

Background: The mammalian TGF-β homologues have highly conserved heterologous motifs that are believed to impart isoform-specific functions.
Results: Exchange of TGF-β1 ligand for TGF-β3 in a mouse reveals functional redundancies/differences in cell signaling.
Conclusion: TGF-β1 and TGF-β3 are only partially interchangeable.
Significance: These findings have relevance for therapeutic approaches aimed at targeting the TGF-β pathway in disease conditions.
Keywords: Immunology, Inflammation, Metabolism, Transforming Growth Factor-β (TGFβ), Transgenic Mice
Abstract
Three homologues of TGF-β exist in mammals as follows: TGF-β1, TGF-β2, and TGF-β3. All three proteins share high homology in their amino acid sequence, yet each TGF-β isoform has unique heterologous motifs that are highly conserved during evolution. Although these TGF-β proteins share similar properties in vitro, isoform-specific properties have been suggested through in vivo studies and by the unique phenotypes for each TGF-β knock-out mouse. To test our hypothesis that each of these homologues has nonredundant functions, and to identify such isoform-specific roles, we genetically exchanged the coding sequence of the mature TGF-β1 ligand with a sequence from TGF-β3 using targeted recombination to create chimeric TGF-β1/3 knock-in mice (TGF-β1Lβ3/Lβ3). In the TGF-β1Lβ3/Lβ3 mouse, localization and activation still occur through the TGF-β1 latent associated peptide, but cell signaling is triggered through the TGF-β3 ligand that binds to TGF-β receptors. Unlike TGF-β1−/− mice, the TGF-β1Lβ3/Lβ3 mice show neither embryonic lethality nor signs of multifocal inflammation, demonstrating that knock-in of the TGF-β3 ligand can prevent the vasculogenesis defects and autoimmunity associated with TGF-β1 deficiency. However, the TGF-β1Lβ3/Lβ3 mice have a shortened life span and display tooth and bone defects, indicating that the TGF-β homologues are not completely interchangeable. Remarkably, the TGF-β1Lβ3/Lβ3 mice display an improved metabolic phenotype with reduced body weight gain and enhanced glucose tolerance by induction of beneficial changes to the white adipose tissue compartment. These findings reveal both redundant and unique nonoverlapping functional diversity in TGF-β isoform signaling that has relevance to the design of therapeutics aimed at targeting the TGF-β pathway in human disease.
Introduction
The mammalian genome contains three related isoforms of the transforming growth factor-β (TGF-β) proteins known as TGF-β1, TGF-β2, and TGF-β3. The TGF-β isoforms are multifunctional cytokines that influence cellular processes such as proliferation, differentiation, and extracellular matrix deposition. Because of these activities, the TGF-β isoforms play important physiological roles in regulating embryonic development, controlling immunity, and maintaining epithelial homeostasis (1, 2). With such potent attributes, TGF-β signaling is tightly regulated where each TGF-β is secreted as a dimer that must undergo further processing to dissociate the LAP2 from the receptor-binding ligand. Upon activation, the mature ligand binds to the type I and type II receptors to induce cellular changes through Smad-dependent or Smad-independent signaling pathways (3). The inhibitory LAP domains display only 40% homology between the three isoforms, but the mature TGF-β ligands, in contrast, share about 80% homology in amino acid sequence and a common hallmark of nine conserved cysteines (4, 5). In the mature region, sequence variation between the TGF-β isoforms is primarily found in the amino-terminal α-helix and in the loops between the β-sheets (6). Although all three TGF-β ligands share a high degree of amino acid homology, each mature protein has unique heterologous motifs that have been maintained throughout evolution. Conservation of these heterologous motifs suggests that each homologue is imbued with some isoform-specific properties.
All three TGF-β homologues signal through the same receptor complexes and appear interchangeable in vitro, yet the activity of these proteins in vivo seems to suggest that each isoform may possess nonredundant functions (7, 8). In the developing embryo, for example, all three TGF-β proteins have distinct cell-specific expression patterns that reflect the differences in the promoters located within each homologue's gene (9, 10). In general, TGF-β1 expression is induced by immediate early genes in response to signals such as injury and stress, whereas TGF-β2 and TGF-β3 appear to be more hormonally and developmentally regulated (8). The distinct pattern of expression for each homologue suggests that the TGF-β isoforms may play differing roles in processes such as development and organogenesis. In agreement with this notion, each TGF-β isoform null mouse has unique phenotypes. The TGF-β1−/− mice have defects in hematopoiesis and vasculogenesis that result in embryonic lethality for 50% of TGF-β1 knock-out mice with a mixed genetic background (11). TGF-β1−/− pups that survive to birth are born developmentally normal but exhibit multifocal autoimmune inflammation by 2 weeks of age that result in organ failure and premature death (12, 13). In contrast to the TGF-β1−/− mice, the TGF-β2−/− mice exhibit perinatal mortality with multiple developmental defects in the heart, lung, and skeleton (14). Targeted deletion of TGF-β3 also displays a unique phenotype wherein TGF-β3−/− mice die shortly after birth with a cleft palate defect and delayed pulmonary development (15, 16). The unique phenotypes for each TGF-β null mouse suggest that the isoforms are not interchangeable, but it is difficult to use the pathological manifestations seen in these knock-out mice to ascribe any nonredundant attributes to each TGF-β homologue, particularly when the loss of an isoform's spatiotemporal expression, in addition to its active/absolute concentrations, contributes to the phenotype of the null mouse.
Although the TGF-β proteins have differing knock-out phenotypes and patterns of expression, some isoform-specific roles have been identified between the ligands with in vivo testing and organ culture. For example, in conjunction with the cleft palate phenotype of TGF-β3 null mice, only TGF-β3 can induce full palate fusion, a function that cannot be rescued by addition of either TGF-β1 or TGF-β2 (17, 18). Additionally, TGF-β3 does not promote scarring like the other two homologues, and it may have a unique role in the orchestration of dermal and epidermal cell motility (19, 20). So, although seemingly similar in vitro, studying the complex physiological activities of each TGF-β in an in vivo setting seems necessary to fully identify any isoform-specific niche for each homologue.
With these previous studies suggesting isoform-specific functions, we exchanged the murine TGF-β1 ligand with its TGF-β3 homologue to fully test the degree of redundancy among these TGF-β isoforms, essentially generating a TGF-β1Lβ3/Lβ3 knock-in mouse. As mentioned, these TGF-β1Lβ3/Lβ3 knock-in mice will provide further insight into the conserved heterology that exists between the TGF-β isoforms, with particular attention paid to the similarities and differences between the well studied TGF-β1 protein and the less characterized isoform TGF-β3. The TGF-β1Lβ3/Lβ3 knock-in mouse will provide a better understanding about the isoform-specific functions linked to TGF-β3, which is needed because this TGF-β homologue, although less studied, still appears to possess significant physiological activity. First, we engineered a chimeric TGF-β1/3 protein wherein the TGF-β1 LAP was combined with an HA-tagged TGF-β3 ligand. This chimeric strategy allows for sequestration and activation via the TGF-β1 LAP in its normal physiological context but for signaling to occur through the TGF-β3 ligand. Once tested for proper folding and latency, the chimeric TGF-β1/3 sequence was knocked into the TGF-β1 genetic locus. The resulting TGF-β1Lβ3/Lβ3 knock-in mice did not manifest any phenotype typical of the TGF-β1−/− mouse, such as embryonic lethality from vasculogenesis defects in utero, nor any signs of multifocal inflammation. Although this appears to show redundancy between the isoforms, physiological alterations in metabolism along with abnormal bone and tooth mineralization suggest that certain physiological functions of TGF-β1 cannot be replaced through knock-in of TGF-β3.
EXPERIMENTAL PROCEDURES
Generation of Chimeric TGF-β1/3 Expression Vector and Targeting Construct
Chimeric TGF-β cDNAs were generated by ligating PCR products of the TGF-β1 LAP with sequence for the mature HA-tagged ligands of murine TGF-β3, murine TGF-β2, and porcine TGF-β1 (21). The resulting chimeric TGF-β cDNAs were subcloned into the pSG5 (Agilent Technologies, Santa Clara, CA) vector to study proper expression and secretion. The chimeric TGF-β1/3 cDNA was selected for further study to test latency and activation. After confirming proper folding latency and activation, TGF-β1 promoter-driven expression vectors were constructed using a clone of the TGF-β1 gene derived from a BALB/c liver genomic library (22). In the E2β1/3 expression vector, the chimeric TGF-β1/3 cDNA was subcloned into a BsaBI site located within exon 2 of the TGF-β1 gene, where placement of the chimeric cDNA would minimize any potential perturbations to the TGF-β1 promoter. The E2β1/3 vector contains about 10.5 kb of the TGF-β1 gene, including both TGF-β1 transcriptional start sites as well as the first exon and intron. For stable transfections, the E2β1/3 expression vector also included a floxed neomycin resistance (Neor) gene downstream of the chimeric cDNA that is driven by the thymidine kinase promoter (gift from H. Gu). The E2β1/3 expression vector was transfected into NIH-3T3 cells that contain active Ras, to induce expression via the TGF-β1 promoter. Stable cell lines were established, and the secreted HA epitope-tagged TGF-β1/3 protein was detected in the supernatants. After confirmation that the chimeric protein could be expressed with the TGF-β1 promoter, a targeting construct was generated wherein the E2β1/3 vector was combined with a 1.8-kb 3′ homology arm from the TGF-β1 genomic clone, as well as the HSV-thymidine kinase gene from pPNT (23), for negative selection.
Cell Culture
COS7 cells were transfected with the pSG5-TGF-β1/3 expression vector to study secretion, latency, and activation of the chimeric protein. COS7 cells were transfected with Lipofectamine LTX (Invitrogen), and after overnight incubation, the medium was replaced with serum-free Opti-MEM supplemented with MITO plus (BD Biosciences). Supernatants were then collected after 48 h and tested with Western blot using both reducing and nonreducing conditions to examine the expression and latency of the chimeric TGF-β1/3 (see below for Western blots). To study the signaling capability of the epitope-tagged TGF-β3 ligand, cell-free supernatants from the transfected COS7 cells were diluted 1:3 with fresh serum-free medium and added onto 5 × 105 HepG2 cells in a 6-cm culture dish. HepG2 cells were tested with either unaltered supernatant or with medium from the transfected cells that had been heated at 80 °C for 5 min, which releases the TGF-β3 ligand from the LAP without denaturing/inactivating the receptor-binding mature domain. The cells were lysed after 30 min, and the lysates were run on a Western blot to determine the level of Smad2 phosphorylation. Before proceeding with a targeting vector, TGF-β1 promoter-driven expression vectors were transfected into NIH-3T3 cells using Lipofectamine. These NIH-3T3 cells were previously infected with Harvey Sarcoma virus and expressed Ha-Ras, an oncogene that would stimulate the TGF-β1 promoter (22). Cells were treated with G418 to derive stable cell lines, and serum-free supernatants were collected within 48 h after plating. With confirmation that the TGF-β1 promoter could drive expression of the chimeric TGF-β1/3, a targeting vector was constructed and electroporated into R1 embryonic stem cells, a cell line derived from a 129/Sv mouse strain. Twenty four hours after electroporation, the cells were treated with G418 and gancyclovir. Individual clones were selected after 10 days of drug treatment. Some stem cells used to generate knock-in mice were allowed to differentiate in culture, and supernatants were collected to test for secretion of the tagged chimeric TGF-β1/3 protein.
