

Significance
During bacterial infection, the eukaryotic innate immune system detects a restricted number of bacterial structures, such as LPSs, and activates signaling pathways conveying an inflammatory reaction aimed at eradication of the pathogen. Shigella spp. are human enteropathogens that invade colonic and rectal mucosa, where they cause deleterious inflammation. Here we show that Shigella drastically modifies the degree of acylation of the lipid A moiety of LPS during host cell invasion. The purified hypoacylated LPS displays a reduced inflammatory potential that allows the bacteria to lower the sensing activity of the immune system and to escape from downstream effector mechanisms.
Keywords: innate immunity, enteric pathogen, immune evasion, PAMPs/PRRs
Abstract
LPS is a potent bacterial effector triggering the activation of the innate immune system following binding with the complex CD14, myeloid differentiation protein 2, and Toll-like receptor 4. The LPS of the enteropathogen Shigella flexneri is a hexa-acylated isoform possessing an optimal inflammatory activity. Symptoms of shigellosis are produced by severe inflammation caused by the invasion process of Shigella in colonic and rectal mucosa. Here we addressed the question of the role played by the Shigella LPS in eliciting a dysregulated inflammatory response of the host. We unveil that (i) Shigella is able to modify the LPS composition, e.g., the lipid A and core domains, during proliferation within epithelial cells; (ii) the LPS of intracellular bacteria (iLPS) and that of bacteria grown in laboratory medium differ in the number of acyl chains in lipid A, with iLPS being the hypoacylated; (iii) the immunopotential of iLPS is dramatically lower than that of bacteria grown in laboratory medium; (iv) both LPS forms mainly signal through the Toll-like receptor 4/myeloid differentiation primary response gene 88 pathway; (v) iLPS down-regulates the inflammasome-mediated release of IL-1β in Shigella-infected macrophages; and (vi) iLPS exhibits a reduced capacity to prime polymorfonuclear cells for an oxidative burst. We propose a working model whereby the two forms of LPS might govern different steps of the invasive process of Shigella. In the first phases, the bacteria, decorated with hypoacylated LPS, are able to lower the immune system surveillance, whereas, in the late phases, shigellae harboring immunopotent LPS are fully recognized by the immune system, which can then successfully resolve the infection.
LPS is a glycolipid located in the outer membrane of Gram-negative bacteria. It is composed of three covalently linked domains: lipid A, which is embedded in the outer membrane; the oligosaccharide core; and the O-polysaccharide or O-antigen, which cover the bacterial surface. During infections sustained by Gram-negative bacteria, detection of LPS initiates an acute inflammatory response as LPS, mainly by the lipid A, which is the real pathogen-associated molecular pattern (PAMP), is sensed by the innate immune system, through the binding to the pattern recognition receptor (PRR) complex of myeloid differentiation protein 2 (MD-2) and Toll-like receptor (TLR) 4 (TLR4) (1–3). The downstream effects of LPS recognition elicit effector mechanisms aimed at pathogen eradication. However, LPS can also elicit an host reaction because it is a major mediator of pathologic processes (4). The strength of the innate immune response to LPS can be modulated by its chemical structure; specifically, a fine tuning of the lipid A structure can significantly affect the immunostimulatory properties of the whole LPS molecule (5, 6). There is a strong correlation between the number of acyl chains of lipid A and the immunological response via the TLR4 pathway. In general, hexaacylated lipid A species are agonists, whereas tetraacylated species are antagonists with a weak inflammatory potential (7). Gram-negative bacteria can synthesize a range of differentially acylated LPSs as a result of the LPS biosynthesis. Changes in lipid A acylation underlie the adaptation of pathogens to different hosts, such as Yersinia pestis (8), or to different phases of pathogenesis such as Salmonella typhimurium (9) or in the establishment of chronic infection such as Pseudomonas aeruginosa (10, 11).
Shigella flexneri is a Gram-negative pathogen that infects humans. The ingestion of as few as 100 bacteria is sufficient to cause bacillary dysentery, a severe rectocolitis caused by the dramatic inflammatory reaction induced by Shigella invasion on the colonic and rectal mucosa (12). Shigella enters epithelial cells by injecting effectors via a type III secretion system (T3SS) (13), escapes from the phagocytic vacuole, and actively proliferates within the cytosol of infected cells (14, 15). Bacterial proliferation is a potent signal to initiate inflammation because intracellular shigellae activate NF-κB following recognition of peptidoglycan (PGN) by the PRR Nod1, leading to IL-8 production (16, 17). IL-8 attracts neutrophils that are required for the clearance of shigellae, but also participates in epithelial barrier destruction (18). In macrophages, Shigella is able to trigger the assembly of the inflammasome, an important defense mechanism that is part of the innate immune system (19). The inflammasome is a multiprotein complex that mediates activation of caspase-1, which promotes the secretion of the proinflammatory cytokines IL-1β and IL-18 as well as a cell death process called pyroptosis (20, 21). Different PRRs, i.e., TLRs and nucleotide-binding oligomerization domain-like receptors (NLRs) contribute to the inflammasome assembly (22). In Shigella-infected macrophages, the activation of the NLRC4-mediated inflammasome triggers cell death and release of IL-1β and IL-18 (19, 23). Indeed, production of IL-1β is a paradigm of shigellosis: the chief role of this cytokine has been highlighted in vivo in several studies (24–26).
In tissues of animals and in ex vivo human samples infected with Shigella (27), a huge amount of LPS is usually observed, reflecting the presence of living bacteria and/or of processed molecules. However, whether, how, and at to what extent this mass of LPS present in Shigella-infected tissues could play a role in the inflammation remains largely unknown.
In 2002, D’Hauteville et al. reported that, in S. flexneri, the lack of msbB genes, msbB1 and msbB2, both encoding the enzyme myristoyl transferase, reduces lipid A acylation degree along with TNF-α production and epithelial lining inflammatory destruction in a rabbit model of Shigella infection (28, 29). This study suggests that LPS composition can greatly influence the degree of inflammation induced by Shigella.
In line with these issues, here we intend to contribute to the understanding of the role played by LPS in Shigella pathogenesis. Hence, we addressed the question of whether Shigella could adapt the LPS structure to the host thereby exploiting the mechanism of LPS modification to hijack the innate immune response. With this aim, we extracted, purified, and analyzed the LPS of shigellae resident in epithelial cells. We detailed the immunopotential of this structure and compared it to that of conventionally grown bacteria. Together our results point to a key role for LPS during the Shigella invasive process.
We report that (i) Shigella is able to modify the LPS composition, e.g., the lipid A and core domains, during proliferation within epithelial cells; (ii) the LPS of intracellular bacteria (iLPS) and that of bacteria conventionally grown (aLPS) differ in the number of acyl chains in lipid A, with iLPS being hypoacylated; (iii) the immunopotential of iLPS is dramatically lower than that of aLPS; (iv) both LPS forms signal mainly through the TLR4/MyD88 pathway; (v) iLPS influences the inflammasome-mediated production of IL-1β in Shigella-infected macrophages; and (vi) iLPS exhibits a reduced capacity to prime PMNs for an oxidative burst.
Results
Analysis of LPSs from Acellular and Intracellular Shigella.
