

Abstract
The issue of G protein–coupled receptor (GPCR) oligomer status has not been resolved. Although many studies have provided evidence in favor of receptor-receptor interactions, there is no consensus as to the exact oligomer size of class A GPCRs. Previous studies have reported monomers, dimers, tetramers, and higher-order oligomers. In the present study, this issue was examined using fluorescence correlation spectroscopy (FCS) with photon counting histogram (PCH) analysis, a sensitive method for monitoring diffusion and oligomer size of plasma membrane proteins. Six different class A GPCRs were selected from the serotonin (5-HT2A), adrenergic (α1b-AR and β2-AR), muscarinic (M1 and M2), and dopamine (D1) receptor families. Each GPCR was C-terminally labeled with green fluorescent protein (GFP) or yellow fluorescent protein (YFP) and expressed in human embryonic kidney 293 cells. FCS provided plasma membrane diffusion coefficients on the order of 7.5 × 10−9 cm2/s. PCH molecular brightness analysis was used to determine the GPCR oligomer size. Known monomeric (CD-86) and dimeric (CD-28) receptors with GFP and YFP tags were used as controls to determine the molecular brightness of monomers and dimers. PCH analysis of fluorescence-tagged GPCRs revealed molecular brightness values that were twice the monomeric controls and similar to the dimeric controls. Reduced χ2 analyses of the PCH data best fit a model for a homogeneous population of homodimers, without tetramers or higher-order oligomers. The homodimer configuration was unaltered by agonist treatment and was stable over a 10-fold range of receptor expression level. The results of this study demonstrate that biogenic amine receptors freely diffusing within the plasma membrane are predominantly homodimers.
Introduction
G protein–coupled receptors (GPCRs) represent one of the largest families of plasma membrane-associated receptors. They are present on virtually every cell in the human body and regulate a wide variety of physiologic responses to light, odorants, hormones, neurotransmitters, and therapeutic agents. Physiologic processes regulated by GPCR activation and blockade have been studied for decades, but there is still great debate as to what constitutes the functional signaling unit: Is it a monomer, dimer, or higher-order oligomer? Although monomeric GPCRs in reconstituted lipid vesicles can activate G proteins (Whorton et al., 2007), GPCR dimers/oligomers have been reported in native tissues and primary cultures (Fotiadis et al., 2003; Rashid et al., 2007; Albizu et al., 2010; Herrick-Davis et al., 2012; Knepp et al., 2012; Teitler and Klein, 2012; Jastrzebska et al., 2013). Although the functional significance of class A GPCR dimerization is still a subject of great debate, GPCR homodimerization and heterodimerization have been reported to regulate ligand binding, second messenger activation, and receptor trafficking (reviewed in Milligan, 2013).
For class A GPCRs, there is no consensus in the published literature as to their monomer/oligomer status. Quantitative studies designed to determine the monomeric/oligomeric composition of class A GPCRs have reported the presence of monomers (James et al., 2006; Meyer et al., 2006; Dorsch et al., 2009; Hern et al., 2010; Kasai et al., 2011), dimers (Mercier et al., 2002; Canals et al., 2004; Goin and Nathanson, 2006; Harikumar et al., 2008; Dorsch et al., 2009; Herrick-Davis et al., 2012; Knepp et al., 2012; Teitler and Klein, 2012; Patowary et al., 2013), tetramers (Fung et al., 2009; Pisterzi et al., 2010; Patowary et al., 2013), and higher-order oligomers (Guo et al., 2008; Dorsch et al., 2009; Albizu et al., 2010; O’Dowd et al., 2011). Coimmunoprecipitation, resonance energy transfer (RET), fluorescence lifetime imaging (FLIM), and bimolecular fluorescence complementation (BiFC) are commonly used methods to evaluate protein–protein interactions. However, immunoprecipitation requires solubilization and disruption of the native GPCR plasma membrane environment, and RET and BiFC are proximity-based assays that monitor the distance between the fluorescent probes. Although these methods can provide evidence consistent with the hypothesis that GPCRs form dimers/oligomers, they do not provide conclusive proof of protein–protein interactions. Recently, purification of photoactivated rhodopsin from native disc membranes using lauryl-maltose-neopentyl-glycol and three-dimensional (3D) projection analysis revealed a rhodopsin dimer in complex with a single G protein (Jastrzebska et al., 2013).
Fluorescence correlation spectroscopy (FCS) provides a good alternative for investigating diffusion and protein interactions in living cells. It requires very low protein expression levels, making it suitable for studying plasma membrane GPCRs at physiologic expression levels. FCS records the fluctuations in fluorescence intensity arising from individual fluorescent molecules, in a temporal manner (Magde et al., 1972). Combining confocal microscopy with FCS led to the development of sensitive methods for monitoring protein dynamics in living cells (Qian and Elson, 1991; Rigler et al., 1993; Pramanik et al., 2001; Digman et al., 2005). FCS has been used to monitor diffusion and ligand binding for ion channels, tyrosine kinase receptors, and GPCRs (reviewed in Briddon and Hill, 2007), to examine neuropeptide Y and β-arrestin interactions (Kilpatrick et al., 2012), and to monitor the oligomer status of various receptors including somatostatin (Patel et al., 2002), epidermal growth factor (Liu et al., 2007), ciliary neurotrophic factor (Neugart et al., 2009), estrogen (Savatier et al., 2010), and serotonin 5-HT1A (Ganguly and Chattopadhyay, 2010) and 5-HT2C receptors (Herrick-Davis et al., 2012).
The oligomer status of a protein cluster can be determined by analyzing the amplitude of the fluctuations in fluorescence intensity measured in an FCS experiment. A photon counting histogram (PCH) can be generated from the FCS data and used to determine the molecular brightness of a fluorescence-tagged protein (Chen et al., 1999). Since the molecular brightness of a cluster of fluorescent molecules is directly proportional to the number of fluorescent molecules present in the cluster, the molecular brightness provides an estimate of the number of fluorescent molecules within the protein complex. Molecular brightness analysis has been used to explore the oligomeric status of nuclear retinoid X receptors (Chen et al., 2003), epidermal growth factor receptors (Saffarian et al., 2007), urokinase plasminogen activator receptors (Malengo et al., 2008), and serotonin 5-HT2C receptors (Herrick-Davis et al., 2012).
FCS combined with confocal microscopy and PCH provide powerful methods for determining the molecular brightness of individual fluorescence-tagged proteins as a measure of their oligomer size. The present study describes the application of FCS and PCH analysis for determining the oligomeric size of biogenic amine serotonin (5-HT2A), adrenergic (α1b-AR and β2-AR), muscarinic (M1 and M2), and dopamine (D1) receptors freely diffusing within the plasma membrane of living cells.
Materials and Methods
Plasmids.
cDNAs encoding the 5-HT2A, α1b-AR, β2-AR, M1-muscarinic, M2-muscarinic, and D1-dopamine receptors were polymerase chain reaction–amplified from human total genomic DNA and cloned into the pEGFP-N1 and pEYFP-N1 vectors (Clonetech Laboratories, Mountain View, CA) at EcoRI/BamHI to create chimeric receptors with fluorescent tags on the C terminus of the receptor. Site-directed mutagenesis (Stratagene, La Jolla, CA) was used to create an A206K mutation in all green fluorescent protein (GFP) constructs and an L221K mutation in the monomeric yellow fluorescent protein (YFP) and dimeric CD-28/YFP constructs to eliminate potential aggregation of the fluorescent tags (Zacharias et al., 2002). BiFC pairs, N- and C-terminal half of yellow fluorescent protein (N-YFP and C-YFP, respectively), were made by site-directed mutagenesis using the β2-AR/YFP cDNA as the starting template. β2-AR/N-YFP was made by inserting a stop codon at amino acid 156 of YFP. β2-AR/C-YFP was made by inserting a BamHI site at amino acid 156 of the YFP, followed by BamHI digest to remove amino acids 1–155 of the YFP, and subsequent re-ligation. The A2a-adenosine BiFC constructs were generously provided by S. Briddon (University of Nottingham). CD-28/YFP was a generous gift from J. Miller (University of Rochester). CD-86/GFP and CD-86/GFP-GFP were generously provided by G. Milligan (University of Glasgow).
