse FGF-2. Despite the cross-reactivity, in situ hybridization, immunohistochemistry and immunofluorescence allowed identification of cells overexpressing FGF-2 (murine plus human) in the TgFGF2 brain.
To the best of our knowledge, no in situ hybridization analysis of FGF-2 mRNA distribution in the normal mouse brain has been previously performed. However, our data in WT mice are consistent with those previously reported for the normal rat (Emoto et al., 1989; Bugra et al., 1994; Gall et al., 1994; Gómez-Pinilla et al., 1995; Simonato et al., 1998). In coronal sections taken at the level of dorsal hippocampus, (1) dense hybridization was found in the CA2 pyramidal layer and in the subiculum; (2) diffuse labeling was found in the hippocampus, cortex and thalamus, suggestive of localization in glial cells; (3) moderate expression of FGF-2 was also observed in the CA1 and CA3 pyramidal layer and in the dentate gyrus granular layer (Fig. 1A). In TgFGF2 mice, a significant increase in FGF-2 mRNA levels was observed in the pyramidal layer of CA1, CA2 and CA3, and in the granular layer of the dentate gyrus, but not in the diffuse labeling in the hippocampus, thalamus and cortex (Fig. 1A).
Figure 1.
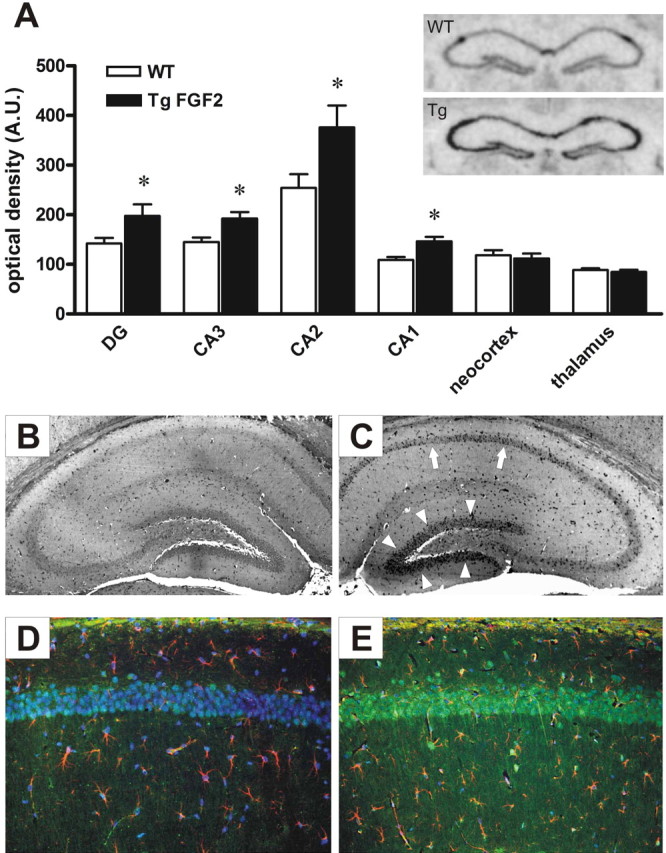
FGF-2 over-expression in the forebrain of TgFGF2 mice. A, Relative optical density values of FGF-2 mRNA hybridization to riboprobes in forebrain regions of WT (open bars) and TgFGF2 mice (solid bars). Data are the mean ± SE of four independent experiments. *p < 0.05 versus WT (Student's t test for nonpaired data). Inset, Anatomic distribution of FGF-2 mRNA expression in the dorsal hippocampus of WT and TgFGF2. Note increased mRNA levels in the CA1, CA2 and CA3 pyramidal layer, and in the dentate gyrus granular layer. B, C, FGF-2-like immunoreactivity (LI) in the dorsal hippocampus of WT (B) and TgFGF2 (C) mice. Shown are representative coronal brain sections at the level of the dorsal hippocampus, exhibiting DAB-labeled FGF-2-LI. Omitting the primary antibody to estimate nonspecific signal yielded completely negative labeling (data not shown). Note increased FGF-2-positive cells in the CA1 (arrows), CA2 and CA3 pyramidal layer, and in the dentate gyrus granular layer (arrowheads). D, E, Double-label immunohistochemistry (CA1 region) for FGF-2 (green) and GFAP (red) in WT (D) and in TgFGF2 (E) mice. Nuclei (stained with DAPI in blue) are of light green color when FGF-2 positive. Note FGF-2 localization in some GFAP-positive cells of both WT and TgFGF2 mice. Also note that only a small fraction of cells in the pyramidal layer (putatively pyramidal neurons) is FGF-2 positive in WT mice, whereas most (if not all) are FGF-2 positive in TgFGF2 animals.
This analysis was refined by using immunohistochemistry. Again, no such analysis of FGF-2 protein distribution in the normal mouse brain has been previously reported, but our findings in WT mice are consistent with the above-described in situ hybridization data and with previous observations in the rat (Gómez-Pinilla et al., 1992; Van Der Wal et al., 1994). Widespread FGF-2-immunoreactive cells were observed throughout the brain in coronal sections taken at the level of dorsal hippocampus. These cells are GFAP-positive astrocytes (see also Gómez-Pinilla et al., 1992), in which FGF-2 labeling was observed within the nucleus and in the perinuclear region. Neuronal elements showing FGF-2 immunoreactivity were essentially restricted to the CA2 area. In these cells, strong labeling was observed in the nucleus and the perinuclear region, with weak staining in the cytoplasm and in processes (presumably dendrites) proximal to the soma. In TgFGF2 mice, consistent with in situ hybridization data, an increase in FGF-2 immunoreactive cells was observed in CA2, and several FGF-2 positive cells were observed in the pyramidal layer of CA1 and CA3, and in the granular layer of the dentate gyrus (Fig. 1C). The pattern of FGF-2 immunoreactivity in these putative neurons was similar to WT CA2 neurons, that is, strong labeling in the nucleus and the perinuclear region, and weak staining in the cytoplasm and in proximal dendrites (Fig. 1E). No alteration in the diffuse astrocytic labeling was apparent in the hippocampus, thalamus and cortex.
To evaluate whether FGF-2 overexpression changed FGF receptor density, we used immunohistochemistry to analyze the distribution of FGFR1, the FGF receptor subtype most expressed in the hippocampus (Ohkubo et al., 2004). Consistent with previous reports in rats and humans (Gonzalez et al., 1995; Weickert et al., 2005), FGFR1 immunoreactivity was found in putative pyramidal neurons of CA1, CA2 and CA3 pyramidal region in WT mice. No alteration in the expression levels or in the distribution of FGFR1 was detected in TgFGF2 mice (data not shown).
Together, these data show that (1) the distribution of FGF-2 mRNA and protein in the WT mouse brain is similar to the one observed in the rat; (2) TgFGF2 mice overexpress FGF-2 mRNA and protein in all hippocampal principal cells (pyramidal neurons and dentate gyrus granule cells); (3) FGF-2 overexpression in TgFGF2 mice does not cause detectable alterations in FGFR1 receptors.