During the invasive process, Shigella multiplies within the cytoplasm of the epithelial cells with a doubling time close to that of bacteria grown in laboratory medium (30). To acquire insights about eventual LPS modification during the residence of bacteria within this cell population, we developed an experimental protocol designed to recover shigellae from infected HeLa cells. At 3 h after infection, HeLa cells containing bacteria were processed to purify the iLPS. iLPS was extracted and analyzed by MALDI-TOF MS and compared with that derived by an equivalent number of bacteria grown under conventional conditions. In preliminary experiments, Shigella was cultured in cell medium, DMEM, to allow a comparison between iLPS and bacteria grown under the same medium as the host cells. However, Shigella cultured under these conditions was submitted to cell lysis making unrealistic to collect bacteria for LPS studies. Therefore, we abandoned this approach and performed an LPS analysis of bacteria grown under standard conditions, i.e., in trypticase soy broth (TSB) (31).
iLPS (in contrast to aLPS) did not exhibit dramatic modifications in the core oligosaccharide or the O-antigen. In fact, the core oligosaccharide region of iLPS possessed the same chemical structure already described by our group (31), the only difference being a phosphate group replacing a 2-amino-ethyl phosphate group linked to the heptose I (Fig. 1, species Oligosaccharide). The O-chain portion was characterized through NMR spectroscopy, and was constituted by the same O-repeating unit already found in other Shigella strains (Fig. S1 and Table S1) (32). On the contrary, the lipid A domain of iLPS exhibited a dramatic change in the acylation pattern (Fig. 1B); in fact, it was highly underacylated and mainly characterized by a very high percentage of tri- and tetraacylated lipid A species and a low amount of pentaacylated species, and only trace amounts of the canonical hexaacylated lipid A were present (Fig. 1B). It is worth noting that this information was achieved directly on the intact LPS molecule; in fact, the lipid A MS spectrum was directly obtained on the intact LPS upon in source fragmentation obtained in our optimized conditions (33); in this way, we can exclude any chemical change in lipid A as a result of chemical treatment. In aLPS, the canonical hexaacylated lipid A species was highly prevalent (Fig. 1B), whereas underacylated species were present in minor amounts. All relevant details of the purification procedures are reported in SI Materials and Methods.
Fig. 1.
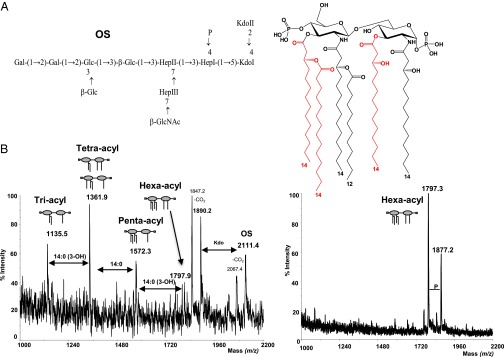
Acylation and glycosylation pattern of iLPS and aLPS of S. flexneri. (A) Structure of the Lipid A (Right) and of the core region (Left) produced by iLPS of S. flexneri. The lipid A is constituted by a mixture of species differing for the acylation pattern; red acyl chains are not stoichiometric. The core is formed by an undecasaccharide carrying a phosphate group on the first heptose residue. (B) Comparison between negative ion MALDI-TOF spectra from iLPS (Left) and aLPS (Right) shows change in the distribution of lipid A species. The relative intensity of the peaks related to the lipid A clearly shows the abundance of species with a low acylation pattern in the iLPS and the presence of a single heptaacylated species in the aLPS and a minor pyrophosphorylated form. In the MALDI TOF mass spectrum of the iLPS, the ion peaks from the core oligosaccharide are also indicated.
The absence of contaminants during the purification steps was assessed through several analyses as GLC-MS (SI Materials and Methods and Fig. S2), showing the presence of sugar residues exclusively ascribable to the LPS moiety.
Activation of NF-κB by LPS Extracted from Acellular and Intracellular Shigellae Correlates with the Degree of Lipid A Acylation.
To ascertain the absence of contamination in the purified LPS, iLPS and aLPS were analyzed in various biological assays (as reported in Fig. S3). The LPS were first analyzed for the presence of bacterial lipoproteins (BLPs; Fig. S3A) and PGN (Fig. S3 B and C) in HEK 293 cells expressing TLR2 (i.e., BLPs) or hNod1 or hNod2 (i.e., PGN) (34). Then, LPSs were assessed in bone marrow-derived macrophages (BMDMs) in the presence or absence of Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (OxPAPC)C (40 μg/mL; Fig. S3D), which inhibits the signaling induced by the bacterial LPS (35).
Changes in the degree of lipid A acylation has a dramatic effect on the binding of this molecule to MD-2 and TLR4. The hexaacylated lipid A species has optimal inflammatory activity. In fact, in the MD-2 pocket, the presence of six acyl chains allows the phosphate groups of glucosamine to interact with positively charged residues of TLR4, thus promoting the dimerization and activation of the receptor complex (36).
Compared with the lipid A with six acyl chains, the lipid A with five lipid chains is ∼100-fold less active, and those with four lipid chains completely lack agonistic activity (37, 38).
Therefore, the biological activity of iLPS was assessed in vitro in the model of HEK 293 cells, which stably express human CD14, MD-2, and TLR4. HEK 293 human (h) TLR4 were exposed to different LPS concentrations (1 and 10 ng/mL). NF-κB activation was measured through the assessment of luciferase activity; il-8 mRNA and release was evaluated via quantitative real-time PCR (qPCR) and ELISA, respectively (SI Materials and Methods). Consistent with its chemical composition, iLPS induced lower NF-κB activation (P < 0.01; Fig. 2A, Upper) and il-8 mRNA production and release than did aLPS (Fig. 2A, Left). Several natural or synthetic hypoacylated lipid A structures have been reported to exhibit a high potential as antagonists for endotoxically active LPS (7). We assessed whether iLPS could interfere with TLR4-mediated signaling induced by hexaacylated lipid A. HEK 293 hTLR4 cells were preincubated 1 h with different amounts (1, 10, and 100 ng/mL) of iLPS or aLPS and then exposed to 100 ng/mL of hexaacyl Escherichia coli LPS for 4 h.
Fig. 2.

TLR4 engagement by iLPS and aLPS. Activation of NF-κB, iL-8 mRNA expression, and IL-8 production in HEK 293 hTLR4/MD2-CD14 stimulated with iLPS, aLPS, and E. coli LPS (A) and TLR4-mediated competition of iLPS and aLPS on E. coli LPS (B). (A) Fold of NF-κB activation upon stimulation of HEK 293 hTLR4/MD2-CD14 with 1 and 10 ng/mL of LPS derived from intracellular shigellae (iLPS) and shigellae grown in culture medium (aLPS) for 4 h. Commercial hexaacylated E. coli LPS was used as a control; qPCR of il-8 mRNA induction after stimulation with the three LPS forms for 4 h; and IL-8 secretion after stimulation for 18 h as described earlier. (B) Fold of NF-κB activation upon stimulation of HEK 293 hTLR4/MD2-CD14 for 1 h with 1, 10, and 100 ng/mL of iLPS or aLPS before exposure to E. coli LPS 100 ng/mL for 4 h. For activation of NF-κB and IL-8 production, mean values (±SEM) of three representative experiments are shown (*P < 0.05, **P < 0.01, and ***P < 0.001). For qPCR, results are normalized to the internal gapdh gene control and are presented as the ratio of gene expression between stimulated and unstimulated HEK 293 cells.
iLPS significantly antagonized hexaacyl LPS-dependent TLR4-mediated NF-κB activation at low concentrations (P < 0.001 at 1 and 10 ng/mL; Fig. 2B). The absence of antagonistic effect at high iLPS doses could like be explained by the increase in the iLPS blend of hexaacylated lipid A species in addition to tri- and tetraacylated forms. No antagonistic effect was observed with aLPS (Fig. 2B, Right).
