

Abstract
Defective responses to DNA single- or double-strand breaks can result in neurological disease, underscoring the critical importance of DNA repair for neural homeostasis. Human DNA repair-deficient syndromes are generally congenital, in which brain pathology reflects the consequences of developmentally incurred DNA damage. Although, it is unclear to what degree DNA strand-break repair defects in mature neural cells contributes to disease pathology. However, DNA single-strand breaks are a relatively common lesion which if not repaired can impact cells via interference with transcription. Thus, this lesion, and probably to a lesser extent DNA double strand breaks, may be particularly relevant to aging in the neural cell population. In this review we will examine the consequences of defective DNA strand break repair towards homeostasis in the brain. Further, we also consider the utility of mouse models as reagents to understand the connection between DNA strand breaks and aging in the brain.
Keywords: DNA damage, DNA repair, nervous system, aging, ATM, AOA1, SCAN1
INTRODUCTION
The mammalian nervous system is formed through continual cycles of proliferation, differentiation and maturation to generate the large number of cell types required for function (Jacobsen 1991). Although the human nervous system forms in a matter of months, neural tissues must be functional for decades of life, and the mature neurons bear the brunt of handling a lifetime of potential threats to the integrity of their DNA. Due to the substantial oxygen requirement for maintenance of CNS tissue, neurons must cope with oxidative and metabolic stress that can result in DNA strand breaks (Lombard et al. 2005; Barzilai 2007; Chen et al. 2007). Accordingly, neurons require efficient DNA strand-break surveillance and repair mechanisms to deal with these types of lesions. Human neurological syndromes resulting from defects in DNA repair highlight the importance of multiple repair pathways for maintaining homeostasis in the brain (Rolig and McKinnon 2000; McKinnon and Caldecott 2007; Subba Rao 2007). Hence, individuals who incur genetic mutations that inactivate these repair pathways show accelerated neuronal death, which can manifest as neurodegenerative disease. As most of these inherited syndromes are congenital, less is know about the effects of DNA repair deficiency during aging. Nonetheless, there are many studies reporting a link between aging and a decline in DNA repair activity (Intano et al. 2003; Lu et al. 2004; Vijg and Calder 2004; Imam et al. 2006; Gorbunova et al. 2007; Rutten et al. 2007; Wilson and Bohr 2007). However, the causal relationship between decreased DNA repair activity, increased mutations and an effect upon aging still has to be thoroughly evaluated. Clearly, understanding the role of DNA repair and aging in the brain will require suitable model systems and careful in vivo assessment of how the spatiotemporal changes in DNA repair capacity affects neural homeostasis.
This review will emphasize the requirement for DNA strand-break repair in the context of neural development. We will consider neurodegenerative diseases that are directly attributable to failure in strand-break responses and the importance of these pathways in the nervous system. We will also discuss the utility of mouse models of DNA repair deficiency as an important tool for understanding the impact of genomic instability in the brain, and how more refined genetic manipulation of the mouse will help us better understand the links between DNA repair deficiency and age-related disease of the CNS.
Neural development
Formation of mature neural tissue requires the expansion and differentiation of precursor cells into a variety of neural cell types that migrate, organize and stratify into distinct CNS structures. Figure 1 illustrates the CNS with expanded views of two representative tissues, the retina and cerebellum, illustrating the laminar structure of these tissues. In many DNA repair syndromes the cerebellum is often a target, and as the cerebellum is responsible for motor coordination, ataxia is associated with these syndromes (Frappart and McKinnon 2006; Lee and McKinnon 2007). Although the cerebellum only comprises 10% of the total brain mass, it contains approximately 50% of the neurons in the brain. The cerebellum is primarily composed of three general neuronal populations, the granule cells, the Purkinje cells and interneurons, with each type found in distinct cell layers; the inner granule layer, the Purkinje cell layer and interneurons which are found throughout the molecular layer and the inner granule layer (Fig. 1). During development, an external germinal layer is present and as cerebellar development progresses, granule neuron precursors generate granule cells that migrate inwards and populate the IGL as the external granule layer gradually diminishes (Goldowitz and Hamre 1998; Wang and Zoghbi 2001). The cell populations of the cerebellum are notable because granule cells are the most numerous neuronal cell type in the brain, while Purkinje cells are amongst the largest neuronal cell type in the brain. The outer molecular layer consists of interneurons (stellate and basket cells) together with Purkinje cell dendrites and parallel fibers arising from granule cells, making the molecular layer a synapse-rich area (Jacobsen 1991; Goldowitz and Hamre 1998). As the cerebellum serves primarily to control sensory-motor function, individuals with cerebellar neurodegenerative disorders, such as spinocerebellar atrophy and ataxia-telangiectasia, present with ataxia (impaired motor coordination) and eye-movement defects and speech disturbance (dysarthria) (Frappart and McKinnon 2006; Limperopoulos and du Plessis 2006).
Figure 1. The adult mammalian CNS.
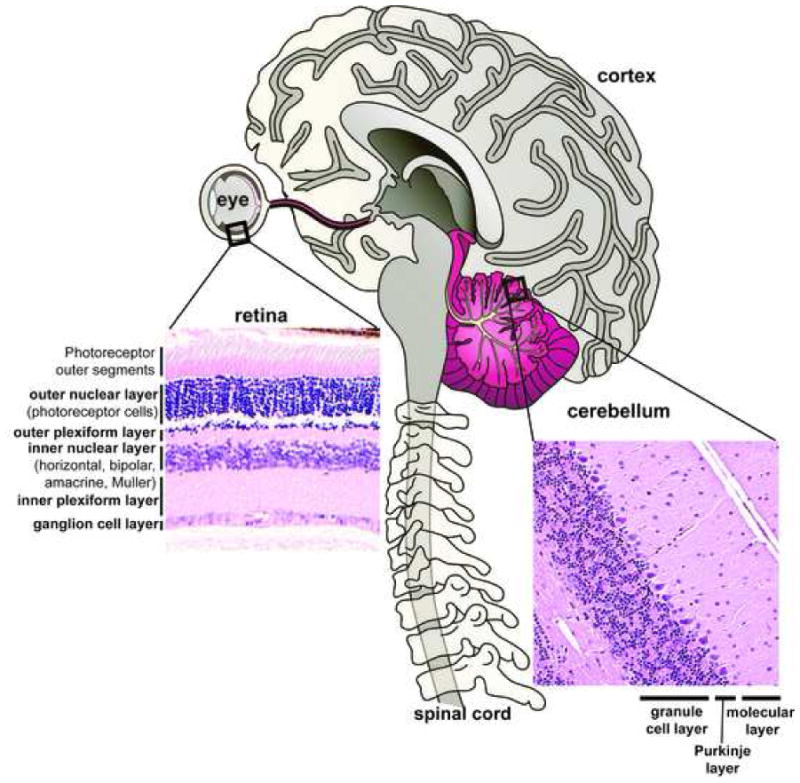
The mature nervous system contains a myriad of different cell types and tissues. DNA repair processes impact substantially during neural development leading to defective neurogenesis and development. However, less is known regarding the requirement for DNA repair processes in mature neural cell. Inset panels are hematoxylin and eosin stained retinal and cerebellar sections that show cell organization in these tissues. The retina is laminar in nature and cell-types are stratified into three distinct nuclear layers: outer, inner and ganglion. The outer nuclear layer contains the photoreceptor (rods and cones) neurons. The inner nuclear layer contains various signal processing cell types: bipolar, horizontal, amacrine, interplexiform and the Müller glia. The ganglion cells carry the visual signal via its axons through the optic nerve and project onto the brain. The cerebellum is stratified into three primary layers: inner granule cell layer, the Purkinje cell layer and the molecular layer. Excitory sensory signals originating from the cerebellum are ultimately transmitted through granule-Purkinje synapses and out of the cerebellum through Purkinje neuron axons to affect normal control of movement.
