

Background: Tamoxifen (TX), a selective estrogen receptor modulator, enhances glutamate transporter (GLT-1) expression in astrocytes.
Results: TX up-regulated GLT-1 expression via the CREB and NF-κB pathways.
Conclusion: TX enhanced GLT-1 expression at the transcriptional level.
Significance: Understanding the mechanisms of TX action on GLT-1 will contribute to the development of neuroprotectants against excitotoxicity.
Keywords: Astrocytes, CREB, Glutamate, NF-κB, TGF-α), EAAT2, Tamoxifen
Abstract
Tamoxifen (TX), a selective estrogen receptor modulator, exerts antagonistic effects on breast tissue and is used to treat breast cancer. Recent evidence also suggests that it may act as an agonist in brain tissue. We reported previously that TX enhanced the expression and function of glutamate transporter 1 (GLT-1) in rat astrocytes, an effect that was mediated by TGF-α. To gain further insight into the mechanisms that mediate TX-induced up-regulation of GLT-1 (EAAT2 in humans), we investigated its effect on GLT-1 at the transcriptional level. TX phosphorylated the cAMP response element-binding protein (CREB) and recruited CREB to the GLT-1 promoter consensus site. The effect of TX on astrocytic GLT-1 was attenuated by the inhibition of PKA, the upstream activator of the CREB pathway. In addition, the effect of TX on GLT-1 promoter activity was abolished by the inhibition of the NF-κB pathway. Furthermore, TX recruited the NF-κB subunits p65 and p50 to the NF-κB binding domain of the GLT-1 promoter. Mutation of NF-κB (triple, −583/-282/-251) or CRE (-308) sites on the GLT-1 promoter led to significant repression of the promoter activity, but neither mutant completely abolished the TX-induced GLT-1 promoter activity. Mutation of both the NF-κB (-583/-282/-251) and CRE (-308) sites led to a complete abrogation of the effect of TX on GLT-1 promoter activity. Taken together, our findings establish that TX regulates GLT-1 via the CREB and NF-κB pathways.
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian CNS, and it has been implicated in numerous pathological disorders such as epilepsy, cerebral ischemia, Alzheimer disease, Parkinson disease, and schizophrenia (1). The astrocytic glutamate transporters play a major role in maintaining glutamate homeostasis by removing excessive synaptic glutamate, thus preventing excitotoxicity (2, 3). Among the five subtypes of human EAATs, two glial subtypes, excitatory amino acid transporter 1 (EAAT1)2 and EAAT2 (glutamate aspartate transporter and GLT-1, respectively, in rodents), are localized primarily in astrocytes and responsible for the clearance of excess glutamate from the synaptic cleft (3–6). Optimal regulation of astrocytic EAATs is essential because the dysfunction of these transporters leads to various neurological diseases (7). Therefore, understanding the regulatory mechanisms of astrocytic glutamate transporters function is crucial for the development of novel therapeutics to treat and prevent neurological diseases.
Glutamate transporter activity is regulated by gene expression, molecular targeting, trafficking, and posttranslational modifications (8). Up-regulation of the glutamate transporter affords neuroprotection in various animal disease models (9–12). Estrogen (estradiol-17β) has long been recognized as a neurotropic agent, affording neuroprotection in several in vitro and in vivo models of injury and neurodegenerative diseases (13, 14). However, the therapeutic efficacy of estrogen is limited because its long term use increases the risk of breast and uterine cancers, coronary heart disease, and stroke (15). Selective estrogen receptor modulators such as tamoxifen (TX) and raloxifene have gained attention as potential alternatives to estrogen therapy. TX affords neuroprotection in animal models of cerebral ischemia (16), stroke (17), and Parkinson disease (18). However, the underlying mechanism(s) of its neuroprotection have yet to be understood. Several studies have attributed the antioxidant and free radical scavenging properties of TX to its neuroprotection, whereas others have shown that TX inhibits the release of excitatory amino acids such as glutamate (19, 20).
Growth factors are known modulators of the expression and function of glutamate transporters (21). TGF-β, which is released from astrocytes in response to estrogen or TX, mediates the neuroprotective effects of both estrogen and TX (22). TGF-α has also been shown to afford protection in cerebral artery occlusion and cerebral ischemia models (23, 24). Both EGF and TGF-α induce GLT-1 expression via NF-κB activation in astrocytes (25). In our previous study, we showed that estrogen and TX induced up-regulation of GLT-1 via TGF-α (26). To further characterize the signaling pathways and molecular mechanisms involved in TX-induced up-regulation of GLT-1, we investigated the role of cAMP response element-binding protein (CREB) and NF-κB in TX-induced GLT-1 up-regulation. Our results demonstrate that both the CREB and NF-κB pathways are critical for TX-induced enhancement of GLT-1 expression.
EXPERIMENTAL PROCEDURES
Materials
Cell culture media and reagents were purchased from Invitrogen. TGF-α was from Peprotech (Rocky Hill, NJ). PP2, pyrrolidine dithiocarbamate, H89, G15, and G1 were obtained from Tocris Bioscience (Ellisville, MO). Estrogen, dibutyryl cAMP (dbcAMP), protease inhibitor mixture, and poly-D-ornithine were purchased from Sigma-Aldrich (St. Louis, MO). GLT-1, TGF-α, CREB, NF-κB, β-actin, and Src antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA) or Cell Signaling Technology (Danvers, MA). The RNA isolation kit was purchased from Qiagen (Valencia, CA). The luciferase reporter assay kit was obtained from Promega (Madison, WI). All chemicals were prepared in Hanks' buffered salt solution, 95% ethanol, or dimethyl sulfoxide according to the instructions of the manufacturer and diluted to final working concentrations in Opti-MEM prior to use.
Primary Cultures of Astrocytes
Astrocyte cultures were prepared as described previously (27). Briefly, after carefully removing the meninges, cerebral cortices of newborn (1-day-old) Sprague-Dawley rats were digested with dispase (Invitrogen). Astrocytes were then recovered by the repeated removal of dissociated cells and plated at a density of 1 × 105 cells/ml. Twenty-four hours after the initial plating, the medium was changed to preserve the adhering astrocytes and to remove neurons, microglia, and oligodendrocytes. The cultures were maintained at 37 °C in a 95% air, 5% CO2 incubator for 3 weeks in minimal essential medium supplemented with 10% horse serum, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. These cultures showed > 95% positive staining for the astrocyte-specific marker glial fibrillary acidic protein. All experiments were performed 3 weeks post-isolation.
Western Blot Analysis
Whole cell lysates for SDS-PAGE were prepared as described previously (28). Astrocytes were treated with the designated compounds for the indicated time periods, followed by two washes with cold Hanks' buffered salt solution. Subsequently, the cells were lysed with radioimmune precipitation assay buffer containing a protease inhibitor mixture. The protein concentration in the lysate was determined with BCA. Next, 30 μg of protein samples were mixed with Laemmli sample buffer containing 5% β-mercaptoethanol and heated at 95 °C for 5 min, followed by 10% SDS-PAGE under reducing conditions. The proteins were then electrophoretically transferred to a nitrocellulose membrane. Western blot analysis was performed using primary antibodies (GLT-1, 1:1000, Millipore or Santa Cruz Biotechnology; β-actin, 1:5000, Sigma-Aldrich), phospho-CREB, CREB (1:1000, Santa Cruz Biotechnology) followed by secondary antibodies (1:3000, anti-rabbit, anti-mouse IgG peroxidase conjugates; Promega). The immunoreactive proteins were detected by an enhanced chemiluminescence Western blotting detection kit (Pierce).