LPS of Intracellular Shigella Modulates Cytokine Release in BMDMs.
We next proceeded to compare the ability of the two LPS forms to activate BMDMs for cytokine production (TNF-α, IL-1β, IL-6, KC, CXCL-10, CCL-5, IL-18, IFN-β, and IL-10). BMDMs were treated with 1 and 10 ng/mL of the two LPS forms as described earlier, and cytokine release was recorded at two time points, 6 h and 18 h (Fig. 3). Hexaacylated E. coli LPS was used in parallel as a control. First, for all three LPS forms (iLPS, aLPS, and E. coli LPS), we observed a low release of IL-1β and a barely detectable release of IL-18, especially at 6 h of stimulation. By comparing the two forms of Shigella LPS (aLPS and iLPS), we found that, at both time points, stimulation with iLPS (especially with 10 ng/mL) caused a dramatic reduction of the yield of cytokine release with respect to the corresponding amounts of aLPS. The only exception was CCL-5 (Fig. 3): the amount of this cytokine was the same with 10 ng/mL of all three LPS forms at 18 h.
Fig. 3.

Modulation of cytokine release in BMDMs stimulated with Shigella iLPS or aLPS. Cytokine release in BMDMs stimulated with Shigella iLPS, aLPS, and E. coli LPS: TNF-α, IL-1β, IL-6, KC, CXCL-10, CCL-5, and IL-18 released by BMDMs after stimulation with 1 and 10 ng/mL of LPS derived from intracellular shigellae (iLPS) and shigellae grown in TSB medium (aLPS) and E. coli LPS, measured by ELISA at 6 h (A) and 18 h (B). For TNF-α, IL-6, KC, CXCL-10, and CCL-5, mean values (±SEM) of three representative experiments are shown; for IL-1β, data shown are mean values (±SEM) of six representative experiments. Significant difference between iLPS-generated values and the corresponding aLPS values are indicated (iLPS vs. aLPS; *P < 0.05, **P < 0.01, and ***P < 0.001).
It must be stressed that TNF-α, which is considered the canonical readout measuring the proinflammatory potency of LPS, was undetectable with 1 ng/mL and barely detectable with 10 ng/mL of iLPS, with the latter values being significantly lower (P < 0.001) with respect to aLPS.
Because IFN-β production was undetectable (Fig. S4 A and B, Left), we proceeded to analyze the mRNA levels for this cytokine. The exposure of 1 ng/mL of all forms of LPS did not result in the expression of ifn-β mRNA; with 10 ng/mL, the expression of IFN-β was similar with aLPS and E. coli LPS, but reduced with iLPS (Fig. S4C).
MyD88 and TRIF Pathways Are Engaged by both Forms of Shigella LPS.
TLR4-mediated LPS signaling engages two pathways, one mediated by the adaptor protein MyD88 leading to the production of proinflammatory cytokines, and the other involving the proteins Mal (also called Tirap), TRIF, and TRAM allowing the expression of genes encoding type 1 of IFN (IFN-1) and IFN-associated genes (39). The first signaling pathway is activated by the TLR4 plasma membrane and is transmitted through the adaptor molecule MyD88. Then, TLR4 is internalized in the endosomal network in which a second signal pathway is triggered through TRIF and TRAM (40). The TRIF pathway leads to the activation of the transcription factor IFN regulatory factor-3 (IRF-3) (41), which regulates expression of IFN-1 and associated genes.
We therefore assessed whether Shigella LPS could engage both signaling pathways and whether the two LPS forms could selectively activate MyD88 or the TRIF-mediated circuit.
iLPS and the aLPS were used to stimulate BMDMs from myd88- and trif-defective mice by applying the same experimental scheme described earlier. In myd88−/− BMDMs, the production of TNF-α was totally abrogated after 6 h and 18 h exposure with all LPS forms tested (Shigella iLPS and aLPS and E. coli LPS; Fig. 4). In trif-defective macrophages, TNF-α was still produced, although with a significant reduction with respect to all conditions applied in WT BMDMs. However, the relative proportions and differences among the three LPS forms observed in WT BMDMs were maintained in trif−/− cells. The low amount of IL-1β observed following LPS stimulation in WT macrophages was abolished and reduced (at 18 h) in myd88−/− and trif−/− cells, respectively (Fig. 4). IL-6 yield was suppressed in both myd88−/− and trif−/− macrophages under all experimental conditions applied (Fig. 4). Likewise, KC was null in myd88−/− macrophages at both time points and reduced in trif−/− BMDMs (Fig. 4).
Fig. 4.

MyD88- and TRIF-mediated signaling is involved in cytokine production upon stimulation with Shigella iLPS and aLPS. WT, myd88−/−, or trif−/− BMDMs stimulated with Shigella iLPS, aLPS, and E. coli LPS at 6 h (A) and 18 h (B). (A and B) TNF-α, IL-1β, IL-6, KC, CXCL-10, and CCL-5 released by BMDMs after stimulation with 1 and 10 ng/mL of LPS purified by intracellular shigellae (iLPS), shigellae grown in TSB (aLPS), and E. coli LPS measured through ELISA at 6 h and 18 h, respectively. Mean values (±SEM) of three representative experiments are shown. Significant difference between values WT cells and the corresponding values in defective cells are indicated (WT vs. defective; *P < 0.05, **P < 0.01, and ***P < 0.001).
CXCL-10 has been described as being under the control of IRF-3 (42). The CXCL-10 release was similar in WT and myd88−/− macrophages at 18 h (Fig. 4). In trif-defective cells, the amount of CXCL-10 was reduced with all LPS tested compared with the corresponding data in WT cells. Production of CCL-5 was similar in WT and myd88−/− cells at both time points, whereas, in trif-defective cells, it was significantly reduced. Low mRNA for ifn-β was noticed particularly in trif−/− cells (Fig. S4C). In summary, both forms of LPS (iLPS and aLPS) seem to signal mainly via the MyD88 pathway with the involvement of TRIF-mediated signaling.
iLPS and aLPS Differently Influence the Macrophage Response to S. flexneri Infection.
The interaction of S. flexneri with macrophages is followed by the death of infected cells. It has been reported that this process mainly depends on a mechanism of pyroptosis involving the formation of the NLRC4-mediated inflammasome, caspase-1 activation, and IL-1β release (23). Macrophage death and subsequent release of IL-1β are hallmarks of Shigella infections and play a central role in eliciting the inflammatory reaction in the gut.
The inflammasome assembly requires a priming signal, often referred as “signal 1,” via TLRs or intracellular receptors, which is necessary to up-regulate the expression of certain inflammasome receptors and the substrate pro–IL-1β. Then, a “signal 2” can promote a complex formation (43) and events downstream of the inflammasome activation. Recently, various reports have proposed that two inflammasome platforms could coexist in infected cells. One, usually called “death complex,” leads to cell death whereby activation of caspase-1 does not necessarily involve its autoproteolytic cleavage. The other governs IL-1β production and release and requires caspase-1 processing (43). Given the relevance of these processes in the context of shigellosis, we decided to analyze whether and to what extent macrophage stimulation with iLPS or aLPS could affect macrophage death and IL-1β release induced by Shigella.