Anatomy and physiology
Next, we examined the function of the transgenic hippocampus. We focused on the dorsal hippocampus because (1) we documented FGF-2 overexpression here in TgFGF2 mice (above); (2) it is a region of interest for epilepsy; (3) it allows comparative analysis of many morphological and physiological variables. Anatomically, no obvious difference between WT and TgFGF2 mice could be detected regarding changes in the size of the hippocampus, the cytoarchitecture and the number of neurons.
FGF-2 has been reported to increase the number of excitatory synaptic contacts in the hippocampus in vitro (Li et al., 2002). Immunohistochemistry was used to assay expression of the glutamatergic vesicular transporter VGLUT1, which reflects the density of glutamate release vesicles (Fadda et al., 2007). VGLUT1 immunoreactivity was intense in many hippocampal areas but most prominent in the strata oriens and radiatum of CA1, in the strata oriens, lucidum and radiatum of CA3 and in the molecular layer and hilus of the dentate gyrus. A clear enhancement of VGLUT1 immunoreactivity was observed in the hippocampus of TgFGF2 mice, compared with WT (Fig. 2). In contrast, no differences were observed between WT and TgFGF2 mice in the density and distribution of VGAT, the vesicular transporter of GABA and a marker of GABAergic terminals (data not shown).
Figure 2.
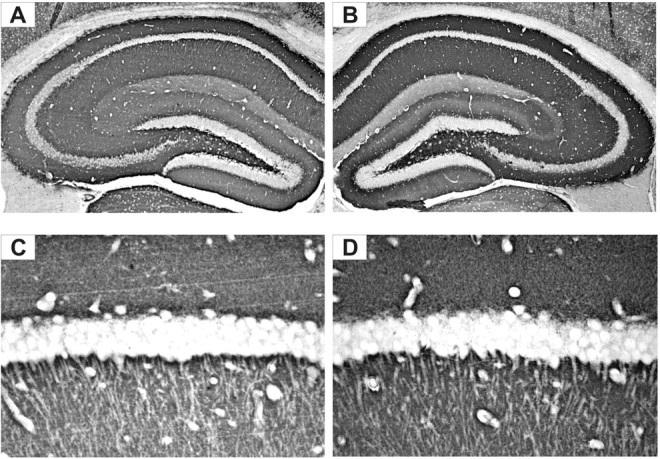
VGLUT1-like immunoreactivity (LI) in the dorsal hippocampus of WT (A, C) and TgFGF2 (B, D) mice. Shown are representative coronal brain sections at the level of the dorsal hippocampus, exhibiting DAB-labeled VGLUT1-LI. A higher magnification of the CA1 area is shown in C and D. Omitting the primary antibody to estimate nonspecific signal yielded completely negative labeling (data not shown). Note a widespread increase in VGLUT1-LI in TgFGF2 mice.
To explore the functional correlates of these observations, we investigated the effect of FGF-2 overexpression on synaptic transmission, comparing the basal properties of the Schaffer collateral-CA1 synapse in hippocampal slices prepared from WT and TgFGF2 mice. Extracellular field potential recordings in the dendritic region of area CA1 were performed to generate input/output (I/O) curves for each mouse group. These curves display the field excitatory postsynaptic potential slope as a function of the stimulation intensity. We found that the I/O curve was shifted leftwards in TgFGF2 (versus WT) mice, indicating increased excitability in the former (Fig. 3). Furthermore, after-depolarizations at supramaximal stimulation intensities were recorded much more frequently in the TgFGF2 than in the WT group.
Figure 3.

Increased excitability of the TgFGF2 hippocampus. Input-output curve at the Schaffer collateral to CA1 synapse in WT (open circles) and TgFGF2 mice (solid circles). Data are the mean ± SE of 9 animals per group (2 slices examined for each animal). Inset, Fold increase in input stimulus intensity necessary for evoking a half-maximal response (see Materials and Methods for details). Data are the mean ± SE of 9 animals per group (2 slices examined for each animal). *p < 0.05 versus WT, Student's t test for nonpaired data.
Standard extracellular stimulations activate large numbers of axons and do not allow to measure variations in EPSC amplitudes resulting from fluctuations in quantal content, because individual responses result from the release of transmitter from many presynaptic fibers. Therefore, to measure changes in neurotransmitter release due to alterations in the quantal content, we used the “minimal extracellular stimulation” technique, that triggers release from only one axon connected to the neuron under recording (Hess et al., 1987; Raastad et al., 1992). In this experimental series, paired-pulse minimal stimulation of Schaffer collaterals was used to evoke EPSCs in CA1 pyramidal neurons. Cells were voltage-clamped at –70 mV; only cells having resting membrane potential more negative than –50 mV were considered for study.
In WT animals, the responses to the first shock showed mean amplitude of 18.0 ± 2.8 pA (n = 16). Although we have not performed a detailed statistical analysis of the EPSC amplitudes, it is generally accepted that their fluctuations result from the release of a randomly varying number of transmitter-containing vesicles, as described at the neuromuscular junction (Fatt and Katz, 1952) and in acute slices (Hess et al., 1987; Malinow, 1991; Jonas et al., 1993; Manabe et al., 1993; Debanne et al., 1996; Stricker et al., 1996).
When pairs of EPSCs were elicited every 60 s at intervals of 50 ms, the mean amplitude of the second EPSC (EPSC2) was larger (43.3 ± 6.1 pA) than EPSC1, an effect known as paired-pulse facilitation (PPF) (Fig. 4). Assuming that the fluctuation in the amplitude of EPSC1 results from changes in its quantal content, then the amplitude of the second response is inversely related to the quantal content of the first response, that is, increased probability of glutamate release results in a decrease of PPF and of failures (Creager et al., 1980; Manabe et al., 1993; Debanne et al., 1996). Accordingly, the dependence of the paired-pulse ratio (EPSC2/EPSC1) on the amplitude of EPSC1 in individual trials was not random, but rather was inversely related to the amplitude of EPSC1 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 4.

Current-clamp recordings in CA1 cells. Paired-pulse facilitation (PPF) in WT and TgFGF2 mice. EPSCs elicited by double-pulse stimulation of the Shaffer collaterals (60 s interval, 50 ms delay) showed an evident PPF both in WT (A, C) and TgFGF2 mice (B, D). E, The ratio (S2/S1) between the amplitudes of response to the second (EPSC2, S2) and to the first (EPSC1, S1) stimulus is reduced in TgFGF2 mice, indicating an augmented release of neurotransmitter (see text for details). Data in C, D and E are the mean ± SE of 10 animals per group. ***p < 0.001 versus WT, Student's t test for nonpaired data.