While the cerebellum is often affected in diseases associated with DNA strand break repair, progressive widespread neurodegeneration also occurs. In many cases these progressive changes are a later event than the effects upon the cerebellum. The likely scenario is that the cerebellum and perhaps granule cells in particular are very susceptible during postnatal neurogenesis to DNA damage. Furthermore, as defects in the cerebellum generally present as ataxia, an obvious movement disorder, this may cause milder cortical defects to be initially overlooked. As will be discussed later, mice with defective DNA strand break repair further reveal the cerebellum as a primary lesion in the nervous system.
Repair of DNA strand breaks
DNA strand breaks can occur as either single-strand breaks (SSBs) or double-strand breaks (DSBs) and biochemically distinct pathways repair these lesions. DNA DSBs are repaired by either non-homologous end-joining (NHEJ) or homologous recombination (HR), while SSBs are repaired by the DNA SSB repair (SSBR) pathway (Caldecott 2003; Lieber et al. 2003; West 2003; Wyman and Kanaar 2006; Helleday et al. 2007). In the nervous system DNA strand breaks can arise endogenously from normal cellular metabolism, during DNA replication or from exogenous agents such as ionizing radiation or chemicals in the environment. In differentiated neurons that do not divide, DNA DSBs clearly occur independently of replication and may result from the formation of adjacent SSBs via oxidative stress.
The choice between HR and NHEJ is related to the availability of a sister chromatid with which to use as an intact template for repair. Therefore, HR is restricted to replicating cells, and in the nervous system these are the progenitor populations that give rise to neurons and glia, and are found in the proliferative ventricular zones. Thus, HR promotes error-free DNA DSBR and is the major DNA repair pathway utilized during early mammalian development, particularly in tissues that give rise to the nervous system (Orii et al. 2006). A large recombinase complex (composed of RAD50, RAD51, RAD54, XRCC2 and XRCC3) is recruited to the DNA break site in a BRCA2-dependent manner to mediate HR (West 2003; Wyman and Kanaar 2006; Helleday et al. 2007). The requirement for an undamaged sister-chromatid restricts usage of HR to the S and G2 phase of the cell cycle. In contrast, NHEJ utilizes end-processing enzymes to facilitate ligation of incompatible DNA ends. Major proteins involved in NHEJ include the Ku heterodimer (Ku70/80), DNA protein kinase (DNA-PKcs) and the XRCC4/LigaseIV/XLF complex (Lees-Miller and Meek 2003; Lieber et al. 2003; Wyman and Kanaar 2006; van Gent and van der Burg 2007). The Ku complex binds to DNA ends and recruits the DNA-PKcs protein kinase to facilitate efficient DNA ligation by XRCC4/LigIV/XLF. In differentiating and post-mitotic neurons, the repair of DSBs occurs via NHEJ (Orii et al. 2006).
Coincident with DNA repair is the activation of DNA damage-induced signaling pathways that serve to halt the cell cycle while DNA repair occurs, or alternatively to activate apoptosis where the damage is extensive (Shiloh 2001; Kastan and Bartek 2004; Ward and Chen 2004). Amongst the earliest signaling events resulting from DNA DSBs is the phosphorylation of histone H2AX (Sedelnikova et al. 2003). This event serves to retain DSB sensing factors at the DNA break sites, thereby promoting efficient DNA repair. Recruitment of the DNA damage sensing Mre11-Rad50-Nbs1 (MRN) complex to the DSBs is important for activating the apical signaling kinase ATM, which effects cell cycle arrest or apoptosis (Carson et al. 2003; Petrini and Stracker 2003; Uziel et al. 2003; Difilippantonio et al. 2005). In the developing nervous system the activation of apoptosis after genotoxic stress is often a preferred course as cell replacement can readily occur from widespread germinal zones throughout the embryonic nervous system and at some postnatal stages such as the maturing cerebellum (Lee and McKinnon 2007). However, defects in many components of the DNA DSB repair machinery can lead to neuropathology (see later).