In preliminary experiments, C57BL/6 BMDMs were first stimulated with different amounts of LPS, as described earlier, and then infected with S. flexneri. We observed that priming the macrophage with 1 ng/mL of LPS (of both Shigella LPS forms and E. coli LPS) did not change responses upon Shigella infection compared with those obtained with untreated and infected cells. Consequently, we performed all of the following experiments mainly by using the optimal LPS concentration of 10 ng/mL. We analyzed parameters associated with the inflammasome in BMDMs stimulated with iLPS, aLPS, or E. coli LPS for 4 h and infected with M90T [multiplicity of infection (MOI) of 10] over 3 h.
First, we evaluated the rate of cell death and cytotoxicity through the analysis of propidium iodide (PI) staining and lactate dehydrogenase (LDH) release (Fig. 5A). Caspase-1 activation and caspase-1 maturation were analyzed through the fluorescent labeled inhibitor of caspases (FLICA) and Western blot approaches, respectively (Fig. 5B). No difference was observed in cell death, LDH release, and caspase-1 activation/recruitment in infected BMDMs that were previously unstimulated or stimulated with aLPS or iLPS. However, in cell lysates, the processing of caspase-1 was significantly reduced upon iLPS treatment with respect to aLPS, whereas the cleaved caspase-1 fragment (p10) was weakly detectable when the M90T-infected macrophages were not previously exposed to LPS (Fig. 5B). Likewise, in cell supernatants, the cleaved fragment of caspase-1 was weakly detectable following pretreatment of the M90T-infected macrophages with iLPS. These results seem to suggest that cell death is a process independent of LPS priming and involving caspase-1 recruitment, and that full cleavage of caspase-1 is not necessary to this process, in accordance with the hypothesis of the death complex.
Fig. 5.

Influence of the iLPS and aLPS on the Shigella-mediated inflammasome in BMDMs. (A) (Left) Representative cytofluorimetric output of PI analysis of unstimulated BMDMs or BMDMs pretreated with 10 ng/mL of iLPS or aLPS or E. coli LPS (4 h) and infected with M90T at an MOI of 10 (3 h). (Right) LDH results of BMDMs stimulated and infected as described earlier. (B) (Upper) Representative cytofluorimetric output of caspase-1 activation through the FLICA approach under conditions as described earlier (caspase-1–negative BMDMs in R1 quadrant; caspase-1–positive BMDMs in the R2 quadrant). (Lower) Immunoblot analysis of caspase-1 maturation in cell lysates and supernatants of M90T-infected BMDMs treated as described earlier. The blot is probed with the protein Hsp70 as a loading control. (C) (Upper) IL-1β (Left) and IL-18 (Right) release of Shigella-infected BMDMs measured through ELISA under conditions as described before. Mean values (±SEM) of three representative experiments are shown. Significant differences between iLPS-generated values and the corresponding aLPS values are indicated (iLPS vs. aLPS; **P < 0.01). (Lower Left) qPCR of il-1β mRNA following stimulation in BMDMs with 10 ng/mL LPS at 3 h as described earlier and in BMDMs unstimulated or pretreated with LPS (4 h) as described earlier and infected with M90T at an MOI of 10 (3 h). Results are normalized to the internal gapdh gene control and are presented as the ratio of gene expression between stimulated and unstimulated BMDMs. (Lower Right) Western immunoblot analysis of IL-1β maturation in cell lysates and supernatant of M90T-infected BMDMs treated as described earlier. The blot is probed with the protein Hsp70 as a loading control.
We proceeded to analyze the production of IL-1β under the conditions described earlier (stimulation plus infection). Stimulation with 10 ng/mL of iLPS produced a release of a significantly lower amount of IL-1β with respect to 10 ng/mL of aLPS (P < 0.001) and E. coli LPS (Fig. 5C, Left). IL-18 secretion (P < 0.001) also followed the trend observed for IL-1 β (Fig. 5C, Right).
To verify whether this result reflected a general limited ability of iLPS to stimulate M90T-infected macrophages to produce inflammatory cytokines, we also measured the release of TNF-α, IL-6, and KC. The trend observed for IL-1β was partially maintained for IL-6 but not for TNF-α and KC, for which the values obtained with iLPS were similar to those recorded with aLPS and E. coli LPS (Fig. S5). As observed in the experiments with LPS stimulation, both LPS-mediated pathways, MyD88 and TRIF, were involved in the signaling leading to the production of IL-1β following LPS stimulation and M90T infection (Fig. S6).
In cell lysates of C57BL/6, the low IL-1β release observed with 10 ng/mL of iLPS was associated with a reduced production of il-1β mRNA (Fig. 5C, Lower Left), also reflected in a level of mature IL-1β that was lower than that produced by aLPS (Fig. 5C, Lower Right).
NLRC4/Ipaf has been reported as the NLR protein mediating the inflammasome assembly in macrophages infected with Shigella (23). In accordance with these data, the secretion of IL-1β and IL-18 in nlrc4 defective BMDMs were abrogated under all conditions tested (Fig. S7).
In summarizing the results, it appears that treatment of iLPS did not affect the cell death rate of BMDMs with respect to the data obtained with aLPS or with untreated M90T-infected macrophages. Caspase-1 recruitment was similar under all conditions, whereas maturation was reduced when BMDMs were treated with iLPS compared with aLPS. Likewise, stimulation with 10 ng/mL of iLPS strongly reduced the expression, processing, and release of IL-1β with respect to aLPS.
iLPS Influences the Neutrophil Response.
Neutrophils play a pivotal, although somewhat complex, role during Shigella infections. Neutrophils are able to efficiently ingest and kill the bacterium, yet, paradoxically, the inflammation that is triggered by PMNs is also necessary for Shigella to colonize successfully. To determine how iLPS may influence neutrophil responses, we compared the ability of iLPS and aLPS to prime neutrophils. Human neutrophils were isolated and primed for 30 min with Shigella iLPS, aLPS, or LPS purified from S. typhimurium (Fig. 6). Neutrophils were then stimulated with 100 nM N-Formyl-Met-Leu-Phe (fMLF) to produce an oxidative burst. Compared with aLPS or S. typhimurium LPS, neutrophils primed with iLPS induced significantly lower levels of reactive oxygen species (ROS) production at all concentrations tested. In contrast, ROS production in neutrophils was similar after aLPS and S. typhimurium LPS priming. These data suggest that hypoacylation of Shigella LPS can reduce priming of NADPH oxidase in human neutrophils, giving the bacterium an additional mechanism to control inflammation.
Fig. 6.

iLPS fails to effectively prime human neutrophils. Human neutrophils were isolated and primed with Shigella aLPS, iLPS, or S. typhimurium S-form TLR-grade LPS at 100, 33, 11, 4, or 1 ng/mL. Neutrophils were then stimulated with 100 nM fMLF, and ROS levels were measured continuously for 15 min by chemiluminescence by using HPR and luminol. Peak ROS production occurred at 1 min 30 seconds after fMLF addition, and these data are the average of triplicate wells from this time point. Data are displayed as relative light units (RLU). The experiment was performed three times. Means and SDs from a representative experiment are shown. Significant differences between iLPS generated values and the corresponding aLPS values are indicated (*P < 0.05, **P < 0.01, and ***P < 0.001).