When the same protocol was applied to TgFGF2 mice (n = 15), the results showed a larger EPSC1 mean amplitude (21.5 ± 2.7 pA) and smaller EPSC2 amplitude (33.4 ± 4.1 pA) compared with WT animals (Fig. 4). The EPSC2 to EPSC1 (S2/S1) ratio of mean values in TgFGF2 tissue was 1.56 ± 0.08, significantly smaller (p < 0.001) than in WT (2.40 ± 0.18) (Fig. 4E); in addition, the number of failures was greater in WT (14.5%) than in TgFGF2 mice (8.1%). These results suggest an increase in the amount of neurotransmitter released in TgFGF2 mice because of an increase of the quantal content in the Schaffer collaterals.
Susceptibility to kainate seizures
Together, functional anatomy and physiology data suggest that FGF-2 overexpression brings about a potentiation of excitatory glutamatergic transmission. One obvious implication could be an increased susceptibility to seizures. To explore this possibility, WT and TgFGF2 littermates were injected with 20 mg/kg i.p. kainate, and their behavior monitored for 2 h thereafter. This dose of kainate was chosen because it causes relatively mild seizures in WT animals, allowing proper observation of possible alterations in the TgFGF2 group. The response to kainate was consistent with a latent epileptic phenotype in TgFGF2 mice. (1) Behavioral seizure scores, based on two distinct scoring systems, were significantly increased in TgFGF2 mice (Fig. 5A). (2) The latency to generalized seizure onset (in the subset of animals displaying generalized seizures) was significantly shorter in TgFGF2 mice (19 ± 2 min versus 31 ± 5 min in WT; p < 0.01, Student's t test for nonpaired data). (3) The percentage of animals displaying generalized seizures (stage 3 or higher) was much higher in the TgFGF2 group: 14 of 26 WT animals (53.8%) versus 28 of 29 kainate-treated TgFGF2 mice (96.6%). During most of the 2 h after kainate administration, the seizure class was indeed higher for the TgFGF2 mice compared with the WT group (Fig. 5B). Furthermore, at the end of the observation period, nearly all WT animals were behavioral seizure-free (3 of 26, i.e., 11.5%, still experiencing generalized seizures), whereas many TgFGF2 mice were still seizing (9 of 29, i.e., 31.0%, with generalized seizures). (4) Lethality was increased: whereas no WT mouse of 26 treated with kainate died, 4 of 29 (13.8%) TgFGF2 died during observation (i.e., in the first 2 h after kainate administration) and another 3 died within the following 3 d (overall mortality 24.1% versus 0% in WT animals).
Figure 5.
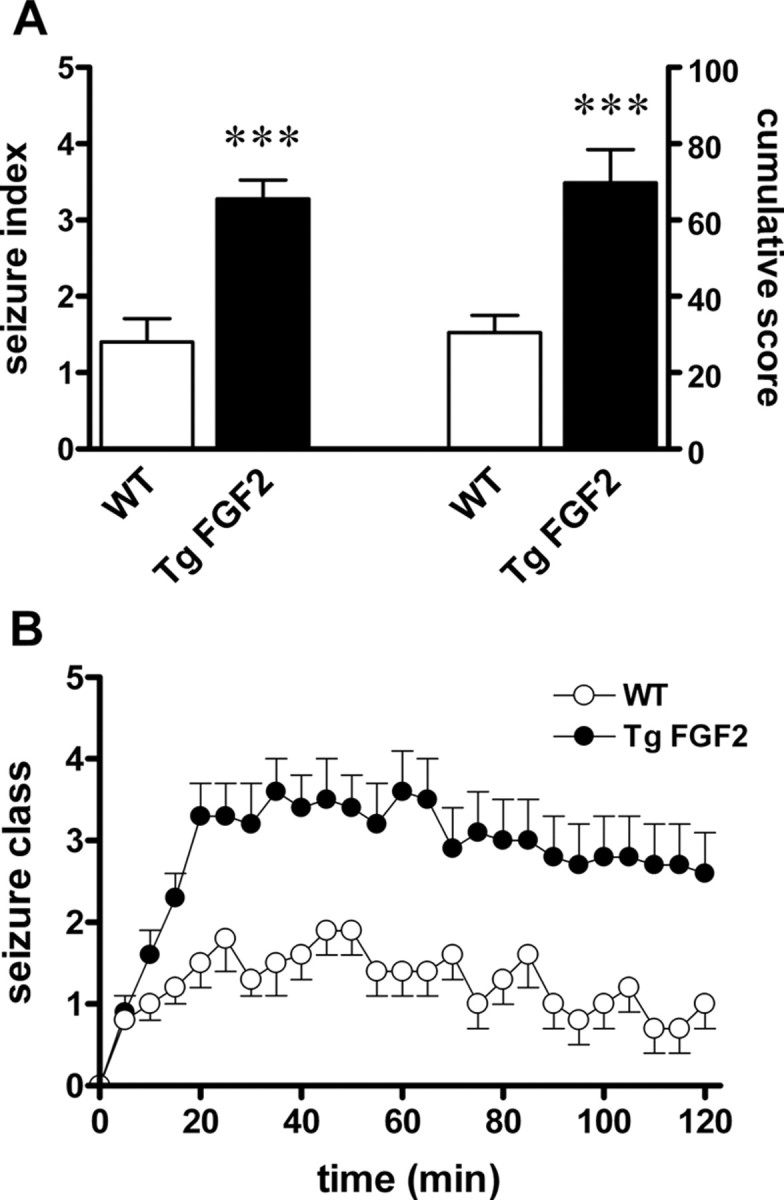
Kainate seizure susceptibility in TgFGF2 mice. A, Seizure severity. Seizure index was rated according to the following arbitrary scale (Bregola et al., 2002): 0, no seizure; 1, 1 severe (class 4–5) seizure; 2, 2–5 severe seizures; 3, 6–10 severe seizures; 4, >10 severe seizures, or very severe seizures (class 6–7); 5, death within 2 h. Cumulative seizure score was rated as the sum of the scores recorded in each 5 min interval of the 2 h after kainate injection. Seizures were scored as described in the Materials and Methods (Janumpalli et al., 1998; Bregola et al., 2002). B, Time course of the seizure severity in the 2 h observation after kainate administration. WT = wild-type mice; TgFGF2 = FGF-2 transgenic mice. Data are the mean ± SE of 26 animals per group. A, B, ***p < 0.001 versus WT, Mann–Whitney U test. C, **p < 0.01 versus WT, Student's t test for nonpaired data.
Pathological sequalae to seizures
Next, we hypothesized that FGF-2 overexpression altered the pathological sequelae of prolonged seizures. At a cellular level, these sequelae include cell death, increased neurogenesis and axonal arborization like sprouting of the mossy fibers (for review, see Pitkänen and Sutula, 2002). To obtain a proper comparison of seizure-induced cellular alterations, WT and TgFGF2 mice were divided in four groups based on the cumulative score, avoiding the bias produced by the higher susceptibility to seizures of TgFGF2 mice.