Derivation and Genotyping of the TGF-β1Lβ3/Lβ3 Mice
Southern blot analysis and PCR were performed to identify which clones had undergone proper homologous recombination in the R1 cells. The DNA from drug-resistant stem cell clones was purified through phenol and chloroform extraction and digested with BamHI. A 0.5-kb TGF-β1 PCR fragment derived from ES cell genomic DNA was used as a flanking probe for the Southern analysis. An 8-kb band was detected for the wild type allele, whereas a 6-kb band appeared if proper homologous recombination occurred. PCR for homologous recombination was performed using a primer located in the Neor marker (GTTCGAGGCCACACGCGTCA) and a primer downstream of any TGF-β1 genetic sequence used in the 3′ homology arm of the targeting construct (GCCTCTGCCTCACAAGCGCA). Two positive clones were injected into C57Bl/6 mouse blastocysts that underwent germ line transmission. The male chimeras were then crossed with C57Bl/6 females to generate F1 offspring. The offspring were bred to homozygosity and were genotyped by PCR using the following primers: In-B3For (GTTTGGTTTTGTTTGAGGCGTG), In-B3Rev (ACACAGCAGTTCTCCTCCAGGTTG), and Ex2 Rev (GGGCTGACCTGTGTGCCACC). The PCR was run for 40 cycles at an annealing temperature of 60 °C. A 1-kb PCR product was generated in this reaction for the knock-in allele, whereas only a 0.6-kb band was produced if the wild type TGF-β1 allele was present. All care to the mice was given in compliance with the National Institutes of Health guidelines on the use of laboratory and experimental animals, and the studies were approved by the NIDCR Animal Care and Use Committee. Food and water were provided ad libitum. Because of defects noticed in the teeth of the mice, animals were supplemented with a transgenic dough diet (Bio Serv, Frenchtown, NJ).
Histology and Immunohistochemistry
Tissues were carefully dissected from the mice and fixed overnight in 10% buffered formalin. The tissues were primarily embedded in paraffin, and 5-μm sections were used for histopathological analysis. Pathology was examined using slides stained with hematoxylin and eosin (H&E); mast cells were detected using toluidine blue staining, and fat accumulation was examined using Oil Red O staining from frozen sections of the liver. For immunohistochemistry, sections were deparaffinized in xylene, rehydrated with descending grades of ethanol, blocked for 30 min, and incubated with primary antibody (see below) prepared in background-reducing antibody diluent (Dako, Carpenteria, CA). Next, the sections were treated with either rabbit on rodent HRP-polymer (Biocare, Concord, CA) or with a biotinylated secondary antibody (Vector Laboratories, Burlingame, CA), followed by treatment with the Vectastain ABC reagent (Vector Laboratories). Antibody complexes were detected using liquid 3,3′-diaminobenzidine (Biogenex, Fremont, CA). For immunohistochemistry of the bone, femurs and tibias of the mice were decalcified using 0.1 m EDTA for 2 weeks. For antigen retrieval, slides were heated overnight at 60 °C in target retrieval solution (Dako). Primary antibody dilutions are as follows: a 1:50 dilution for F4/80 (Invitrogen); 1:150 anti-Smad2, phospho-specific (Ser-465/467) (AB3849 Millipore, Billerica, MA); 1:200 TGF-β3 (24), anti-Smad3, phospho-specific (Ser-423/425) (9520 Cell Signaling, Danvers, MA); 1:1000 phospho-TAK1 (Thr-187) (ab79583 Abcam Cambridge, MA) and 1:2000 Osteocalcin (Takara Otsu, Shiga, Japan).
Western Blot Analysis
Protein lysates were generated from cultured cells and mouse tissues using T-PER (Thermo Scientific Pierce) with a complete mini protease inhibitor mixture (Roche Applied Science). To study secretion of the TGF-β proteins, serum-free medium from the cultured cell lines was collected, treated with 200 mm PMSF (Sigma), and concentrated using an Amicon ultracentrifugal filter device (Millipore). Activation of the chimeric TGF-β1/3 protein was achieved by heating supernatants at 80 °C for 5 min or through incubation with two known TGF-β activators, either plasmin (0.001 unit/ml, Sigma) or thrombospondin (0.04 mg/ml, Sigma) at 37 °C for 2 h. For Western blot, about 50 μg of protein were denatured and separated with electrophoresis on NuPAGE 4–12% BisTris precast gels using XCell SureLock Mini-Cell (Invitrogen). Nonreducing Western blot was performed using conditions specified for the NativePAGE Novex BisTris gel system (Invitrogen). Proteins were then transferred onto a nitrocellulose membrane, blocked for 1 h, and incubated with primary antibodies overnight. The bound primary antibodies were then visualized using a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) followed by chemiluminescence detection (Pierce). The following primary antibody dilutions were used: 1:1000 anti-actin (Millipore MAB1501); 1:100 anti-glyceraldehyde-3-phosphate dehydrogenase (Abcam ab9485); 1:500 TGF-β1 (Promega G1221); 1:500 TGF-β2 (Santa Cruz Biotechnology); 1:500 TGF-β3 (Santa Cruz Biotechnology); 1:1000 anti-HA (Cell Signaling); 1:200 TGF-β1 LAP (R&D Systems); 1:500 anti-Smad2 (Invitrogen 51-1300); and 1:500 anti-Smad2, phospho-specific (Ser-465/467) (Millipore AB3849).
Preparation of RNA and RT-PCR
RNA was extracted from tissues of the mice using miRNeasy (Qiagen) according to the manufacturer's protocol. Using random primers (Invitrogen), about 500 ng of RNA were reverse-transcribed into cDNA through Super Script III reverse transcriptase (Invitrogen). To examine for any TGF-β1 expression, 40 cycles of RT-PCR were performed using TGF-β1 primers from exon 2 to 7 (25). RT-PCR for GAPDH (TGCACCACCAACTGCTTAG and GATGCAGGGATGATGTTC) was used as a control. For gene expression analysis of white adipose tissue, total RNA was isolated from epididymal WAT by using the RNeasy kit, and cDNA was transcribed using high capacity cDNA reverse transcription kit (Invitrogen catalog no. 4368814) according to the manufacturer's protocol. Quantitative gene expression was measured by real time PCR in Applied Biosystems 7500 Fast Real Time PCR System using Fast SYBR® Green Master Mix (catalog no. 4385612). The expression of several adipose tissue- and lipid-related gene markers were determined by performing 40 cycles as follows: Cidea, AGCTCGCCCTTTTCGAGTTTC and GGCCAGTTGTGATGACTAAGAC; PRDM16, TGCTGACGGATACAGAGGTGT and CCACGCAGAACTTCTCGCTAC; Cox8b, TGTGGGGATCTCAGCCATAGT and AGTGGGCTAAGACCCATCCTG; DiO2, ACACTGGAATTGGGAGCATC and ATGCTGACCTCAGAAGGGCT; UCP-1, AGGCTTCCAGTACCATTAGGT and CTGAGTGAGGCAAAGCTGATTT; PGC1a, TATGGAGTGACATAGAGTGTGCT and CCACTTCAATCCACCCAGAAAG; ELOVL3, CCGCGTTCTCATGTAGGTCT and CTTAAGGCCCTTTTTGGAGG; Tgfbr1, CAACCCAGGTCCTTCCTAAA and GGAGAGCCCTGGATACCAAC; Tgfbr2, CCGCTGCATATCGTCCTGTG and AGTGGATGGATGGTCCTATTACA; ID1, GGTGAACGTCCTGCTCTACGA and AGACTCCGAGTTCAGCTCCAG; Jun-B, TGCGGACGGTTTTGTCAA and GCGTCACGTGGTTCATCTTG; Serpine1, CAGAGCAACAAGTTCAACTACACTGA and CAGCGATGAACATGCTGAGG; Smad7, GGCCGGATCTCAGGCATTC and TTGGGTATCTGGAGTAAGGAGG.
Gene expression was analyzed by adopting the standard ΔΔCT method of real time PCR, and results were expressed as a relative fold change in gene expression compared with wild type/control.
Analysis of TGF-β1 Levels in Serum by ELISA
Mice were euthanized with CO2, and blood samples were collected by intracardiac puncture into microtainer tubes. Blood samples were left on ice for 15 min and spun at 3500 × g for 25 min in an Eppendorf centrifuge at room temperature. Serum was collected, aliquoted, and stored at −70 °C. The level of TGF-β1 in the serum was determined by ELISA using the mouse/rat/porcine TGF-β1 immunoassay kit (R&D Systems). Serum was diluted 60-fold after acid activation, and total TGF-β1 was determined according to the manufacturer's instructions.
Metabolic Studies of the TGF-β1Lβ3/Lβ3 Mice
For the glucose tolerance test (26), mice underwent a 12-h fast, and the next morning they were injected with intraperitoneal glucose (2 mg/kg body weight). Tail blood was collected at given time intervals for automated glucose measurement using Alphatrak glucometer and glucose strips (catalog no. 32006-06). For adipokine measurement, mice were fasted for 12 h; a tail bleed was performed, and serum was analyzed using the Mouse Adipokine Panel- 7 Plex (Millipore MADPK-71K-07) according to the manufacturer's instructions. For food intake, 10–4-week-old female TGF-β1Lβ3/Lβ3 and TGF-β1Lβ3/+ mice were housed separately and supplied standard pellet food and water. The amount of food consumed along with the weight of the mice was measured three times a week for month.
Microradiography and Microcomputed Tomography Analysis
Skulls from the mice were radiographed using a Faxitron MX20 Specimen Radiography System (Faxitron X-Ray Corp., Wheeling, IL) for 20 s at 20 kV. The images were captured on Kodak MIN-R film. Using ImageJ software (National Institutes of Health), the mean gray values were measured to calculate the difference in opacities of the molars/incisors for both the wild type and the knock-in mice. To examine kyphosis, mice were radiographed for 40 s at 30 kV, and images were captured using Kodak PPL X-Ray film. With microcomputed tomography, femurs were dissected from the TGF-β1Lβ3/Lβ3 mice, fixed overnight in 10% buffered formalin, and changed to 70% ethanol. Femurs were analyzed using the SkyScan 1172 (Bruker microCT Kontich, Belgium) at 40 kV.
Flow Cytometry Analysis
Cells from spleen, mesenteric lymph nodes, and peripheral lymph node organs were removed and prepared as described previously (27). Briefly, cells were stained with the indicated fluorochrome-conjugated antibodies against surface markers. For Foxp3 staining, cells were subsequently stained using the Foxp3 staining set (eBioscience, San Diego) according to the manufacturer's instructions. To detect cytokine production ex vivo, cell suspensions were stimulated with 25 ng/ml phorbol 12-myristate 13-acetate (Sigma) and 2.5 μg/ml ionomycin (Sigma) in the presence of Golgi stop (BD Biosciences) for 4 h. After stimulation, cell surface markers were stained before cells were fixed in Cytofix/Cytoperm (BD Biosciences). Cells were stained with fluorochrome-conjugated antibodies against cytokines for 40 min in Permeabilization Buffer (BD Biosciences). Cell acquisition was performed on an LSRII or Calibur (both BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Ashland, OR). Mice used for FACS analysis were aged between 18 and 28 weeks, and data were pooled from four female TGF-β1Lβ3/Lβ3 mice along with the corresponding littermate controls. Two male mice aged 29–33 weeks were also analyzed.
Statistical Analysis
The data were analyzed for statistical significance using GraphPad Prism version 5.00 for Windows (Graph-Pad Software Inc., La Jolla, CA). Survival curves were generated using the Kaplan-Meier method and evaluated using the log-rank (Mantel-Cox) test. A two-tailed Student's unpaired t test was used to determine statistical significance for all other comparisons. Values of p ≤ 0.05 were considered statistically significant.