Discussion
In this study, we describe a powerful mechanism, based on changes in lipid A acylation, developed by Shigella, to dampen immune surveillance and to interfere with the processes of pathogen recognition and eradication. Our results show that Shigella remodels LPS during residence in epithelial cells by drastically reducing the acylation rate of lipid A and slightly modifying the inner core moiety. Shigella is an intracellular pathogen whose interaction with infected cells often results in the death of the majority of them. Macrophages are rapidly killed through different mechanisms (19, 44–46); likewise, dendritic cells quickly die upon Shigella exposure (47), whereas survival of neutrophils, following bacterial ingestion, is still unclear. Indeed, bacteria do not proliferate within these cell populations, and they likely survive for a while. Shigella-infected epithelial cells also activate death programs (27, 48, 49). However, at the same time, these cells initiate a prosurvival response (27, 50) allowing the bacteria to multiply and to colonize the microenvironment up until the death of the cells (51). Bacterial multiplication is necessary for LPS modifications, as modified LPS are exposed on the bacterial surface only upon bacterial growth. Therefore, LPS modifications of shigellae proliferating in HeLa cells might reproduce what occurs in infected epithelial cells during natural shigellosis. Within cytosol, bacteria could sense one or more host triggers that signal the need to lower the recognition of immune cells that are attracted to the site of infection via the activation of PRRs such as Nod1. Lipid A hypoacylation is a potent immune evasion strategy applied by various pathogens and resulting in dramatic consequences on the outcome of the infection. Y. pestis, the causative agent of plague, alters the degree of LPS acylation according to host temperature, producing hexaacylated lipid A at temperatures between 21 and 27 °C (flea temperature) and tetraacylated lipid A at 37 °C (host temperature) (8). Genetically modified Y. pestis harboring fully potent LPS is completely avirulent, suggesting that the evasion of LPS-TLR4–mediated inflammation is critical for Y. pestis virulence (52, 53). Very recently, two reports have highlighted in S. flexneri serotype two adaptation mechanisms involving LPS modifications and amount, respectively (54, 55). LPSs contribute to the capacity of Shigella to resist acid conditions, like those encountered in the stomach, with modifications in the lipid A region, which is added with a phosphoethanolamine residue to decrease the negative charge at the bacterial surface (54). Furthermore, LPS layer from Shigella grown at 30 °C appeared thicker than that of cells grown at 37 °C. The decreased amount of LPS grown at higher temperature could be a regulatory mechanism, likely to favor the assembly of the T3SS needle at 37 °C, which is a mechanism requiring a less abundant LPS coat (55).
D’Hauteville et al. (28) reported that WT S. flexneri serotype 5 (M90T, the same strain as in the present study) possessed a predominantly hexaacylated lipid A (93%). MALDI TOF/TOF MS performed on LPS of a S. flexneri 5 M90T revealed the presence of lipid A isoforms with the major component being the hexaacylated lipid A, with minor peaks being attributed to pentaacylated and tetraacylated species (33).
Two research groups dealt with S. flexneri 2a LPS analysis. The first report (56) described in S. flexneri 2a lipid A a mixture of species in which the pentaacyl component was the most abundant, whereas the second study (57) described the tetraacylated species as prevailing on the other forms. In lipid A of M90T (S. flexneri 5, the same strain as in the present study) lacking the two copies of msbB (msbB1 and msbB2) genes, both of which encode the enzyme myristoyl transferase, only pentaacylated (86%) and tetraacylated (14%) lipid A species were present, whereas the hexaacylated form was undetectable (28). Likewise, the LPS of intracellular shigellae is hypoacylated; it is composed of a blend in which the tetra- and triacylated forms are prevalent. As expected, Shigella hypoacylated iLPSs display a low ability to engage the TLR4-mediated signaling, and exhibit a TLR4-mediated antagonistic activity toward hexaacylated LPS, as already described for synthetic or natural hypoacylated LPS against E. coli hexaacylated LPS (58, 59). In accordance with data of TLR4 activation, in BMDMs, iLPS elicits a significantly lower amount of inflammatory cytokines such as TNF-α, KC, IL-6, Il-1β, CXCL-10, and CXCL-5, and of ifn-β mRNA, than aLPS. The LPS of M90T ΔmsbB1msbB2 also shows a low ability to signal through TLR4 and elicits poor cytokine release, approximately similar to that induced by the iLPS (Fig. S8). It must be stressed that the LPS purified from M90T ΔmsbB1msbB2 arises from a population of mutant bacteria definitely lacking myristoyl transferase functions, whereas iLPS derives from a WT bacterial population in which the genes involved in lipid A modifications are still present and, likely, not homogenously expressed/repressed. In LPS-TLR4 signaling, the MyD88–Mal pathway leads to early activation of NF-κB and release of several proinflammatory mediators, including TNF-α and IL-6 (41), whereas, in the MyD88-independent pathway, TRAM and TRIF mediate activation of IRF-3, release of IFN-1 and the late activation of NF-κB. We found that both Shigella LPS variants, iLPS and aLPS, mainly signal via MyD88-mediated pathway and that TRIF signaling is involved in early and late cytokine production. These results are consistent with the ability of this adaptor to interact with TRAF6, a component of the MyD88-dependent pathway that activates NF-κB and controls the expression of genes such as TNF-α and IL-6 (60).
The pivotal role of IL-1β in shigellosis and the key contribution of the inflammasome have been highlighted by various studies in vitro and in vivo (23–26). By using casp1−/− mice reconstituted with recombinant IL-1β and intranasally infected with Shigella, Sansonetti et al. (26) showed that the administration of IL-1β enhanced the inflammatory reaction. Likewise, in IL-1β KO animals, the severity of the bacterial infection was reduced. Usually, the inflammasome assembly needs two signals: the first signal is often provided by TLR upon PAMP or damage-associated molecular pattern activation, and the second one is based on triggers eliciting the activation of one or more NLRs (43). In Shigella-infected macrophages, the inflammasome assembly and IL-1β release is dependent on the activation of NLR NLRC4 through a bacterial trigger not yet identified, but likely belonging to T3SS (23, 61, 62). Here, we found that the activity of MyD88 and the downstream pathways are necessary for the release of IL-1β in infected macrophages. Likewise, in the absence of LPS stimulation, Shigella-infected BMDMs produce very low il-1β expression and IL-1β release (Fig. 5C). These findings suggest that LPS may play a key role as a “first” signal in triggering the inflammasome activation in Shigella-infected macrophages. This process could be extremely relevant under conditions of natural infection, when a huge amount of LPS arising from bacterial processing/lysis would act as a trigger for the immune cells committed to mounting an inflammasome-mediated response.
Priming of Shigella-infected macrophages with iLPS results in a limited yield of IL-1β mRNA and IL-1β cleaved protein associated with low levels of mature caspase-1. In contrast to these results, cell death seems to be independent of the presence of LPS and apparently minimally dependent on the cleavage of caspase-1. Conclusively, under our experimental conditions, the yield of IL-1β produced by Shigella-infected macrophages is strictly dependent on the LPS used as first signal for the inflammasome assembly.
The role of LPS in inducing subepithelial PMN emigration and enhancing PMN–epithelium interactions before and during subsequent Shigella-induced transepithelial migration was reported several years ago (63), and the contribution of this cell population in Shigella infection has been elucidated (64, 65). In addition to the influence of iLPS on the inflammasome efficiency, we found that Shigella hypoacylated LPS was poorly able to eradicate Shigella.