Cell death
Seizure-induced cell damage was initially explored using the FJB labeling, a marker of cell death. No FJB-positive cells were observed under basal conditions in either WT or TgFGF2 mice (Fig. 6A,B). After administration of kainate, WT animals showed FJB labeling in the dorsal hippocampus with the typical pattern of neurodegeneration associated with kainate seizures, involving different subfields (CA1, CA3, hilus of the dentate gyrus) (Fig. 6C). In contrast, in TgFGF2 mice that underwent severe kainate seizures, FJB-positive neurons were observed only in the hilus of the dentate gyrus (Fig. 6D). Kainate seizures also induced damage in the ventral hippocampus and in extrahippocampal regions, most prominently in the amygdala, in the dorsal nuclei of the thalamus, in the enthorinal cortex, in the neocortex. Also in these regions, the degree of damage was substantially lower in TgFGF2 mice (supplemental Table 1, supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Degeneration patterns observed with FJB were confirmed using silver impregnation. Data in the dorsal hippocampus have been quantified by using a scoring system (“degeneration index”) and a digital analysis (percentage of FJB-positive pixels in the hippocampus) after stratifying the animals on the basis of the cumulative seizure score: seizures of similar grade produced a significantly greater cell damage in WT than in TgFGF2 mice (Fig. 6E,F).
Figure 6.

Kainate-induced cell damage in the hippocampus. Shown are dorsal hippocampal, FJB-stained sections prepared from WT (A, C) and TgFGF2 (B, D) mice, killed 3 d after treatment with vehicle (A, B) or kainate (B, D). Kainate administration evoked seizures of equal severity in the WT and TgFGF2 mice of C and D: cumulative seizure scores of 80 (C) and 84 (D). Note that seizures of identical grade induce more numerous FJB-positive cells in WT than in TgFGF2 animals. Moreover, positive cells are only present in the hilus of the TgFGF2 brain whereas postive cells were found in the hilus as well as in the CA3 and CA1 pyramidal layer of the WT. C, Inset, Adjacent section (CA1 area) stained with active caspase 3, a marker of apoptosis. Note that many FJB-positive calls are also active caspase 3-positive, indicating that kainate-induced neurodegeneration is produced by both necrosis and apoptosis. (E, F) Quantification of neurodegeneration as a function of seizure severity. The degeneration index (E) and the percentage of FJB-positive pixels in the hippocampus (F) in WT (open bars) and TgFGF2 mice (solid bars) are plotted as a function of seizure severity, by dividing kainate-treated animals in four groups based on the cumulative score recorded after administration of the convulsant agent. Data are the mean ± SE of 3–6 animals per group. *p < 0.05, **p < 0.01 versus WT; Mann–Whitney U test.
To explore the contribution of apoptosis to the observed cell death, sections adjacent to those used for FJB and for silver impregnation staining were used for active caspase 3 immunohistochemistry. Active caspase 3-positive cells were found to be a subgroup of those positive for FJB and silver impregnation, both in WT and in TgFGF2 animals (data not shown). Thus, kainate-induced cell damage appears to be produced by both necrotic and apoptotic mechanisms in both experimental groups.
These seizure-induced cell death data contrast with excitability and seizure susceptibility data and, therefore, it can be hypothesized the FGF-2 activates distinct mechanisms to increase excitability and to protect neurons. Based on our data, the increased excitability appears to depend on increased excitatory neurotransmission. Possible mechanisms of FGF-2-dependent neuroprotection include potentiation of Ca2+-dependent inactivation of NMDA currents (Boxer et al., 1999) and/or induction of ActA, a cytokine belonging to the transforming growth factor-β superfamily (Mattson, 2000; Tretter et al., 2000). Thus, we began to explore these possibilities.
To examine a possible effect on Ca2+-dependent inactivation of NMDA currents, 50 μm NMDA was applied during patch recording on CA1 pyramidal cells in the presence of 20 μm glycine and 2 mm Ca2+, with the membrane voltage clamped at −70 mV. We found no significant difference between WT and TgFGF2 animals in the amplitude and time course of the NMDA-evoked currents (Fig. 7A,B). Immunohistochemistry was used to explore the possible induction of ActA in WT and TgFGF2 mice, both under control conditions (naive animals) and 24 h after kainate administration (as per Tretter et al., 2000). Both WT and TgFGF2 naive mice showed a low number of weakly ActA-positive cells in the CA3 and CA1 pyramidal layer. Although the number of ActA-positive cells was increased after kainate in WT mice, in TgFGF2 mice this increased expression was highly potentiated: a higher number of more strongly positive cells was observed in the CA3 and CA1 pyramidal layer (14.2 ± 1.5% of total pyramidal layer cells in WT, 63.0 ± 3.3% in TgFGF2; n = 4 per group; p < 0.01, Mann–Whitney U test; Fig. 7C–F). These data indicate that induction of ActA is a plausible mechanism for the neuroprotection observed in TgFGF2 mice.
Figure 7.
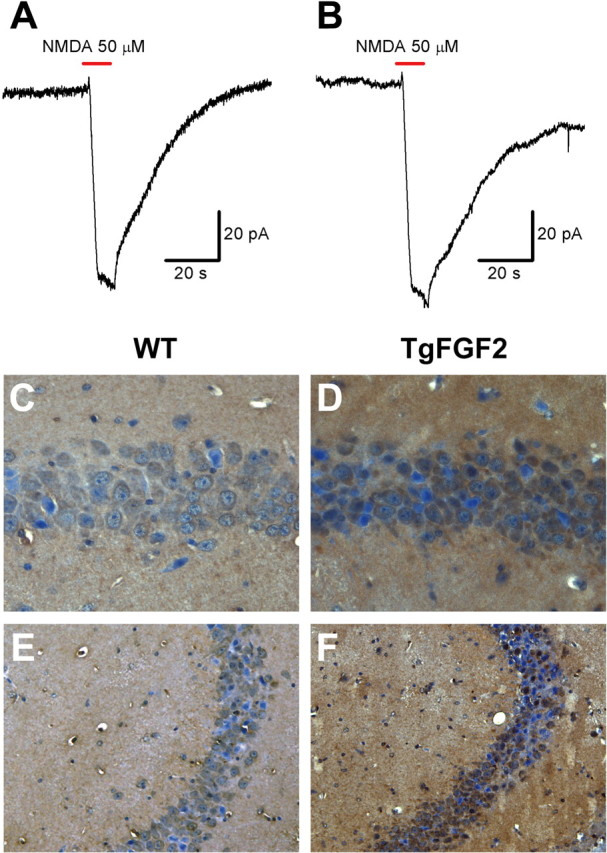
A, B, Ca2+-dependent NMDA-evoked currents in naive WT (A) and TgFGF2 mice (B). No significant difference was observed between WT and TgFGF2 mice in the amplitude and time course of the evoked NMDA currents: amplitude (pA): 81.3 ± 11.1 in WT, 89.7 ± 13.6 in TgFGF2; rise time from 10 to 90% (s): 2.16 ± 0.31 in WT, 2.41 ± 0.43 in TgFGF2; decay time constant (s): 22.4 ± 4.1 in WT, 25.3 ± 4.7 in TgFGF2 (mean ± SE of 4 animals per group). C–E, ActA-like immunoreactivity (LI) in the dorsal hippocampus CA1 (C, D) and CA3 (E, F) area of WT (C, E) and TgFGF2 (D, F) mice killed 24 h after kainate seizures of similar grade (cumulative scores: 55 in WT, 61 in TgFGF2). Shown are representative sections exhibiting DAB-labeled ActA-LI. Omitting the primary antibody to estimate nonspecific signal yielded completely negative labeling. Note the greater number of ActA-positive cells in the pyramidal layer of the TgFGF2 and the more intense ActA signal in these positive cells.