RESULTS
Development and Testing of the Chimeric TGF-β1/3 Protein
Chimeric TGF-β constructs were generated where the sequence from the TGF-β1 LAP was fused with HA-tagged versions of the TGF-β ligands, including murine TGF-β3, murine TGF-β2, and the porcine TGF-β1 as a control (21). The strategy behind making the chimeric constructs is demonstrated in Fig. 1A with the chimeric TGF-β1/3 protein as an example. In this case, cell signaling occurs via the TGF-β3 ligand, but the TGF-β1 LAP determines localization, bioavailability, and activation of the protein. Expression vectors for each chimeric TGF-β construct were transfected into COS7 cells, and the supernatants were collected and concentrated. Fig. 1B shows the efficient expression of each chimeric TGF-β protein. Because both TGF-β1 and TGF-β3 share similar binding affinities to the TGF-β receptor TβR-II (28), the chimeric TGF-β1/3 protein was chosen for further analysis. Although TGF-β1 LAP has previously been shown to bind to recombinant TGF-β3 ligand, rendering it inactive (29), Western blot using nonreducing conditions was nevertheless performed to show the formation of both a small and large latent complex (Fig. 1C). With high heat, these complexes could also be dissociated to cause release of the active TGF-β3 ligand. Dissociation of the TGF-β1/3 latent complexes could also occur via the known TGF-β activation mechanisms of plasmin-mediated proteolysis and thrombospondin-induced conformational change (Fig. 1D) (30, 31). Formation of a latent complex was further confirmed via immunoprecipitation, wherein the HA-tagged TGF-β3 ligand was pulled down using a TGF-β1 LAP antibody (Fig. 1E). Untagged full-length human TGF-β3 was used as a control. Finally, increased TGF-β associated phosphorylation of Smad2 occurred primarily upon heat activation of the chimeric TGF-β1/3, further confirming the formation of a latent complex (Fig. 1F). Although these TGF-β isoforms share a high degree of homology, the proper folding and binding of these two protein domains into a latent yet activatable chimeric complex needed to be ensured, because uncontrolled TGF-β signaling would be deleterious.
FIGURE 1.

A, schematic depicting the strategy behind the chimeric TGF-β1/3 knock-in. With this approach, the TGF-β1 LAP is maintained for localization and activation, but cell signaling occurs through the TGF-β3 ligand. B, chimeric TGF-β cDNA expression vectors were transfected into COS7 cells. The secretion of all three HA-tagged TGF-β ligands along with the accompanying TGF-β1 LAP is seen in the cultured supernatants by Western blot. C, Western blot using nonreducing conditions that demonstrate the formation of small and large latent chimeric TGF-β1/3 complexes. The lower molecular mass band corresponds to the small latent complex (SLC), which is ∼100 kDa. The upper band is the large latent complex (LLC) that has a molecular mass of ∼290 kDa (the large latent complex is composed of the small latent complex linked with the latent TGF-β-binding proteins). The latent TGF-β1/3 complexes can be dissociated by heat (ΔH), where an incubation of 80 °C for 5 min is able to induce the release of the active TGF-β3 ligand. D, additional Western blot under nonreducing conditions to show the activation of the chimeric TGF-β1/3 via plasmin and thrombospondin. Dissociation of the LAP is needed for the release of the TGF-β3 ligand to allow for cell signaling. E, immunoprecipitation (IP) demonstrates the formation of a latent TGF-β1/3 complex as anti-TGF-β1 LAP antibody is capable of pulling down the HA-tagged TGF-β3 ligand in the supernatant from transfected COS7 cells. A vector for the untagged human TGF-β3 cDNA was used as control. IB, immunoblot. F, Smad2 phosphorylation is effectively increased only after the TGF-β3 ligand was released from the TGF-β1 LAP by heat to trigger signaling, validating the formation of a latent chimeric TGF-β1/3 complex. G, before designing a knock-in targeting construct, TGF-β1 promoter expression vectors were tested using NIH-3T3 cells that have active Ha-ras to drive promoter activity. Efficient expression of the TGF-β1/3 protein was seen using a vector, E2β1/3, where the chimeric cDNA was fused into exon 2 of a TGF-β1 genomic clone (∼10.5 kb of upstream genomic sequence). Another expression vector with both a shorter promoter of only ∼1.2 kb and placement of the chimeric TGF-β1/3 in exon 1 did not work as efficiently as the E2β1/3 construct. An SV40 promoter-driven vector was used as a positive control while a GFP vector was a negative control.
Generation of a TGF-β1/3 Targeting Vector
Before generating a targeting construct, expression of the chimeric TGF-β1/3 protein by the TGF-β1 promoter was tested. For this purpose, TGF-β1 promoter-driven expression vectors were transfected into NIH-3T3 cells expressing Ha-ras to activate TGF-β production (Fig. 1G). Fusing the chimeric TGF-β1/3 construct into exon 2 of a TGF-β1 genomic clone not only seemed to show the best results for driving expression by the TGF-β1 promoter but also minimized any perturbation to the transcriptional and translational elements in the promoter. This strategy was used to develop the targeting construct (Fig. 2A). The 3′ homology arm of the targeting vector consisted of a sequence downstream of exon 2 that included all of exon 3. A floxed Neor was cloned after the chimeric TGF-β1/3 cDNA for positive selection of integrated constructs, whereas the thymidine kinase gene was added downstream of the 3′ homology arm to select against random integrants. The chimeric TGF-β1/3 targeting vector was electroporated into R1 cells, an embryonic stem cell line derived from a 129/Sv mouse strain. Of the clones collected after drug selection, about 1 in 20 showed proper homologous recombination as determined by both Southern blot (Fig. 2B) and PCR analyses (Fig. 2, C and D). Targeted clones were injected into C57Bl/6 blastocysts for derivation of the knock-in mice. Following injection, some of the targeted stem cell clones, along with the parent R1 cells, were then allowed to differentiate in culture, and supernatants were collected. As seen in Fig. 3A, the parent R1 cells actively secrete TGF-β1, although its expression is diminished in the recombined clone that has one targeted knock-in allele. The TGF-β1 promoter in the recombined allele, however, was able to drive expression of the HA-tagged TGF-β3 ligand that is absent in the unaltered parent R1 cells.
FIGURE 2.

A, diagram showing the chimeric TGF-β1/3 targeting vector, the wild type TGF-β1 allele, and the properly recombined allele generated upon homologous recombination. The location of all the primers (arrows) used for genotyping are also depicted along with the Southern blot strategy used to identify proper homologous recombination. For the Southern blot, a BamHI digest will generate an 8-kb Southern blot genomic fragment for the WT allele (WT Band), whereas a 6-kb genomic fragment is derived from the insertion of the targeting vector (Lβ3 Band). These BamHI Southern blot fragments (dashed gray lines) are detected through the 0.5-kb radiolabeled probe, shown as a solid gray line. B, Southern blot shows proper homologous recombination in the ES cell clones isolated after drug selection with the expected BamHI digest fragments from the wild type and targeted allele generated as schematically predicted above. C, PCR confirms targeted recombination through a forward primer (Neo ◮) in the Neor gene of the inserted targeting vector and a reverse primer (3′ ◭) located within intronic sequence that is 3′ downstream of sequence in the homology arm. A PCR product of about 3 kb in length is generated when proper homologous recombination occurs. D, genotyping PCR product of 1 kb in length is generated with the targeted Lβ3 allele through a forward primer in exon 1 TGF-β1 genomic locus (For ◮) and a reverse primer located in the inserted TGF-β3 cDNA (B3 ◭). For detection of the wild type TGF-β1 allele, a 0.6-kb PCR product is generated using the same forward primer and a reverse primer (Ex2 ◭) matching sequence from exon 2 that is lost in the targeted allele due to homologous recombination.
FIGURE 3.

A, targeted ES cells express the HA-tagged TGF-β3 protein but secrete reduced amounts of TGF-β1 as compared with the naive ES cells. B, TGF-β1 was undetectable in the serum of the TGF-β1Lβ3/Lβ3 knock-in mice (n = 5) through ELISA. Serum TGF-β1 levels from TGF-β1Lβ3/Lβ3 mice were compared with wild type (n = 2) and TGF-β1−/− (n = 2) mice. C, TGF-β1 expression was not detected in the TGF-β1Lβ3/Lβ3 mice by RT-PCR. Tissues from the TGF-β1Lβ3/Lβ3 and TGF-β1+/Lβ3 mice were examined using both TGF-β1 and GAPDH primers. D, Western blot reveals expression of the HA-tagged TGF-β3 ligand in the spleen and lung from both TGF-β1+/Lβ3 and TGF-β1Lβ3/Lβ3 mice but not in the TGF-β1+/+ animals. E, intense TGF-β3 staining is seen in tissues from 3-week-old TGF-β1Lβ3/Lβ3 mice. The increased staining in the TGF-β1Lβ3/Lβ3 mice versus the TGF-β1−/− mice suggests that the TGF-β3 expression is driven by the recombined allele and is not a result of targeted deletion of TGF-β1.
Replacement of TGF-β1 Ligand with TGF-β3 in a Genetically Engineered Mouse
Germ line transmission was achieved with two targeted stem cell clones and mice were bred to homozygosity. The resulting TGF-β1Lβ3/Lβ3 mice had no measurable TGF-β1 present in the serum, as measured through ELISA (Fig. 3B). Additionally, RT-PCR with primers from exon 2 to 7 was unable to detect any TGF-β1 mRNA transcript in tissues from the TGF-β1Lβ3/Lβ3 mice (Fig. 3C). An HA-tagged TGF-β3 protein, however, was detected by Western blot in tissues from the TGF-β1Lβ3/Lβ3 mice such as the spleen and lung (Fig. 3D). Finally, strong TGF-β3 staining was seen in the TGF-β1Lβ3/Lβ3 tissues as compared with either the wild type controls or the TGF-β1−/− mice (Fig. 3E). The stronger TGF-β3 staining seen in the TGF-β1Lβ3/Lβ3 mice versus the TGF-β1−/− mice suggests that the TGF-β3 detected is expressed by the knock-in allele and is probably not due to compensatory TGF-β3 expression resulting from a lack of TGF-β1. In addition, no changes in TGF-β2 staining were apparent between the TGF-β1Lβ3/Lβ3 mice and either the TGF-β+/+ or the TGF-β1−/− mice (Fig. 4A). The genetically engineered targeted recombination in the TGF-β1Lβ3/Lβ3 mice therefore appears to have caused disruption of the TGF-β1 gene while allowing for generation of the chimeric TGF-β1/3 protein by the TGF-β1 promoter. Smad-dependent and -independent signaling also seems to be unchanged overall between the TGF-β1Lβ3/Lβ3 and the wild type mice in tissues such as the lung and kidneys (Fig. 4, B–D).
FIGURE 4.

A, levels of TGF-β2 are unaffected by targeted replacement of TGF-β1 with TGF-β3. Similar TGF-β2 staining is seen between TGF-β1Lβ3/Lβ3 mice and tissues from either TGF-β1+/+ or TGF-β1−/− controls. B–D, TGF-β-mediated signaling was unaffected by targeted replacement of TGF-β1 with TGF-β3. No difference was seen between TGF-β1Lβ3/Lβ3 mice and wild type controls with either Smad-dependent signaling via Smad2 (A) and Smad3 phosphorylation (B) nor by Smad-independent signaling through TAK1 phosphorylation (C).