In conclusion, the mechanism of LPS modification of intracellular bacteria is likely to be defined by two phases of the invasion process of Shigella. In the initial phase, bacteria that have escaped from epithelial cells, and are thus decorated with hypoacylated lipid A, are less competent in alerting the immune system through the interaction of the poorly immunogenic form of LPS with TLR4 on different cell populations. This inefficient immune surveillance allows the bacteria to spread and to disseminate in the gut microenvironment. In contrast, in the late phase of infection, when the epithelial lining of the microenvironment initially invaded is destroyed, shigellae proliferating freely in tissues and equipped with fully immunopotent LPS reacquire the ability to stimulate the immune system effectively, which can successfully challenge the bacterial infection, but at the price of deleterious inflammation.
Materials and Methods
Bacterial Strains and Growth Conditions.
The S. flexneri 5a strains used are M90T streptomycin-resistant, and the derivative M90T transformed with the plasmid pILL1101 encoding an afimbrial adhesin of uropathogenic E. coli (Afa E) (66) M90T ΔmsbB1msbB2 has been described by D’Hauteville et al. (28). Bacteria were grown in TSB (BBL; Becton Dickinson) and maintained on agar (TSA). Streptomycin and spectinomycin were added to cultures at 100 mg/mL.
Recovery of LPS from Intracellular Bacteria and Preparation of Bacterial Cultures for LPS Isolation.
To collect and purify the LPS from intracellular Shigella, 2 g bacteria were necessary. To maximize the collection of intracellular bacteria, HeLa cells seeded in 120-mm-diameter plates were infected with S. flexneri M90T pILL1101 (66) at an MOI of 50. After 30 min of incubation at room temperature and an additional 30 min at 37 °C, the acellular bacteria were removed by extensive washing with PBS solution. A total of 10 mL of fresh medium containing gentamicin (60 μg/mL) was added to each plate, and infected cells were incubated at 37 °C for 2 h. At this time, after three washes with PBS solution, the cell monolayers were detached with trypsin and then subjected to sonication and enzymatic hydrolysis to recover intracellular LPS (SI Materials and Methods). Approximately 400–500 infections in 120-mm-diameter plates provided 20 g of infected HeLa cells, from which ∼2 g of intracellular bacteria were recovered. To isolate LPS of bacterial grown in TSB, M90T pILL1101 and M90T ΔmsbB1msbB2 pILL1101 were grown in in a shake flask culture of 4 L under constant aeration at 37 °C for 12 h. A preculture in the same medium was used to inoculate the flask. These conditions resulted in approximately 0.5 g⋅L−1 of dried cells. The culture was checked for purity at the end of the cell cycle. Bacterial culture was then centrifuged and briefly washed in PBS solution, and the pellet dried, yielding approximately 2 g of dried bacteria for LPS isolation and purification (SI Materials and Methods). The full experimental scheme (including the LPS analysis) was repeated twice.
HEK 293 hTLR4/CD14/MD2 Cell Culture, Transfection, and Stimulation.
Stably transfected HEK 293 hTLR4/MD2-CD14 cell lines (InvivoGen) were seeded into 96-well plates at the concentration of 3 × 105 cells per milliliter. For NF-κB studies, cells were transfected with Firefly luciferase reporter constructs, pGL3.ELAM.tk, and Renilla luciferase reporter plasmid, pRLTK, as published previously (34). HEK 293 hTLR4/MD2/CD14 were exposed to different concentrations of Shigella iLPS or aLPS or E. coli LPS (LPS-EB ultrapure; InvivoGen; 1 and 10 ng/mL) and stimulation was prolonged for 4 h or 18 h. For the competition assays, the cells were primed with Shigella iLPS (1, 10, and 100 ng/mL) for 1 h and then restimulated with 100 ng/mL of E. coli LPS (LPS-EB ultrapure; InvivoGen) for 4 h. To assess the absence of contamination (Fig. S3) in LPS preparations, HEK 293 hTLR2 and HEK 293 expressing hNod1 or hNod2 (all cell lines from InvivoGen) were stimulated (hTLR2) and transfected (hNod1 or Nod2) (34) with Shigella and E. coli LPS. Sonicated E. coli PGN (1 μg/mL) and L-Ala-γ-D-Glu-mDAP (TriDAP) (1 μg/mL) and MDP (1 μg/mL) were used as controls in HEK hNod1 and Nod2 studies, whereas Pam3CSK4 (InvivoGen) was used in hTLR2.
NF-κB–dependent luciferase activity was measured by using the Dual-Luciferase Reporter Assay System (Promega) as reported previously (34), and il-8 mRNA was analyzed through qPCR. IL-8 production was quantified after 18 h of stimulation by ELISA.
BMDM Culture and Infection and Stimulation Assays.
All animals were on pure C57BL/6 background and were maintained in a specific pathogen-free animal facility; all experiments were performed in accordance with the guidelines established in the Principles of Laboratory Animal Care (directive 86/609/EEC) and approved by the Italian Ministry of Health. BMDMs were derived from bone marrow cells collected from 5-wk-old WT C57BL/6 (Charles River Laboratories) (67), myd88−/−, trif−/−, ipaf (nlrc4)−/−, or tlr2−/− female mice (gifts from M. Rescigno, FIRC Institute of Molecular Oncology Foundation-European Istitute of Oncology Campus, Milan, Italy; F. Granucci, Università degli Studi di Milano–Bicocca, Milan, Italy; Mathias Chamaillard, Université Lille Nord de France–Institut Pasteur de Lille, Lille, France; and Anna Zumsteg, Max Planck Institute for Infection Biology).
Stimulation assays.
For stimulation assays, BMDMs were seeded into 24-well plates (5 × 105 cells per milliliter) and exposed to different concentrations of Shigella iLPS, aLPS, and commercial E. coli LPS (1 and 10 ng/mL), and stimulation was carried out for 6 and 18 h. Poly(I:C) (5 µg/mL; InvivoGen) was used as a control for the activation of the TRIF-mediated pathway. Cell supernatants were recovered and processed for ELISA. Pro il-1β and ifn-β mRNA were evaluated at 3 h. To evaluate the contribution of TLR4 to the release of cytokines in BMDMs, the LPS stimulation was also performed in the presence of OxPAPC inhibitor (InvivoGen; Fig. S3).
Infections.
For infections, BMDMs were primed for 4 h with Shigella iLPS or aLPS or E. coli LPS (10 ng/mL) and then infected with M90T at an MOI of 10. Infected BMDMs were incubated at 37 °C for 1 h, washed twice with 1× PBS solution, and treated with gentamicin (60 μg/mL) for 3 h. Supernatants were recovered for ELISA analysis, and the infected monolayers were processed for qPCR (pro–il-1β and ifn-β) and for the study of parameters associated with cell death. Unstimulated, infected BMDMs as described earlier and uninfected cells were processed in parallel and used as controls.
Neutrophil Isolation and ROS Measurements.
All human subjects are informed on admission that tissue samples may be used for research purposes, through an appropriate informed consent form approved by the Ethics Committee of the Max Planck Institute (Berlin). Human neutrophils were isolated from blood obtained from the blood bank in a protocol approved by the ethics committee of the Charité Hospital (Berlin, Germany). Neutrophils were purified by Histopaque/Percoll (68). After isolation, neutrophils were resuspended in Hanks balanced salt solution (Gibco) containing cations supplemented with 5% FCS and seeded at 7.5 × 104 neutrophils per well in a 96-well plate and allowed to rest for 30 min. Neutrophils were then primed for 30 min with aLPS, iLPS, or S. typhimurium S-form TLR-grade (Enzo Life Sciences). After priming, neutrophils were stimulated with 100 nM fMLF (Sigma). ROS formation was measured over time by chemiluminescence (69) by using 50 μM luminal and 1.2 U/mL HRP (Calbiochem). Chemiluminescence was detected by using a Victor Light 1420 counter (Perkin-Elmer), and data are displayed as relative light units.