Mutagenesis
The GLT-1 (EAAT2) promoter containing −954 to +44 bp (954) sequences subcloned into the pGL3 basic plasmid vector (9) was used as the original template for mutations. The NF-κB triple mutant (-583/-272/-251) was a gift from Dr. Sitcheran (Texas A&M Health Sciences Center). To generate a mutation at the −308 CREB binding site, the sequence 5′-GCC CGG CGC GGG TGA TGT CAG CTC TCG ACG AAA-3′ was changed to 5′-GCC CGG CGC GGG TAA TGA CAG CTC TCG ACG AAA-3′ by PCR-based mutagenesis using a site-directed mutagenesis kit (Stratagene). Clones were sequenced to confirm the mutations.
Construction of the Deletion Mutants of the EAAT2 Promoter
Deletion constructs were generated from the EAAT2–954 (-954/+44) pGL3-basic vector. PCR was performed for 35 cycles at 94 °C for 1 min, at 58 °C for 1 min, and at 74 °C for 1 min, with a final extension at 74 °C for 10 min using the following primers attached with the XhoI and HindIII sites: 5′-ACT ATT CTC GAG GGA GAC GGA GGG GCA TCC CGG-3′ for the −590/+44 deleted construct (EAAT2–590), 5′-ATT ACC CTC GAG CGG CGC GGG TGA TGT CAG CTC-3′ for the −315/+44 deleted construct (EAAT2–315), 5′-GCC CTC GAG CGA AAA TAG AGA GGG ATC GCC-3′ for the −290/+44 deleted construct (EAAT2–290), 5′-ACC CTC GAG CTG CAA ATC CCC AGC TCC GGC-3′ for the −270/+44 deleted construct (EAAT2–270), and 5′-5-GCC GCC AAG CTT TGG CTT CCC CGA GAG AGC GAT-3′ for a reverse primer. The amplified EAAT2 DNA fragments were subcloned into the XhoI and HindIII sites of the pGL3-basic vector. The sequences of the new constructs were confirmed by DNA sequencing.
Cloning the TGF-α Promoter-Luciferase Vector
A 990-bp DNA fragment from −950 to +40 of the human TGF-α gene was generated by PCR using human genomic DNA. Primer sets were designed as follows: 5′-ATT CTC GAG GCA CCA GGG GCC ACC TCA GAG-3′ for sense containing the XhoI site and 5′-GCC AAG CTT GCT CTC CAG CCT CCT GCC CTA-3′ for antisense containing the HindIII site. The amplified TGF-α DNA fragment was digested with XhoI and HindIII, and the fragment was purified according to the instructions of the manufacturer (gel extraction system, Qiagen, Valencia, CA). The purified TGFα promoter was subcloned into the XhoI and HindIII sites of the pGL3-basic vector (Promega). The sequence of the TGF-α promoter construct was confirmed by DNA sequencing.
Transient Transfections and Luciferase Assay
Astrocytes cultured in 500 μl of growth medium in 24-well plates for 2–3 days were transfected overnight with 0.5 μg of the pGL3 basic luciferase reporter vector containing EAAT2Pro-954 (EAAT2, provided by Dr. Fisher, Virginia Commonwealth University), various mutants of EAAT2, or TGF-α with Lipofectamine 2000 transfection reagent (Invitrogen) in minimal essential medium containing 5% FBS. For some experiments, astrocytes were cotransfected with the luciferase vector and expression vectors (100 ng) such as pRC/RSV-CREB, pCMV-A-CREB, pcDNA-p50, pRC/RSV-p65, and pCMV-IκB-α. Empty vectors of the corresponding expression vectors were used as controls. Luciferase reporter vectors for NF-κB and CRE were purchased from Clontech. After transfection, astrocytes were treated with the designated compounds in Opti-MEM for 24 h. Luciferase activity was measured with the Bright-Glo luciferase kit (Promega) according to the instructions of the manufacturer and normalized to the protein content (Bradford protein assay, Bio-Rad). Normalization with protein content was verified by cotransfection of astrocytes with luciferase vectors and firefly reporter pGL4.75 plasmids (Promega) carrying the Renilla luciferase reporter gene.
Real-time PCR
The total RNA extraction from astrocytes was performed with the RNeasy mini RNA isolation kit (Qiagen). 2 μg of total RNA was transcribed with poly(dT) oligonucleotides. The primers for GLT-1 and TGF-α were designed as follows: 5′-CCT CAT GAG GAT GCT GAA GA-3′ (GLT-1 forward) and 5′-TCC AGG AAG GCA TCC AGG CTG-3′ (GLT-1 reverse); 5′-ACG GTC ACT GCT GTC ATT G-3′ (TGF-α forward) and 5′-GCG CTG GGC TTC TCG TG-3′ (TGF-α reverse); and 5′-TCC CTC AAG ATT GTC AGC AA-3′ (GAPDH forward) and 5′-AGA TCC ACA ACG GAT ACA TT-3′ (GAPDH reverse). The CFX96 real-time PCR detection system (Bio-Rad) was used to amplify TGF-α. The reactions were carried out in a total volume of 25 μl containing 1 μg of cDNA template of each sample, 0.4 μm of the appropriate primers, and RT2 SYBR Green qPCR Master Mix (SABiosciences/Qiagen). The PCR protocol consisted of one cycle at 95 °C for 10 min and 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. All samples were normalized relative to GAPDH. A web-based PCR array data analysis (SA Biosciences/Qiagen) was used for data analysis.
ChIP Assays
ChIP analysis was performed with the EZ-ChIP chromatin immunoprecipitation kit (Millipore) following the instructions of the manufacturer. Briefly, protein-DNA complexes were cross-linked with formaldehyde (10 min at room temperature) and sonicated to lengths of 100–500 bp. Next, 100 μl of supernatant was mixed with 900 μl of ChIP dilution buffer. After preclearing, 1% of the reaction was saved for PCR amplification as input. The remainder was incubated overnight at 4 °C with the CREB antibody (Cell Signaling), p50 (Cell Signaling Technology), p65 (Santa Cruz Biotechnology), or control rabbit IgG (Millipore). After isolation and washing of antibody-containing complexes, DNA was extracted. PCRs were performed using the following primers: CREB, 5′-GGG ACA ACA GAA GAG GGA CA-3′ (forward) and 5′-AGG GAT TGC AAG GTT TAG CC-3′ (reverse); NF-κB, 5′-CTG TGG GAC TCC CCA TCC CAG-3′ (forward) and 5′-TGG CAG GAT CCC AGG GTC TAA-3′ (reverse). PCR products were resolved on 1% agarose gel and visualized under UV light.
EMSA
EMSA was performed using the LightShift chemiluminescent kit from Thermo Scientific (Rockford, IL) following the instructions of the manufacturer. Briefly, 5 μg of nuclear extract from control or treated astrocytes was incubated for 20 min on ice with biotin-labeled oligos containing NF-κB or CREB binding sites of the EAAT2 promoter. In some reactions, 10 μg of antibody was added prior to the reaction. Next, DNA-protein complexes were resolved in 8% non-denaturing DNA polyacrylamide gels and transferred to a nylon membrane. The complexes were detected with the chemiluminescent nucleic acid detection module from Thermo Scientific. The primer pairs used for NF-κB (EAAT2 −583) were as follows: 5′-GGA GAC GGA GGG GCA TCC CGG GTC TCC GC-3′ and 5′-GCG GAG ACC CGG GAT GCC CCT CCG TCT CC-3′. The primer pairs used for CRE (EAAT2 −308) were as follows: 5′-CGG CGC GGG TGA TGT CAG CTC TCG ACG AAA-3′ and 5′-TTT CGT CGA GAG CTG ACA TCA CCC GCG CCG-3′. All oligos were HPLC-purified and biotin-labeled (Operon Technologies).