Cell Culture, Transfection, and Drug Treatment.
Human embryonic kidney (HEK) 293 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s minimal essential medium (Cellgro, Manassas, VA) with 10% fetal bovine serum (HyClone, Logan, UT) in a humidified chamber at 37°C, 5% CO2. HEK293 cells were plated in 6-well plates fitted with 25-mm poly-d-lysine coated glass coverslips (Thermo Fisher Scientific, Waltham, MA) at a density of 5 × 105 cells per coverslip and transfected with 50 ng of the indicated plasmid DNA using lipofectamine reagent (Invitrogen, Carlsbad, CA) for 5 hours. After transfection, cells were cultured in minimal essential medium (MEM) (without phenol red) with 10% charcoal stripped serum (Gibco, Carlsbad, CA) for 20 hours at 37°C, 5% CO2. In our hands, this transfection protocol typically yields plasma membrane receptor expression levels on the order of 1 pmol/mg protein, by radioligand binding analysis. For drug treatment, isoproterenol and carbachol were diluted in HEPES-buffered MEM (without phenol red) and added directly to the viewing chamber to achieve a final concentration of 0.1 µM. FCS measurements were initiated 1 minute after the addition of ligand. We previously determined this time point to be sufficient for receptor activation, measured as β-arrestin recruitment to the plasma membrane (Herrick-Davis et al., 2007, 2012).
FCS.
For FCS measurements, cells were washed twice with HEPES-buffered Krebs-ringer (without glucose) and the coverslip was placed in a viewing chamber with 1 ml HEPES-buffered MEM (without phenol red). FCS measurements were made using a Zeiss LSM-780 confocal microscope equipped with gallium arsenide phosphide photon detectors (Carl Zeiss, Jena, Germany). One-photon excitation with a continuous argon ion laser was performed using a 40× (numerical aperture 1.2) C-apochromat water immersion objective to create an observation volume on the order of 10−15 liters. Since the observation volume is not illuminated homogenously, optimal positioning of the plasma membrane within the center of the observation volume is critical for accurate determination of the molecular brightness of fluorescent-tagged membrane proteins. FCS measurements were made on the apical plasma membrane, directly above the cell nucleus of HEK293 cells transfected with the indicated fluorescence-tagged receptor. Positioning of the plasma membrane in the center of the observation volume was achieved by monitoring the photon counts per molecule in real time (by opening the interactive counts/molecule window in the Zeiss software menu) while simultaneously focusing upward through the plasma membrane to identify the focal plane corresponding to the maximal photon counts per molecule. FCS measurements were recorded at 23°C in HEPES-buffered MEM (without phenol red) for 100 seconds, as 10 consecutive 10-second intervals. As fluorescent-tagged receptors enter and diffuse through the observation volume they are excited by the laser. GFP and YFP were excited at 488 and 514 nm, respectively, with a laser intensity of 0.1%. It is critical to use the lowest laser power possible, while still maintaining a good signal to noise ratio, because higher laser powers will result in photobleaching of the fluorescent probe. The time-dependent fluctuations in fluorescence intensity were recorded on gallium arsenide phosphide detectors as follows: emitted fluorescence captured by the objective is passed through an appropriate band pass filter and focused onto the detector using a pinhole of one airy unit. FCS recordings were analyzed by a digital temporal correlator (using nonlinear least-squares minimization; Zeiss Aim 4.2 software) to calculate the autocorrelation function G(τ), which represents the time-dependent decay in fluorescence fluctuation intensity as in Eq. 1,
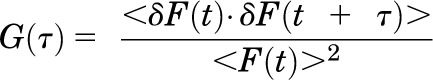 |
(1) |
where G(τ) is the <time average> of the change in fluorescence fluctuation intensity (δF) at some time point (t) and at a time interval later (t + τ), divided by the square of the average fluorescence intensity. Autocorrelation analyses were performed using the Zeiss Aim 4.2 software package with an autocorrelation bin time of 0.2 μs. FCS data were fit to a two-dimensional (2D) model (for lateral diffusion within the plasma membrane) with two components as in Eq. 2,
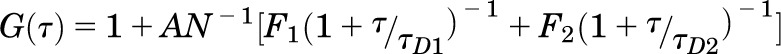 |
(2) |
where N is the number of molecules in the observation volume. F1 and F2 as well as τD1 and τD2 represent the respective fractions and diffusion times of the two components. A pre-exponential term is included to account for photophysical properties (blinking) of the fluorescent probe 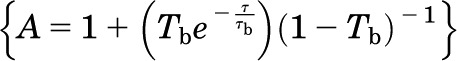 , where Tb and τb represent the blinking fraction and relaxation time, respectively. It should be noted that individual GFP and YFP molecules are not always fluorescent. They can exhibit blinking, exist in a prolonged dark state, or be immature and nonfluorescent (Ulbrich and Isacoff, 2007). The resulting autocorrelation curve depicts the fluorescence intensity fluctuations as a function of particle number and diffusion time. The average dwell time of the fluorescent species within the observation volume (τD) is calculated from the mid-point of the autocorrelation curve. The diffusion coefficient (D) for lateral diffusion of fluorescence-tagged GPCRs within the plasma membrane can then be calculated as in Eq. 3, where ω0 is the radial waist of the observation volume.
, where Tb and τb represent the blinking fraction and relaxation time, respectively. It should be noted that individual GFP and YFP molecules are not always fluorescent. They can exhibit blinking, exist in a prolonged dark state, or be immature and nonfluorescent (Ulbrich and Isacoff, 2007). The resulting autocorrelation curve depicts the fluorescence intensity fluctuations as a function of particle number and diffusion time. The average dwell time of the fluorescent species within the observation volume (τD) is calculated from the mid-point of the autocorrelation curve. The diffusion coefficient (D) for lateral diffusion of fluorescence-tagged GPCRs within the plasma membrane can then be calculated as in Eq. 3, where ω0 is the radial waist of the observation volume.
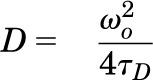 |
(3) |
The radial waist was determined experimentally from the full width at half maximum of a Gaussian distribution fitted to the image of subresolution fluorescent beads (0.1 μm, FluoSpheres; Invitrogen) as described by Cole et al. (2011). In our experimental set-up, the radial waist was determined to be 0.30 μm.
The amplitude of the autocorrelation function G(0) (equal to the y-intercept) is inversely related to the number of molecules in the observation volume (NPSF) as in Eq. 4,
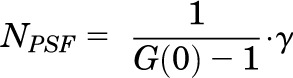 |
(4) |
where γ is the point spread function (PSF) that describes the shape of the observation volume. The numerical value of γ differs depending on the model selected for analysis and is 0.5 for 2D FCS analysis and 0.35 for a 3D Gaussian model used for PCH analysis. Although the absolute numerical values of molecular brightness will vary depending on whether a 2D FCS or 3D PCH model is selected (due to the different numerical value of γ used in Eq. 4), the overall conclusions will remain the same, because the molecular brightness of a dimer will still be twice that of a monomer regardless of the selected model.
The average fluorescence intensity or average photon count rate (k) recorded for a given sample is determined by the number of fluorescent molecules (NPSF) and their molecular brightness (ε), as described in Eq. 5.
| (5) |
Thus, dividing the count rate (k) by the number of molecules (NPSF) provides an estimate of the molecular brightness (ε) of the sample.
PCH.