Cell Death/Cytotoxicity Studies and Caspase Activity.
Cell death/cytotoxicity were assessed by using PI staining (Apoptosis Detection kit; BD Pharmingen) and LDH release (Cytotox96 Cytotoxicity Assay; Promega), according to the manufacturers’ instructions. Caspase-1 activity was evaluated through carboxyfluorescein FLICA Apoptosis Detection kit caspase assay (Immunochemistry Technologies). Cell death parameters and caspase-1 activity were analyzed by using a flow cytometric analysis on a FACSCalibur cytometer (Becton Dickinson). Data acquisition (104 events for each sample) was performed by using CellQuest software (Becton Dickinson). Analysis was performed with FlowJo software (TreeStar). LDH release was quantified on a LT4000 ELISA reader at a wavelength of 450 nm (Labtech International).
Western Blot Analysis.
Total protein extracts obtained through cell lysis and Western blot procedures were carried out as described (27). Protein extraction from cell culture supernatants was performed as reported (70). Monoclonal antibody to caspase-1 (IMG-50 28; Imgenex/Histo-Line Laboratories) was used to recognize full-length caspase. Caspase-1 and IL-1β processing was determined by using rabbit anti–mouse caspase-1 p10 antibody (sc514; Santa Cruz Biotechnology) and goat anti-mouse IL-1β antibody (401-NA; R&D Systems), respectively; mouse monoclonal anti-hsp70 (SMC-164 C/D; Stress Marq Biosciences) was used to ensure equal loading.
Statistical Analysis.
Data are reported as means ± SD, and the numbers of independent experiments are indicated in the legends of each of the figures. Statistical calculations and tests were performed by using the Student t test. A P value of 0.05 was considered statistically significant. A P value of 0.001 was considered extremely significant.
Supplementary Material
Acknowledgments
We thank Philippe Sansonetti for the gift of the strain M90T ΔmsbB1msbB2; Maria Rescigno, Elena Zagato, Francesca Granucci, and Mathias Chamaillard for providing the defective mice; Arturo Zychlinsky for useful discussions; and Alison Abbott for critical reading of the manuscript. I.P., L.L.-F., and G.N. were fellows of the Istituto Pasteur–Fondazione Cenci Bolognetti. This work was supported by grants from International Fondo per gli Investimenti della Ricerca di Base (FIRB). The research leading to these results has received funding from the European Union Seventh Framework Programme FP7/2007-2013 under Grant Agreement 261472-STOPENTERICS.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303641110/-/DCSupplemental.
References
- 1.Hoshino K, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752. [PubMed] [Google Scholar]
- 2.Shimazu R, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeda K, Akira S. Microbial recognition by Toll-like receptors. J Dermatol Sci. 2004;34(2):73–82. doi: 10.1016/j.jdermsci.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Schletter J, Heine H, Ulmer AJ, Rietschel ET. Molecular mechanisms of endotoxin activity. Arch Microbiol. 1995;164(6):383–389. doi: 10.1007/BF02529735. [DOI] [PubMed] [Google Scholar]
- 5.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Quinn PJ. Endotoxins: Lipopolysaccharides of gram-negative bacteria. Subcell Biochem. 2010;53:3–25. doi: 10.1007/978-90-481-9078-2_1. [DOI] [PubMed] [Google Scholar]
- 7.DeMarco ML, Woods RJ. From agonist to antagonist: Structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol Immunol. 2011;49(1-2):124–133. doi: 10.1016/j.molimm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahara K, Tsukano H, Watanabe H, Lindner B, Matsuura M. Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect Immun. 2002;70(8):4092–4098. doi: 10.1128/IAI.70.8.4092-4098.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo L, et al. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science. 1997;276(5310):250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 10.Pier GB. Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol. 2007;297(5):277–295. doi: 10.1016/j.ijmm.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cigana C, et al. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PloS One. 2009;4(12):e8439. doi: 10.1371/journal.pone.0008439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuPont HL, Levine MM, Hornick RB, Formal SB. Inoculum size in shigellosis and implications for expected mode of transmission. J Infect Dis. 1989;159(6):1126–1128. doi: 10.1093/infdis/159.6.1126. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa M, Handa Y, Ashida H, Suzuki M, Sasakawa C. The versatility of Shigella effectors. Nat Rev Microbiol. 2008;6(1):11–16. doi: 10.1038/nrmicro1814. [DOI] [PubMed] [Google Scholar]
- 14.Clerc PJ, Ryter A, Mounier J, Sansonetti PJ. Plasmid-mediated intracellular multiplication of Shigella flexneri. Ann Inst Pasteur Microbiol. 1986;137A(3):315–320. doi: 10.1016/s0769-2609(86)80041-2. [DOI] [PubMed] [Google Scholar]
- 15.Sansonetti PJ, Ryter A, Clerc P, Maurelli AT, Mounier J. Multiplication of Shigella flexneri within HeLa cells: Lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect Immun. 1986;51(2):461–469. doi: 10.1128/iai.51.2.461-469.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Philpott DJ, Yamaoka S, Israël A, Sansonetti PJ. Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000;165(2):903–914. doi: 10.4049/jimmunol.165.2.903. [DOI] [PubMed] [Google Scholar]
- 17.Girardin SE, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2(8):736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sansonetti PJ, Tran Van Nhieu G, Egile C. Rupture of the intestinal epithelial barrier and mucosal invasion by Shigella flexneri. Clin Infect Dis. 1999;28(3):466–475. doi: 10.1086/515150. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T, et al. A novel caspase-1/toll-like receptor 4-independent pathway of cell death induced by cytosolic Shigella in infected macrophages. J Biol Chem. 2005;280(14):14042–14050. doi: 10.1074/jbc.M414671200. [DOI] [PubMed] [Google Scholar]
- 20.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 21.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PloS Pathog. 2007;3(8):e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sansonetti PJ, Arondel J, Cavaillon JM, Huerre M. Role of interleukin-1 in the pathogenesis of experimental shigellosis. J Clin Invest. 1995;96(2):884–892. doi: 10.1172/JCI118135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arondel J, Singer M, Matsukawa A, Zychlinsky A, Sansonetti PJ. Increased interleukin-1 (IL-1) and imbalance between IL-1 and IL-1 receptor antagonist during acute inflammation in experimental Shigellosis. Infect Immun. 1999;67(11):6056–6066. doi: 10.1128/iai.67.11.6056-6066.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sansonetti PJ, et al. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12(5):581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 27.Lembo-Fazio L, et al. Gadd45α activity is the principal effector of Shigella mitochondria-dependent epithelial cell death in vitro and ex vivo. Cell Death Dis. 2011;2:e122. doi: 10.1038/cddis.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Hauteville H, et al. Two msbB genes encoding maximal acylation of lipid A are required for invasive Shigella flexneri to mediate inflammatory rupture and destruction of the intestinal epithelium. J Immunol. 2002;168(10):5240–5251. doi: 10.4049/jimmunol.168.10.5240. [DOI] [PubMed] [Google Scholar]