DNA Affinity Purification Assay
A DNA affinity purification assay was performed with a Factor Finder kit from Miltenyi Biotec, Inc. (Auburn, CA). Briefly, 1.5 μg of biotinylated oligos (the same sequences as for EMSA) were incubated with 50 μg of nuclear extract in binding buffer for 20 min. Next, 100 μl of streptavidin microbeads were added and further incubated for 10 min. The reaction mixture was applied onto the microcolumn, which was equilibrated with binding buffer and set in the magnetic field. After washing four times with 100 μl of low-salt and high-salt buffers each, bound proteins were eluted with 30 μl of elution buffer and analyzed by Western blotting.
Statistical Analysis
The mean and S.E. were determined for each data set. One-way analysis of variance (ANOVA) followed by Tukey post hoc test was used to determine significance. Student's t test was used for two sets of data to determine significance. p < 0.05 was considered to represent statistically significant differences.
RESULTS
Tamoxifen Increases Astrocytic GLT-1 and TGF-α Expression
Several growth factors, including EGF and basic fibroblast growth factor, increase GLT-1 expression in astrocytes (29, 30). Similarly, estrogen increases GLT-1 and glutamate aspartate transporter expression, thereby enhancing astrocytic glutamate uptake (31, 32). Our previous study established that both estrogen- and TX-induced astrocytic GLT-1 expression was mediated by TGF-α signaling upon 24 h exposure (26). Given that EGF or neuron-dependent activation require a prolonged period of time to induce GLT-1 expression (33), this study assessed the effects of TX on astrocytic GLT-1 protein levels after 6-day treatment. As shown in Fig. 1A, both TX and TGF-α induced a significant increase of astrocytic GLT-1 protein expression at this time point. dbcAMP was used as a positive control. The time course study revealed that TX induced GLT-1 protein expression as early as 30 min post-treatment and continuously for 6 days (data not shown). TX also increased GLT-1 mRNA (Fig. 1B) and TGF-α mRNA (Fig. 1C) levels, showing a significant increase within 4 h of incubation, in agreement with our previous findings (26). Analogous to the TX-induced GLT-1 protein increase, GLT-1 mRNA was also continuously increased up to 6 days post-treatment (data not shown). TX also significantly increased TGF-α promoter activity (p < 0.05, Fig. 1D). Moreover, blocking the TGF-α receptor, EGF receptor (EGFR), with AG1478 completely abrogated the TX-induced GLT-1 promoter activity, suggesting a possible role of TGF-α in this process (Fig. 1E).
FIGURE 1.

TX increases expression of GLT-1 and TGF-α in astrocytes. A, rat astrocytes were treated with TGF-α (50 ng/ml), dbcAMP (250 μm), or TX (1 μm) for 6 days, and the cell lysates were analyzed for GLT-1 protein expression by Western blotting (C, control). B and C, GLT-1 mRNA and TGF-α mRNA levels were measured after TX treatment (0–4 h) by real-time PCR (normalized to GAPDH). D, astrocytes were transfected overnight with the TGF-α promoter vector and treated with TX for 24 h. TGF-α promoter activity was measured by luciferase assay. E, astrocytes were treated with 1 μm TX, 10 μm AG1478 (AG), or AG1478 plus TX for 24 h, followed by the luciferase assay. *, p < 0.05; ***, p < 0.001, compared with the control; ANOVA followed by Tukey's post hoc test; n = 3.
CREB Is Involved in TX-induced GLT-1 Up-regulation
The CREB-regulated transcriptional machinery provides neuroprotection against ischemia (34). Interestingly, both the GLT-1 and TGF-α promoters contain one CRE consensus site, and CRE-mutation on the GLT-1 promoter decreases its activity (28). On the basis of these observations, we tested whether the CREB pathway plays a role in TX-induced regulation of GLT-1. As shown in Fig. 2A, both TX and TGF-α increased CRE reporter activity to levels comparable with dbcAMP (a positive control). The activation of CREB is triggered by PKA, and estrogen has been shown to induce this pathway (35). Accordingly, we tested whether the PKA pathway is involved in TX-induced CREB activation using H89, a PKA inhibitor. Inhibition of PKA significantly attenuated the effect of TX or TGF-α on EAAT2 promoter activity (Fig. 2, B and C). Overexpression of CREB significantly increased EAAT2 promoter activity (Fig. 2D). Previous studies reported that NF-κB played a critical role in the positive regulation of EAAT2 promoter activity (36). Thus, we tested whether CREB can still activate the EAAT2 promoter when all three NF-κB sites were mutated and determine its role in EAAT2 regulation independently of NF-κB. The results showed that CREB overexpression significantly increased EAAT2 promoter activity in the NF-κB triple mutant of the EAAT2 promoter (Fig. 2D), indicating that, in addition to NF-κB, the CREB pathway also independently plays a critical role in EAAT2 regulation. CREB overexpression by the transient transfection of the CREB expression plasmid vector (pRcRSV served as a control empty vector) was confirmed by Western blot analysis (Fig. 2D).
FIGURE 2.
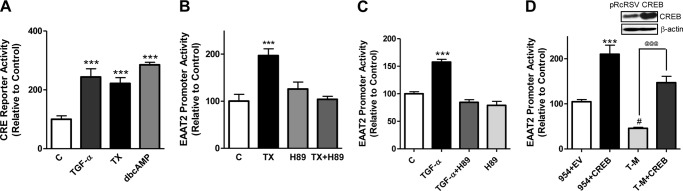
The CREB pathway is involved in TX-induced GLT-1 regulation. A, astrocytes were transfected overnight with the CRE reporter vector, followed by 24 h treatment with 50 ng/ml TGF-α, 250 μm dbcAMP, or 1 μm TX and subsequent assayed for EAAT2 promoter activity by luciferase assay (C, control). B and C, post-transfected astrocytes were incubated for 1 h with 10 μm H89 prior to 24-h treatment with TX (B) or TGF-α (C), followed by luciferase assay. D, astrocytes were cotransfected for 24 h with the wild-type (954, containing −954 to +44 bp of the EAAT2 promoter) or triple NF-κB mutant (T-M) of EAAT2 promoter vectors with empty vector (EV) pRcRSV or CREB expression vector (CREB), followed by luciferase assay. ***, p < 0.001; #, p < 0.05, compared with the control; @@@, p < 0.001; ANOVA followed by Tukey's post hoc test; n = 3.
TX Induces CREB Phosphorylation and CREB Binding to the EAAT2 Promoter in Cultured Astrocytes (in Vivo)
The phosphorylation of CREB is essential for its role as a transcriptional regulator (37). Phosphorylation of astrocytic CREB was noted as early as 5 min after TX treatment and peaked at 15 min, followed by a gradual decline to basal levels (Fig. 3A). Estrogen also increased phospho-CREB but showed a delayed effect, increasing CREB phosphorylation 1 h after treatment (Fig. 3B). The total CREB proteins were reprobed from the same blot to ensure the presence of an equal amount of unphosphorylated CREB proteins in the control and all treated groups. EAAT2 contains a CRE consensus site in the region of −308 of its promoter. Therefore, we performed a ChIP assay to examine whether TX modulates CREB binding to the EAAT2 promoter in vivo using cultured primary astrocytes. As shown in Fig. 3, C and D, TX induced a significant increase in CREB binding to its consensus site on the EAAT2 promoter. Estrogen also significantly increased CREB binding to the EAAT2 promoter, albeit to a lesser extent than TX (Fig. 3, C and D).
FIGURE 3.

TX activates the CREB signaling pathway. A and B, astrocytes were treated with 1 μm TX (A) or 10 nm of estrogen (E2) (B) for the indicated time periods, and the cell lysates were analyzed for phospho-CREB and CREB proteins by Western blot analysis (C, control). C and D, astrocytes were treated with TX or estrogen for 24 h, followed by the analysis of CREB binding to its consensus site in the EAAT2 promoter by ChIP assay. *, p < 0.05; ***, p < 0.001, compared with the control; ANOVA followed by Tukey's post hoc test; n = 3. Control groups were treated with vehicle (ethanol) and IgG groups were treated with TX or E2 but immunoprecipitated with IgG instead of CREB antibody for negative control.