In contrast to DNA DSBs, a more common strand break is a DNA SSB. Tens of thousands of DNA SSBs are estimated to occur daily within a cell due to oxidative stress, and it is this type of lesion that may well underpin much of the source of DNA damage in the brain (Caldecott 2003; Rass et al. 2007). Mammals are oxygen-consuming organisms, and reactive oxygen species (ROS) occur via normal cellular metabolic processes. ROS production represents the single greatest genotoxin to neurons as ROS-mediated attack of DNA results in direct DNA SSBs. Resolution of DNA SSBs utilizes a distinct DNA SSBR pathway (Caldecott 2003; McKinnon and Caldecott 2007). Detection of SSBs is mediated by poly(ADP-ribose) polymerase (PARP) which acts to recruit the XRCC1 scaffolding protein accompanied with a variety of enzymes involved in DNA processing (TDP1, APTX, APE1, PKNP), gap-filling (DNA polymerase β) and ligation (ligase III) (McKinnon and Caldecott 2007; Wilson and Bohr 2007). Depending on the chemistry of the nucleophilic attack on the DNA backbone, DNA SSBs incur a multitude of 3′- and 5′- end-modifications, which must be processed by one or many end-processing enzymes prior to DNA, nick resealing (McKinnon and Caldecott 2007; Wilson 2007; Wilson and Mattson 2007). These processing enzymes are important in neural homeostasis as various neurodegenerative syndromes arise when germline mutations inactivate these enzymes. The inability to properly repair or process a SSB can lead to a variety of genotoxic consequences, including interference with DNA transcriptional machinery, formation of a DNA DSB upon encounter with the DNA replication machinery, or formation of either DNA SSB or DSB as a product of abortive Top1-DNA cleavage intermediates during DNA replication (Saxowsky and Doetsch 2006; el-Khamisy and Caldecott 2007; Katyal and McKinnon 2007). Notably, SSBs can be converted to DSBs in replicating cells via collision with replication forks, where HR is available as an adjunct to SSBR. However, such back-up mechanisms do not exist in post-mitotic neurons, and so the ability of these cells to repair SSBs will be very important for their survival (El-Khamisy et al. 2005; Katyal and McKinnon 2007).
Neurodegenerative disease and DNA strand break repair
Specific neuropathology is often present in syndromes involving DNA repair deficiency, highlighting the essential need for responding to genotoxic stress in the nervous system. As mentioned, the cerebellum is generally susceptible early in disease progression to the effects of DNA repair deficiency. This relative sensitivity may reflect the tremendous levels of neurogenesis that occur postnatally in this organ. Furthermore, the fact that neurons are post-mitotic, have critical higher-order functions and have demanding oxygen requirements makes them particularly sensitive to deficiency in resolving genotoxic stress. This section will consider the basis for the neurological defects in individuals with lesions associated with sensing or repairing DNA strand breaks. A representative list of syndromes associated with defective DNA strand break repair is presented in Table 1.
Table 1.
DNA repair Syndromes resulting from DNA strand break deficiency.
| Disease/Syndrome | Gene | Neurological | Extra-neurological | Mouse mutant(s) | Phenotype | References |
|---|---|---|---|---|---|---|
| DNA DSB Sensing and Repair | ||||||
| Ataxia-telangiectasia | ATM | ataxia, neurodegeneration telangiectasia, dysarthria, |
immunological defects, malignancy, sterility |
Atm−/− | growth retardation, infertility, immunological defects, radiosensitivity, malignancy |
(Barlow et al. 1996) (Elson et al. 1996) (Xu and Baltimore 1996) (Herzog et al. 1998) (Spring et al. 2001) (Borghesani et al. 2000) |
| Seckel syndrome |
ATR PCNT |
microcephaly, mental retardation | growth defects, abnormal facial features |
Atr−/− none reported |
embryonic lethal (<E7.5) | (Brown and Baltimore 2003) |
| Nijmegen breakage syndrome | NBS1 | microcephaly | immunological defects, lymphoid malignancy |
Nbs1−/− Nbs1ΔB/ΔB Nbs1ins-6/Δ6 Tg-Mx-Cre Nbs1Δ6/Δ6 nestin-Cre Nbs1ΔC/ΔC |
embryonic lethal (<E7.5) immunological defects, lymphoid malignancy defects in lymphogenesis, marrow, spleen, thymus ataxia, cerebellar and growth defects, microcephaly defective apoptotic response after IR |
(Zhu et al. 2001) (Williams et al. 2002) (Demuth et al. 2004) (Frappart et al. 2005) (Stracker et al. 2007) (Difilippantonio et al. 2005) |
| Ataxia-telangiectasia-like disorder | MRE11 | ataxia, neurodegeneration dysarthria, oculomotor apraxia | mild immunological defects |
Mre11−/− Mre11ATLD/ATLD |
embryonic lethal reduced embryonic viability |
(Xiao and Weaver 1997) (Theunissen et al. 2003) |
| LIG4 syndrome | LIG4 | microcephaly | immunodeficiency, lymphoma, developmental/growth delay |
LIG4−/− | embryonic lethal (<E16.5): neuronal apoptosis and defective lymphogenesis |
(Frank et al. 2000) (Barnes et al. 1998) |
| Fanconi anemia | BRCA2 | medulloblastoma | bone marrow and congenital defects | Brca2loxP/loxP nestin-Cre | defects in neurogenesis, tumor prone | (Frappart et al. 2007) |
| Human Immunodeficiency with microcephaly | XLF | microcephaly | immunological defects, infection developmental/growth delay |
Xlf−/− ES cells only none reported |
genomic instability, chromosomal translocations | (Zha et al. 2007) |
| DNA SSB Repair | ||||||
| Spinocerebellar ataxia with axonal neuropathy | TDP1 | ataxia, neurodegeneration muscle weakness |
hypercholesterolemia, hypoalbuminemia |
Tdp1−/− | mild cerebellar atrophy, hematopoeitic/intestinal sensitivity to topoisomerase-1 inhibitors (Topotecan) |
(Katyal et al. 2007) (Hirano et al. 2007) |
| Ataxia with oculomotor apraxia 1 | APTX | ataxia, neurodegeneration oculomotor apraxia |
hypercholesterolemia, hypoalbuminemia |
Aptx−/− | Defective resolution of 5′-end abortive ligation intermediates, murine analysis in progress |
(Ahel et al. 2006) |
Ataxia-telangiectasia and related diseases
Ataxia telangiectasia (A-T) is amongst the best studied of the human neurological syndromes arising from DNA damage response defects. A-T results from mutation of ATM (ataxia telangiectasia, mutated) a large nuclear phosphoinositol-3-kinase-related serine/threonine protein kinase (Shiloh 2003). DNA DSBs activate ATM resulting in a multi-faceted signaling response involving chromatin remodeling, DNA repair, cell cycle checkpoint activation and initiation of apoptotic pathways (Shiloh 2003; Lavin and Kozlov 2007; Matsuoka et al. 2007). Individuals with A-T develop profound ataxia and are confined to a wheelchair before the first decade of life. Dysarthria, oculomotor apraxia, cerebellar atrophy and various extra-neurological features such as immunodeficiency, radiosensitivity and a predisposition to malignancy also feature in this disease (McKinnon 2004; Frappart and McKinnon 2006; McKinnon and Caldecott 2007). The DNA damage-response defects in A-T strongly affect the cerebellum as MRI and autopsy analysis reveal widespread loss of cerebellar Purkinje cell and granule neurons in A-T brains, that are subsequently accompanied by other cerebral and spinal defects (Frappart and McKinnon 2006).