Neurogenesis
Despite the fact that FGF-2 is a potent mitogenic factor for hippocampal neural stem cells (Temple, 2001), its overexpression in TgFGF2 mice did not appear to lead to increased neurogenesis. Under basal conditions, the number of BrdU immunoreactive cells in the dentate gyrus (an index of neural stem and progenitor cell proliferation), did not differ between TgFGF2 mice and their WT littermates (Fig. 8A, vehicle). Kainate seizures increased cellular proliferation in a seizure severity-dependent manner, whereby the number of BrdU-positive cells increased as a function of seizure severity. However, there was no difference between the increases in BrdU-positive cells for the TgFGF2 and WT groups when the seizures were similar in grade (Fig. 8A).
Figure 8.

A, Seizure-induced upregulation of cell proliferation in the dorsal hippocampus dentate gyrus of WT and TgFGF2 mice. The number of proliferating cells in the dentate gyrus (BrdU-positive cells, see Materials and Methods for details) in WT (open bars) and TgFGF2 mice (solid bars) is plotted as a function of seizure severity, by dividing kainate-treated animals in four groups based on the cumulative score recorded after administration of the convulsant agent. These animals were administered a series of four injections of 50 mg/kg i.p. BrdU 3 d after kainate and killed 24 h after the last injection. Data are the mean ± SE of 3–6 animals per group. No significant difference is observed between WT and TgFGF2 mice. B, C, Seizure-induced neurogenesis and ectopic localization of newly produced cells. Quantification of BrdU (B)- and DCX-positive cells (C) in the dentate gyrus of WT (open bars) and TgFGF2 mice (solid bars). Animals in this experiment were selected on the basis of a cumulative seizure score of 50–75. They were then administered five injections of 50 mg/kg i.p. BrdU 3–7 d after kainate and killed 9 d after kainate. Data are the mean ± SE of 6 animals per group. No significant difference is observed between WT and TgFGF2 mice. Total: total cell counts in the dentate gyrus; ectopic: number of cells identified in the hilus of the dentate gyrus. D, E, Double-label immunofluorescence for DCX (green) and BrdU (red) in WT (D) and in TgFGF2 (E) mice. Nuclei are stained with DAPI (blue). Note colocalization of BrdU and DCX in many cells (22 ± 5% in WT and 26 ± 6% in TgFGF2 mice). Also note ectopic cells in the hilus in both groups.
To determine whether FGF-2 overexpression led to changes in the differentiation of these newly produced cells, we employed an early marker of neuronal differentiation (DCX), alone and in combination with BrdU. Animals were treated with a single injection of BrdU for 5 d from day 3 after kainate and killed at day 9 (Jessberger et al., 2005). Six WT and six TgFGF2 animals with cumulative seizure scores in the 50–75 range were used for this experiment. The total number of BrdU-positive, DCX-positive and BrdU-DCX-double positive cells in the dentate gyrus was not significantly different in WT compared with TgFGF2 mice (Fig. 8B–E). Seizure-induced neurogenesis leads to ectopic location in the hilus of many newly generated cells, and it is thought that these ectopic cells may contribute to epileptogenesis (Parent et al., 2006). Thus, we asked whether FGF-2 overexpression leads to alterations in the ectopic migration of seizure-induced cells. As shown in Figure 8, B and C, however, no difference was observed between WT and TgFGF2 mice in this respect.
Sprouting of the mossy fibers
Epileptic seizures can cause long-term synaptic rearrangements, including mossy fiber sprouting (for review, see Nadler, 2003). These morphological alterations may depend on neurotrophic factors, and on FGF-2 in particular (Patel and McNamara, 1995). Timm staining showed that, under basal condition, neither WT nor TgFGF2 mice displayed sprouting of the mossy fibers. Thirty days after kainate treatment, sprouting was observed in both WT and in TgFGF2 groups, but there was no significant difference (Fig. 9). Thus, the overexpression of FGF-2 protects from seizure-induced damage but does not seem to be sufficient to influence seizure-induced neurogenesis and mossy fiber sprouting.
Figure 9.

Sprouting of the mossy fibers in WT and TgFGF2 mice. The Timm index was calculated as described in the Materials and Methods. A subset of kainate-treated animals with cumulative scores in the 25–50 range (average 33 ± 4 for the WT, 33 ± 7 for the TgFGF2) was used for this experiment. Data are the mean ± SE of 4 animals per group. No significant difference is observed between WT and TgFGF2 mice.
Discussion
Main findings
Two main findings emerge from this study. First: FGF-2 overexpression in the hippocampus (CA3 and CA1 pyramidal cells and dentate gyrus granule cells) brings about an increase in excitatory inputs. This leads to a condition of latent hyperexcitability, made apparent as an increased susceptibility to chemoconvulsant-induced status epilepticus. Second: however, status epilepticus-induced cell damage, but not other sequelae of prolonged seizures, is highly reduced by FGF-2 overexpression. Neither neurogenesis or mossy fiber sprouting were significantly different in the two groups. In contrast, cell damage, assessed with FJB, silver impregnation and anti-caspase 3 immunohistochemistry, was significantly lower in TgFGF2 mice, especially in areas of FGF-2 overexpression, indicating reduction of seizure-induced necrosis and apoptosis. These data suggest that FGF-2 overexpression exerts pleiotropic effects by favoring ictogenesis while reducing seizure-induced cell death. Characterizing the molecular mechanisms for these results is the next step for this project. Below, we will discuss these main findings and their possible mechanisms.
Hyperexcitability and epileptic phenotype
The biological effects of FGF-2 overexpression during development and in maturity may induce plastic modifications of the brain network that account for hyperexcitability. Based on our data, the latent epileptic phenotype of TgFGF2 mice cannot be explained on the basis of FGF-2 effects on neurogenesis (not altered in the mutant mice), cell death (not altered under basal conditions, and actually reduced after seizures), or sprouting of the mossy fibers (not altered). Another alternative hypothesis suggests that this phenotype depends on the capability of FGF-2 to increase the number of excitatory synapses (Li et al., 2002). In vitro studies indicate that FGF-2 may increase excitatory synapses in cultured hippocampal neurons, and that these synapses are normal in both their morphology and their neurotransmitter release function (Li et al., 2002). Indeed, areas of FGF-2 overexpression showed an increased density of glutamatergic, but not GABAergic, synaptic vesicles. We also found that the quantal content of glutamate released by stimulation of a single axon of the Schaffer collaterals was increased. This observation suggests that FGF-2 transgenesis either increased the number of vesicles per glutamatergic terminal or increased the number of terminals per axon (i.e., induced axonal sprouting). Our data do not allow distinction between these two possibilities; however, the above-mentioned in vitro evidence (Li et al., 2002) strongly supports the latter. Together, these data suggest that an anomalously increased excitatory synaptic input may cause an increased propensity to seizures.