Breeding heterozygous mice to generate the TGF-β1Lβ3/Lβ3 mice resulted in the pups being born close to the expected genotypic ratio (30% TGF-β1Lβ3/Lβ3, 50% TGF-β1Lβ3/+, and 20% TGF-β1+/+; n = 141 mice). This finding differs from the TGF-β1−/− mouse, wherein over half of the pups in a genetically mixed background die in utero because of vasculogenesis defects (11). With no signs of embryonic lethality, the knock-in of TGF-β3 appears able to compensate for the lack of TGF-β1 in terms of early developmental vasculogenesis. The TGF-β1Lβ3/Lβ3 embryos, however, are not completely devoid of any TGF-β1 during development because TGF-β1 can cross the placenta from the TGF-β1Lβ3/+ mothers (32). The TGF-β1Lβ3/Lβ3 female mice appear so far to be infertile, so a complete lack of developmental TGF-β1 could not be tested in this animal model.
Survival and Weight Gain in the TGF-β1Lβ3/Lβ3 Mice
The TGF-β1Lβ3/Lβ3 mice are born developmentally normal and show no signs of any congenital defects. The life span of the TGF-β1Lβ3/Lβ3 mice, however, was shortened to a median of 30 weeks of age as compared with the controls (Fig. 5A). A shorter life span shows that all the defects associated with targeted disruption of TGF-β1 could not be completely rescued by knock-in of TGF-β3 and that these two TGF-β isoforms are not totally interchangeable. Although the life spans of the TGF-β1Lβ3/Lβ3 mice are reduced, their survival is still dramatically longer when compared with the TGF-β1−/− mice that only live at most to 3–4 weeks of age and generally die from multifocal inflammation. A longer life span seems to suggest that the knock-in of the TGF-β3 may have prevented some of the severe autoimmunity typical to TGF-β1 deficiency. Overall, the TGF-β1Lβ3/Lβ3 mice are smaller in stature than their control littermates. The TGF-β1Lβ3/Lβ3 mice keep pace developmentally with the littermate controls until about 7–8 weeks of age when the weight of the TGF-β1Lβ3/Lβ3 mice generally plateaus (Fig. 5B). These size differences became more noticeable as the TGF-β1Lβ3/Lβ3 animals age (Fig. 5C).
FIGURE 5.

A, improved survival of TGF-β1Lβ3/Lβ3 mice (median age = 30. 5 weeks) versus the TGF-β1−/− mice (median age = 3 weeks) demonstrating rescue from the severe autoimmunity inherent with TGF-β1 deficiency. The life span of the TGF-β1Lβ3/Lβ3 mice is still less than the controls, suggesting that TGF-β1 and TGF-β3 are not completely interchangeable (p ≤ 0.0001; log rank (Mantel-Cox) test). B, reduced body weight of male TGF-β1Lβ3/Lβ3 mice, which plateaus starting around 5–6 weeks of age. C, photos of B6129 female mice showing the reduced size and kyphosis in the TGF-β1Lβ3/Lβ3 mice over time. D, faxitron images and photos of a pair of male mice at 27 weeks of age illustrating the hunchback posture of the TGF-β1Lβ3/Lβ3 mouse caused by curvature of the thoracic vertebrae. E, teeth of TGF-β1Lβ3/Lβ3 mice show signs of undermineralization and chipping. The adult mice shown are between 8 and 32 weeks of age. Severe malocclussion was also detected in some of the TGF-β1Lβ3/Lβ3 mice. F, decreased mineral density and dental attrition in the teeth of the TGF-β1Lβ3/Lβ3 mice as shown by faxitron (mice are 15 weeks of age). Using the faxitron images of the teeth, the mineral density of both molars and incisors was measured in the TGF-β1Lβ3/Lβ3 mice (n = 6) and compared with TGF-β1+/+ mice (n = 6). The quantitation in TGF-β1Lβ3/Lβ3 mice incisor opacity revealed a reduction of opacity by about 28% in the upper incisors (TGF-β1+/+, 63.7 ± 14.1 au (arbitrary units); TGF-β1Lβ3/Lβ3, 45.9 ± 16 au, and a reduction by 31% in the lower incisors (TGF-β1+/+, 111.8 ± 23 au; TGF-β1Lβ3/Lβ3, 76.8 ± 14.5 au (*, p ≤ 0.05, mean ± S.E.)). In the molars, the reduction in opacity was less significant but still present. For the upper molars, there was a 6% reduction in opacity (TGF-β1+/+, 126.8 ± 12.7 au; TGF-β1Lβ3/Lβ3, 119.2 ± 19.6 au) and an 18% reduction in the lower molars (TGF-β1+/+, 119.2 ± 2 au; TGF-β1Lβ3/Lβ3, 97.6 ± 11.2 au).
TGF-β3 Does Not Fully Compensate for the Lack of TGF-β1 in Dental Homeostasis
Kyphosis was commonly seen in most older TGF-β1Lβ3/Lβ3 mice (Fig. 5D), and tooth defects were observed in the TGF-β1Lβ3/Lβ3 mice, with the incisors appearing aberrantly white, undermineralized, and chipped (Fig. 5E). A few mice (3 out of 34) displayed severe malocclusions of the teeth, whereas none of the controls had such dental defects. Because of these tooth defects, mice were supplemented with a high fat dough diet. The size and weight of the TGF-β1Lβ3/Lβ3 mice, however, remained unchanged even with the addition of the dough diet, suggesting that the tooth defects were not responsible for the differences in weight and did not hinder normal food consumption. Microradiography was nevertheless performed using the skulls of the mice, and the images confirmed that the teeth of the TGF-β1Lβ3/Lβ3 were undermineralized. The lower incisors specifically displayed a significant reduction in relative mineral opacity as compared with the controls (Fig. 5F). The molars of the TGF-β1Lβ3/Lβ3 mice also displayed signs of attrition, particularly wearing of the molar cuspids. In all the TGF-β1Lβ3/Lβ3 mice examined, the upper and lower incisors were offset and striking onto one another rather than self-sharpening like the wild type mice. The TGF-β1Lβ3/Lβ3 mice additionally showed indications of atrophy in the ameloblasts, along with abnormal enamel. Dental attrition has been reported in TGF-β1−/− mice, which suggests an isoform-specific role for TGF-β1 in tooth mineralization (33). The tooth defects seen in the TGF-β1Lβ3/Lβ3 mice, although not as severe as in the TGF-β1−/− mice, still share enough similarities to indicate that the knock-in of TGF-β3 may not be able to fully compensate for the lack of TGF-β1 in terms of dental homeostasis.
Isoform-specific Effects on Bone Formation in TGF-β1Lβ3/Lβ3 Mice
The defects in tooth mineralization, along with the aberrantly small stature of some of the TGF-β1Lβ3/Lβ3 mice, point to possible skeletal defects in these animals as well. Bones from the knock-in mice were therefore examined histologically. Defects in secondary ossification were present in the long bones of the older TGF-β1Lβ3/Lβ3 mice (Fig. 6, A and B). Tang et al. (34) have reported that in TGF-β1−/− Rag2−/− mice the osteoblasts are absent on the trabecular bone, which matches with TGF-β1's critical role in guiding osteogenic bone mesenchymal stem cells (BMSCs) to the bone resorptive surfaces. In bone sections from the TGF-β1Lβ3/Lβ3 mice, we also observed reduced numbers of osteoblasts on the trabecular bone by staining for the osteoblast marker osteocalcin (Fig. 6C). Tang et al. (34) showed that TGF-β3 seems to lack the same potent migratory effects on BMSCs as TGF-β1, where neutralization of TGF-β1 from bone resorption-conditioned medium stops BMSC migration, but inhibition of TGF-β3, in contrast, has no effect. The bone defects in the TGF-β1Lβ3/Lβ3 mice may therefore reflect these migratory differences between the two TGF-β isoforms. Any bone defects in the mice, however, would probably not explain such differences in weight when compared with the controls.
FIGURE 6.

A, histology of the femoral condyle shows defects in secondary ossification in a “40-week old” TGF-β1Lβ3/Lβ3 mouse. B, representative microcomputed tomography colored images from younger TGF-β1Lβ3/Lβ3 and wild type mice (14 weeks old) confirm the defects seen in the histology. The intensity of color in the images is indicative of mineral density where the color scale represents the grayscale pixel density. C, osteocalcin staining for osteoblasts. Tibias from the wild type mice show recruitment of osteoblasts to the trabecular bone, whereas the numbers of osteoblasts is decreased on the trabecular bone of the TGF-β1Lβ3/Lβ3 mice. Mice were about 40 weeks old. D, palate development in 1-day-old pups is unaffected by the additional copies of TGF-β3 gained through the knock-in allele of the TGF-β1Lβ3/Lβ3 mice.
Finally, TGF-β3 is commonly known to be involved in palate fusion (15–18). We therefore examined the palate of 1-day-old pups to determine whether doubling the alleles that express TGF-β3 would result in any palate defects. The palates from a TGF-β1Lβ3/Lβ3 and a TGF-β1+/+ mouse were essentially indistinguishable, so no defects in palate development arose with additional copies of TGF-β3 (Fig. 6D).
Knock-in of TGF-β3 Prevents Multifocal Inflammation Typical to TGF-β1 Deficiency
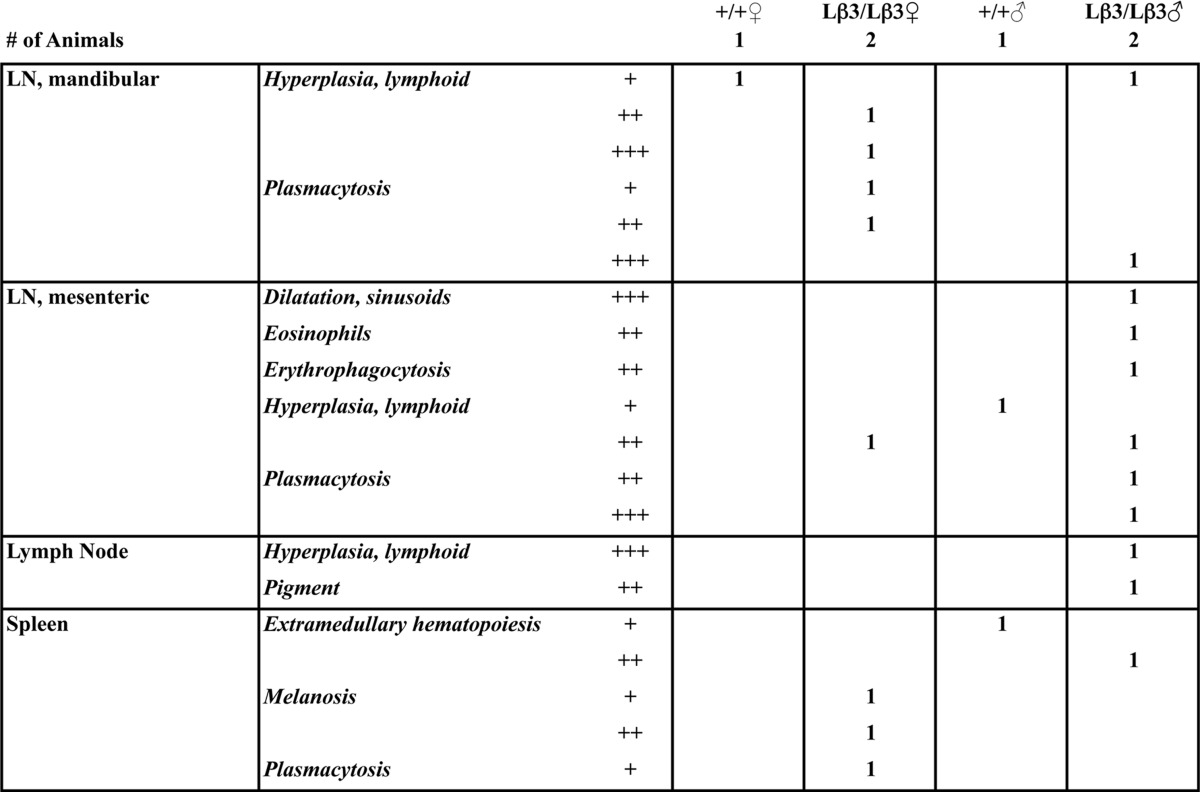
A characteristic of the TGF-β1−/− mice is multifocal inflammation that results in a wasting syndrome (12, 13), and inhibition of the TGF-β signaling with a pan-specific antibody also leads to decreased food intake and weight loss through inflammation of the tongue and esophagus (35). To determine whether inflammation is responsible for decreased survival and reduced weight of the knock-in mice, we examined tissues from the TGF-β1Lβ3/Lβ3 mice at a time point when the TGF-β1−/− mice generally develop multifocal inflammation. Unlike the TGF-β1−/− mice, the tissues from the TGF-β1Lβ3/Lβ3 mice are indistinguishable from the TGF-β1Lβ3/+ controls and show no major signs of infiltration and inflammation (Fig. 7A and Tables 1 and 2). No change in the number and localization of macrophages was seen in the TGF-β1Lβ3/Lβ3 mice through F4/80 staining, whereas the numbers of mast cells detected in dermis of the skin were increased but not significantly (Fig. 7, B and C). Inflammation also did not appear to be a factor in the survival of the TGF-β1Lβ3/Lβ3 mice, as tissues taken perimortem still lacked any signs of widespread autoimmunity. The TGF-β1Lβ3/Lβ3 mice also showed a higher monocyte blood count compared with the controls (0.38 ± 0.15 TGF-β1Lβ3/Lβ3 versus 0.14 ± 0.01 K/μl for TGF-β1+/+ mice). Although high monocyte counts are often indicative of inflammation, the exact underlying cause for this increase is not known.