Mutation of two members of the MRN complex have also been linked to human disease. Patients with hypomorphic mutation of Mre11 develop a syndrome called ataxia telangiectasia like syndrome (A-TLD) and show neurological symptoms similar to A-T including ataxia, oculomotor apraxia and dysarthria (Petrini and Stracker 2003; Taylor et al. 2004). In contrast, hypomorphic mutation of NBS1, lead to Nijmegen breakage syndrome (NBS) which is characterized by immunodeficiency and lymphoid malignancy and in the nervous system microcephaly rather than neurodegeneration is present (Weemaes et al. 1981; Demuth and Digweed 2007). MRE11 has DNA processing activity, while NBS1 contains specific protein interaction domains (BRCT and FHA) that mediate critical MRN interactions. Like A-T, cells from A-TLD and NBS patients show substantial radiosensitivity and increased genomic instability (Shiloh 1997; Kobayashi et al. 2004). Interestingly, symptoms pertaining to both A-TLD and NBS seem to make up the breadth of those found in A-T, confirming biochemical analyses that the MRN complex is important for ATM activity. Mre11 and Nbs1 mutations result in defective localization of MRN and ATM to DSBs, reduced ATM activation and defective ATM-dependent phosphorylation of downstream substrates (Carson et al. 2003; Petrini and Stracker 2003; Uziel et al. 2003; Kitagawa et al. 2004; Difilippantonio et al. 2005). However, most, if not all of the pronounced neurological phenotypes in A-T, A-TLD and NBS can be attributed to DSB processing defects during neural development. Further, it is also uncertain if the progressive decline of the nervous system in A-T solely reflects the consequence of a lack of this protein during development. As yet it’s quite unclear as to what extent ATM, MRE11 and NBS1 are functionally important in the mature brain or as the brain ages.
DNA repair deficiency and microcephaly
In addition to neurodegeneration are DNA repair syndromes that lead to congenital brain development abnormalities resulting in microcephaly (when the brain size is 2 standard deviations below normal). Some individuals with Seckel syndrome, caused by hypomorphic ATR (ATM and Rad3-related) mutations, display severe growth retardation, microcephaly, mental retardation and “bird-like” facial features (Goodship et al. 2000; O’Driscoll et al. 2004; O’Driscoll and Jeggo 2006). Like ATM, ATR is a large PI3K-like protein kinase involved in the DNA damage response (Shiloh 2001; Paulsen and Cimprich 2007). Although ATM and ATR have similar downstream substrates, ATR is primarily involved in monitoring DNA replication stress during cell proliferation, such as stalled replication forks (Paulsen and Cimprich 2007). Therefore, the functional requirement for ATR in terminally differentiated or aging neural cells is unclear. It is conceivable that after proliferation has ceased ATR is no longer important, with perhaps ATM performing genomic surveillance functions.
Mutation of the centrosomal protein, pericentrin, has also been recently linked to the pathogenesis of Seckel syndrome (Griffith et al. 2007). These patients, like those with ATR-related Seckel syndrome, also show pronounced microcephaly. Mutant pericentrin results in its mislocalization from the centrosome resulting in replication fork defects. Disruption of this structural chromatin protein also results in defective DNA repair responses including those dependent on ATR (Griffith et al. 2007).
Patients with hypomorphic mutations in the essential NHEJ protein LIG4 show microcephaly, developmental and growth delay and immunological defects including lymphoid malignancy (O’Driscoll et al. 2004). The severity of Lig4 syndrome correlates with the extent of residual LIG4 activity (Girard et al. 2004). Inactivation of mouse Lig4 results in early embryonic lethality associated with widespread neuronal apoptosis and defective lymphogenesis (Barnes et al. 1998; Gao et al. 1998). Additionally, mutation in another NHEJ factor cernunnos/XLF results in Human Immunodeficiency with Microcephaly (HIM), an autosomal recessive childhood disease characterized by microcephaly, developmental and growth delay, autoimmune defects and recurrent infection (Ahnesorg et al. 2006; Buck et al. 2006). XLF functions in NHEJ via association with the XRCC4/LIG4 complex (Andres et al. 2007; Li et al. 2008). Many other DNA repair deficient syndromes also show microcephaly including Cockayne’s syndrome in which transcription coupled repair is defective and also NBS described above, indicating this brain defect probably occurs as a result of cell loss during development from various types DNA damage.
While the above syndromes involve DNA repair or damage response factors, the syndromes are congenital and so the exact requirements for any of these factors during aging in the brain remain unclear. In fact one unresolved issue in hereditary DNA repair syndromes is determining the portion of the collective phenotype that is attributable to the DNA repair deficiency during aging. For example, as mentioned above, is the progressive neurodegeneration associated with A-T solely a result of developmentally incurred defects, or is there a component of aging involved; i.e. is the ATM pathway important in mature neural cells? Assessing the requirement for ATM and ATR in the mature nervous system will be important for understanding the relative roles ATM and ATR contribute to the DNA damage response and how this impacts neural homeostasis. Other DNA damage signaling factors such as TopBP1 (Kumagai et al. 2006) have also been implicated in modulating ATM and ATR function, and again it will be informative to determine how these factors modulate the DNA damage response in postmitotic cells.
Determining the specific requirements for the DNA damage response system during the life of neural cells is an area that warrants further attention. As discussed later, more refined animal models for DNA repair deficiency will be important for understanding the role of DNA DSB repair in mature neurons. In contrast however, DNA SSBs are likely to affect terminally differentiated neural cells as they occur at much greater frequency than DSBs and are likely to impact transcription (El-Khamisy et al. 2005; Saxowsky and Doetsch 2006; Katyal and McKinnon 2007; McKinnon and Caldecott 2007), perhaps pointing to a greater need for activity of this pathway in the aging neural population. Additionally, the potential interrelationships between DNA DSBR and SSBR (Clements et al. 2004) suggest a more complex scenario, and imply that cross-talk between pathways may also be important for neural homeostasis. In the following we consider the importance of DNA SSBR in the nervous system.