- 29.Ranallo RT, et al. Virulence, inflammatory potential, and adaptive immunity induced by Shigella flexneri msbB mutants. Infect Immun. 2010;78(1):400–412. doi: 10.1128/IAI.00533-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cersini A, Salvia AM, Bernardini ML. Intracellular multiplication and virulence of Shigella flexneri auxotrophic mutants. Infect Immun. 1998;66(2):549–557. doi: 10.1128/iai.66.2.549-557.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Castro C, Parrilli M, Holst O, Molinaro A. Microbe-associated molecular patterns in innate immunity: Extraction and chemical analysis of gram-negative bacterial lipopolysaccharides. Methods Enzymol. 2010;480:89–115. doi: 10.1016/S0076-6879(10)80005-9. [DOI] [PubMed] [Google Scholar]
- 32.Molinaro A, et al. Full structural characterization of Shigella flexneri M90T serotype 5 wild-type R-LPS and its delta galU mutant: Glycine residue location in the inner core of the lipopolysaccharide. Glycobiology. 2008;18(3):260–269. doi: 10.1093/glycob/cwm140. [DOI] [PubMed] [Google Scholar]
- 33.Sturiale L, et al. Reflectron MALDI TOF and MALDI TOF/TOF mass spectrometry reveal novel structural details of native lipooligosaccharides. J Mass Spectrom. 2011;46(11):1135–1142. doi: 10.1002/jms.2000. [DOI] [PubMed] [Google Scholar]
- 34.Nigro G, et al. Muramylpeptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol. 2008;10(3):682–695. doi: 10.1111/j.1462-5822.2007.01075.x. [DOI] [PubMed] [Google Scholar]
- 35.Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283(36):24748–24759. doi: 10.1074/jbc.M800352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 37.Kim HM, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130(5) issue 5:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 38.Ohto U, Fukase K, Miyake K, Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science. 2007;316(5831):1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 39.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125(5):943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 40.Zanoni I, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147(4):868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 42.Carrigan SO, et al. IFN regulatory factor 3 contributes to the host response during Pseudomonas aeruginosa lung infection in mice. J Immunol. 2010;185(6):3602–3609. doi: 10.4049/jimmunol.0903429. [DOI] [PubMed] [Google Scholar]
- 43.Broz P, Monack DM. Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev. 2011;243(1):174–190. doi: 10.1111/j.1600-065X.2011.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willingham SB, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2(3):147–159. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koterski JF, Nahvi M, Venkatesan MM, Haimovich B. Virulent Shigella flexneri causes damage to mitochondria and triggers necrosis in infected human monocyte-derived macrophages. Infect Immun. 2005;73(1):504–513. doi: 10.1128/IAI.73.1.504-513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nonaka T, Kuwae A, Sasakawa C, Imajoh-Ohmi S. Shigella flexneri YSH6000 induces two types of cell death, apoptosis and oncosis, in the differentiated human monoblastic cell line U937. FEMS Microbiol Lett. 1999;174(1):89–95. doi: 10.1111/j.1574-6968.1999.tb13553.x. [DOI] [PubMed] [Google Scholar]
- 47.Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity and interleukin-1beta processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol. 2002;32(5):1464–1471. doi: 10.1002/1521-4141(200205)32:5<1464::AID-IMMU1464>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 48.Carneiro LA, et al. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe. 2009;5(2):123–136. doi: 10.1016/j.chom.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Tattoli I, et al. Intracellular bacteriolysis triggers a massive apoptotic cell death in Shigella-infected epithelial cells. Microbes Infect. 2008;10(10-11):1114–1123. doi: 10.1016/j.micinf.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Clark CS, Maurelli AT. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect Immun. 2007;75(5):2531–2539. doi: 10.1128/IAI.01866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sansonetti PJ. [Molecular and cellular bases of Shigella flexneri virulence] Bull Acad Natl Med. 1991;175(6):803–809. French. [PubMed] [Google Scholar]
- 52.Montminy SW, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006;7(10):1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 53.Airhart CL, et al. Induction of innate immunity by lipid A mimetics increases survival from pneumonic plague. Microbiology. 2008;154(pt 7):2131–2138. doi: 10.1099/mic.0.2008/017566-0. [DOI] [PubMed] [Google Scholar]
- 54.Martinić M, Hoare A, Contreras I, Alvarez SA. Contribution of the lipopolysaccharide to resistance of Shigella flexneri 2a to extreme acidity. PLoS ONE. 2011;6(10):e25557. doi: 10.1371/journal.pone.0025557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niu C, et al. Analysis of Soluble protein complexes in Shigella flexneri reveals the influence of temperature on the amount of lipopolysaccharide. Mol Cell Proteomics. 2013;12(5):1250–1258. doi: 10.1074/mcp.M112.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan S, Reinhold VN. Detailed structural characterization of lipid A: Electrospray ionization coupled with tandem mass spectrometry. Anal Biochem. 1994;218(1):63–73. doi: 10.1006/abio.1994.1141. [DOI] [PubMed] [Google Scholar]
- 57.Rallabhandi P, et al. Differential activation of human TLR4 by Escherichia coli and Shigella flexneri 2a lipopolysaccharide: combined effects of lipid A acylation state and TLR4 polymorphisms on signaling. J Immunol. 2008;180(2):1139–1147. doi: 10.4049/jimmunol.180.2.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flad HD, Loppnow H, Rietschel ET, Ulmer AJ. Agonists and antagonists for lipopolysaccharide-induced cytokines. Immunobiology. 1993;187(3-5):303–316. doi: 10.1016/S0171-2985(11)80346-3. [DOI] [PubMed] [Google Scholar]
- 59.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato S, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171(8):4304–4310. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- 61.Miao EA, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. 2010;107(7):3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Senerovic L, et al. Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 2012;3(9):e384. doi: 10.1038/cddis.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beatty WL, Sansonetti PJ. Role of lipopolysaccharide in signaling to subepithelial polymorphonuclear leukocytes. Infect Immun. 1997;65(11):4395–4404. doi: 10.1128/iai.65.11.4395-4404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ingersoll MA, Zychlinsky A. ShiA abrogates the innate T-cell response to Shigella flexneri infection. Infect Immun. 2006;74(4):2317–2327. doi: 10.1128/IAI.74.4.2317-2327.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandic-Mulec I, Weiss J, Zychlinsky A. Shigella flexneri is trapped in polymorphonuclear leukocyte vacuoles and efficiently killed. Infect Immun. 1997;65(1):110–115. doi: 10.1128/iai.65.1.110-115.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garcia MI, Labigne A, Le Bouguenec C. Nucleotide sequence of the afimbrial-adhesin-encoding afa-3 gene cluster and its translocation via flanking IS1 insertion sequences. J Bacteriol. 1994;176(24):7601–7613. doi: 10.1128/jb.176.24.7601-7613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marim FM, Silveira TN, Lima DS, Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PloS One. 2010;5(12):e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ermert D, Zychlinsky A, Urban C. Fungal and bacterial killing by neutrophils. Methods Mol Biol. 2009;470:293–312. doi: 10.1007/978-1-59745-204-5_21. [DOI] [PubMed] [Google Scholar]
- 69.Liu L, Dahlgren C, Elwing H, Lundqvist H. A simple chemiluminescence assay for the determination of reactive oxygen species produced by human neutrophils. J Immunol Methods. 1996;192(1-2):173–178. doi: 10.1016/0022-1759(96)00049-x. [DOI] [PubMed] [Google Scholar]
- 70.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.