TX Induces NF-κB Binding to the EAAT2 Promoter in a ChIP Assay
The NF-κB signaling pathway plays a critical role in EGF- and ceftriaxone-induced GLT-1 expression (38, 39). Accordingly, we tested whether the NF-κB pathway also mediates the effect of TX on EAAT2 expression. As shown in Fig. 4A, TX significantly increased NF-κB luciferase reporter activity, and inhibition of the NF-κB pathway with pyrrolidine dithiocarbamate abrogated the TX-induced EAAT2 promoter activity (Fig. 4B). Next, we performed a ChIP assay to determine whether TX induced the binding of NF-κB to the EAAT2 promoter. Estrogen was also tested as an endogenous molecule related to TX. As shown in Fig. 4C, estrogen increased the binding of the NF-κB subunits p65 and p50 to the EAAT2 promoter. TX also induced significant recruitment of both p65 and p50 (Fig. 4, D and E), although the preference of NF-κB subunit recruitment to the EAAT2 promoter by TX is unclear in this study.
FIGURE 4.

The NF-κB pathway is involved in TX-induced GLT-1 regulation. A, after overnight transfection with the EAAT2 promoter vector, astrocytes were incubated with 50 μm of pyrrolidine dithiocarbamate (PDTC) for 30 min prior to treatment with 1 μm of TX for 24 h, followed by luciferase assay (C, control). B, after overnight transfection with the NF-κB reporter vector, astrocytes were treated with TX for 24 h, followed by luciferase assay. C and D, astrocytes were incubated with estrogen (E2) (C) or TX (D), and binding of NF-κB p50 or p65 to its consensus sites in the EAAT2 promoter was determined by ChIP assay. Control groups were treated with vehicle (ethanol) and IgG groups were treated with TX or E2 but immunoprecipitated with IgG instead of CREB antibody for negative control. E, the intensity of TX-induced binding was quantified. ***, p < 0.001; compared with the control; @, p < 0.05; ANOVA followed by Tukey's post hoc test; n = 3.
TX Induces NF-κB and CRE Binding to the EAAT2 Promoter in Nuclear Extracts (in Vitro)
Because TX induced both CREB and NF-κB binding to the EAAT2 promoter in vivo in cultured astrocytes, as shown in the ChIP assay, we next performed both EMSA and DNA affinity purification assay experiments to confirm that TX induces their binding to the EAAT2 promoter in vitro using nuclear extracts. As shown in Fig. 5, A and B, TX enhanced the binding of both the p65 and p50 subunits of NF-κB and CRE to the EAAT2 promoter. The EMSA results show that TX induced specific NF-κB p65 binding to its consensus oligos because an excess amount of unlabeled oligos completely blocked the binding. Interestingly, the NF-κB p65 antibody did not cause a supershift of these complexes. Nonetheless, the experiments corroborate that TX induced specific NF-κB p65 binding to its consensus oligos (Fig. 5A). The TX-induced binding of CRE consensus oligos to the CREB protein was also specific because excess unlabeled CRE oligos competed with biotinylated CRE oligos, inhibiting the formation of a protein-DNA complex, and coincubation with CREB antibody induced the supershift of the complexes (Fig. 5B). Moreover, DNA affinity purification assay experiments showed that TX induced strong and specific bindings of NF-κB and CREB to the EAAT2 promoter (Fig. 5, C and D).
FIGURE 5.

TX induces binding of NF-κB and CREB to the EAAT2 promoter. A and B, astrocytes were treated with TX for 24 h, followed by nuclear extraction. Next, nuclear extracts were incubated with biotinylated oligos containing NF-κB and CRE binding sequences of EAAT2 promoter for 20 min. EMSA was carried out to determine complex formation. An excess (100 ×) of non-biotinylated oligos was used to determine whether the protein-DNA complex was specific for the designated oligos. Antibodies for p65 or CREB were also added to some reactions to determine the supershift of the complexes. Two sets of TX groups are shown in A, and dbcAMP was used as a positive control. C and D, astrocytes were treated with TX for 24 h, and isolated nuclear extracts were incubated with biotinylated oligos of the NF-κB and CRE binding sequences of the EAAT2 promoter, followed by elution from the magnetic beads in the microcolumn. The eluents were subjected to Western blot analysis to detect p65 and p50 (C) or CREB (D). In both groups, 10 μg of nuclear extracts was loaded as input (C, control).
Both the CREB and NF-κB Binding Sites Are Critical for TX Regulation on EAAT2
Activation of CREB signaling mediates the effect of growth factors, such as EGF and TGF-α, on GLT-1 protein expression (40). Nevertheless, there is a dearth of information regarding the mechanism by which CREB regulates GLT-1 (EAAT2) at the transcriptional levels. Because, in addition to the significant role of CREB through the CRE consensus site at −308 (28), NF-κB also serves as a critical regulator of the EAAT2 promoter, we used the mutant construct of all three NF-κB sites (-251/-272/-583) of the EAAT2 promoter. As shown in Fig. 6, A and B, either CRE or the triple NF-κB mutation (TM) significantly decreased the EAAT2 promoter activity, confirming that both the CREB and NF-κB pathways are critical in EAAT2 regulation. Moreover, TX and TGF-α still significantly increased EAAT2 promoter activity in these mutants (Fig. 6, A and B), indicating that inactivation of one of the CRE or NF-κB sites is insufficient to completely block the effect of TX/TGF-α on EAAT2. We also generated deletion constructs of the EAAT2 promoter to determine the sites responsible for the effects of TX. As shown in Fig. 6C, each deletion construct containing at least one of the NF-κB (−251, −272, or −583) or CRE (-308) sites significantly decreased promoter activity compared with the wild-type EAAT2, but the TX-induced stimulatory effect remained significant. Indeed, the EAAT2 mutant construct that was inactivated for both the CRE and triple NF-κB sites completely abrogated the effect of TX on EAAT2 promoter activity (Fig. 6D), indicating that these two pathways are critical for TX-induced EAAT2 regulation.
FIGURE 6.

The role of the CRE and NF-κB binding sites in TX regulation on EAAT2 promoter activity. A and B, after overnight transfection with the wild type, the CRE mutant (-308), or the triple NF-κB mutant (-583/-272/-251, TM) of the EAAT2 promoter vector, astrocytes were treated with 1 μm TX (A) or 50 ng/ml TGF-α (B) for 24 h. EAAT2 promoter activity was measured by luciferase assay (C, control). C, after overnight transfection with the wild type (EAAT2–954, −954 to + 44 bp) or various deletion mutants (270, −270 to +44; 290, −290 to +44; 315, −315 to +44, and 590, −590 to + 44 bp of the EAAT2 promoter), astrocytes were treated with TX for 24 h, followed by luciferase assay. D, after overnight transfection with various mutant constructs of the EAAT2 promoter vectors and single or multiple site mutants of CRE or NF-κB, astrocytes were treated with TX for 24 h, followed by luciferase assay. *, p < 0.05; **, p < 0.01; ###, p < 0.001; compared with the control; @, p < 0.05; ANOVA followed by Tukey's post hoc test; n = 3.
CREB and NF-κB Also Play a Critical Role in TGF-α Regulation
Because the EAAT2 (or GLT-1) promoter does not contain an estrogen response element consensus sequence, the regulation of GLT-1 expression by TX is likely mediated via TGF-α (Fig. 1). Interestingly, both the TGF-α and EAAT2 promoters contain one CRE and three NF-κB consensus sites. Our results show that overexpression of CREB increased, whereas repression of CREB with mutant CREB (ACREB), the dominant negative CREB inhibitor, decreased TGF-α promoter activity (Fig. 7, A and B, respectively). Moreover, activation of NF-κB p65 or p50 by overexpression also significantly increased TGF-α promoter activity, but repression of NF-κB by overexpression of IκB-α significantly decreased TGF-α promoter activity (Fig. 7, C–E, respectively). These results indicate that both the CREB and NF-κB pathways play a critical role in regulating TGF-α promoter activity.