However, as discussed below, TgFGF2 mice not only display a significantly higher susceptibility to kainate seizures with respect to their WT littermates, but they also show a reduction in seizure-induced cell damage. Interestingly, these epileptologic characteristics are similar to those of young brains (Haut et al., 2004), and FGF-2 expression during postnatal development is highest in the first weeks of life (Monfils et al., 2006).
Susceptibility to the consequences of seizures
In this study, seizure-associated plasticity was characterized in terms of neurogenesis, mossy fiber axonal sprouting and cell damage.
We found that TgFGF2 mice were protected from seizure-induced damage compared with the WT. These results are consistent with data showing that chronic infusion of low FGF-2 doses into the cerebral ventricles prevents seizure-induced hippocampal cell loss (Liu et al., 1993). Neuroprotective effects of FGF-2 supplementation have been also reported in stroke models (Ikeda et al., 2005). Examining the long-term consequences of this effect was beyond the aims of this study. These observations, however, suggests that FGF-2, by attenuating the neurodegeneration occurring after kainate-induced acute seizures, might thwart the development and the progression of chronic epilepsy.
Alternative mechanisms may account for the neuroprotective action of FGF-2. First, FGF-2 mediated neuroprotection may depend on inactivation of NMDA receptors, because FGF-2 chronic treatments have been reported to potentiate Ca2+-dependent inactivation of NMDA currents in hippocampal neurons (Boxer et al., 1999). However, we did not find any significant difference in the amplitude and time course of the NMDA-evoked currents in hippocampal slices prepared from WT and TgFGF2 mice. Another possible mechanism for this protective effect may be the induction of ActA. When coinjected with kainate in the hippocampus, FGF-2 prevents neuronal loss (Tretter et al., 2000). In mice treated with kainate and FGF-2, much more than in those treated with kainate alone, ActA-immunoreactivity was found to be high in CA3 and CA1 pyramidal neurons (Tretter et al., 2000). Moreover, FGF-2 fails to protect CA3 neurons against kainate-induced death in the presence of the ActA-neutralizing protein follistatin (Tretter et al., 2000). Thus, it has been hypothesized that FGF-2 can enhance the production of ActA in CA1 and CA3 neurons, protecting these cell types from injury (Mattson, 2000). Accordingly, in TgFGF2 mice we observed the preservation from degeneration of cells overproducing FGF-2 (CA1 and CA3 pyramidal neurons), but not of those not overproducing FGF-2 (interneurons in the hilus of the dentate gyrus). These hippocampal regions, CA1 and CA3, were also those in which we observed increased ActA expression. These data suggest that the neuroprotective effect of FGF-2 on kainate seizures-induced damage is ActA-mediated. However, further studies will be needed to conclusively validate this hypothesis.
Other sequalae of status epilepticus were not affected by FGF-2 overexpression. For example, FGF-2 overexpression did not increase seizure-induced neural stem and progenitor cell proliferation and neurogenesis. Seizure-induced neurogenesis is reportedly abolished in FGF-2 null mice (Yoshimura et al., 2001). Together, these data suggest that FGF-2 is necessary, but not alone sufficient, for seizure-induced neurogenesis. We also found that FGF-2 overexpression does not increase seizure-induced mossy fiber sprouting, a finding apparently in contrast with previous in vitro reports (Aoyagi et al., 1994; Patel and McNamara, 1995; Szebenyi et al., 2001). Differences in the in vitro conditions compared with the in vivo conditions of this study may account for this discrepancy. However, differences in the cell types examined and in their age may also be important. As discussed above, in fact, our data on the Schaffer collaterals suggest (even if they do not directly demonstrate) sprouting of these other axons. Thus, the specific environment to which specific axons are exposed, including availability of FGF-2, may dictate the characteristics and the extent of ramification and formation of synaptic contacts.
Distinct mechanisms for FGF-2-induced hyperexcitability and neuroprotection?
How might FGF-2 activate the distinct pathways implicated in hyperexcitability and in neuroprotection? FGF-2 can produce many different (pleiotropic) effects, based on differential isoform-specific (HMW vs LMW) actions and/or on differential signaling through the multiple FGF receptors. Specific HMW and LMW effects were found using transgenic mice that specifically overexpress either HMW or LMW (Xiao et al., 2003). The HMW isoforms have intracellular (intracrine) actions on proliferation, differentiation and survival (Sørensen et al., 2006). Only the LMW FGF-2 can be secreted to putatively interact, in an autocrine or paracrine manner, with FGF receptors on the cell surface. Therefore, increased availability of HMW FGF-2 in TgFGF2 mice may primarily account for the neuroprotective effects, whereas the LMW isoform may be responsible for hyperexcitability and susceptibility to seizures. Given the availability of transgenic mice that specifically overexpress either HMW or LMW, this hypothesis is now amenable to experimental challenge.
In conclusion, FGF-2 was found here to exert two different, apparently contrasting effects: increased seizure susceptibility and reduced cell death. Verification of this conclusion (that is based on FGF-2 overexpression across development and adulthood) by determining the effects of transient FGF-2 overexpression in the adult, and accurately dissecting out the mechanisms underlying these contrasting effects, will be critical in pursuing therapeutic goals (for example, controlling seizures and their deleterious consequences) through modulation of the FGF-2 system.
Footnotes
This work was supported by grants from the University of Ferrara (STAMINA, to M.S. and O.B.), the Italian Ministry for the University and Scientific Research (Prin 2005 to M.S.; Prin 2006 to A.M.), and the European Community [LSH-CT-2006–037315 (EPICURE), thematic priority LIFESCIHEALTH, to M.S.].