FIGURE 7.

A, tissues from TGF-β1Lβ3/Lβ3 mice are histologically similar to the littermate controls and lack the inflammatory infiltrates typical of the TGF-β1−/− mice. The TGF-β1Lβ3/Lβ3 mice do not develop signs of severe autoimmunity throughout their life span, even perimortem. B, no difference in the localization or number of macrophages was seen with F4/80 staining of tissues from the TGF-β1Lβ3/Lβ3 and TGF-β1+/+ mice. Macrophages were principally seen in the spleen of the mice and were not seen infiltrating other tissues such as the heart. C, toluidine blue staining shows mast cells in both the dermis of the skin and within the oral mucosa of the tongue. The numbers of mast cells in the dermis were counted within a ×400 magnification field of view, and 10 counts were performed per skin section. The numbers of mast cells were increased in the skin of the TGF-β1Lβ3/Lβ3 mice, but there was no significant difference (51.25 ± 9.187 TGF-β1+/+ mice (n = 4) versus 80.80 ± 9.303 TGF-β1Lβ3/Lβ3 mice (n = 5)). D–G, spleen (SPL), peripheral lymph nodes (PLN), and mesenteric lymph nodes (MLN) were taken from female mice (18–28 weeks old), and cell populations were examined by flow cytometry. D–F, graphs show frequencies of T cells (D), CD4+ and CD8+ T cells (E), Tregs in indicated organs (F). G, representative plots from spleen and mesenteric lymph nodes showing cytokine production from CD4+ T cells following 4 h of stimulation with phorbol myristate acetate and ionomycin. (n = 4 TGF-β1+/+ mice and n = 4 TGF-β1Lβ3/Lβ3 mice.) Error bars represent mean ± S.E. *, p ≤ 0.05 (unpaired two-tailed Student's t test).
TABLE 1.
Pathology report
The grade (severity) is as follows: +, minimal; ++, mild; +++, moderate; ++++, severe.
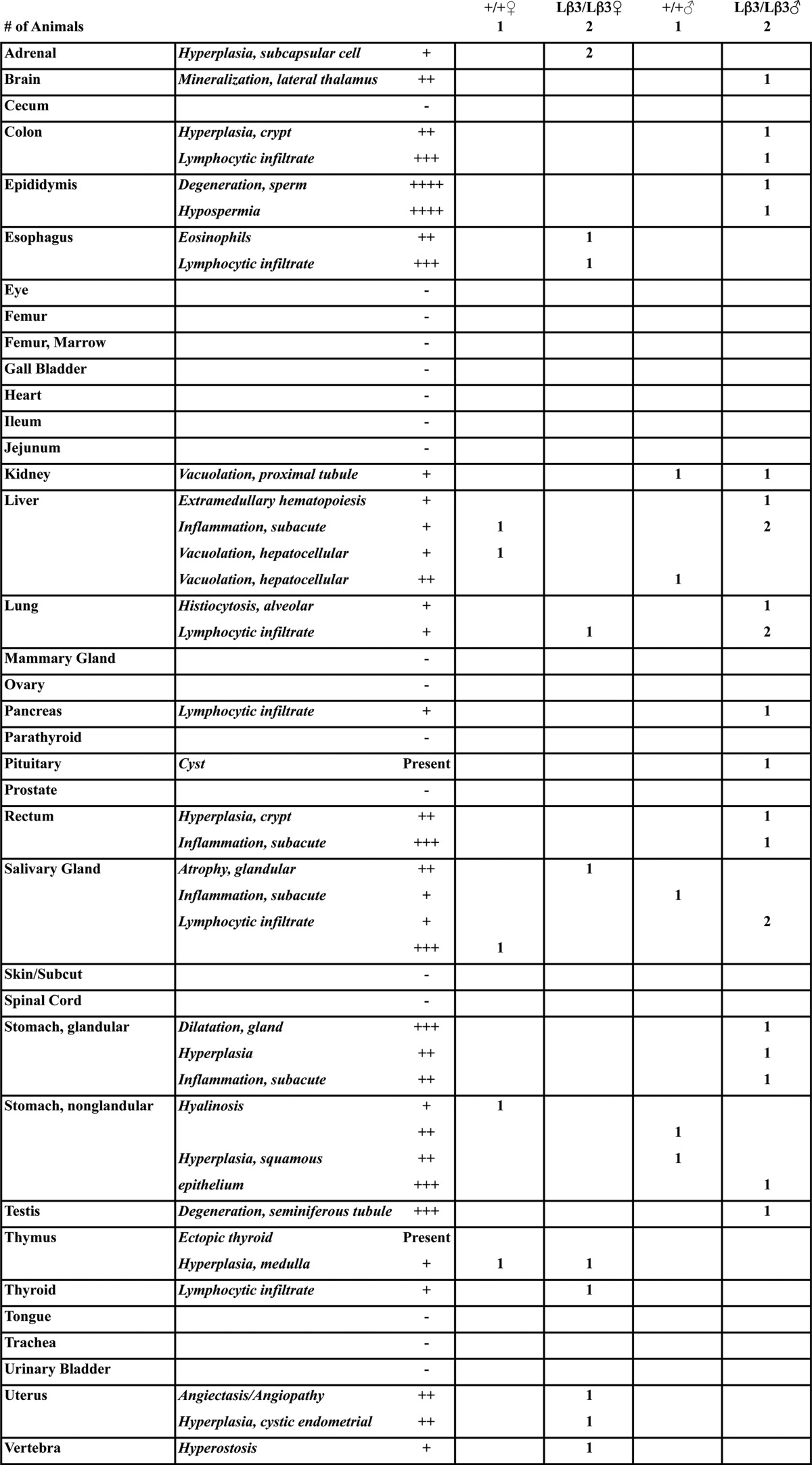
TABLE 2.
Pathology report for lymphoid tissues
The grade (severity) is as follows: +, minimal; ++, mild; +++, moderate; ++++, severe.
To further study the immunological effects of replacing TGF-β1 with TGF-β3, FACS analysis was performed to examine the immune profile of T cells in the TGF-β1Lβ3/Lβ3 mice, as TGF-β1−/− mice exhibit extreme lymphoproliferation and overactivation of T cells. Analysis of four female mice (aged 5–7 months) showed no significant difference in either the percent of T cell receptor β+ lymphocytes between TGF-β1Lβ3/Lβ3 and TGF-β1+/+ mice (Fig. 7D) or the numbers of activated CD44hi and naive CD62Lhi T cells (data not shown). However, there was a slight alteration in the CD4+/CD8+ ratio of the TGF-β1Lβ3/Lβ3 mice, but this was not as dramatic as that seen in the TGF-β1−/− mice (Fig. 7E). Because TGF-β1 expression is known to regulate the development of Tregs (27), we examined the frequencies of Foxp3+ Tregs in lymphoid tissues. In the peripheral lymph nodes of the TGF-β1Lβ3/Lβ3 mice, there was a significant increase in the percent of Foxp3+ regulatory T cells (Fig. 7F), but this increase was not see in any other location. Inflammatory cytokines, however, were also analyzed, and no difference could be detected in the production of IFN-γ, IL-17, IL-10, or IL-9 (Fig. 7G and data not shown). Overall, these findings hint at the possibility that TGF-β3 has immunoregulatory properties similar to TGF-β1, as there were no signs in TGF-β1Lβ3/Lβ3 mice of the uncontrolled autoimmunity that is associated with TGF-β1−/− mice. In addition, multifocal inflammation was not responsible for the reduced weight of these mice when compared with the controls.
Improved Glucose Homeostasis in TGF-β1Lβ3/Lβ3 Mice
TGF-β signaling is known to play a key role in glucose homeostasis. We have previously reported that a loss of the TGF-β signal transducer Smad3 results in improved whole body glucose homeostasis via beneficial effects on pancreatic islet beta cells (36) and the WAT (26). Smad3−/− WAT exhibits phenotypic and functional characteristics typically seen in brown adipose tissue. Moreover, Smad3−/− mice are resistant to the obesogenic and diabetic effects of a high fat diet challenge. Finally, levels of TGF-β1 are elevated as a function of obesity, and a TGF-β-neutralization antibody (1D11) protects mice from obesity and diabetes.
We therefore evaluated the metabolic phenotype of TGF-β1Lβ3/Lβ3 mice to determine whether it contributed to the reduced weight gain. As mentioned, the TGF-β1Lβ3/Lβ3 mice were mainly fed a dough diet as a means of offsetting any weight loss that could be caused by tooth defects, but instead this diet only exaggerated the discrepancies in weight gain (Figs. 5B and 8A), as the male TGF-β1Lβ3/Lβ3 mice differed from the controls by as much as 20 g. Both monitored groups ate about the same daily amount of food, eliminating the possibility that a difference in energy intake could explain the reduced body weight. However, when the weight of the mice was factored in, the food intake in the TGF-β1Lβ3/Lβ3 mice was actually significantly higher than the controls (Fig. 8B). The TGF-β1Lβ3/Lβ3 mice displayed reduced fat mass compared with that seen in control littermates, and fat accumulation in all the fat depots was significantly less in TGF-β1Lβ3/Lβ3 mice, and the layer of fat between the muscle and the dermis was reduced (Fig. 8C). Furthermore, we observed reduced hepatic steatosis, a condition associated with fat accumulation in the liver (Fig. 8D). Adipocytes from the TGF-β1Lβ3/Lβ3 mouse epididymal WAT appeared significantly smaller (Fig. 8E), as confirmed by quantification of the adipocyte diameter (Fig. 8F). Morphologically, these smaller adipocyte cells were multilocular and similar to brown adipocytes, compared with normal white adipocytes that are characterized by unilocular lipid droplets.