FIGURE 7.
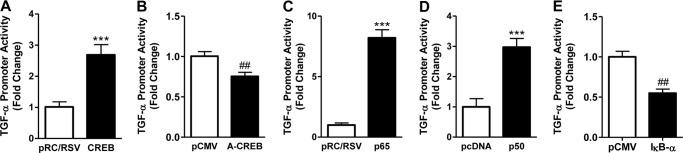
The role of CREB and NF-κB in TGF-α regulation. A and B, astrocytes were cotransfected for 24 h with the TGF-α promoter vector and expression vectors, CREB (A), or dominant negative CREB (ACREB) (B). C–E, astrocytes were cotransfected with the TGF-α promoter vector and expression vectors NF-κB p65, NF-κB p50, or dominant negative NF-κB (IκB-α mutant) with their corresponding control vectors, respectively, followed by measurement of TGF-α promoter activity by luciferase assay. ***, p < 0.001; ##, p < 0.01, compared with the control; unpaired Student's t test; n = 3.
TX Enhances GLT-1 via Estrogen Receptor (ER)-α, ER-β, and G Protein-coupled Receptor 30 (GPR30)
In addition to the conventional nuclear ERs (ER-α and ER-β), GPR30 also mediates the biological effects of estrogen via transactivation of growth factor receptors (41). We used several modulators of the ERs to gain better understanding on the ER mechanism/s associated with TX-induced GLT-1 regulation. Our results show that G1, an agonist of GPR30, increased GLT-1 protein expression to a similar extent as TX (Fig. 8A). ICI 182,780, a non-selective antagonist of ER-α and ER-β, did not block the TX-induced increase in GLT-1 protein expression, whereas G15, a GPR30 antagonist, partially, but significantly reduced the TX-induced enhancement of GLT-1 protein expression. These results indicate that GPR30 may be, at least in part, responsible for TX-induced GLT-1 regulation. Interestingly, ICI 182,780 alone also increased GLT-1 expression, possibly acting as an agonist of GPR30 (Fig. 8A). To further confirm the role of GPR30 in TX-induced GLT-1 regulation, we tested whether G15, a GPR30 antagonist, blocks the TX-induced EAAT2 promoter activity. G15 significantly, but not completely, abolished the effect of TX on EAAT2 promoter activity (Fig. 8B). Next, we examined whether ER-α or ER-β play a role in TX-induced EAAT2 promoter activity by overexpression of ER-α or ER-β. As shown in Fig. 8, C and D, TX activated both ER-α and ER-β, leading to a significant increase in EAAT2 promoter activity. These results indicate that TX regulates GLT-1 function via all ERs, including ER-α, ER-β, and GPR30.
FIGURE 8.

TX regulates GLT-1 via ER-α, ER-β, and GPR30. A, astrocytes were treated with 1 μm TX and various ER modulators such as 1 μm G1, 1 μm G15, and 1 μm ICI 182,780 (ICI), alone or in combination, followed by detection of GLT-1 expression in cell lysates by Western blot analysis and quantification (C, control). B, after overnight transfection with the EAAT2 promoter vector, astrocytes were pre-treated with 1 μm of G15 for 1 h followed by 24-h incubation 1 μm TX. EAAT2 promoter activity was measured by luciferase assay. C and D, after overnight cotransfection with the EAAT2 promoter vector and expression vectors, including control vector eGFP ER-α, or ER-β, astrocytes were treated for 24 h with 1 μm TX. EAAT2 promoter activity was determined by luciferase assay. **, p < 0.01; ***, p < 0.001; compared with the control; @, p < 0.05; ANOVA followed by Tukey's post hoc test; n = 3.
Inhibition of Src Family Kinases Partially Blocks TX-induced GLT-1 Expression
As shown in Fig. 9, A and B, inhibition of Src family tyrosine kinases with 10 μm PP2 abrogated both the TX-induced effect on GLT-1 protein expression and EAAT2 promoter activity. PP2 completely abrogated G1-induced GLT-1 protein expression but only partially (p < 0.01) suppressed the effect of TX on GLT-1.
FIGURE 9.
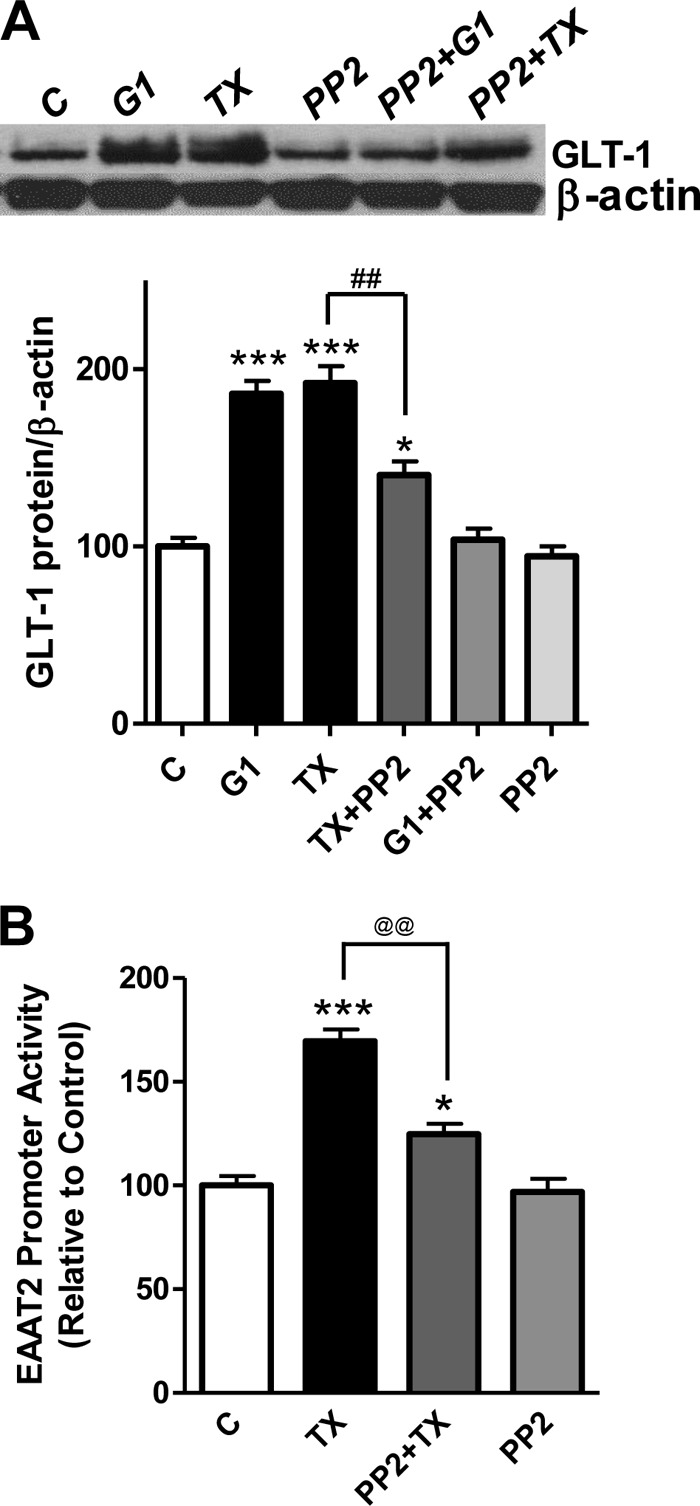
Inhibition of the Src family tyrosine kinases abolishes TX-induced GLT-1 expression via GPR30. A, astrocytes were incubated with the Src family tyrosine kinase inhibitor PP2 (10 μm, 1 h) prior to treatment with 1 μm of TX or G1 for 24 h, and cell lysates were analyzed for the detection of GLT-1 protein expression by Western blot analysis (β-actin served as a loading control). C, control. B, astrocytes were incubated with PP2 for 1 h prior to 1 μm TX treatment and subsequent measurement of the EAAT2 promoter activity by luciferase assay. *, p < 0.05; ***, p < 0.001; ##, p < 0.01, compared with the control; @@, p < 0.01; ANOVA followed by Tukey's post hoc test; n = 3.