References
- Adams B, Vaccarella L, Fahnestock M, Racine RJ. The cholinergic system modulates kindling and kindling-induced mossy fiber sprouting. Synapse. 2002;44:132–138. doi: 10.1002/syn.10064. [DOI] [PubMed] [Google Scholar]
- Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL. The LTP program: a data acquisition program for on-line analysis of long-term potentiation and other synaptic events. J Neurosci Methods. 2001;108:71–83. doi: 10.1016/s0165-0270(01)00374-0. [DOI] [PubMed] [Google Scholar]
- Aoyagi A, Nishikawa K, Saito H, Abe K. Characterization of basic fibroblast growth factor-mediated acceleration of axonal branching in cultured rat hippocampal neurons. Brain Res. 1994;661:117–126. doi: 10.1016/0006-8993(94)91188-6. [DOI] [PubMed] [Google Scholar]
- Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- Böhlen P, Baird A, Esch F, Ling N, Gospodarowicz D. Isolation and partial molecular characterization of pituary fibroblast growth factor. Proc Natl Acad Sci U S A. 1984;81:5364–5368. doi: 10.1073/pnas.81.17.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Moreno H, Rudy B, Ziff EB. FGF-2 potentiates Ca2+-dependent inactivation of NMDA receptor currents in hippocampal neurons. J Neurophysiol. 1999;82:3367–3377. doi: 10.1152/jn.1999.82.6.3367. [DOI] [PubMed] [Google Scholar]
- Bregola G, Zucchini S, Rodi D, Binaschi A, D'Addario C, Landuzzi D, Reinscheid R, Candeletti S, Romualdi P, Simonato M. Involvement of the neuropeptide nociceptin/orphanin FQ in kainate seizures. J Neurosci. 2002;22:10030–10038. doi: 10.1523/JNEUROSCI.22-22-10030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugra K, Pollard H, Charton G, Moreau J, Ben-Ari Y, Khrestchatisky M. aFGF, bFGF and flg mRNAs show distinct patterns of induction in the hippocampus following kainate-induced seizures. Eur J Neurosci. 1994;6:58–66. doi: 10.1111/j.1460-9568.1994.tb00247.x. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression and performance. J Neurosci. 1991;11:2795–2803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JD, Florkiewicz RZ, Neumann J, Mort-Hopkins T, Dorn GW, 2nd, Lightfoot P, German R, Howles PN, Kier A, O'Toole BA, Sasse J, Gonzalez AM, Baird A, Doetschman T. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol Biol Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager R, Dunwiddie T, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol. 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto N, Gonzalez AM, Walicke PA, Wada E, Simmons DM, Shimasaki S, Baird A. Basic fibroblast growth factor (FGF) in the central nervous system: identification of specific loci of basic FGF expression in the rat brain. Growth Factors. 1989;2:21–29. doi: 10.3109/08977198909069078. [DOI] [PubMed] [Google Scholar]
- Fadda P, Bedogni F, Fresu A, Collu M, Racagni G, Riva MA. Reduction of corticostriatal glutamatergic fibers in basic fibroblast growth factor deficient mice is associated with hyperactivity and enhanced dopaminergic transmission. Biol Psychiatry. 2007;62:235–242. doi: 10.1016/j.biopsych.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol. 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: Three initiate translation from non-AUG codons. Proc Natl Acad Sci U S A. 1989;86:3978–3981. doi: 10.1073/pnas.86.11.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Baird A, Gonzalez AM. Multiple forms of basic fibroblast growth factor: differential nuclear and cell surface localization. Growth Factors. 1991;4:265–275. doi: 10.3109/08977199109043912. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Sancini G, Buzzi A, Zucchini S, Paradiso B, Magnaghi G, Frassoni C, Chikhladze M, Avanzini G, Simonato M. A pathogenetic hypothesis of Unverricht-Lundborg disease onset and progression. Neurobiol Dis. 2007;25:675–685. doi: 10.1016/j.nbd.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, Kivi A, Zeller M, Meencke HJ, Cavalheiro EA, Heinemann U, Lehmann TN. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci. 2004;24:10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall CM, Berschauer R, Isackson PJ. Seizures increase basic fibroblast growth factor mRNA in adult rat forebrain neurons and glia. Mol Brain Res. 1994;21:190–205. doi: 10.1016/0169-328x(94)90250-x. [DOI] [PubMed] [Google Scholar]
- Gallyas F, Wolff JR, Böttcher H, Zaborszky L. A reliable and sensitive method to localize terminal degeneration and lysosomes in the central nervous system. Stain Technol. 1980;55:299–306. doi: 10.3109/10520298009067258. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Lee JW, Cotman CW. Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria-fornix transection. J Neurosci. 1992;12:345–355. doi: 10.1523/JNEUROSCI.12-01-00345.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pinilla F, van der Wal EA, Cotman CW. Possible coordinated gene expressions for FGF receptor, FGF-5, and FGF-2 following seizures. Exp Neurol. 1995;133:164–174. doi: 10.1006/exnr.1995.1019. [DOI] [PubMed] [Google Scholar]
- Gonzalez AM, Berry M, Maher PA, Logan A, Baird A. A comprehensive analysis of the distribution of FGF-2 and FGFR1 in the rat brain. Brain Res. 1995;701:201–226. doi: 10.1016/0006-8993(95)01002-x. [DOI] [PubMed] [Google Scholar]
- Gritti A, Frölichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF, Bjornson CR, Vescovi AL. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci. 1999;19:3287–3297. doi: 10.1523/JNEUROSCI.19-09-03287.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe C, Wewetzer K. Fibroblast growth factor and its implications for developing and regenerating neurons. Int J Dev Biol. 1996;40:403–410. [PubMed] [Google Scholar]
- Haut SR, Velísková J, Moshé SL. Susceptibility of immature and adult brains to seizure effects. Lancet Neurol. 2004;3:608–617. doi: 10.1016/S1474-4422(04)00881-6. [DOI] [PubMed] [Google Scholar]
- Hess G, Kuhnt U, Voronin LL. Quantal analysis of paired-pulse facilitation in guinea pig hippocampal slices. Neurosci Lett. 1987;77:187–192. doi: 10.1016/0304-3940(87)90584-2. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
- Ikeda N, Nonoguchi N, Zhao MZ, Watanabe T, Kajimoto Y, Furutama D, Kimura F, Dezawa M, Coffin RS, Otsuki Y, Kuroiwa T, Miyatake S. Bone marrow stromal cells that enhanced fibroblast growth factor-2 secretion by herpes simplex virus vector improve neurological outcome after transient focal cerebral ischemia in rats. Stroke. 2005;36:2725–2730. doi: 10.1161/01.STR.0000190006.88896.d3. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the FGF and FGFR gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Janumpalli S, Butler LS, MacMillan LB, Limbird LE, McNamara JO. A point mutation (D79N) of the alpha2A adrenergic receptor abolishes the antiepileptogenic action of endogenous norepinephrine. J Neurosci. 1998;18:2004–2008. doi: 10.1523/JNEUROSCI.18-06-02004.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Römer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol. 2005;196:342–351. doi: 10.1016/j.expneurol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci. 1999;4:D165–D177. doi: 10.2741/klint. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Albuquerque C, Gu J, MacDermott AB. Ca2+-dependent inactivation of NMDA receptors: fast kinetics and high Ca2+ sensitivity in rat dorsal horn neurons. J Physiol. 1996;495:449–463. doi: 10.1113/jphysiol.1996.sp021606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AJ, Suzuki S, Suzuki M, Mizukoshi E, Imamura T. Fibroblast growth factor-2 increases functional excitatory synapses on hippocampal neurons. Eur J Neurosci. 2002;16:1313–1324. doi: 10.1046/j.1460-9568.2002.02193.x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Holmes GL. Basic fibroblast growth-induced seizures in rats. Neurosci Lett. 1997;233:85–88. doi: 10.1016/s0304-3940(97)00627-7. [DOI] [PubMed] [Google Scholar]