FIGURE 8.

A, B6129 TGF-β1Lβ3/Lβ3 mice remain small and thin although the control littermate mice show weight gain on a high fat dough diet. Typical male mice are shown at 33 weeks of age. B, daily amount of pellet food consumed by the female mice was measured and equilibrated using the body weight to obtain the food intake. The TGF-β1Lβ3/Lβ3 mice show significantly higher food intake when compared with the TGF-β1+/Lβ3 mice (***, p ≤ 0.001, mean ± S.E.). C, adipose tissue underlying the skin dermis is reduced in an aged “15-week-old” TGF-β1Lβ3/Lβ3 mouse. D, TGF-β1Lβ3/Lβ3 mice do not develop steatosis like TGF-β1+/+ mice as seen with H&E (40 weeks) and Oil Red O (17 weeks) staining. E and F, adipocyte size is reduced in the TGF-β1Lβ3/Lβ3 mice (****, p ≤ 0.0001, mean ± S.E.).
With the histological findings seen in the TGF-β1Lβ3/Lβ3 mice, we next examined gene expression from the epididymal WAT. Gene expression analysis revealed elevated mRNA transcripts of genes that represent brown adipocytes such as UCP-1, PGC1-α, and Cidea (Fig. 9A) (26). Because of the brown adipocyte-specific gene signature, glucose homeostasis in the TGF-β1Lβ3/Lβ3 mice was then assessed where we observed increased glucose tolerance in the TGF-β1Lβ3/Lβ3 mice (Fig. 9B). Fasting insulin levels were significantly lower in TGF-β1Lβ3/Lβ3 mice (Fig. 9C). The adipokine leptin was also significantly lower in TGF-β1Lβ3/Lβ3 mice, consistent with the improved glucose metabolism in the TGF-β1Lβ3/Lβ3 mice (Fig. 9C). Taken together, these results suggest that improved metabolic phenotypes in the form of reduced body weight and improved glucose homeostasis can be attributed to the appearance of brown-like cells in the WAT of TGF-β1Lβ3/Lβ3 mice.
FIGURE 9.

A, gene expression analysis from white adipose tissue that shows elevated mRNA transcripts of genes that represent brown adipocytes. B, TGF-β1Lβ3/Lβ3 mice display increased glucose tolerance as compared with the wild type controls (**, p ≤ 0.01; *, p ≤ 0.05, mean ± S.E.). C, serum levels of insulin and leptin are significantly reduced in the TGF-β1Lβ3/Lβ3 mice (*, p ≤ 0.05, mean ± S.E.). B and C, metabolic studies were performed on “12–16-week old” mice after an overnight fast. D, gene expression analysis shows reduced expression of the TGF β receptor 1 from white adipose tissue of the TGF-β1Lβ3/Lβ3 mice (**, p ≤ 0.01, mean ± S.E.). E, white adipose tissue from the TGF-β1Lβ3/Lβ3 mice also concurrently display decreased expression in TGF-β target genes (*, p ≤ 0.05, mean ± S.E.).
These findings mimic the metabolic phenotype of the Smad3−/− mouse, yet the TGF-β1Lβ3/Lβ3 mice do not share the same immune or skeletal phenotypes as these mice (26, 37, 38). In addition, inhibition of TGF-β-mediated signaling through an anti-TGF-β neutralizing antibody also promotes improved glucose tolerance and reduced weight gain in mouse models of obesity (26). Because of the parallels between the TGF-β1Lβ3/Lβ3 mice and these mouse models of impaired TGF-β signaling, we decided to examine the WAT from the mice for any shifts associated with the cellular mechanisms mediating TGF-β signaling. In the WAT of the TGF-β1Lβ3/Lβ3 mice, the expression of the level of the TGF-β receptor I was significantly reduced, which, in turn, could lead to a decrease in the capacity of adipocytes to respond to TGF-β signaling (Fig. 9D). This correlated with a trend toward increased levels of PGC-1α in the WAT of the TGF-β1Lβ3/Lβ3 mice (Fig. 9A), as the expression of PGC-1α is known to be inhibited by Smad3 (26). The effects of reduced receptor I expression in the WAT was further confirmed by reductions of other known TGF-β target genes that are normally induced with TGF-β signaling such as Smad7 and JunB (Fig. 9E). Isoform differences in regulating the expression of the TGF-β receptors have been reported where TGF-β1 was shown to induce more receptor I levels than TGF-β3 with no difference in phosphorylated Smad2 or Smad3, yet this effect involved mouse lung tissue (39). Reduced TGF-β-mediated signaling, however, may be one possible explanation why the adipose tissue in the TGF-β1Lβ3/Lβ3 mice displays characteristics similar with brown fat.
DISCUSSION
Even though the TGF-β isoforms share a high degree of homology, the heterology between each isoform is also evolutionarily well conserved so that the sequence identity for each protein has been well maintained in the mammals. Deciphering the nature of these conserved heterologous motifs will help to further characterize the physiological roles of these important cytokines. TGF-β1, the prototype member for the TGF-β superfamily, is the most abundant circulating isoform in plasma (40), is present in the bone matrix at 200 μg kg−1 (34), and is well characterized for its role in regulating immune homeostasis (41). We therefore decided to target the TGF-β1 genetic locus for replacement with another TGF-β homologue to identify any functional role that is unique to this homologue. TGF-β3, an isoform prominently known for its involvement in palate fusion, was selected to replace TGF-β1 because both homologues share similar binding affinities to the TGF-β receptor TβR-II (28). We successfully generated a knock-in of a chimeric TGF-β1/3 protein, and these TGF-β1Lβ3/Lβ3 mice provide a good animal model to study the isoform-specific properties between the TGF-β proteins.
In the adult mouse, TGF-β3 expression is prominent in brain, heart, adipose tissue, and testis but, in general, TGF-β3 levels are less abundant than the other two TGF-β homologues (42, 43). Consequently, TGF-β3 is not as well understood as the two other isoforms with regard to its role in physiology and disease states (44, 45). TGF-β3, however, should be comprehensively studied, as it is sure to possess a wide range of biological activities like the other TGF-β proteins. The defects in palate fusion and lung morphogenesis in the TGF-β3−/− mice, for example, suggest that TGF-β3 plays an important role in epithelial-mesenchymal interactions (15, 16). In the heart, genetic mutations in human Tgfb3 have been correlated to arrhythmogeneic right ventricular dysplasia type 1 (46), whereas a gene polymorphism has been linked to hypertension (47). Genetic polymorphisms in the Tgfb3 gene have also been associated with cleft palate (48) and with pulmonary fibrosis in sarcoidosis patients (49), both of which correlate well with TGF-β3's known roles in palate fusion and scarring. In addition, increased levels of TGF-β3 are seen in patients with HELLP syndrome (hemolysis-elevated liver enzymes-low platelet count) (50). In cases of cancer, however, TGF-β3 expression is thought to have protective effects against tumorigenesis throughout a range of tissues (45). Unlike TGF-β1, TGF-β3 also helps to protect keratinocytes against the death-inducing effects of agents like 12-O-tetradecanoylphorbol-13-acetate, as TGF-β3 contrasts with TGF-β1 by suppressing 12-O-tetradecanoylphorbol-13-acetate-induced Jun kinase activity in keratinocytes (51). Finally, TGF-β3 appears to hold promise for use in tissue engineering (52). The TGF-β1Lβ3/Lβ3 mice will therefore allow for further characterization of TGF-β3 and will particularly contribute to our understanding of the conserved heterology that exists between the TGF-β isoforms.
In TGF-β1Lβ3/Lβ3 mice, the TGF-β1 LAP is maintained, whereas only the receptor-binding ligand is switched to TGF-β3. The TGF-β1 LAP domain contains key determinants that affect sequestration and activation, such as an RGD sequence that is needed for integrin-mediated activation (53). The strategy of the chimeric TGF-β1/3 protein is to use the TGF-β1 LAP for localization and activation and for signaling to occur via the TGF-β3 ligand, allowing for true direct comparison of the inherent functions of each ligand.
The TGF-β1Lβ3/Lβ3 mice were born at the expected genotypic ratio without complications in yolk sac vasculogenesis typically linked to defects that interrupt TGF-β1 signaling (11). TGF-β3 has been shown to be partially redundant with TGF-β1, at least in terms of brain vascular morphogenesis (54). Such similarities between the two isoforms in terms of vascular development may account for this lack of embryonic lethality. The TGF-β1Lβ3/Lβ3 pups were also born without any congenital birth defects. The TGF-β1Lβ3/Lβ3 females, however, produced no viable pups. Under these circumstances with the TGF-β1Lβ3/+ mothers, TGF-β1 will essentially cross the placenta to the embryos and, additionally, will be present in the mother's milk, thus obscuring any analysis of the isoform-specific roles of these homologues in terms of understanding development (32). Nevertheless, physiological homeostasis was maintained over time, as most of the organs examined were essentially normal with no signs of any pathological disorders. Excess circulating TGF-β1 is known to be associated with chronic fibrotic disorders that include glomerulonephritis and renal failure (55), but the knocked in tagged TGF-β3 did not appear to cause any excess TGF-β signaling, fibrosis, or other complications in the liver or kidney.
Multifocal inflammation is another hallmark associated with targeted disruption of the TGF-β1 gene, but the severe autoimmunity phenotype seen in the TGF-β1−/− mice was also essentially rescued by knock-in of TGF-β3. Because TGF-β3 is found in low concentrations in the plasma (<0.1 ng/ml) (40), its role in regulating immunity has not been as extensively studied as the role of TGF-β1. Using TGF-β3 fate-reporter mice, TGF-β3 expression was detected in CD4+ T cells, CD8+ T cells, γδ T cells, and B cells but not in cells of the myeloid lineage (56). In terms of immunoregulation, TGF-β3 has been shown to be more functionally potent than TGF-β1 at inducing chemotaxis of human mast cells but is less effective at inhibiting proliferation of these cells (28). In addition, TGF-β3 has also been reported to be secreted by resting B cells to promote the expansion of CD4+CD25+ Foxp3+ cells (57). However, combining resting B cells with purified CD4+CD25− T cells did not result in de novo development of Tregs. Finally, unlike TGF-β1, TGF-β3 may promote Th17 cells to become pathogenic (56), where the substitution of TGF-β1 for TGF-β3 during Th17 cell induction essentially drove differentiation toward an alternative phenotype that expressed a more pathogenic molecular signature. These TGF-β3-induced Th17 cells also had less phosphorylated Smad2 and Smad3 than TGF-β1-induced Th17 cells, suggesting a difference between these isoforms in terms of cell signaling. Despite the reported generation of Th17 cells with a more pathogenic phenotype, our TGF-β1Lβ3/Lβ3 mice appear to exhibit no gross immunological abnormalities and no significant differences in the production of IL-17 and other cytokines such as IFN-γ, IL-10, or IL-9. Essentially, the TGF-β1Lβ3/Lβ3 mice showed no signs of multifocal inflammation, and there were no significant differences in the percentage of T cell receptor β+ lymphocytes. Slight alterations were seen in CD4+/CD8+ ratios, but these were not as severe as the TGF-β1−/− mice. Finally, the percentage of Foxp3+ regulatory T cells was significantly increased in the peripheral lymph nodes of the TGF-β1Lβ3/Lβ3 mice, possibly due to the aforementioned proliferative properties of TGF-β3 on Tregs. Taken together, the immunological findings learned from the TGF-β1Lβ3/Lβ3 mice demonstrate that TGF-β3 has immunomodulatory properties similar to TGF-β1.