Fluorescence fluctuation data recorded during an FCS experiment can be used to generate PCHs, which provide quantitative information about the number of fluorescent molecules and the number of photon counts per molecule (Chen et al., 1999). PCH analysis uses a 3D Gaussian approximation of the laser beam profile. In the present study, cells transfected with fluorescence-tagged receptors were selected with an average plasma membrane photon count rate ranging from 50 to 250 kHz, corresponding to plasma membrane receptor expression levels in the nanomolar range. Membrane regions containing ruffles, filopodia, and high concentrations of fluorescent proteins (>250 kHz) were avoided. Ten measurements were made on the upper plasma membrane of each cell by monitoring the photon count rate for 100 seconds, as 10 consecutive 10-second observation periods. While the laser intensity was set to 0.1% to minimize photobleaching, photobleaching was apparent during the first 10-second observation period. Therefore, the average molecular brightness from the second through tenth observation periods was calculated and reported as the molecular brightness for that cell. Segments of the fluorescence intensity trace that showed large spikes or drifts in fluorescence intensity (due to cell movement) were excluded from the analysis. To generate a histogram, each 10-second observation period was broken down into 1 million intervals or bins (PCH bin time = 10 μs). Histograms were constructed using the PCH module in the Zeiss Aim 4.2 software in which the number of 10-μs bins was plotted on the y-axis and photon counts on the x-axis. The resulting histogram depicts the number of bins that registered 1,2,3 photon counts and so forth during one 10-second observation period. Since a constant intensity light source produces a photon count distribution that follows Poisson statistics, as fluorescent molecules enter and diffuse through the nonhomogenously illuminated observation volume, the fluctuations in fluorescence intensity result in a broadening of the Poisson distribution. This super-Poisson characteristic is observed in the tail of the PCH curve. Initially, PCH data were fit to a one-component model in which concentration and molecular brightness were allowed to be free (and the first-order correction was fixed at zero) to determine the average molecular brightness of the sample.
Multicomponent PCH Analysis.
The PCH data were subjected to a multicomponent analysis to test for the presence of a mixture of monomers, dimers, and tetramers. Fitting the PCH data to a multicomponent model was performed as described by Müller et al. (2000) using the PCH module in the Zeiss Aim4.2 analysis software. Molecular brightness values were fixed based on the control values for monomers and dimers obtained from the initial one-component fit of the data (Table 1). Reduced χ2 analysis was used to determine the goodness of fit to both one-component and multicomponent models.
TABLE 1.
FCS and PCH analysis of HEK293 cells expressing YFP- and GFP-tagged biogenic amine receptors
Known monomeric (CD-86) and dimeric (CD-28) receptors are included as controls. M1/GFP and β2-AR/GFP were coexpressed with a 3-fold excess of untagged/nonfluorescent receptors (e.g., M1/GFP + M1/no tag 1:3). FCS τD2 values for GPCR diffusion within the plasma membrane are reported in milliseconds and represent the average dwell time of the receptor in the observation volume. Diffusion coefficients (μm2/s) were calculated using Eq. 3 (in the Materials and Methods). The 2D FCS molecular brightness values were determined by dividing the photon count rate by the number of molecules (as in Eqs. 4 and 5 in the Materials and Methods). The 3D PCH analysis was performed using the PCH module in the Zeiss Aim 4.2 software package by fitting the data to a one-component model (for a single population of fluorescent species) and the resulting reduced χ2 values are reported. Data represent the mean ± S.E.M. for the number of cells as indicated.
| Fluorescent Receptor | Diffusion | Molecular Brightness |
Reduced χ2 | n | ||
|---|---|---|---|---|---|---|
| 2D FCS | 3D PCH | |||||
| ms | μm2/s | |||||
| Dimeric CD-28/YFP | 29.1 ± 1.7 | 0.77 ± 0.03 | 12,728 ± 372 | 17,819 ± 484 | 1.07 ± 0.08 | 20 |
| 5-HT2A/YFP | 31.1 ± 0.6 | 0.72 ± 0.01 | 12,714 ± 255 | 17,927 ± 332 | 1.13 ± 0.04 | 25 |
| α1b-AR/YFP | 32.5 ± 0.6 | 0.69 ± 0.02 | 12,560 ± 606 | 17,709 ± 849 | 1.37 ± 0.13 | 15 |
| D1/YFP | 30.8 ± 0.6 | 0.73 ± 0.01 | 12,716 ± 509 | 17,930 ± 712 | 1.07 ± 0.04 | 15 |
| M2/YFP | 30.1 ± 0.5 | 0.75 ± 0.01 | 13,295 ± 410 | 18,480 ± 578 | 1.06 ± 0.07 | 10 |
| β2-AR/YFP | 30.3 ± 1.4 | 0.74 ± 0.03 | 13,201 ± 866 | 18,279 ± 1311 | 1.11 ± 0.10 | 12 |
| β2-AR/YFP BiFC | 31.3 ± 1.1 | 0.72 ± 0.02 | 6854 ± 343 | 9596 ± 480 | 1.10 ± 0.06 | 13 |
| Monomeric CD-86/GFP | 32.7 ± 1.4 | 0.69 ± 0.03 | 7088 ± 135 | 9364 ± 390 | 1.04 ± 0.10 | 12 |
| CD-86/GFP-GFP | 32.5 ± 1.1 | 0.69 ± 0.02 | 13,131 ± 450 | 18,777 ± 565 | 1.19 ± 0.08 | 10 |
| M1/GFP | 33.7 ± 2.1 | 0.67 ± 0.04 | 12,594 ± 295 | 17,852 ± 410 | 1.04 ± 0.06 | 20 |
| M1/GFP + carbachol | 36.0 ± 2.0 | 0.63 ± 0.04 | 12,878 ± 390 | 18,029 ± 546 | 1.12 ± 0.08 | 10 |
| M1/GFP + M1/no tag (1:3) | 36.5 ± 0.9 | 0.62 ± 0.02 | 6980 ± 334 | 9912 ± 434 | 1.08 ± 0.08 | 10 |
| β2-AR/GFP | 32.1 ± 1.0 | 0.70 ± 0.02 | 12,558 ± 572 | 18,117 ± 790 | 1.23 ± 0.05 | 15 |
| β2-AR/GFP + isoproterenol | 37.8 ± 1.7 | 0.60 ± 0.03 | 12,681 ± 564 | 17,602 ± 369 | 1.12 ± 0.09 | 12 |
| β2-AR/GFP + β2-AR/no tag (1:3) | 32.0 ± 1.1 | 0.70 ± 0.02 | 7087 ± 216 | 10,564 ± 300 | 1.05 ± 0.05 | 13 |
| β2-AR/GFP + M1/no tag (1:3) | 35.2 ± 1.7 | 0.63 ± 0.03 | 12,506 ± 227 | 17,950 ± 351 | 1.11 ± 0.09 | 10 |
Controls for Molecular Brightness Analysis.
Five different plasma membrane controls were used to decode the molecular brightness of GFP- and YFP-tagged biogenic amine receptors. Known monomeric (CD-86) and dimeric (CD-28) plasma membrane receptors with C-terminal GFP (CD-86/GFP and CD-86/GFP-GFP) and YFP (CD-28/YFP) were used to determine the molecular brightness of GFP and YFP monomers and dimers. The β2-AR/YFP BiFC pair and coexpression of β2-AR/GFP or M1/GFP with a 3-fold excess of untagged, nonfluorescent receptors were used as additional controls. To eliminate self-aggregation of GFP or YFP (Zacharias et al., 2002), all GFP constructs contained an A206K mutation and the CD-28/YFP construct contained an L221K mutation. To determine the contribution of background autofluorescence from cytoplasmic proteins, a dilute solution of purified monomeric GFP was evaluated. The molecular brightness of GFP in solution [8268 counts per second per molecule (CPSM)] was similar to GFP from pEGFP plasmid expressed in the cytosol of HEK293 cells (8508 CPSM), indicating that background autofluorescence from cytoplasmic proteins was minimal (approximately 3%) in our experimental set-up.
2D FCS Analysis of Molecular Brightness with Varying Receptor Expression Levels.
To investigate the relationship between molecular brightness and receptor expression level, 2D FCS analysis was used to determine the number of fluorescent molecules in the observation volume (as in Eq. 4) and the molecular brightness (as in Eq. 5). The area of plasma membrane in the observation volume was calculated using πω02, where ω0 is the radius of the observation volume (0.30 μm), determined experimentally as described in Eq. 3. The total surface area of an HEK293 cell, determined to be 2591 μm2 (Sommerhage et al., 2008), was used to estimate the total number of receptors per cell.