DNA SSB repair deficiency in the nervous system
The importance of DNA SSB repair in maintaining neural homeostasis has recently been highlighted by the identification of defective repair enzymes as the underlying cause for several neurodegenerative syndromes. In contrast to DNA DSBs, phenotypes associated with SSBR deficient syndromes are relatively specific to the nervous system. Two such diseases are spinocerebellar ataxia with axonal neuropathy (SCAN1) and ataxia with oculomotor apraxia (AOA1). These are inherited autosomal recessive syndromes with neurodegenerative phenotypes similar to A-T, including cerebellar atrophy and ataxia, dysarthria and oculomotor apraxia (only in AOA1) (Date et al. 2001; Moreira et al. 2001; Takashima et al. 2002). While SCAN1 is quite rare, AOA1 is among the most common autosomal recessive ataxic disorders in Japan and Portugal. AOA1 patients show loss of cerebellar Purkinje neurons, while MRI analysis of SCAN1 patients has shown reduced cerebellar size (Takashima et al. 2002; Sugawara et al. 2008). SCAN1 and AOA1 result from heritable mutations in the 3′-end processing enzyme tyrosyl DNA phosphodiesterase 1 (TDP1) and the 5′-end processing enzyme aprataxin (APTX), respectively (Date et al. 2001; Moreira et al. 2001; Takashima et al. 2002). TDP1 is involved in processing of a variety of damaged DNA ends, including 3′-phosphoglycolate, 3′-Top1 and other types of non-ligateable 3′-DNA ends generated after DNA oxidation, DNA replication or other genotoxic stress (El-Khamisy et al. 2005). APTX is a nucleotide hydrolase and studies using APTX−/− cells have shown a failure to cleave a 5′-adenylate intermediate prior to sealing the nick, implicating a role for APTX in processing abortive ligation intermediates (Ahel et al. 2006; Rass et al. 2007). While the ability of HR to provide a backup repair pathway in proliferative tissues can circumvent SSBR defects, the specific impact toward the nervous system in defective SSBR syndromes probably reflects the high oxygen consumption of this tissue and the associated increased levels of oxidative damage (El-Khamisy et al. 2005; Katyal and McKinnon 2007; Rass et al. 2007).
Retinitis Pigmentosa (RP)
Retinitis pigmentosa (RP) is a heterogeneous neurodegenerative condition resulting in a gradual loss of photoreceptor neurons, leading to progressive constriction of the visual field and night blindness. Many RP gene groups and forms exist: including autosomal recessive, autosomal dominant and X-linked forms. However, mutation of RP2 accounts for the second most frequent cause of X-linked RP (Sharon et al. 2003). RP2 shares homology to nucleotide diphosphate kinases and in vitro oligo nuclease studies implicate RP2 as a novel 3′-5′ exonuclease involved in SSBR. A recent study describes nuclear relocalization from the membrane of RP2 protein upon exposure to oxidative damage (Yoon et al. 2006). While a number of vision disorders are attributed to retinal degeneration including RP, age-related macular degeneration and cone-rod dystrophy, links to DNA repair deficiency have not been made in these diseases.
Utilizing mouse models to understand DNA damage in the nervous system
Human DNA repair syndromes reveal the requirement for DNA repair during neural development, although these are less revealing about repair requirements in the mature nervous system. Detailed analysis of these processes will depend upon suitable experimental systems that are amenable to experimental manipulation. Perhaps the most significant model system that will directly expand our understanding of the biology of DNA repair in the brain is the mouse. Important information has been obtained using germline disruption of DNA repair factors, and more recently tissue specific gene ablation has helped refine our understanding to a much greater degree (Friedberg and Meira 2006). However, there are also limitations to the ability of the mouse to recapitulate human neurodegenerative disease phenotypes. For example, while Atm−/− mice have been important for understanding many aspects of A-T, the substantial neuropathology present in humans is absent in Atm-null animals (Frappart and McKinnon 2006). The lack of discernable neuropathology suggests some fundamental differences between the response of the nervous system in mice and humans. It is not certain why the difference is pronounced in the nervous system, when in other physiological compartments there is equivalency after ATM loss between mouse and man. Similarly, while murine inactivation of either Mre11 or Nbs1 results in early embryonic lethality (Xiao and Weaver 1997; Zhu et al. 2001; Dumon-Jones et al. 2003), hypomorphic murine mutations that mimic the human gene mutation in these syndromes reflect the cellular and extra-neurological characteristics of the human disease, but not the neurological symptoms (Kang et al. 2002; Williams et al. 2002; Theunissen et al. 2003; Difilippantonio et al. 2005). Two potential reasons are that the mouse lifespan is insufficient to manifest age-accumulated stochastic lesions, or that the mouse nervous system is more resistant to DNA damage. Nonetheless, important lessons have been learnt from the mouse regarding ATM signaling in the nervous system. Atm−/− mice have established that ATM is required for the induction of neuronal apoptosis selectively in immature, postmitotic neural cells after DNA damage (Herzog et al. 1998), suggesting that progressive neurodegeneration in A-T results from faulty neurons remaining integrated in neural tissue.
Additionally, Tdp1−/− mice have been created in an effort to understand the role of TDP1 in preventing SCAN1. Cerebellar neurons from these mice have deficient DNA SSBR activity after damage induced by oxidative stress, ionizing radiation and Top1 inhibitors (Hirano et al. 2007; Katyal et al. 2007). Notably, in contrast to mouse models of DNA DSB response deficiency, Tdp1−/− mice exhibit late-onset cerebellar atrophy, presumably due to accumulation of DNA lesions as result of neuronal repair deficiency (Katyal et al. 2007). These mice are also extremely sensitive to Top1 inhibitors, particularly in hematopoietic and intestinal tissue.
These knockout mouse models highlight the potential differences in outcome of repair enzyme loss between mouse and human, underscoring relevant concerns in considering the mouse as a suitable model for studies of aging in the brain (Zahn et al. 2007). While this may be an issue for generating faithful disease models, the mouse is nonetheless the best currently available system for garnering a comparative understanding of the biological role of DNA repair pathways in the developing and mature brain. In particular, valuable data has been generated from mouse models in which inactivation of DNA repair factors show profound tissue-specific effects, particularly within the nervous system (Gao et al. 1998; Frappart et al. 2005; Orii et al. 2006; Frappart et al. 2007).