Although the key pathological phenotypes linked to the TGF-β1−/− knock-out appear to be compensated for by the knock-in of TGF-β3, the TGF-β1Lβ3/Lβ3 mice still have a shorter life span compared with the littermate controls, and so the two isoforms are not totally interchangeable. The TGF-β1Lβ3/Lβ3 mice also have skeletal and tooth defects that suggest an incomplete rescue by TGF-β3 for all of the functions of TGF-β1. For mouse limb bud mesenchymal cells, chondrogenesis is differentially affected by the TGF-β isoforms where micromass cultures of these cells are more responsive to TGF-β1 than TGF-β3 (58). In addition, TGF-β3 appears to inhibit osteogenic differentiation of mesenchymal stem cells, although TGF-β1 and TGF-β2 have been reported to stimulate osteogenesis (59). No congenital skeletal defects were seen in the TGF-β1Lβ3/Lβ3 pups, but maternal transfer of TGF-β1 could mask any developmental problems (32). In the TGF-β1Lβ3/Lβ3 mice, skeletal defects such as kyphosis were not prominent in the young mice but became more visible over time. In the TGF-β1−/− knock-out mice, bone is less elastic and more fragile; the width of the tibial growth plate is reduced, and osteoblasts are absent on the trabecular bone (34, 60, 61). The teeth also show dental attrition (33) resulting from a lack of TGF-β1 expression. The TGF-β1Lβ3/Lβ3 mice share similarities with the TGF-β1−/− knock-out mice in terms of skeletal and tooth defects. In bone, latent TGF-β1 is normally sequestered in the bone matrix where it can become activated and released by osteoclasts during bone resorption. Secretion of active TGF-β1 leads, in turn, to the recruitment of osteoblasts for new bone formation. Interestingly, bone resorption conditioned medium obtained from osteoclasts promotes the migration of BMSCs, a precursor of osteoblasts, and this process is dependent upon the release of active TGF-β1 (34). Unlike TGF-β1, however, depletion of TGF-β3 with neutralizing antibodies had no effect on BMSC migration. An altered balance in bone resorption versus formation could account for some of the bone defects seen in the TGF-β1Lβ3/Lβ3 mice.
The most surprising phenotype of the TGF-β1Lβ3/Lβ3 mice is the reduced weight gain when compared with the controls, which may result from a possible isoform-specific difference in metabolism regulation. TGF-β1 expression has been previously linked to adiposity, wherein elevated plasma levels of this isoform are correlated with an increased body mass index in both humans and mice (26). Adipose tissue in obese mouse models has also been shown to contain increased expression of TGF-β1 (62). Finally, TGF-β1 can promote the secretion of cytokines such as PAI-1 from adipocytes, and elevated plasma levels of PAI-1 have been linked to obesity (62). In our TGF-β1Lβ3/Lβ3 mice, the targeted exchange of the TGF-β isoforms actually resulted in reduced weight gain and suppressed fat accumulation. Inhibition of TGF-β signaling through the pan-specific anti-TGF-β antibody, 1D11, suppressed body weight gain in obese mice, in agreement with TGF-β1's association with adiposity, yet a high dose of this same antibody resulted in hyperplasia and inflammation of the tongue and esophagus in various strains of mice (26, 35). TGF-β1Lβ3/Lβ3 mice lacked any major signs of inflammation or hyperplasia over the course of their life spans. Expression of TGF-β3 by the knock-in allele seems to provide sufficient immunoregulatory signaling, yet the TGF-β1Lβ3/Lβ3 mice still exhibit an altered metabolic phenotype that suggests a possible isoform-specific difference in terms of adipocyte regulation. Adipocyte size is smaller in the TGF-β1Lβ3/Lβ3 mice and expresses a brown adipocyte-specific gene signature indicative of a probable switch to a more brown fat-like phenotype wherein excess energy is dissipated as heat. The TGF-β1Lβ3/Lβ3 mice also show more glucose tolerance and lower levels of serum insulin and leptin, which is associated with the phenotypic change in adipocyte morphology. Results presented here are consistent with previous findings where it was reported that disruption of TGF-β signaling, via deletion of Smad3, provides protection from obesity and diabetes (26). The findings in the TGF-β1Lβ3/Lβ3 mice demonstrate that some TGF-β functions, such as immune regulation, are shared between these two isoforms, although other roles are isoform-specific, as in the case of metabolism and bone resorption and formation. These two TGF-β homologues may have some similarities to bone morphogenetic proteins, growth factors in the TGF-β superfamily, which can play differential roles in adipogenesis, whereas BMP2 and BMP4 promote white fat differentiation, and BMP7 induces brown fat adipogenesis (63).
TGF-β1 and TGF-β3 use the same cell receptors and downstream signaling pathways, yet the phenotype of the TGF-β1Lβ3/Lβ3 mice suggests that some nonoverlapping functions exist between these two isoforms. With the TGF-β ligands, dissimilarity in aggregation properties and interactions with other physiological molecules such as heparin could potentially contribute to some of the isoform-specific traits identified in these mice (64, 65). Another possible explanation for differing cellular responses may have to do with the conformational shape of the ligands when binding to the receptors. TGF-β3, for example, is known to adopt a more open conformation, whereas TGF-β1 is more closed (66). The genetically engineered TGF-β ligand exchange in the TGF-β1Lβ3/Lβ3 mice provides a good method for studying both redundancies and dissimilarities between these isoforms in vivo. Our preliminary investigation into the phenotype of the TGF-β1Lβ3/Lβ3 mice has provided new insights into the physiological roles of the TGF-β homologues. Some overlap appears with immunoregulation in the mice, but the conserved heterology in the ligands seems to confer some isoform-specific activities in terms of regulating metabolism and mineralization. Further analysis of the TGF-β1Lβ3/Lβ3 mice will be performed to determine the mechanisms behind the isoform-specific differences and to detect any novel biological activities that may be distinct to either TGF-β homologue. Moreover, these findings, taken together, are relevant to the design and development of anti-TGF-β isoform-specific therapies for human diseases with deregulated TGF-β signaling.
Acknowledgments
We thank Drs. Miriam Anver, Yansong Bian, Naoto Haruyama, Kenn Holmbeck, Mina Mina, Alfredo Molinolo, and Lalage M. Wakefield for their helpful discussions during the course of these studies. We are also thankful to Andrew Cho for work with bone and teeth, Danielle Donahue for assistance with the microcomputed tomography, Shelagh Johnson for expert editorial assistance, and Ruth Yaskovich for help with histology. In addition, we thank the Gene Transfer Core for the generation of this mouse model.
This work was supported, in whole or in part, by National Institutes of Health Grant ZO1 DE000698-13 and grants from Divisions of Intramural Research, NIDCR, NCI, and NIDDK.
- LAP
- latency associated peptide
- WAT
- white adipose tissue
- au
- arbitrary unit
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BMSC
- bone mesenchymal stem cell.
REFERENCES
- 1. Massagué J. (1987) The TGF-β family of growth and differentiation factors. Cell 49, 437–438 [DOI] [PubMed] [Google Scholar]
- 2. Sporn M. B., Roberts A. B., Wakefield L. M., de Crombrugghe B. (1987) Some recent advances in the chemistry and biology of transforming growth factor-β. J. Cell Biol. 105, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 4. Mittl P. R., Priestle J. P., Cox D. A., McMaster G., Cerletti N., Grütter M. G. (1996) The crystal structure of TGF-β3 and comparison to TGF-β2: Implications for receptor binding. Protein Sci. 5, 1261–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. ten Dijke P., Hansen P., Iwata K. K., Pieler C., Foulkes J. G. (1988) Identification of another member of the transforming growth factor type β gene family. Proc. Natl. Acad. Sci. U.S.A. 85, 4715–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kingsley D. M. (1994) The TGF-β superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 8, 133–146 [DOI] [PubMed] [Google Scholar]
- 7. Graycar J. L., Miller D. A., Arrick B. A., Lyons R. M., Moses H. L., Derynck R. (1989) Human transforming growth factor-β3: recombinant expression, purification, and biological activities in comparison with transforming growth factors-β1 and -β2. Mol. Endocrinol. 3, 1977–1986 [DOI] [PubMed] [Google Scholar]
- 8. Roberts A. B., Sporn M. B. (1992) Differential expression of the TGF-β isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol. Reprod. Dev. 32, 91–98 [DOI] [PubMed] [Google Scholar]
- 9. Millan F. A., Denhez F., Kondaiah P., Akhurst R. J. (1991) Embryonic gene expression patterns of TGF-β1, β2, and β3 suggest different developmental function in vivo. Development 111, 131–143 [DOI] [PubMed] [Google Scholar]
- 10. Pelton R. W., Saxena B., Jones M., Moses H. L., Gold L. I. (1991) Immunohistochemical localization of TGFβ1, TGFβ2, and TGFβ3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J. Cell Biol. 115, 1091–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dickson M. C., Martin J. S., Cousins F. M., Kulkarni A. B., Karlsson S., Akhurst R. J. (1995) Defective haematopoiesis and vasculogenesis in transforming growth factor β1 knock out mice. Development 121, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 12. Shull M. M., Ormsby I., Kier A. B., Pawlowski S., Diebold R. J., Yin M., Allen R., Sidman C., Proetzel G. (1992) Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 359, 693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kulkarni A. B., Huh C. G., Becker D., Geiser A., Lyght M., Flanders K. C., Roberts A. B., Sporn M. B., Ward J. M., Karlsson S. (1993) Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A. 90, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T. (1997) TGF-β2 knockout mice have multiple developmental defects that are non-overlapping with other TGF-β knockout phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Proetzel G., Pawlowski S. A., Wiles M. V., Yin M., Boivin G. P., Howles P. N., Ding J., Ferguson M. W., Doetschman T. (1995) Transforming growth factor-β3 is required for secondary palate fusion. Nat. Genet. 11, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaartinen V., Voncken J. W., Shuler C., Warburton D., Bu D., Heisterkamp N., Groffen J. (1995) Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415–421 [DOI] [PubMed] [Google Scholar]
- 17. Brunet C. L., Sharpe P. M., Ferguson M. W. (1995) Inhibition of TGF-β3 (but not TGF-β1 or TGF-β2) activity prevents normal mouse embryonic palate fusion. Int. J. Dev. Biol. 39, 345–355 [PubMed] [Google Scholar]
- 18. Yang L. T., Kaartinen V. (2007) Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 312, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah M., Foreman D., Ferguson M. (1995) Neutralisation of TGF-β1 and TGF-β2 or exogenous addition of TGF-β3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 108, 985–1002 [DOI] [PubMed] [Google Scholar]