Results
FCS and PCH analyses were applied to determine the diffusion dynamics and oligomer status of biogenic amine GPCRs selected from the serotonin (5-HT2A), adrenergic (α1b-AR and β2-AR), muscarinic (M1 and M2), and dopamine (D1) receptor families. Fluorescent probes (GFP or YFP) were attached to the C terminus of each GPCR. In the present study, proper plasma membrane targeting of fluorescence-tagged GPCRs was confirmed by confocal microscopy (Fig. 1). FCS measurements were made on the upper plasma membrane of transfected HEK293 cells (as shown in Fig. 1D).
Fig. 1.
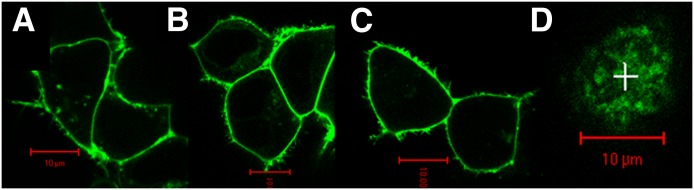
Confocal microscopy of transfected HEK293 cells showing plasma membrane localization of YFP-tagged proteins. (A) β2-AR/YFP. (B) D1/YFP. (C) Dimeric CD-28/YFP. (D) The upper plasma membrane of an HEK293 cell expressing β2-AR/YFP showing the location (marked by +) where an FCS recording was made. Red scale bar, 10 μm.
In an FCS experiment, a high numerical aperture objective is used to focus a laser beam into a small diffraction-limited spot, creating a detection or observation volume on the order of 10−15 liters. The upper plasma membrane of HEK293 cells expressing fluorescence-tagged GPCRs was positioned within the laser-illuminated observation volume by focusing upward from the middle of the cell to the top, while simultaneously monitoring the photon counts per molecule. Optimal positioning of the plasma membrane within the center of the observation volume is critical for accurate FCS/PCH analysis of molecular brightness because the detected photon counts decrease as fluorescent molecules travel away from the center of the observation volume. The fluorescence-tagged GPCRs, freely diffusing within the plasma membrane, pass through the observation volume where they are excited by a laser and give off bursts of photons. The fluctuations in fluorescence, produced by fluorescence-tagged GPCRs entering and leaving the observation volume, are recorded in real time, as shown in the fluorescence intensity traces in Fig. 2A.
Fig. 2.

FCS recordings from the plasma membrane of HEK293 cells expressing YFP-tagged GPCRs. (A) Fluorescence intensity traces for one 10-second observation period. (B) Autocorrelation analysis of the fluorescence intensity traces. The red line represents the autocorrelation of the observed fluorescence signal and the green line represents the fit to a two-component model. The fast component (measured in microseconds) is related to the photophysical properties of the fluorescent probe, whereas the slower component (measured in milliseconds) represents the translational diffusion of fluorescence-tagged receptors in the plasma membrane. Dividing the average photon count rate (kHz) determined from the fluorescence intensity trace shown in A by the number of fluorescent molecules determined from the autocorrelation curve shown in B (calculated as in Eq. 4) predicts the average molecular brightness of the sample expressed as CPSM.
Autocorrelation analyses of the fluorescence intensity traces (from Fig. 2A) are performed using a nonlinear least-squares fitting routine that graphically represents the autocorrelation function G(τ) on the ordinate and diffusion time on the abscissa (Fig. 2B). The rate at which GPCRs diffuse within the plasma membrane is reported as the average dwell time (τD) of the fluorescence-tagged GPCRs within the observation volume and is calculated from the mid-point of the autocorrelation decay curve. The biphasic autocorrelation curves shown in Fig. 2B are best fit by a two-component model with a very fast component characteristic of the photophysical properties of the fluorescent probe (τD1) and a slower component representing the translational diffusion of the GPCR within the plasma membrane (τD2).
Diffusion coefficients for fluorescence-tagged GPCRs in HEK293 cell plasma membranes are reported in Table 1. All diffusion data best fit a two-component model with τD1 values related to the photophysical properties of the fluorescent probe (50–100 μs for YFP and 250–300 μs for GFP) and τD2 values (representing the average dwell time of the GPCR within the observation volume) on the order of 30 milliseconds, yielding a diffusion coefficient on the order of 7.5 × 10−9cm2/s. Since FCS measures the mobile fraction of freely diffusing proteins, fluorescence-tagged GPCRs that are sequestered into microdomains with reduced mobility (Day and Kenworthy, 2009) or associated with cytoskeletal or extracellular matrix proteins could photobleach during an FCS recording. In the present study, approximately 40–50% of the initial fluorescence signal was photobleached during the first 10 seconds of each FCS recording (data not shown). This phenomenon, observed for all six GPCRs tested, is consistent with previous results obtained using fluorescence recovery after photobleach (FRAP) to monitor GPCR diffusion (Dorsch et al., 2009; Fonseca and Lambert, 2009).
Figure 2 shows representative FCS autocorrelation graphs for YFP-tagged 5-HT2A, β2-AR, D1-dopamine, M2-muscarinic, dimeric CD-28, and the β2-AR BiFC pair (β2-AR/N-YFP + β2-AR/C-YFP). Inspection of the fluorescence intensity traces in Fig. 2A reveals similar average photon count rates for the GPCR, and the dimeric CD-28/YFP control. The amplitude (y-intercept) of the autocorrelation curve (Fig. 2B) is inversely related to the number of fluorescent proteins or complexes present in the observation volume and is similar for the GPCR and CD-28/YFP. The molecular brightness of the YFP-tagged GPCRs can be determined from the FCS data by dividing the average photon count rate obtained from the fluorescence intensity traces in Fig. 2A (130 kHz on average for the GPCR and for the dimeric CD-28) by the number of fluorescent molecules calculated from the amplitude of the autocorrelation curves in Fig. 2B (using Eq. 4 in the Materials and Methods with a 2D FCS model yields and average N = 10). This provides an approximate molecular brightness of 13,000 CPSM for the GPCR and dimeric CD-28, consistent with a dimeric structure for the GPCR. The molecular brightness of the β2-AR BiFC pair, estimated as 42 kHz (Fig. 2A) divided by 6.3 (the number of molecules determined from Fig. 2B using Eq. 4) is 6667 CPSM, approximately half that of the YFP-tagged GPCR and dimeric CD-28.
PCH analysis uses a 3D Gaussian approximation of the laser beam profile and Poisson statistics to predict what the molecular brightness of the fluorescent particle would be when it is at the center of the observation volume (Chen et al., 1999). PCHs were generated from the FCS data presented in Fig. 2. The shape of the histogram is a function of the number of fluorescent molecules and their molecular brightness, and was the same for the GPCR and dimeric CD-28 (Fig. 3). To generate a histogram, each 10-second fluorescence intensity trace (as in Fig. 2A) was broken down into 1 million 10-µs intervals or bins (PCH bin time = 10 μs). Histograms were constructed in which the number of 10-μs bins was plotted on the y-axis and photon counts were plotted on the x-axis. The resulting histogram depicts the number of bins that registered 1,2,3…n photon counts during one 10-second observation period.
Fig. 3.

PCHs of the corresponding FCS recordings shown in Fig. 2. To generate the histograms, each 10-second fluorescence intensity trace (shown in Fig. 2A) was broken down into 1 million 10-µs intervals or bins (PCH bin time = 10 μs). The number of bins is plotted on the y-axis and photon counts on the x-axis. The resulting histogram depicts the number of bins that registered 1,2,3…n photon counts during one 10-second observation period. The residuals of the curve fit (shown in the lower panels) plot the number of bins on the y-axis and photon counts on the x-axis. The data were fit to a one-component model for a single homogenous population of homodimers. The residuals of the curve fit are <2 S.D. and are randomly distributed about zero, indicating that the data are a good fit for the selected model, with reduced χ2 equal to unity.
PCHs for the biogenic amine GPCR and dimeric CD-28 (Fig. 3) show an average number of photon counts per 10-µs bin time of 1.25, equivalent to 125,000 counts per second. Dividing by the average number of molecules calculated from the amplitude of the autocorrelation curves in Fig. 2B (using Eq. 4 with a 3D PCH model where N = 7) yields an average molecular brightness of 17,857 CPSM for the GPCR and dimeric CD-28. For the β2-AR BiFC pair, the PCH predicts 0.4 photon counts per 10-µs bin time, equivalent to 40,000 counts per second. Dividing by the average number of molecules calculated from the amplitude of the autocorrelation curve in Fig. 2B (using Eq. 4 with a 3D PCH model where N = 4.3) yields an average molecular brightness of 9302 CPSM.