Refined mouse models to study DNA repair in the nervous system
The increasing availability of reagents for tissue specific gene inactivation has provided a more refined approach with which to selectively manipulate gene inactivation (Orban et al. 1992; Jonkers and Berns 2002). Tissue specific deletion of DNA repair genes in the nervous system has already provided valuable insights into the role of these factors during neural development (Garcia and Mills 2002; Gaveriaux-Ruff and Kieffer 2007). A standard approach to gene deletion in the nervous system via the Cre/LoxP system utilizes the Nestin promoter to drive the cre recombinase. Nestin is expressed at E10.5 a point at which neural development is commencing. As a result, gene deletion via nestin-driven Cre expression will result in gene deletion throughout CNS tissue (Bates et al. 1999). For example, while inactivation of Brca2 or Nbs1 leads to early embryonic lethality, selective ablation of these genes in the nervous system using Nestin-cre results in a viable animal, providing important insight into the respective roles of these genes in this tissue (Frappart et al. 2005; Frappart et al. 2007). A wide array of neural-specific Cre lines have been developed which exhibit Cre recombinase activation at specific stages of development of tissues (A comprehensive database is accessible at: http://nagy.mshri.on.ca/cre/index.php or www.jax.org). Depending on the spatiotemporal expression pattern of any given gene promoter, Cre recombinase activity can be pan-neuronal, CNS tissue- or cell-type specific. In addition, inducible tissue-specific Cre lines been developed to allow increased temporal control of Cre expression (Kuhn et al. 1995; Feil et al. 1996; St-Onge et al. 1996; Garcia and Mills 2002).
Perspective & Conclusion
Neurodegenerative syndromes highlight the importance of responding to DNA strand breaks in the nervous system. However, the precise requirement for DNA repair factors during aging of the brain is not necessarily revealed by the phenotype of human DNA repair syndromes. While progeria is associated with some DNA repair syndromes (e.g. Werner’s), it is not strongly associated with DNA strand break repair deficiency syndromes. DNA DSB syndromes appear to primarily affect developing tissues, and most if not all of the neurological phenotype can be attributed to defects associated with development. In comparison however, the DNA SSBR deficiency syndromes may reflect a greater link to the process of aging. In particular, the high frequency of this lesion and the ability of the damage to impact transcription point to this lesion as one that is likely to be important during aging, perhaps in a synergistic manner with other common age-related neurological diseases. The exact requirements for DNA DSBR and SSBR in the aging brain is not clear, but as outlined above mouse models will be extremely important for generating a basic understanding of how various repair pathways participate in cellular homeostasis in the brain.
A general reduction in DNA repair activity has been associated with brain aging in humans and rodents raising the possibility that this is a normal aspect of aging (Intano et al. 2003; Lu et al. 2004; Imam et al. 2006; Gorbunova et al. 2007; Rao 2007; Rutten et al. 2007). However, a correlative relationship between repair activity and aging doesn’t necessarily imply that an assayable decrease in enzyme activity would be associated with increased DNA lesions or accumulation of mutations. Interestingly, relative to the liver the brain was found to be resistant to accumulated DNA mutations (Dolle et al. 1997; Stuart et al. 2000).
Defective DNA repair is being increasingly linked with diseases with striking neuropathology that are associated with aging, such as Alzheimer’s and Parkinson’s disease (Nouspikel and Hanawalt 2003; Bender et al. 2006; Hegde et al. 2006; Kraytsberg et al. 2006; Shackelford 2006; Weissman et al. 2007). Studies of DNA repair from brains of Alzheimer’s individuals have shown reduced DNA DSBR during NHEJ that was attributed to reduced DNA-PKCS levels, while protein levels of the MRN complex have also been reported to be diminished (Jacobsen et al. 2004; Shackelford 2006). Reduced base excision repair capacity was also found in Alzheimer’s disease brains compared to age-matched controls (Weissman et al. 2007). In Parkinson’s disease (PD) patients, increased DNA damage was also found in the mitochondria in substantia nigra neurons (a neuronal population lost in PD), implying a relationship between PD and mitochondrial DNA damage (Bender et al. 2006; Kraytsberg et al. 2006; Reeve et al. 2008).
Other considerations for aging in the nervous system includes the requirement for DNA repair in neural stem cells, perhaps those that potentially contribute towards new neurogenesis in regions of the mature brain (Toni et al. 2007; Zhang et al. 2008). Certainly, recent data points to a strong requirement of DNA DSBR in some stem cell populations (Nijnik et al. 2007; Rossi et al. 2007). However, the functional role for stem cells in the aging brain is unclear, as is the requirement for new neurogenesis as a component of normal brain function.
While DNA repair syndromes highlight the importance of maintaining genomic integrity in the brain, the role of DNA damage in normal aging processes is less clear. As mentioned previously, reports documenting a loss of repair capacity with age suggest that DNA damage may be involved in aging, although many other reports find no clear defects in DNA repair associated with normal aging. Perhaps, assessment of other DNA damage parameters may be more informative for understanding the contribution of DNA damage to aging? For example, telomeres feature prominently in eliciting a DNA damage response when disrupted, and telomere erosion is a consequence of cellular proliferation (Verdun and Karlseder 2007). Telomeres are structures present at the end of chromosomes, and serve a critical function to ensure that DNA ends of chromosomes are not detected as DNA breaks (DePinho and Wong 2003; Blasco 2005). Intriguingly, telomere shortening has been found in cells from individuals experiencing emotional stresses often associated with aging, suggesting a strong environmental link to maintenance of genome integrity (Epel et al. 2004; Sapolsky 2004; Epel et al. 2006). However, the need for telomere maintenance in non-replicating tissue such as the nervous system or the potential impact of loss of function of telomere protection factors has not yet been carefully assessed in vivo. Many new insights to address these questions will be forthcoming as new mouse models of DNA repair deficiency are generated. These insights will be important guides as we plan therapeutic strategies to potentially alleviate neuropathology associated with DNA repair deficiency and aging.