- 20. Bandyopadhyay B., Fan J., Guan S., Li Y., Chen M., Woodley D. T., Li W. (2006) A “traffic control” role for TGFβ3: orchestrating dermal and epidermal cell motility during wound healing. J. Cell Biol. 172, 1093–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wolfraim L. A., Alkemade G. M., Alex B., Sharpe S., Parks W. T., Letterio J. J. (2002) Development and application of fully functional epitope-tagged forms of transforming growth factor-β. J. Immunol. Methods 266, 7–18 [DOI] [PubMed] [Google Scholar]
- 22. Geiser A., Kim S., Roberts A., Sporn M. (1991) Characterization of the mouse transforming growth factor-β1 promoter and activation by the Ha-ras oncogene. Mol. Cell. Biol. 11, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tybulewicz V. L., Crawford C. E., Jackson P. K., Bronson R. T., Mulligan R. C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 24. Figueroa J. D., Flanders K. C., Garcia-Closas M., Anderson W. F., Yang X. R., Matsuno R. K., Duggan M. A., Pfeiffer R. M., Ooshima A., Cornelison R., Gierach G. L., Brinton L. A., Lissowska J., Peplonska B., Wakefield L. M., Sherman M. E. (2010) Expression of TGF-β signaling factors in invasive breast cancers: relationships with age at diagnosis and tumor characteristics. Breast Cancer Res. Treat. 121, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jakowlew S., Moody T., You L., Mariano J. (1998) Transforming growth factor-β expression in mouse lung carcinogenesis. Exp. Lung Res. 24, 579–593 [DOI] [PubMed] [Google Scholar]
- 26. Yadav H., Quijano C., Kamaraju A. K., Gavrilova O., Malek R., Chen W., Zerfas P., Zhigang D., Wright E. C., Stuelten C., Sun P., Lonning S., Skarulis M., Sumner A. E., Finkel T., Rane S. G. (2011) Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 14, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y., Zhang P., Li J., Kulkarni A. B., Perruche S., Chen W. 2008. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 9, 632–640 [DOI] [PubMed] [Google Scholar]
- 28. Olsson N., Piek E., ten Dijke P., Nilsson G. (2000) Human mast cell migration in response to members of the transforming growth factor-β family. J. Leukocyte Biol. 67, 350–356 [DOI] [PubMed] [Google Scholar]
- 29. Böttinger E. P., Factor V. M., Tsang M. L., Weatherbee J. A, Kopp J. B., Qian S. W., Wakefield L. M., Roberts A. B., Thorgeirsson S. S., Sporn M. B. (1996) The recombinant proregion of transforming growth factor β1 (latency-associated peptide) inhibits active transforming growth factor β1 in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 5877–5882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyons R., Gentry L., Purchio A., Moses H. (1990) Mechanism of activation of latent recombinant growth factor β1 by plasmin. J. Cell Biol. 110, 1361–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schultz-Cherry S., Murphy-Ullich J. E. (1993) Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J. Cell Biol. 122, 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Letterio J., Geiser A., Kulkarni A., Roche N., Sporn M., Roberts A. (1994) Maternal rescue of transforming growth factor-β1 null mice. Science 264, 1936–1938 [DOI] [PubMed] [Google Scholar]
- 33. D'Souza R. N., Cavender A., Dickinson D., Roberts A., Letterio J. (1998) TGF-β1 is essential for the homeostasis of the dentin-pulp complex. Eur. J. Oral Sci. 106, Suppl. 1, 185–191 [DOI] [PubMed] [Google Scholar]
- 34. Tang Y., Wu X., Lei W., Pang L., Wan C., Shi Z., Zhao L., Nagy T. R., Peng X., Hu J., Feng X., Van Hul W., Wan M., Cao X. (2009) TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vitsky A., Waire J., Pawliuk R., Bond A., Matthews D., Lacasse E., Hawes M. L., Nelson C., Richards S., Piepenhagen P. A., Garman R. D., Andrews L., Thurberg B. L., Lonning S., Ledbetter S., Ruzek M. C. (2009) Homeostatic role of transforming growth factor-β in the oral cavity and esophagus of mice and its expression by mast cells in these tissues. Am. J. Pathol. 174, 2137–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin H. M., Lee J. H., Yadav H., Kamaraju A. K., Liu E., Zhigang D., Vieira A., Kim S. J., Collins H., Matschinsky F., Harlan D. M., Roberts A. B., Rane S. G. (2009) Transforming growth factor-β/Smad3 signaling regulates insulin gene transcription and pancreatic islet β-cell function. J. Biol. Chem. 284, 12246–12257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Datto M. B., Frederick J. P., Pan L., Borton A. J., Zhuang Y., Wang X. F. (1999) Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol. Cell. Biol. 19, 2495–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang X., Letterio J. J., Lechleider R. J., Chen L., Hayman R., Gu H., Roberts A. B., Deng C. (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18, 1280–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ask K., Bonniaud P., Maass K., Eickelberg O., Margetts P. J., Warburton D., Groffen J., Gauldie J., Kolb M. (2008) Progressive pulmonary fibrosis is mediated by TGF-β isoform 1 but not TGF-β3. Int. J. Biochem. Cell Biol. 40, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wakefield L. M., Letterio J. J., Chen T., Danielpour D., Allison R. S., Pai L. H., Denicoff A. M., Noone M. H., Cowan K. H., O'Shaughnessy J. A., et al. (1995) Transforming growth factor-β1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin. Cancer Res. 1, 129–136 [PubMed] [Google Scholar]
- 41. Li M. O., Wan Y. Y., Sanjabi S., Robertson A. K., Flavell R. A. (2006) Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 [DOI] [PubMed] [Google Scholar]
- 42. Miller D. A., Lee A., Matsui Y., Chen E. Y., Moses H. L., Derynck R. (1989) Complementary DNA cloning of the murine transforming growth factor-β3 (TGFβ3) precursor and the comparative expression of TGFβ3 and TGFβ1 messenger RNA in murine embryos and adult tissues. Mol. Endocrinol. 3, 1926–1934 [DOI] [PubMed] [Google Scholar]
- 43. Roberts A., Sporn M. (1991) in Peptide Growth Factors and their Receptors I (Sporn M., Roberts A. eds) Springer-Verlag, New York [Google Scholar]
- 44. Doetschman T., Georgieva T., Li H., Reed T. D., Grisham C., Friel J., Estabrook M. A., Gard C., Sanford L. P., Azhar M. (2012) Generation of mice with a conditional allele for the transforming growth factor β3 gene. Genesis 50, 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laverty H. G., Wakefield L. M., Occleston N. L., O'Kane S., Ferguson M. W. (2009) TGF-β3 and cancer: a review. Cytokine Growth Factor Rev. 20, 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beffagna G., Occhi G., Nava A., Vitiello L., Ditadi A., Basso C., Bauce B., Carraro G., Thiene G., Towbin J. A., Danieli G. A., Rampazzo A. (2005) Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 65, 366–373 [DOI] [PubMed] [Google Scholar]
- 47. Hu B. C., Li L., Sun R. H., Gao P. J., Zhu D. L., Wang J. G., Chu S. L. (2010) The association between transforming growth factor β3 polymorphisms and left ventricular structure in hypertensive subjects. Clin. Chim. Acta 411, 558–562 [DOI] [PubMed] [Google Scholar]
- 48. Saleem S., Rajendran R., Moinak B., Anna J., Pramod B. J. (2012) Evidence for transforming growth factor-β3 gene polymorphism in non-syndromic cleft lip and palate patients from Indian sub-continent. Med. Oral Patol. Oral Cir. Bucal. 17, e197–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kruit A., Grutters J. C., Ruven H. J., van Moorsel C. H., Weiskirchen R., Mengsteab S., van den Bosch J. M. (2006) Transforming growth factor-β gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest 129, 1584–1591 [DOI] [PubMed] [Google Scholar]
- 50. Emanuelli M., Giannubilo S. R., Landi B., Sartini D., Pierella F., Corradetti A., Tranquilli A. L. (2008) Placental overexpression of transforming growth factor-β3 in the HELLP syndrome. Gynecol. Obstet. Invest. 65, 1–5 [DOI] [PubMed] [Google Scholar]
- 51. Li J., Foitzik K., Calautti E., Baden H., Doetschman T., Dotto G. P. (1999) TGF-β3, but not TGF-β1, protects keratinocytes against 12-O-tetradecanoylphorbol-13-acetate-induced cell death in vitro and in vivo. J. Biol. Chem. 274, 4213–4219 [DOI] [PubMed] [Google Scholar]
- 52. Hao J., Varshney R. R., Wang D. A. (2008) TGF-β3: A promising growth factor in engineered organogenesis. Expert Opin. Biol. Ther. 8, 1485–1493 [DOI] [PubMed] [Google Scholar]
- 53. Yang Z., Mu Z., Dabovic B., Jurukovski V., Yu D., Sung J., Xiong X., Munger J. S. (2007) Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 176, 787–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mu Z., Yang Z., Yu D., Zhao Z., Munger J. S. (2008) TGFβ1 and TGFβ3 are partially redundant effectors in brain vascular morphogenesis. Mech. Dev. 125, 508–516 [DOI] [PubMed] [Google Scholar]
- 55. Sanderson N., Factor V., Nagy P., Kopp J., Kondaiah P., Wakefield L., Roberts A. B., Sporn M. B., Thorgeirsson S. S. (1995) Hepatic expression of mature transforming growth factor β1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. U.S.A. 92, 2572–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee Y., Awasthi A., Yosef N., Quintana F. J., Xiao S., Peters A., Wu C., Kleinewietfeld M., Kunder S., Hafler D. A., Sobel R. A., Regev A., Kuchroo V. K. (2012) Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shah S., Qiao L. (2008) Resting B cells expand a CD4+CD25+Foxp3+ Treg population via TGF-β3. Eur. J. Immunol. 389, 2488–2498 [DOI] [PubMed] [Google Scholar]
- 58. Chimal-Monroy J., Díaz de León L. (1997) Differential effects of transforming growth factors β1, β2, β3, and β5 on chondrogenesis in mouse limb bud mesenchymal cells. Int. J. Dev. Biol. 41, 91–102 [PubMed] [Google Scholar]
- 59. Moioli E. K., Hong L., Mao J. J. (2007) Inhibition of osteogenic differentiation of human mesenchymal stem cells. Wound Repair Regen. 15, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Janssens K., ten Dijke P., Janssens S., Van Hul W. (2005) Transforming growth factor-β1 to the bone. Endocr. Rev. 26, 743–774 [DOI] [PubMed] [Google Scholar]
- 61. Geiser A. G., Zeng Q. Q., Sato M., Helvering L. M., Hirano T., Turner C. H. (1998) Decreased bone mass and bone elasticity in mice lacking the transforming growth factor-β1 gene. Bone 23, 87–93 [DOI] [PubMed] [Google Scholar]
- 62. Samad F., Yamamoto K., Pandey M., Loskutoff D. J. (1997) Elevated expression of transforming growth factor-β in adipose tissue from obese mice. Mol. Med. 3, 37–48 [PMC free article] [PubMed] [Google Scholar]
- 63. Tseng Y. H., Kokkotou E., Schulz T. J., Huang T. L., Winnay J. N., Taniguchi C. M., Tran T. T., Suzuki R., Espinoza D. O., Yamamoto Y., Ahrens M. J., Dudley A. T., Norris A. W., Kulkarni R. N., Kahn C. R. (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 45, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pellaud J., Schote U., Arvinte T., Seelig J. (1999) Conformation and self-association of human recombinant transforming growth factor-β3 in aqueous solutions. J. Biol. Chem. 274, 7699–7704 [DOI] [PubMed] [Google Scholar]
- 65. Lyon M., Rushton G., Gallagher J. (1997) The interaction of the transforming growth factor-βs with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 272, 18000–18006 [DOI] [PubMed] [Google Scholar]
- 66. Hart P. J., Deep S., Taylor A. B., Shu Z., Hinck C. S., Hinck A. P. (2002) Crystal structure of the human TβR2 ectodomain–TGF-β3 complex. Nat. Struct. Biol. 9, 203–208 [DOI] [PubMed] [Google Scholar]