DISCUSSION
We investigated the mechanism of TX-induced transcriptional regulation of GLT-1 in rat primary astrocytes. Our findings established that TX enhanced GLT-1 expression by activating both the CREB and NF-κB pathways. Finally, the effect of TX was mediated via GPR30 as well as ER-α and ER-β (Fig. 10).
FIGURE 10.

Proposed mechanism for the regulation of GLT-1 by TX. TX activates GPR30 as well as ER-α and ER-β, leading to the up-regulation of TGF-α via CREB and NF-κB. TGF-α is released into the extracellular compartment where it triggers the activation of EGFR, resulting in increased expression and activity of GLT-1 via both the CREB and NF-κB signaling pathways.
Excessive glutamate-induced excitotoxicity has been linked to various neurological disorders (38). The astrocytic glutamate transporter GLT-1 (EAAT2 in humans) is primarily responsible for synaptic glutamate uptake (∼90%) (42). Thus, delineating the molecular regulatory mechanisms of GLT-1 is critical for the development of efficacious therapeutics to increase GLT-1 expression and reduce synaptic glutamate concentrations. Several studies have reported that increased EAAT2 protein levels effectively reduce excitotoxicity, leading to neuroprotection in a number of neurodegenerative disease models (9, 10, 12). TX is routinely used to treat breast cancer because of its antagonistic effect on breast tissue (43). In recent years, it has also gained attention for its agonistic effects in the CNS as a neuroprotective agent (44). TX is neuroprotective in several experimental models by inducing antioxidative and antiapoptotic functions (45, 46). Our finding that TX enhances GLT-1 expression provides novel information regarding potential molecular targets to mitigate excitotoxic injury.
Because identification of molecular targets for TX-induced GLT-1 expression could be vital for neuroprotection, our finding that both CREB and NF-κB signaling play a critical role in the TX-induced enhancement of GLT-1 expression provides valuable information. We have shown previously that TX enhanced expression of GLT-1 mRNA and protein in astrocytes that was mediated by TGF-α (26). Given the mediating role of TGF-α in TX-induced GLT-1 expression, we observed that TX increased GLT-1 mRNA levels before the TGF-α mRNA increase (30 min versus 1 h). TX-induced GLT-1 increase prior to TGF-α might be due to TX-induced transactivation on EGFR via GPR30. It has been reported that estrogen induced transactivation of EGFR through GPR30 activation (47). The estrogen binding to GPR30 induced transactivation of the EGFR, leading to the rapid activation of intracellular signaling cascades within 5 min (48). This could be the case for a TX-induced rapid increase of GLT-1 prior to TGF-α production. This notion is also supported by our earlier study demonstrating that G1, a selective agonist of GPR30, rapidly increased GLT-1 expression within 30 min (28). In addition, the result that inhibition of the EGFR or TGF-α knockdown with siRNA led to abrogation of the effect of TX on GLT-1 expression provides evidence that TGF-α plays a major role in mediating the effect of TX on GLT-1 expression.
The GLT-1 promoter contains one CRE and three NF-κB consensus sites (Fig. 10) (33). Regulation of the CREB pathway may be critical in affording neuroprotection because CREB activation leads to the expression of genes that encode neuroprotective molecules (49, 50). In the rat hippocampus, TX regulates the expression of synaptophysin via CREB (51). TX-induced CREB activation is also involved in improving acute mania (52). Our finding shows that the NF-κB pathway plays a critical role in TX-induced GLT-1 expression (Figs. 4 and 5). The role of NF-κB in TX-induced neuroprotection is supported by observations that TX protected human coronary artery endothelial cells from hypoxic injury via NF-κB activation (53). NF-κB signaling is also crucial for the enhancement of GLT-1 expression induced by growth factors such as EGF and TGF-α (36, 39). There are three NF-κB consensus sites in the GLT-1 promoter, −583, −272, and −251. Several compounds are known to activate the NF-κB sites specific to the GLT-1 promoter. EGF enhances GLT-1 expression mainly by activation of the −583 NF-κB site of the EAAT2 promoter (39). The β-lactam antibiotic ceftriaxone has been shown to enhance GLT-1 expression via NF-κB activation (27) exclusively by activation of the −272 NF-κB site of the GLT-1 promoter. Neuronal regulation of GLT-1 expression specifically targets the −583 and −251 NF-κB sites of the GLT-1 promoter (14). Our results indicate that TX-induced NF-κB activation was not specific to the −583 or −272 sites because mutation of these sites on the GLT-1 promoter failed to completely abrogate the TX-enhancing GLT-1 promoter activity (Fig. 6D). This likely reflects the ability of TX to activate all of the NF-κB sites and the CRE site, indicating that both the CREB and NF-κB pathways are involved in TX-induced enhancement of GLT-1 expression.
The results from this study reveal that the TX-induced transcriptional regulation of TGF-α expression also involves CREB and NF-κB. The transcription factors p53 and AP-2 are known to positively regulate TGF-α promoter activity (54). Our findings suggest that both NF-κB and CREB activation are critical for the enhancement of TGF-α promoter activity. On the basis of our results, TX may directly activate the NF-κB and CREB pathways to induce GLT-1 expression. However, our observation that inhibition of the TGF-α receptor EGFR or knockdown of TGF-α with siRNA abolished the stimulatory effect of TX on GLT-1 suggests that TGF-α is an essential mediator of the effects of TX on GLT (26). The most likely scenario is that TX may utilize NF-κB and CREB signaling to generate TGF-α, in turn activating EGFR in an autocrine mode and, thus, leading to increased GLT-1 expression by NF-κB and CREB (Fig. 10). Further investigation is warranted to determine the exact role of TGF-α in TX-induced GLT-1 up-regulation.
TX-induced neuroprotection involves multiple mechanisms, including the activation of intracellular signaling proteins such as MAPK/ERK and PI3K/Akt (31, 55). We reported previously that activation of MAPK/ERK, PI3K/Akt, and EGFR is involved in TX regulation of GLT-1 (31). Src signaling is involved in estrogen-induced neuroprotection against glutamate toxicity (56). Estrogen stimulates the c-fos gene via GPR30 through activation of both Src and EGFR signaling in breast cancer cells (57). TX activates GPR30 to stimulate cell growth activation via Src and EGFR signaling in TX-resistant breast cancer MCF-7 cells (58). Likewise, TX may activate the Src family tyrosine kinases via GPR30 to induce GLT-1 expression in primary astrocytes (Fig. 9).
Estrogen exerts its cellular effects via the nuclear receptors ER-α and ER-β as well as GPR30, a G protein-coupled ER (59, 60). Our finding that TX-induced GLT-1 expression is partially inhibited by G15, an antagonist of GPR30, indicates that GPR30 is not the sole mechanism that mediates the effects of TX on GLT-1 expression. Indeed, ER-α and ER-β also contributed to the TX regulation of GLT-1 expression (Fig. 8, C and D). ER-α and ER-β, localized to the plasma membrane, are known to induce rapid estrogen signaling (61). These ERs may also trigger cross-talk with membrane-associated proteins, such as EGFR. It is unclear at present whether TX-induced activation of ER-α and ER-β is via membrane-bound ER-α/β or nuclear ER-α/β.