FCS and PCH molecular brightness values for YFP- and GFP-tagged receptors are reported in Table 1. Known monomeric (CD-86) and dimeric (CD-28) plasma membrane receptors with C-terminal GFP (CD-86/GFP and CD-86/GFP-GFP) and YFP (CD-28/YFP) were used to determine the molecular brightness of GFP and YFP monomers and dimers. Molecular brightness values for all GPCRs tested were similar to one another and were similar to dimeric CD-28/YFP and tandem GFP attached to monomeric CD-86 (CD-86/GFP-GFP). The monomeric CD-86/GFP control was half the brightness of the dimeric control (Fig. 4) and of the GPCR.
Fig. 4.

FCS recordings from the plasma membrane of HEK293 cells expressing CD-86/GFP and CD-86/GFP-GFP. (A) Fluorescence intensity traces for one 10-second observation period. (B) Autocorrelation analysis of the fluorescence intensity traces. The red line represents the autocorrelation of the observed fluorescence signal and the green line represents the fit to a two-component model. The fast component (measured in microseconds) is related to the photophysical properties of the fluorescent probe, whereas the slower component (measured in milliseconds) represents the translational diffusion of the fluorescence-tagged receptors in the plasma membrane. (C) PCHs of the corresponding FCS recordings yield molecular brightness values of 9693 CPSM for CD-86/GFP and 18,194 CPSM for CD-86/GFP-GFP. (D) Residuals of the curve fit. The data were fit to a one-component model for a single homogenous population of fluorescence-tagged receptors.
An experimental paradigm using coexpression of fluorescence-tagged GPCRs with an excess of untagged/nonfluorescent receptor was used as an additional measure of GPCR oligomer size. In this paradigm, HEK293 cells were cotransfected with GFP-tagged GPCRs and a 3-fold excess of untagged, nonfluorescent GPCRs. These studies were performed using β2-AR and M1-muscarinic receptors. For example, if β2-AR are monomeric, then coexpression of GFP-tagged β2-AR with an excess of nonfluorescent β2-AR would have no effect on the observed molecular brightness. However, if the receptors form homodimers, then this paradigm would produce dimers composed of one GFP-tagged protomer and one nonfluorescent protomer. In this case, the observed molecular brightness would be reduced by half. On the other hand, if the receptors are capable of forming tetramers, then coexpression with a 3-fold excess of untagged receptor should reduce the molecular brightness by greater than 50%. Coexpression of β2-AR/GFP with excess untagged β2-AR or M1/GFP with the excess untagged M1 receptor reduced the molecular brightness by approximately half. In contrast, coexpression of β2-AR/GFP with the untagged M1 receptor had no effect on the molecular brightness of β2-AR/GFP. These results are consistent with a homodimeric structure for β2-AR and M1-muscarinic receptors.
BiFC between the N- and C-YFP attached to the β2-AR was utilized as an additional control. BiFC involves the recombination of two nonfluorescent halves of a protein, such as the N-YFP and the C-YFP. When two proteins (one with N-YFP and one with C-YFP) are in close proximity, N-YFP and C-YFP can recombine to reconstitute YFP fluorescence. In the present study, YFP BiFC pairs were created using the β2-AR and A2a-adenosine receptors. Coexpression of β2-AR/N-YFP with β2-AR/C-YFP in HEK293 cells results in fluorescence complementation and the generation of YFP fluorescence on the plasma membrane of transfected HEK293 cells (Fig. 5). The specificity of the positive BiFC signal is demonstrated by reduced fluorescence after coexpression of β2-AR/N-YFP with A2a-adenosine/C-YFP, and restoration of fluorescence in cells coexpressing A2a-adenosine/N-YFP with A2a-adenosine/C-YFP.
Fig. 5.

BiFC. The N- and C-terminal halves of YFP were attached to the C-terminal end of β2-AR or A2a-adenosine receptors. (Top) HEK293 cells coexpressing β2-AR/N-YFP with β2-AR/C-YFP show plasma membrane YFP fluorescence 20 hour post-transfection (left). The middle panel shows the differential interference contrast image and the right panel shows the merged image. (Middle) HEK293 cells coexpressing β2-AR/N-YFP and A2a-adenosine/C-YFP show minimal fluorescence complementation. (Bottom) Restoration of plasma membrane fluorescence complementation in HEK293 cells coexpressing A2a-adenosine/N-YFP and A2a-adenosine/C-YFP. Red scale bar, 10 μm.
Plasma membrane FCS analysis of cells coexpressing β2-AR/N-YFP and β2-AR/C-YFP yielded diffusion coefficients similar to those observed for the parent β2-AR/YFP complex (Table 1). If β2-AR form homodimers, then the molecular brightness of the β2-AR BiFC pair would be expected to be approximately half that observed for β2-AR/YFP. FCS and PCH analyses revealed molecular brightness values for the β2-AR BiFC pair that were approximately half that observed for β2-AR/YFP and the dimeric CD-28/YFP control (Table 1), again predicting a homodimeric structure for the β2-AR.
Molecular brightness values equivalent to a homodimer, as reported in Table 1, could be produced by a homogeneous population of homodimers or by a mixture of monomers, dimers and tetramers. Since FCS analysis would yield similar results in both cases (Meseth et al., 1999), PCH and reduced χ2 analyses were used to determine the goodness of fit to both one and multicomponent models (Müller et al., 2000). Reduced χ2 values were close to unity when the PCH data were fit to a one-component model for a homogeneous population of homodimers (Table 1). Multicomponent modeling of the PCH data to test for a mixture of monomers, dimers and tetramers did not provide a better fit of the data. Modeling the data using fixed monomer/dimer/tetramer ratios of 20%/70%/10% and 40%/40%/20% (ratios predicted to give an average molecular brightness equivalent to a dimer) produced reduced χ2 values greater than 3 and 10, respectively (Figs. 6 and 7).
Fig. 6.

Single-component and multicomponent analyses for HEK293 cells expressing β2-AR/GFP, 5-HT2A/YFP, or D1/YFP. PCHs and residuals of the curve fit for the following: (A) a one-component fit of the data for a single population of fluorescence-tagged receptors; (B) a mixture of monomers/dimers/tetramers in a fixed 20%/70%/10% ratio; and (C) a mixture of monomers/dimers/tetramers in a fixed 40%/40%/20% ratio. Reduced χ2 values for the one-component and the multicomponent 20%70%/10% and 40%/40%/20% ratios were 0.8, 4.7, and 18, respectively, for β2-AR/GFP; 1.1, 4.3, and 17, respectively, for 5-HT2A/YFP; and 1.0, 4.6, and 18, respectively, for D1/YFP.
Fig. 7.

Single-component and multicomponent analyses for HEK293 cells expressing M1/GFP, M2/YFP or α1b-AR/YFP. PCH and residuals of the curve fit for the following: (A) a one-component fit of the data for a single population of fluorescence-tagged receptors; (B) a mixture of monomers/dimers/tetramers in a fixed 20%/70%/10% ratio; and (C) a mixture of monomers/dimers/tetramers in a fixed 40%/40%/20% ratio. Reduced χ2 values for the one-component and the multicomponent 20%/70%/10% and 40%/40%/20% ratios were 0.7, 7.0 and 23, respectively, for M1/GFP; 1.1, 3.2 and 16, respectively, for M2/YFP; and 1.3, 6.0 and 23, respectively, for α1b-AR/YFP.
To determine the effect of agonist treatment on receptor diffusion rate and on GPCR oligomer status, transfected cells expressing GFP-tagged β2-AR or M1-muscarinic receptors were treated with 0.1 μM isoproterenol or carbachol, respectively, for 1 minute prior to FCS recording. As shown in Table 2, diffusion rates and molecular brightness values were not altered after treatment with agonist, similar to our previous findings with serotonin 5-HT2C receptors (Herrick-Davis et al., 2012).
TABLE 2.