- Liu Z, D'Amore PA, Mikati M, Gatt A, Holmes GL. Neuroprotective effect of chronic infusion of basic fibroblast growth factor on seizure-associated hippocampal damage. Brain Res. 1993;626:335–338. doi: 10.1016/0006-8993(93)90598-h. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Arsenault L. The effects of growth factors on the survival and differentiation of cultured dentate gyrus neurons. J Neurosci. 1996;16:1759–1769. doi: 10.1523/JNEUROSCI.16-05-01759.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R. Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science. 1991;252:722–724. doi: 10.1126/science.1850871. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Marconi P, Simonato M, Zucchini S, Bregola G, Argnani R, Krisky D, Glorioso JC, Manservigi R. Replication-defective herpes simplex virus vectors for neurotrophic factor gene transfer in vitro and in vivo. Gene Therapy. 1999;6:904–912. doi: 10.1038/sj.gt.3300882. [DOI] [PubMed] [Google Scholar]
- Marconi P, Zucchini S, Berto E, Bozac A, Paradiso B, Bregola G, Grassi C, Volpi I, Argnani R, Marzola A, Manservigi R, Simonato M. Effects of defective herpes simplex vectors expressing neurotrophic factors on the proliferation and differentiation of nervous cells in vivo. Gene Therapy. 2005;12:559–569. doi: 10.1038/sj.gt.3302438. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Activin to the rescue for overexcited neurons. Nat Med. 2000;6:739–741. doi: 10.1038/77453. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Driscoll I, Melvin NR, Kolb B. Differential expression of basic fibroblast growth factor-2 in the developing rat brain. Neuroscience. 2006;141:213–221. doi: 10.1016/j.neuroscience.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Nadler JV. The recurrent mossy fiber pathway of epileptic brain. Neurochem Res. 2003;28:1649–1658. doi: 10.1023/a:1026004904199. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci. 2004;24:6057–6069. doi: 10.1523/JNEUROSCI.1140-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol. 2006;59:81–91. doi: 10.1002/ana.20699. [DOI] [PubMed] [Google Scholar]
- Patel MN, McNamara JO. Selective enhancement of axonal branching of cultured dentate gyrus neurons by neurotrophic factors. Neuroscience. 1995;69:763–770. doi: 10.1016/0306-4522(95)00281-m. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain. New York: Academic; 2001. [Google Scholar]
- Pitkänen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002;1:173–181. doi: 10.1016/s1474-4422(02)00073-x. [DOI] [PubMed] [Google Scholar]
- Quarto N, Finger FP, Rifkin DB. The NH2-terminal extension of high molecular weight bFGF is a nuclear targeting signal. J Cell Physiol. 1991;147:311–318. doi: 10.1002/jcp.1041470217. [DOI] [PubMed] [Google Scholar]
- Raastad M, Storm JF, Andersen P. Putative single quantum and single fibre excitatory postsynaptic currents show similar amplitude range and variability in rat hippocampal slices. Eur J Neurosci. 1992;4:113–117. doi: 10.1111/j.1460-9568.1992.tb00114.x. [DOI] [PubMed] [Google Scholar]
- Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- Simonato M, Bregola G, Donatini A, Bianchi C, Beani L, Ferri S, Romualdi P. Kindled seizure-induced c-fos and prodynorphin mRNA expressions are unrelated in the rat brain. Eur J Neurosci. 1996;8:2064–2067. doi: 10.1111/j.1460-9568.1996.tb00726.x. [DOI] [PubMed] [Google Scholar]
- Simonato M, Molteni R, Bregola G, Muzzolini A, Piffanelli M, Beani L, Racagni G, Riva M. Different patterns of induction of FGF-2, FGF-1 and BDNF mRNAs during kindling epileptogenesis in the rat. Eur J Neurosci. 1998;10:955–963. doi: 10.1046/j.1460-9568.1998.00105.x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Sørensen V, Nilsen T, Wiedlocha A. Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays. 2006;28:504–514. doi: 10.1002/bies.20405. [DOI] [PubMed] [Google Scholar]
- Stricker C, Field AC, Redman SJ. Statistical analysis of amplitude fluctuations in EPSCs evoked in rat CA1 pyramidal neurones in vitro. J Physiol. 1996;490:419–441. doi: 10.1113/jphysiol.1996.sp021155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Dent EW, Callaway JL, Seys C, Lueth H, Kalil K. Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. doi: 10.1523/JNEUROSCI.21-11-03932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, Roderick TH, Stewart CL, Lilly F, Hansen CT, Overbeek PA. FVB/N: An inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci U S A. 1991;88:2065–2069. doi: 10.1073/pnas.88.6.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- Tretter YP, Hertel M, Munz B, ten Bruggencate G, Werner S, Alzheimer C. Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat Med. 2000;6:812–815. doi: 10.1038/77548. [DOI] [PubMed] [Google Scholar]
- Van Der Wal EA, Gómez-Pinilla F, Cotman CW. Seizure-associated induction of basic fibroblast growth factor and its receptor in the rat brain. Neuroscience. 1994;60:311–323. doi: 10.1016/0306-4522(94)90245-3. [DOI] [PubMed] [Google Scholar]
- Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. doi: 10.1523/JNEUROSCI.19-14-06006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Kittell DA, Saunders RC, Herman MM, Horlick RA, Kleinman JE, Hyde TM. Basic fibroblast growth factor and fibroblast growth factor receptor-1 in the human hippocampal formation. Neuroscience. 2005;131:219–233. doi: 10.1016/j.neuroscience.2004.09.070. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Cole TB, Born DE, Schwartzkroin PA, Palmiter RD. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl Acad Sci U S A. 1997;94:12676–12681. doi: 10.1073/pnas.94.23.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Liu P, Sobue T, Lichtler A, Coffin JD, Hurley MM. Effect of overexpressing fibroblast growth factor 2 protein isoforms in osteoblastic ROS 17/2.8 cells. J Cell Biochem. 2003;89:1291–1301. doi: 10.1002/jcb.10589. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Takagi Y, Harada J, Teramoto T, Thomas SS, Waeber C, Bakowska JC, Breakefield XO, Moskowitz MA. FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc Natl Acad Sci U S A. 2001;98:5874–5879. doi: 10.1073/pnas.101034998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini S, Buzzi A, Bergamaschi M, Pietra C, Villetti G, Simonato M. Neuroprotective activity of CHF3381, a putative N-methyl-D-aspartate receptor antagonist. Neuroreport. 2002;13:2071–2074. doi: 10.1097/00001756-200211150-00016. [DOI] [PubMed] [Google Scholar]