Taken together, the results presented here support the following model for the mechanism of TX-induced GLT-1 regulation (Fig. 10). TX activates GPR30 as well as ER-α and ER-β, resulting in up-regulation of TGF-α, which is released into the extracellular milieu in an autocrine fashion. TGF-α, in turn, activates EGFR and increases GLT-1 expression via the CREB and NF-κB pathways. Activated GPR30 may also directly transactivate EGFR, leading to CREB and NF-κB activation as well as increased GLT-1 expression. Because the up-regulation of GLT-1 attenuates excitotoxicity, pharmacological agents that increase GLT-1 expression and glutamate transporter function may be beneficial for the treatment of neurodegenerative diseases with an inherent excitotoxic etiology. Therefore, targeting specific sites along the scheme (Fig. 10) may lead to the development of novel therapeutic modalities to mitigate neurological disorders associated with impairment of glutamate transporters.
This work was supported, in whole or in part, by NIGMS, National Institutes of Health Grant SC1 089630; NIAID, National Institutes of Health Grant SC1 I089073; and NIEHS, National Institutes of Health Grant R01 10563.
- EAAT1
- excitatory amino acid transporter 1
- TX
- tamoxifen
- CREB
- cAMP response element-binding protein
- dbcAMP
- dibutyryl cAMP
- ANOVA
- analysis of variance
- EGFR
- EGF receptor
- CRE
- cAMP response element
- ER
- estrogen receptor.
REFERENCES
- 1. Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105 [DOI] [PubMed] [Google Scholar]
- 2. Gillessen T., Budd S. L., Lipton S. A. (2002) Excitatory amino acid neurotoxicity. Adv. Exp. Med. Biol. 513, 3–40 [DOI] [PubMed] [Google Scholar]
- 3. Amara S. G., Fontana A. C. (2002) Excitatory amino acid transporters. Keeping up with glutamate. Neurochem. Int. 41, 313–318 [DOI] [PubMed] [Google Scholar]
- 4. Pines G., Danbolt N. C., Bjørås M., Zhang Y., Bendahan A., Eide L., Koepsell H., Storm-Mathisen J., Seeberg E., Kanner B. I. (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360, 464–467 [DOI] [PubMed] [Google Scholar]
- 5. Storck T., Schulte S., Hofmann K., Stoffel W. (1992) Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. U.S.A. 89, 10955–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Danbolt N. C., Chaudhry F. A., Dehnes Y., Lehre K. P., Levy L. M., Ullensvang K., Storm-Mathisen J. (1998) Properties and localization of glutamate transporters. Prog. Brain Res. 116, 23–43 [DOI] [PubMed] [Google Scholar]
- 7. Sattler R., Rothstein J. D. (2006) Regulation and dysregulation of glutamate transporters. Handb. Exp. Pharmacol. 175, 277–303 [DOI] [PubMed] [Google Scholar]
- 8. Sheldon A. L., Robinson M. B. (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 51, 333–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo H., Lai L., Butchbach M. E., Stockinger M. P., Shan X., Bishop G. A., Lin C. L. (2003) Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 12, 2519–2532 [DOI] [PubMed] [Google Scholar]
- 10. Ganel R., Ho T., Maragakis N. J., Jackson M., Steiner J. P., Rothstein J. D. (2006) Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol. Dis. 21, 556–567 [DOI] [PubMed] [Google Scholar]
- 11. Wisman L. A., van Muiswinkel F. L., de Graan P. N., Hol E. M., Bär P. R. (2003) Cells over-expressing EAAT2 protect motoneurons from excitotoxic death in vitro. Neuroreport 14, 1967–1970 [DOI] [PubMed] [Google Scholar]
- 12. Chu K., Lee S. T., Sinn D. I., Ko S. Y., Kim E. H., Kim J. M., Kim S. J., Park D. K., Jung K. H., Song E. C., Lee S. K., Kim M., Roh J. K. (2007) Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke 38, 177–182 [DOI] [PubMed] [Google Scholar]
- 13. Merchenthaler I., Dellovade T. L., Shughrue P. J. (2003) Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. N.Y. Acad. Sci. 1007, 89–100 [DOI] [PubMed] [Google Scholar]
- 14. Brann D. W., Dhandapani K., Wakade C., Mahesh V. B., Khan M. M. (2007) Neurotrophic and neuroprotective actions of estrogen. Basic mechanisms and clinical implications. Steroids 72, 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shao B., Cheng Y., Jin K. (2012) Estrogen, neuroprotection and neurogenesis after ischemic stroke. Curr. Drug Targets 13, 188–198 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Jin Y., Behr M. J., Feustel P. J., Morrison J. P., Kimelberg H. K. (2005) Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp Neurol 196, 41–46 [DOI] [PubMed] [Google Scholar]
- 17. Mehta S. H., Dhandapani K. M., De Sevilla L. M., Webb R. C., Mahesh V. B., Brann D. W. (2003) Tamoxifen, a selective estrogen receptor modulator, reduces ischemic damage caused by middle cerebral artery occlusion in the ovariectomized female rat. Neuroendocrinology 77, 44–50 [DOI] [PubMed] [Google Scholar]
- 18. Dluzen D. E., McDermott J. L., Anderson L. I. (2001) Tamoxifen diminishes methamphetamine-induced striatal dopamine depletion in intact female and male mice. J Neuroendocrinology 13, 618–624 [DOI] [PubMed] [Google Scholar]
- 19. Kimelberg H. K., Feustel P. J., Jin Y., Paquette J., Boulos A., Keller R. W., Jr., Tranmer B. I. (2000) Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 11, 2675–2679 [DOI] [PubMed] [Google Scholar]
- 20. Osuka K., Feustel P. J., Mongin A. A., Tranmer B. I., Kimelberg H. K. (2001) Tamoxifen inhibits nitrotyrosine formation after reversible middle cerebral artery occlusion in the rat. J. Neurochem. 76, 1842–1850 [DOI] [PubMed] [Google Scholar]
- 21. Koeberle P. D., Bähr M. (2008) The upregulation of GLAST-1 is an indirect antiapoptotic mechanism of GDNF and neurturin in the adult CNS. Cell Death Differ. 15, 471–483 [DOI] [PubMed] [Google Scholar]
- 22. Dhandapani K. M., Wade F. M., Mahesh V. B., Brann D. W. (2005) Astrocyte-derived transforming growth factor-β mediates the neuroprotective effects of 17β-estradiol. Involvement of nonclassical genomic signaling pathways. Endocrinology 146, 2749–2759 [DOI] [PubMed] [Google Scholar]
- 23. Justicia C., Pérez-Asensio F. J., Burguete M. C., Salom J. B., Planas A. M. (2001) Administration of transforming growth factor-α reduces infarct volume after transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 21, 1097–1104 [DOI] [PubMed] [Google Scholar]
- 24. Justicia C., Planas A. M. (1999) Transforming growth factor-α acting at the epidermal growth factor receptor reduces infarct volume after permanent middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 19, 128–132 [DOI] [PubMed] [Google Scholar]
- 25. Zelenaia O., Schlag B. D., Gochenauer G. E., Ganel R., Song W., Beesley J. S., Grinspan J. B., Rothstein J. D., Robinson M. B. (2000) Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-κB. Mol. Pharmacol. 57, 667–678 [DOI] [PubMed] [Google Scholar]
- 26. Lee E., Sidoryk-Wegrzynowicz M., Yin Z., Webb A., Son D. S., Aschner M. (2012) Transforming growth factor-α mediates estrogen-induced upregulation of glutamate transporter GLT-1 in rat primary astrocytes. Glia 60, 1024–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aschner M., Gannon M., Kimelberg H. K. (1992) Manganese uptake and efflux in cultured rat astrocytes. J. Neurochem. 58, 730–735 [DOI] [PubMed] [Google Scholar]