Relationship between molecular brightness and receptor expression level
Plasma membrane 2D FCS molecular brightness analysis of HEK293 cells expressing different levels of GFP-tagged M1-muscarinic receptors (M1/GFP), β2-adrenergic receptors (β2-AR/GFP), and monomeric CD-86 labeled with two GFP tags (CD-86/GFP-GFP). The photon count rate (kHz) is a measure of the overall fluorescence intensity of the region of plasma membrane in the observation volume and n is the number of receptor dimers in the observation volume (calculated as in Eq. 4). Molecular brightness values are reported as photon counts per molecule. Data represent the mean ± S.E.M. from three cells with similar photon count rates (kHz). The area of plasma membrane in the observation volume was calculated using πω02, where ω0 is the radius of the observation volume (calculated as in the Materials and Methods), and was used to determine the number of dimers per µm2 of plasma membrane. An HEK293 cell total surface area equivalent to 2591 μm2 (Sommerhage et al., 2008) was used to estimate the total number of dimers per cell.
| Receptor | Count Rate | n | Molecular Brightness | Dimers | |
|---|---|---|---|---|---|
| kHz | per µm2 | per cell | |||
| M1/GFP | 37 ± 1.7 | 3.0 ± 0.2 | 12,525 ± 244 | 10 | 2.6 × 104 |
| β2-AR/GFP | 46 ± 4.4 | 3.5 ± 0.5 | 13,083 ± 941 | 12 | 3.1 × 104 |
| CD-86/GFP-GFP | 69 ± 12 | 5.6 ± 1.2 | 12,537 ± 595 | 20 | 5.2 × 104 |
| M1/GFP | 113 ± 2.9 | 8.9 ± 0.4 | 12,797 ± 446 | 31 | 8.0 × 104 |
| β2-AR/GFP | 192 ± 8.8 | 15 ± 0.4 | 12,579 ± 465 | 53 | 1.4 × 105 |
| M1/GFP | 206 ± 7.4 | 16 ± 0.2 | 12,460 ± 704 | 56 | 1.5 × 105 |
| CD-86/GFP-GFP | 223 ± 12 | 18 ± 0.7 | 12,750 ± 233 | 63 | 1.6 × 105 |
| β2-AR/GFP | 311 ± 26 | 26 ± 2.7 | 12,137 ± 324 | 91 | 2.4 × 105 |
| M1/GFP | 358 ± 4.5 | 29 ± 1.2 | 12,496 ± 707 | 102 | 2.6 × 105 |
| β2-AR/GFP | 413 ± 19 | 34 ± 1.0 | 11,970 ± 285 | 119 | 3.1 × 105 |
The FCS data presented in Table 2 evaluate the relationship between molecular brightness and receptor expression level for β2-AR and M1-muscarinic receptors. The surface area of plasma membrane in the observation volume and the number of dimers per cell were calculated as described in the Materials and Methods. There was no evidence of dimers dissociating into monomers or associating into tetramers over a 10-fold range of receptor expression level. Similar results were obtained for all GPCRs tested in this study.
Discussion
The present study was preformed to shed light on the ongoing controversy related to class A GPCR oligomer size. Confocal microscopy-based FCS was chosen as the method of analysis based on its sensitivity and applicability to live cell membranes. FCS records the fluctuations in fluorescence intensity arising from individual fluorescent molecules, or in this case fluorescence-tagged GPCRs, as they diffuse through the plasma membrane. Two different fluorescent probes were used to ensure that the results were not due to a unique photophysical property of the fluorescent probe and to determine reproducibility of results. Diffusion coefficients for the biogenic amine receptors on the order of 7.5 × 10−9 cm2/s are similar to previously published diffusion coefficients for class A GPCRs (reviewed in Briddon and Hill, 2007). The diffusion rate of a membrane protein is related to the cubic root of the protein’s mass, such that an 8-fold change in mass would be required to produce a 2-fold change in the diffusion rate. To be resolved by FCS, the diffusion rates of two proteins must differ by a factor of 1.6 or greater (Meseth et al., 1999). Therefore, GPCR monomers, dimers, and tetramers cannot be distinguished from one another based on their diffusion coefficients alone.
Information about a protein’s oligomer size can be obtained by analyzing the amplitude of the fluorescence intensity fluctuations recorded during an FCS experiment and generating a PCH to determine the molecular brightness (Chen et al., 1999). Since the molecular brightness is proportional to the number of fluorescent molecules traveling together within a protein complex, a GPCR monomer with a single fluorescent tag would have a molecular brightness of x, a dimer carrying two fluorescent tags would be 2x, a tetramer would be 4x, and so forth. PCH analysis revealed similar molecular brightness values for all six GPCRs tested. Molecular brightness values were similar to the dimeric controls and twice the monomeric controls, consistent with a dimeric structure for the biogenic amine GPCR. Two additional plasma membrane controls were utilized using BiFC constructs and coexpression with excess untagged/nonfluorescent receptors. In both cases, the results were consistent with a homodimeric structure for the biogenic amine GPCR examined in this study.
Previous studies designed to assess the oligomer status of the biogenic amine GPCR have utilized a variety of techniques, including RET, FLIM, FRAP, total internal reflection fluorescence (TIRF), and FCS. These methods differ in sensitivity and the population of receptors that they examine. RET and FLIM are proximity assays, whereas TIRF and FCS have near single molecule sensitivity. TIRF, FRAP, and FCS measure the mobile fraction of receptors and allow discrete regions of plasma membrane to be evaluated, whereas BRET measures the entire pool of receptors. In terms of determining oligomer number, the above-described methods have produced conflicting results even within the same biogenic amine receptor subfamily. For example, RET, FRAP, and TIRF studies of M1-, M2-, and M3-muscarinic receptors have reported monomers (Hern et al., 2010), dimers (Goin and Nathanson, 2006; Hern et al., 2010; Patowary et al., 2013), and tetramers (Pisterzi et al., 2010; McMillin et al., 2011; Patowary et al., 2013). RET and FRAP studies of β1-AR and β2-AR have reported monomers (James et al., 2006; Dorsch et al., 2009), dimers (Mercier et al., 2002; Dorsch et al., 2009), and higher-order oligomers (Dorsch et al., 2009; Fung et al., 2009). D1- and D2-dopamine receptors have been reported to form homodimers and higher-order oligomers assayed by RET (Guo et al., 2008) and using a nuclear translocation assay (O’Dowd et al., 2011). RET studies of serotonin 5-HT1A receptors have reported homodimers (Kobe et al., 2008) and higher-order oligomers (Ganguly et al., 2011), whereas 5-HT2A, 5-HT2C, 5-HT4, and 5-HT7 appear to be predominantly dimeric (Herrick-Davis et al., 2005, 2012; Brea et al., 2009; Pellissier et al., 2011; Teitler and Klein, 2012). To further complicate the issue, FRAP and TIRF studies suggested that β1-AR and M1-muscarinic receptors may exist in equilibrium between monomeric and dimeric states (Dorsch et al., 2009; Hern et al., 2010). In addition, mixed populations of homodimers and higher-order oligomers for D1- and D2-dopamine and M3-muscarinic receptors have been reported (Fonseca and Lambert, 2009; O’Dowd et al., 2011; Patowary et al., 2013). Our FCS/PCH studies did not reveal the presence of tetramers or higher-order oligomers of biogenic amine receptors on the plasma membrane. PCH provides an estimate of the average molecular brightness of all fluorescent species present in the sample (Müller et al., 2000); thus, if the GPCRs tested in this study exist in monomer-dimer or dimer-tetramer equilibrium, then the observed molecular brightness values would be an average based on the monomer-dimer or dimer-tetramer composition of the sample. Reduced χ2 analysis of a multicomponent fit of the PCH data, testing for the presence of a mixture of monomers, dimers, and tetramers, did not provide a better fit of the data than a one-component analysis for a homogeneous population of homodimers.