Acknowledgments
We acknowledge research support from the National Institutes of Health (NIH) and the American Lebanese and Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital. We apologize to colleagues whose primary research references could not be cited due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443(7112):713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Molecular cell. 2007;28(6):1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86(1):159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8(25):1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- Barzilai A. The Contribution of the DNA Damage Response to Neuronal Viability. Antioxid Redox Signal. 2007 doi: 10.1089/ars.2007.9.211. [DOI] [PubMed] [Google Scholar]
- Bates B, Rios M, Trumpp A, Chen C, Fan G, Bishop JM, Jaenisch R. Neurotrophin-3 is required for proper cerebellar development. Nature neuroscience. 1999;2(2):115–117. doi: 10.1038/5669. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature genetics. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6(8):611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A. 2000;97(7):3336–3341. doi: 10.1073/pnas.050584897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17(5):615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. XRCC1 and DNA strand break repair. DNA repair. 2003;2(9):955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. The EMBO journal. 2003;22(24):6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lee HM, Greeley GH, Jr, Englander EW. Accumulation of oxidatively generated DNA damage in the brain: a mechanism of neurotoxicity. Free radical biology & medicine. 2007;42(3):385–393. doi: 10.1016/j.freeradbiomed.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA repair. 2004;3(11):1493–1502. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- Demuth I, Digweed M. The clinical manifestation of a defective response to DNA double-strand breaks as exemplified by Nijmegen breakage syndrome. Oncogene. 2007;26(56):7792–7798. doi: 10.1038/sj.onc.1210876. [DOI] [PubMed] [Google Scholar]
- Demuth I, Frappart PO, Hildebrand G, Melchers A, Lobitz S, Stockl L, Varon R, Herceg Z, Sperling K, Wang ZQ, Digweed M. An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum Mol Genet. 2004;13(20):2385–2397. doi: 10.1093/hmg/ddh278. [DOI] [PubMed] [Google Scholar]
- DePinho RA, Wong KK. The age of cancer: telomeres, checkpoints, and longevity. The Journal of clinical investigation. 2003;111(7):S9–14. [PubMed] [Google Scholar]
- Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, Manova K, Kruhlak M, Camerini-Otero RD, Sharan S, Nussenzweig M, Nussenzweig A. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005;7(7):675–685. doi: 10.1038/ncb1270. [DOI] [PubMed] [Google Scholar]
- Dolle ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nature genetics. 1997;17(4):431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- Dumon-Jones V, Frappart PO, Tong WM, Sajithlal G, Hulla W, Schmid G, Herceg Z, Digweed M, Wang ZQ. Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res. 2003;63(21):7263–7269. [PubMed] [Google Scholar]
- el-Khamisy SF, Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia with axonal neuropathy-1. Neuroscience. 2007;145(4):1260–1266. doi: 10.1016/j.neuroscience.2006.08.048. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434(7029):108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A. 1996;93(23):13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101(49):17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel ES, Lin J, Wilhelm FH, Wolkowitz OM, Cawthon R, Adler NE, Dolbier C, Mendes WB, Blackburn EH. Cell aging in relation to stress arousal and cardiovascular disease risk factors. Psychoneuroendocrinology. 2006;31(3):277–287. doi: 10.1016/j.psyneuen.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Molecular cell. 2000;5(6):993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Frappart PO, Lee Y, Lamont J, McKinnon PJ. BRCA2 is required for neurogenesis and suppression of medulloblastoma. The EMBO journal. 2007;26(11):2732–2742. doi: 10.1038/sj.emboj.7601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, McKinnon PJ. Ataxia-telangiectasia and related diseases. Neuromolecular Med. 2006;8(4):495–511. doi: 10.1385/NMM:8:4:495. [DOI] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med. 2005;11(5):538–544. doi: 10.1038/nm1228. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA repair. 2006;5(2):189–209. doi: 10.1016/j.dnarep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, Zhu C, Chaudhuri J, Davidson L, Ferrini R, Stamato T, Orkin SH, Greenberg ME, Alt FW. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95(7):891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Garcia EL, Mills AA. Getting around lethality with inducible Cre-mediated excision. Seminars in cell & developmental biology. 2002;13(2):151–158. doi: 10.1016/s1084-9521(02)00019-8. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Conditional gene targeting in the mouse nervous system: Insights into brain function and diseases. Pharmacology & therapeutics. 2007;113(3):619–634. doi: 10.1016/j.pharmthera.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13(20):2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends in neurosciences. 1998;21(9):375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Goodship J, Gill H, Carter J, Jackson A, Splitt M, Wright M. Autozygosity mapping of a seckel syndrome locus to chromosome 3q22. 1-q24. American journal of human genetics. 2000;67(2):498–503. doi: 10.1086/303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Mao Z, Hine C. Changes in DNA repair during aging. Nucleic Acids Res. 2007;35(22):7466–7474. doi: 10.1093/nar/gkm756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Sanna NA, Saggar A, Hamel B, Earnshaw WC, Jeggo PA, Jackson AP, O’Driscoll M. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nature genetics. 2007 doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, Subba Rao K, Jagannatha Rao KS. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Archives of biochemistry and biophysics. 2006;449(1–2):143–156. doi: 10.1016/j.abb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA repair. 2007;6(7):923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280(5366):1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, Northrop JL, Salih MA, Inoue K, Armstrong DL, Champoux JJ, Takashima H, Boerkoel CF. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? The EMBO journal. 2007;26(22):4732–4743. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27(8):1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Intano GW, Cho EJ, McMahan CA, Walter CA. Age-related base excision repair activity in mouse brain and liver nuclear extracts. The journals of gerontology. 2003;58(3):205–211. doi: 10.1093/gerona/58.3.b205. [DOI] [PubMed] [Google Scholar]
- Jacobsen E, Beach T, Shen Y, Li R, Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain research. 2004;128(1):1–7. doi: 10.1016/j.molbrainres.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Jacobsen M. Developmental Neurobiology. Plenum Press; New York: 1991. [Google Scholar]
- Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2(4):251–265. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. The EMBO journal. 2002;21(6):1447–1455. doi: 10.1093/emboj/21.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. The EMBO journal. 2007;26(22):4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, McKinnon PJ. DNA Repair Deficiency and Neurodegeneration. Cell Cycle. 2007;6(19) doi: 10.4161/cc.6.19.4757. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004:181423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J, Antoccia A, Tauchi H, Matsuura S, Komatsu K. NBS1 and its functional role in the DNA damage response. DNA repair. 2004;3(8–9):855–861. doi: 10.1016/j.dnarep.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature genetics. 2006;38(5):518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124(5):943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6(8):931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- Lee Y, McKinnon PJ. Responding to DNA double strand breaks in the nervous system. Neuroscience. 2007;145(4):1365–1374. doi: 10.1016/j.neuroscience.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie. 2003;85(11):1161–1173. doi: 10.1016/j.biochi.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Li Y, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, Ahnesorg P, Jackson SP, Blundell TL. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. The EMBO journal. 2008;27(1):290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nature reviews. 2003;4(9):712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- Limperopoulos C, du Plessis AJ. Disorders of cerebellar growth and development. Current opinion in pediatrics. 2006;18(6):621–627. doi: 10.1097/MOP.0b013e32801080e8. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429(6994):883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5(8):772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ, Caldecott KW. DNA Strand Break Repair and Human Genetic Disease. Annu Rev Genomics Hum Genet. 2007:837–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nature genetics. 2001;29(2):189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, Enver T, Bell JI, Slijepcevic P, Goodnow CC, Jeggo PA, Cornall RJ. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447(7145):686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer’s disease. Bioessays. 2003;25(2):168–173. doi: 10.1002/bies.10227. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA repair. 2004;3(8–9):1227–1235. doi: 10.1016/j.dnarep.2004.03.025. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7(1):45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci U S A. 1992;89(15):6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orii KE, Lee Y, Kondo N, McKinnon PJ. Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci U S A. 2006;103(26):10017–10022. doi: 10.1073/pnas.0602436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA repair. 2007;6(7):953–966. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13(9):458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- Rao KS. DNA repair in aging rat neurons. Neuroscience. 2007;145(4):1330–1340. doi: 10.1016/j.neuroscience.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130(6):991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Elson JL, Morris CM, Bender A, Lightowlers RN, Turnbull DM. Nature of mitochondrial DNA deletions in substantia nigra neurons. American journal of human genetics. 2008;82(1):228–235. doi: 10.1016/j.ajhg.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends in neurosciences. 2000;23(9):417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- Rutten BP, Schmitz C, Gerlach OH, Oyen HM, de Mesquita EB, Steinbusch HW, Korr H. The aging brain: accumulation of DNA damage or neuron loss? Neurobiol Aging. 2007;28(1):91–98. doi: 10.1016/j.neurobiolaging.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Organismal stress and telomeric aging: an unexpected connection. Proc Natl Acad Sci U S A. 2004;101(50):17323–17324. doi: 10.1073/pnas.0408041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxowsky TT, Doetsch PW. RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev. 2006;106(2):474–488. doi: 10.1021/cr040466q. [DOI] [PubMed] [Google Scholar]
- Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther. 2003;2(3):233–235. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- Shackelford DA. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol Aging. 2006;27(4):596–605. doi: 10.1016/j.neurobiolaging.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73(5):1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annual review of genetics. 1997:31635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11(1):71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3(3):155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Spring K, Cross S, Li C, Watters D, Ben-Senior L, Waring P, Ahangari F, Lu SL, Chen P, Misko I, Paterson C, Kay G, Smorodinsky NI, Shiloh Y, Lavin MF. Atm knock-in mice harboring an in-frame deletion corresponding to the human ATM 7636del9 common mutation exhibit a variant phenotype. Cancer Res. 2001;61(11):4561–4568. [PubMed] [Google Scholar]
- St-Onge L, Furth PA, Gruss P. Temporal control of the Cre recombinase in transgenic mice by a tetracycline responsive promoter. Nucleic Acids Res. 1996;24(19):3875–3877. doi: 10.1093/nar/24.19.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Morales M, Couto SS, Hussein H, Petrini JH. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature. 2007;447(7141):218–221. doi: 10.1038/nature05740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GR, Oda Y, de Boer JG, Glickman BW. Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice. Genetics. 2000;154(3):1291–1300. doi: 10.1093/genetics/154.3.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subba Rao K. Mechanisms of disease: DNA repair defects and neurological disease. Nature clinical practice. 2007;3(3):162–172. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- Sugawara M, Wada C, Okawa S, Kobayashi M, Sageshima M, Imota T, Toyoshima I. Purkinje cell loss in the cerebellar flocculus in patients with ataxia with ocular motor apraxia type 1/early-onset ataxia with ocular motor apraxia and hypoalbuminemia. Eur Neurol. 2008;59(1–2):18–23. doi: 10.1159/000109256. [DOI] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature genetics. 2002;32(2):267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA repair. 2004;3(8–9):1219–1225. doi: 10.1016/j.dnarep.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JH. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell. 2003;12(6):1511–1523. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
- Toni N, Teng EM, Bushong EA, Aimone JB, Zhao C, Consiglio A, van Praag H, Martone ME, Ellisman MH, Gage FH. Synapse formation on neurons born in the adult hippocampus. Nature neuroscience. 2007;10(6):727–734. doi: 10.1038/nn1908. [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. The EMBO journal. 2003;22(20):5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene. 2007;26(56):7731–7740. doi: 10.1038/sj.onc.1210871. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Karlseder J. Replication and protection of telomeres. Nature. 2007;447(7147):924–931. doi: 10.1038/nature05976. [DOI] [PubMed] [Google Scholar]
- Vijg J, Calder RB. Transcripts of aging. Trends Genet. 2004;20(6):221–224. doi: 10.1016/j.tig.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2(7):484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Ward I, Chen J. Early events in the DNA damage response. Current topics in developmental biology. 2004:631–35. doi: 10.1016/S0070-2153(04)63001-8. [DOI] [PubMed] [Google Scholar]
- Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand. 1981;70(4):557–564. doi: 10.1111/j.1651-2227.1981.tb05740.x. [DOI] [PubMed] [Google Scholar]
- Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35(16):5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC. Molecular views of recombination proteins and their control. Nature reviews. 2003;4(6):435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol. 2002;12(8):648–653. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- Wilson DM., 3rd Processing of nonconventional DNA strand break ends. Environmental and molecular mutagenesis. 2007;48(9):772–782. doi: 10.1002/em.20346. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA repair. 2007;6(4):544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, Mattson MP. Neurodegeneration: nicked to death. Curr Biol. 2007;17(2):R55–58. doi: 10.1016/j.cub.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annual review of genetics. 2006:40363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25(15):2985–2991. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev. 1996;10(19):2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Qiu J, Cai S, Chen Y, Cheetham ME, Shen B, Pfeifer GP. The retinitis pigmentosa-mutated RP2 protein exhibits exonuclease activity and translocates to the nucleus in response to DNA damage. Exp Cell Res. 2006;312(8):1323–1334. doi: 10.1016/j.yexcr.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, Lakatta EG, Boheler KR, Xu X, Mattson MP, Falco G, Ko MS, Schlessinger D, Firman J, Kummerfeld SK, Wood WH, 3rd, Zonderman AB, Kim SK, Becker KG. AGEMAP: a gene expression database for aging in mice. PLoS genetics. 2007;3(11):e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Alt FW, Cheng HL, Brush JW, Li G. Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci U S A. 2007;104(11):4518–4523. doi: 10.1073/pnas.0611734104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Zou Y, He W, Gage FH, Evans RM. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008 doi: 10.1038/nature06562. [DOI] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11(2):105–109. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]