- 28. Lee E., Sidoryk-Wêgrzynowicz M., Wang N., Webb A., Son D. S., Lee K., Aschner M. (2012) GPR30 regulates glutamate transporter GLT-1 expression in rat primary astrocytes. J. Biol. Chem. 287, 26817–26828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller S., Romano C., Cotman C. W. (1995) Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. J. Neurosci. 15, 6103–6109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vermeiren C., Najimi M., Vanhoutte N., Tilleux S., de Hemptinne I., Maloteaux J. M., Hermans E. (2005) Acute up-regulation of glutamate uptake mediated by mGluR5a in reactive astrocytes. J. Neurochem. 94, 405–416 [DOI] [PubMed] [Google Scholar]
- 31. Lee E. S., Sidoryk M., Jiang H., Yin Z., Aschner M. (2009) Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J. Neurochem. 110, 530–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pawlak J., Brito V., Küppers E., Beyer C. (2005) Regulation of glutamate transporter GLAST and GLT-1 expression in astrocytes by estrogen. Brain Res. Mol. Brain Res. 138, 1–7 [DOI] [PubMed] [Google Scholar]
- 33. Ghosh M., Yang Y., Rothstein J. D., Robinson M. B. (2011) Nuclear factor-κB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J. Neurosci. 31, 9159–9169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hara T., Hamada J., Yano S., Morioka M., Kai Y., Ushio Y. (2003) CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region. J. Neurochem. 86, 805–814 [DOI] [PubMed] [Google Scholar]
- 35. Lee B., Butcher G. Q., Hoyt K. R., Impey S., Obrietan K. (2005) Activity-dependent neuroprotection and cAMP response element-binding protein (CREB). Kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J. Neurosci. 25, 1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Su Z. Z., Leszczyniecka M., Kang D. C., Sarkar D., Chao W., Volsky D. J., Fisher P. B. (2003) Insights into glutamate transport regulation in human astrocytes. Cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc. Natl. Acad. Sci. U.S.A. 100, 1955–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayr B., Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 38. Lee S. G., Su Z. Z., Emdad L., Gupta P., Sarkar D., Borjabad A., Volsky D. J., Fisher P. B. (2008) Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 283, 13116–13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sitcheran R., Gupta P., Fisher P. B., Baldwin A. S. (2005) Positive and negative regulation of EAAT2 by NF-κB. A role for N-myc in TNFα-controlled repression. EMBO J. 24, 510–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlüter K., Figiel M., Rozyczka J., Engele J. (2002) CNS region-specific regulation of glial glutamate transporter expression. Eur. J. Neurosci. 16, 836–842 [DOI] [PubMed] [Google Scholar]
- 41. Carmeci C., Thompson D. A., Ring H. Z., Francke U., Weigel R. J. (1997) Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45, 607–617 [DOI] [PubMed] [Google Scholar]
- 42. Kim K., Lee S. G., Kegelman T. P., Su Z. Z., Das S. K., Dash R., Dasgupta S., Barral P. M., Hedvat M., Diaz P., Reed J. C., Stebbins J. L., Pellecchia M., Sarkar D., Fisher P. B. (2011) Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration. Opportunities for developing novel therapeutics. J. Cell. Physiol. 226, 2484–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sawka C. A., Pritchard K. I., Shelley W., DeBoer G., Paterson A. H., Meakin J. W., Sutherland D. J. (1997) A randomized crossover trial of tamoxifen versus ovarian ablation for metastatic breast cancer in premenopausal women. A report of the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG) trial MA. 1. Breast Cancer Res. Treat. 44, 211–215 [DOI] [PubMed] [Google Scholar]
- 44. Arevalo M. A., Santos-Galindo M., Lagunas N., Azcoitia I., Garcia-Segura L. M. (2011) Selective estrogen receptor modulators as brain therapeutic agents. J. Mol. Endocrinol. 46, R1–9 [DOI] [PubMed] [Google Scholar]
- 45. Zhang Y., Milatovic D., Aschner M., Feustel P. J., Kimelberg H. K. (2007) Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Exp. Neurol. 204, 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharma K., Mehra R. D. (2008) Long-term administration of estrogen or tamoxifen to ovariectomized rats affords neuroprotection to hippocampal neurons by modulating the expression of Bcl-2 and Bax. Brain Res. 1204, 1–15 [DOI] [PubMed] [Google Scholar]
- 47. Filardo E. J. (2002) Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Biol. 80, 231–238 [DOI] [PubMed] [Google Scholar]
- 48. Lappano R., De Marco P., De Francesco E. M., Chimento A., Pezzi V., Maggiolini M. (2013) Cross-talk between GPER and growth factor signaling. J. Steroid Biochem. Mol. Biol. [DOI] [PubMed] [Google Scholar]
- 49. Kitagawa K. (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 274, 3210–3217 [DOI] [PubMed] [Google Scholar]
- 50. Bell K. F., Bent R. J., Meese-Tamuri S., Ali A., Forder J. P., Aarts M. M. (2013) Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J. Neurochem. 126, 274–287 [DOI] [PubMed] [Google Scholar]
- 51. Sharma K., Mehra R. D., Dhar P., Vij U. (2007) Chronic exposure to estrogen and tamoxifen regulates synaptophysin and phosphorylated cAMP response element-binding (CREB) protein expression in CA1 of ovariectomized rat hippocampus. Brain Res. 1132, 10–19 [DOI] [PubMed] [Google Scholar]
- 52. Cechinel-Recco K., Valvassori S. S., Varela R. B., Resende W. R., Arent C. O., Vitto M. F., Luz G., de Souza C. T., Quevedo J. (2012) Lithium and tamoxifen modulate cellular plasticity cascades in animal model of mania. J. Psychopharmacol. 26, 1594–1604 [DOI] [PubMed] [Google Scholar]
- 53. Stice J. P., Mbai F. N., Chen L., Knowlton A. A. (2012) Rapid activation of nuclear factor κB by 17β-estradiol and selective estrogen receptor modulators. Pathways mediating cellular protection. Shock 38, 128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang D., Shin T. H., Kudlow J. E. (1997) Transcription factor AP-2 controls transcription of the human transforming growth factor-α gene. J. Biol. Chem. 272, 14244–14250 [DOI] [PubMed] [Google Scholar]
- 55. Du B., Ohmichi M., Takahashi K., Kawagoe J., Ohshima C., Igarashi H., Mori-Abe A., Saitoh M., Ohta T., Ohishi A., Doshida M., Tezuka N., Takahashi T., Kurachi H. (2004) Both estrogen and raloxifene protect against β-amyloid-induced neurotoxicity in estrogen receptor α-transfected PC12 cells by activation of telomerase activity via Akt cascade. J. Endocrinol. 183, 605–615 [DOI] [PubMed] [Google Scholar]
- 56. Nilsen J., Brinton R. D. (2002) Impact of progestins on estrogen-induced neuroprotection. Synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143, 205–212 [DOI] [PubMed] [Google Scholar]
- 57. Maggiolini M., Vivacqua A., Fasanella G., Recchia A. G., Sisci D., Pezzi V., Montanaro D., Musti A. M., Picard D., Andò S. (2004) The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 279, 27008–27016 [DOI] [PubMed] [Google Scholar]
- 58. Ignatov A., Ignatov T., Roessner A., Costa S. D., Kalinski T. (2010) Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res. Treat. 123, 87–96 [DOI] [PubMed] [Google Scholar]
- 59. Chakrabarti S., Davidge S. T. (2012) G-protein coupled receptor 30 (GPR30). A novel regulator of endothelial inflammation. PloS ONE 7, e52357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lappano R., Rosano C., Santolla M. F., Pupo M., De Francesco E. M., De Marco P., Ponassi M., Spallarossa A., Ranise A., Maggiolini M. (2012) Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 12, 531–542 [DOI] [PubMed] [Google Scholar]
- 61. Levin E. R. (2009) Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 20, 477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]