Attempts to reconcile differences in GPCR oligomer status reported by the different methods used in the literature have led to the suggestion that receptor expression level may influence GPCR monomer-dimer and/or dimer-tetramer states on the plasma membrane, with low expression levels favoring monomeric forms and higher expression levels favoring association of dimers into tetramers (Hern et al., 2010; Lambert, 2010; Patowary et al., 2013). In the present study, we compared the molecular brightness of fluorescence-tagged GPCRs over a 10-fold range of receptor expression levels. FCS analysis of the mobile fraction of plasma membrane receptors demonstrates that the homodimer configuration is maintained from 26,000 dimers per cell to 310,000 dimers per cell. Because the FCS technique is best suited for studying proteins at low expression levels, we cannot rule out the possibility that significantly higher expression levels may lead to receptor clustering. It is recognized that palmitoylation promotes sequestration of GPCRs into discrete microdomains (Kobe et al., 2008; Day and Kenworthy, 2009; Woehler et al., 2009), and that the majority of class A GPCRs have at least one palmitoylation site in the C terminus following helix 8. Receptor clustering into membrane microdomains would increase local receptor concentrations, complicating the interpretation of RET-based studies that monitor protein proximity, because positive RET is suggestive of but does not demonstrate protein–protein interaction. Further complicating the issue, stochastic RET has been reported for receptors clustered in membrane microdomains (Meyer et al., 2006; Kobe et al., 2008; Woehler et al., 2009). In one study, serotonin 5-HT1A receptors clustered in membrane microdomains gave RET values consistent with an oligomer number greater than 2, whereas nonpalmitoylated mutant receptors that were excluded from microdomains gave an oligomer number of 2 (Woehler et al., 2009). It is interesting to note that studies employing techniques with near single molecule sensitivity (TIRF and FCS) have only reported monomers or dimers, without tetramers or higher-order oligomers (Hern et al., 2010; Kasai et al., 2011; Herrick-Davis et al., 2012), as observed in the present study.
Studies examining the effect of ligands on GPCR oligomer status have produced differing results ranging from no effect to dissociation or association of dimers into oligomers. fluorescence RET/FLIM studies of serotonin 5-HT1A receptors (Kobe et al., 2008) and FCS studies of 5-HT2C receptors (Herrick-Davis et al., 2012), along with FRAP studies of β1-AR and β2-AR (Dorsch et al., 2009) and fluorescence RET studies of M1- and M2-muscarinic receptors (Goin et al., 2006), report no effect of agonist on GPCR homodimer/oligomer status in intact cells. On the other hand, studies of 5-HT1A (Ganguly et al., 2011), β2-AR (Fung et al., 2009), M3-muscarinic (Alvarez-Curto et al., 2010), and D1- and D2-dopamine receptors (Guo et al., 2008; O’Dowd et al., 2011) suggest that these receptors form tetramers or higher-order oligomers that are differentially regulated by treatment with various ligands. It is probable that the reported changes in GPCR dimer/oligomer status after ligand treatment are method, ligand, and receptor dependent. In our FCS studies, treatment with isoproterenol or carbachol did not result in β2-AR or M1-muscarinic homodimer dissociation or association into higher-order oligomers. Since FCS measures mobile proteins, we cannot exclude the possibility that agonist binding enhances GPCR sequestration and possible clustering within membrane microdomains with reduced mobility (Day and Kenworthy, 2009). GPCR trafficking between mobile and less mobile fractions could potentially account for some of the variability obtained with different techniques that monitor different fractions or subpopulations of receptors within the plasma membrane.
In summary, the functional significance of homodimerization for class A GPCRs is still a subject of great debate. It is possible that homodimerization is an essential step in protein folding required for exit from the endoplasmic reticulum (Salahpour et al., 2004; Herrick-Davis et al., 2006; Lopez-Gimenez et al., 2007) because the dimer may represent the minimal functional signaling unit. In the present study, the enhanced sensitivity of FCS over proximity-based assays provides conclusive demonstration of the presence of biogenic amine homodimers, and lack of tetramers or higher-order complexes, freely diffusing within the plasma membrane of living cells. The homodimer structure was not altered by receptor expression level or after agonist binding, consistent with the hypothesis that the homodimer represents the basic signaling unit.
Abbreviations
- 2D
two-dimensional
- 3D
three-dimensional
- AR
adrenergic receptor
- BiFC
bimolecular fluorescence complementation
- CPSM
counts per second per molecule
- C-YFP
C-terminal half of yellow fluorescent protein
- FCS
fluorescence correlation spectroscopy
- FLIM
fluorescence lifetime imaging
- FRAP
fluorescence recovery after photobleach
- GFP
green fluorescent protein
- GPCR
G protein–coupled receptor
- MEM
minimal essential medium
- N-YFP
N-terminal half of yellow fluorescent protein
- PCH
photon counting histogram
- PSF
point spread function
- RET
resonance energy transfer
- TIRF
total internal reflection fluorescence
- YFP
yellow fluorescent protein
Authorship Contributions
Participated in research design: Herrick-Davis, Grinde, Cowan, Mazurkiewicz.
Conducted experiments: Herrick-Davis, Grinde, Mazurkiewicz.
Contributed new reagents or analytic tools: Cowan.
Performed data analysis: Herrick-Davis, Mazurkiewicz.
Wrote or contributed to the writing of the manuscript: Herrick-Davis, Grinde, Cowan, Mazurkiewicz.
Footnotes
This work was supported by the National Institutes of Health National Institute of Mental Health [Grant R21-MH086796 (to K.H.-D.)].
References
- Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, et al. (2010) Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol 6:587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Curto E, Ward RJ, Pediani JD, Milligan G. (2010) Ligand regulation of the quaternary organization of cell surface M3 muscarinic acetylcholine receptors analyzed by fluorescence resonance energy transfer (FRET) imaging and homogeneous time-resolved FRET. J Biol Chem 285:23318–23330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brea J, Castro M, Giraldo J, López-Giménez JF, Padín JF, Quintián F, Cadavid MI, Vilaró MT, Mengod G, Berg KA, et al. (2009) Evidence for distinct antagonist-revealed functional states of 5-hydroxytryptamine(2A) receptor homodimers. Mol Pharmacol 75:1380–1391 [DOI] [PubMed] [Google Scholar]
- Briddon SJ, Hill SJ. (2007) Pharmacology under the microscope: the use of fluorescence correlation spectroscopy to determine the properties of ligand-receptor complexes. Trends Pharmacol Sci 28:637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Burgueño J, Marcellino D, Cabello N, Canela EI, Mallol J, Agnati L, Ferré S, Bouvier M, Fuxe K, et al. (2004) Homodimerization of adenosine A2A receptors: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Neurochem 88:726–734 [DOI] [PubMed] [Google Scholar]
- Chen Y, Müller JD, So PT, Gratton E. (1999) The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys J 77:553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wei LN, Müller JD. (2003) Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc Natl Acad Sci USA 100:15492–15497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RW, Jinadasa T, Brown CM. (2011) Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nat Protoc 6:1929–1941 [DOI] [PubMed] [Google Scholar]
- Day CA, Kenworthy AK. (2009) Tracking microdomain dynamics in cell membranes. Biochim Biophys Acta 1788:245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Brown CM, Sengupta P, Wiseman PW, Horwitz AR, Gratton E. (2005) Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys J 89:1317–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bünemann M. (2009) Analysis of receptor oligomerization by FRAP microscopy. Nat Methods 6:225–230 [DOI] [PubMed] [Google Scholar]
- Fonseca JM, Lambert NA. (2009) Instability of a class a G protein-coupled receptor oligomer interface. Mol Pharmacol 75:1296–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. (2003) Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 421:127–128 [DOI] [PubMed] [Google Scholar]
- Fung JJ, Deupi X, Pardo L, Yao XJ, Velez-Ruiz GA, Devree BT, Sunahara RK, Kobilka BK. (2009) Ligand-regulated oligomerization of beta(2)-adrenoceptors in a model lipid bilayer. EMBO J 28:3315–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly S, Chattopadhyay A. (2010) Cholesterol depletion mimics the effect of cytoskeletal destabilization on membrane dynamics of the serotonin1A receptor: A zFCS study. Biophys J 99:1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly S, Clayton AH, Chattopadhyay A. (2011) Organization of higher-order oligomers of the serotonin₁(A) receptor explored utilizing homo-FRET in live cells. Biophys J 100:361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goin JC, Nathanson NM. (2006) Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells: regulation of receptor down-regulation by heterodimerization. J Biol Chem 281:5416–5425 [DOI] [PubMed] [Google Scholar]
- Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, Javitch JA. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J 27:2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Happs RM, and Miller LJ (2008) Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochim Biophys Acta 1778:2555–2563. [DOI] [PMC free article] [PubMed]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JET, Lazareno S, Molloy JE, Birdsall NJM. (2010) Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci USA 107:2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz JE. (2005) Inhibition of serotonin 5-hydroxytryptamine2c receptor function through heterodimerization: receptor dimers bind two molecules of ligand and one G-protein. J Biol Chem 280:40144–40151 [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Weaver BA, Grinde E, Mazurkiewicz JE. (2006) Serotonin 5-HT2C receptor homodimer biogenesis in the endoplasmic reticulum: real-time visualization with confocal fluorescence resonance energy transfer. J Biol Chem 281:27109–27116 [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Weaver BA. (2007) Serotonin 5-HT(2C) receptor homodimerization is not regulated by agonist or inverse agonist treatment. Eur J Pharmacol 568:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Lindsley T, Cowan A, Mazurkiewicz JE. (2012) Oligomer size of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor revealed by fluorescence correlation spectroscopy with photon counting histogram analysis: evidence for homodimers without monomers or tetramers. J Biol Chem 287:23604–23614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. (2006) A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods 3:1001–1006 [DOI] [PubMed] [Google Scholar]
- Jastrzebska B, Ringler P, Palczewski K, Engel A. (2013) The rhodopsin-transducin complex houses two distinct rhodopsin molecules. J Struct Biol 182:164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai RS, Suzuki KGN, Prossnitz ER, Koyama-Honda I, Nakada C, Fujiwara TK, Kusumi A. (2011) Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J Cell Biol 192:463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick LE, Briddon SJ, Holliday ND. (2012) Fluorescence correlation spectroscopy, combined with bimolecular fluorescence complementation, reveals the effects of β-arrestin complexes and endocytic targeting on the membrane mobility of neuropeptide Y receptors. Biochim Biophys Acta 1823:1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe F, Renner U, Woehler A, Wlodarczyk J, Papusheva E, Bao G, Zeug A, Richter DW, Neher E, Ponimaskin E. (2008) Stimulation- and palmitoylation-dependent changes in oligomeric conformation of serotonin 5-HT1A receptors. Biochim Biophys Acta 1783:1503–1516 [DOI] [PubMed] [Google Scholar]
- Knepp AM, Periole X, Marrink SJ, Sakmar TP, Huber T. (2012) Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51:1819–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA. (2010) GPCR dimers fall apart. Sci Signal 3:pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Sudhaharan T, Koh RM, Hwang LC, Ahmed S, Maruyama IN, Wohland T. (2007) Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys J 93:684–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gimenez JF, Canals M, Pediani JD, and Milligan G (2007) The alpha1b-adrenoceptor exists as a higher-order oligomer: effective oligomerization is required for receptor maturation, surface delivery, and function. Mol Pharmacol 71:1015–1029. [DOI] [PubMed]
- Malengo G, Andolfo A, Sidenius N, Gratton E, Zamai M, Caiolfa VR. (2008) Fluorescence correlation spectroscopy and photon counting histogram on membrane proteins: functional dynamics of the glycosylphosphatidylinositol-anchored urokinase plasminogen activator receptor. J Biomed Opt 13:031215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde D, Elson E, Webb WW. (1972) Thermodynamic fluctuations in a reacting system: measurement by fluorescence correlation spectroscopy. Phys Rev Lett 29:705–708 [Google Scholar]
- McMillin SM, Heusel M, Liu T, Costanzi S, Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J Biol Chem 286:28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. (2002) Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem 277:44925–44931 [DOI] [PubMed] [Google Scholar]
- Meseth U, Wohland T, Rigler R, Vogel H. (1999) Resolution of fluorescence correlation measurements. Biophys J 76:1619–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BH, Segura JM, Martinez KL, Hovius R, George N, Johnsson K, Vogel H. (2006) FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc Natl Acad Sci USA 103:2138–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. (2013) The prevalence, maintenance, and relevance of g protein-coupled receptor oligomerization. Mol Pharmacol 84:158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller JD, Chen Y, Gratton E. (2000) Resolving heterogeneity on the single molecular level with the photon-counting histogram. Biophys J 78:474–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugart F, Zappe A, Buk DM, Ziegler I, Steinert S, Schumacher M, Schopf E, Bessey R, Wurster K, Tietz C, et al. (2009) Detection of ligand-induced CNTF receptor dimers in living cells by fluorescence cross correlation spectroscopy. Biochim Biophys Acta 1788:1890–1900 [DOI] [PubMed] [Google Scholar]
- O’Dowd BF, Ji X, Alijaniaram M, Nguyen T, George SR. (2011) Separation and reformation of cell surface dopamine receptor oligomers visualized in cells. Eur J Pharmacol 658:74–83 [DOI] [PubMed] [Google Scholar]
- Patel RC, Kumar U, Lamb DC, Eid JS, Rocheville M, Grant M, Rani A, Hazlett T, Patel SC, Gratton E, et al. (2002) Ligand binding to somatostatin receptors induces receptor-specific oligomer formation in live cells. Proc Natl Acad Sci USA 99:3294–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patowary S, Alvarez-Curto E, Xu TR, Holz JD, Oliver JA, Milligan G, Raicu V. (2013) The muscarinic M3 acetylcholine receptor exists as two differently sized complexes at the plasma membrane. Biochem J 452:303–312 [DOI] [PubMed] [Google Scholar]
- Pellissier LP, Barthet G, Gaven F, Cassier E, Trinquet E, Pin JP, Marin P, Dumuis A, Bockaert J, Banères JL, et al. (2011) G protein activation by serotonin type 4 receptor dimers: evidence that turning on two protomers is more efficient. J Biol Chem 286:9985–9997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisterzi LF, Jansma DB, Georgiou J, Woodside MJ, Chou JT, Angers S, Raizu V, and Wells JW (2010) Oligomeric size of the M2 muscarinic receptor in live cells as determined by quantitative fluorescence resonance energy transfer. J Biol Chem 285:16723–16738. [DOI] [PMC free article] [PubMed]
- Pramanik A, Olsson M, Langel U, Bartfai T, Rigler R. (2001) Fluorescence correlation spectroscopy detects galanin receptor diversity on insulinoma cells. Biochemistry 40:10839–10845 [DOI] [PubMed] [Google Scholar]
- Qian H, Elson EL. (1991) Analysis of confocal laser-microscope optics for 3-D fluorescence correlation spectroscopy. Appl Opt 30:1185–1195 [DOI] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, George SR. (2007) D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA 104:654–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler R, Mets U, Widengren J, Kask P. (1993) Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur Biophys J 22:169–175 [Google Scholar]
- Saffarian S, Li Y, Elson EL, Pike LJ. (2007) Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys J 93:1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Mercier JF, Lagacé M, Marullo S, Bouvier M. (2004) Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem 279:33390–33397 [DOI] [PubMed] [Google Scholar]
- Savatier J, Jalaguier S, Ferguson ML, Cavaillès V, Royer CA. (2010) Estrogen receptor interactions and dynamics monitored in live cells by fluorescence cross-correlation spectroscopy. Biochemistry 49:772–781 [DOI] [PubMed] [Google Scholar]
- Sommerhage F, Helpenstein R, Rauf A, Wrobel G, Offenhäusser A, Ingebrandt S. (2008) Membrane allocation profiling: a method to characterize three-dimensional cell shape and attachment based on surface reconstruction. Biomaterials 29:3927–3935 [DOI] [PubMed] [Google Scholar]
- Teitler M, Klein MT. (2012) A new approach for studying GPCR dimers: drug-induced inactivation and reactivation to reveal GPCR dimer function in vitro, in primary culture, and in vivo. Pharmacol Ther 133:205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. (2007) Subunit counting in membrane-bound proteins. Nat Methods 4:319–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehler A, Wlodarczyk J, Ponimaskin EG. (2009) Specific oligomerization of the 5-HT1A receptor in the plasma membrane. Glycoconj J 26:749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]