

Background: The independent functions of the glycerol-3-phosphate acyltransferase (GPAT) isoforms are unknown.
Results: When compared with Gpat4−/−, Gpat1−/− hepatocytes oxidized more fatty acids (FA) and were unable to incorporate de novo synthesized FA into triacylglycerol.
Conclusion: GPAT1, but not GPAT4, metabolizes FA synthesized de novo from [14C]acetate and diverts FA away from mitochondrial oxidation.
Significance: GPAT1 and GPAT4 use different cellular pools of FA.
Keywords: Beta-Oxidation, Fatty Acid Metabolism, Lipogenesis, Liver, Phospholipid, Triacylglycerol, Acylcarnitine, Acyltransferase
Abstract
Four glycerol-3-phosphate acyltransferase (GPAT) isoforms, each encoded by a separate gene, catalyze the initial step in glycerolipid synthesis; in liver, the major isoforms are GPAT1 and GPAT4. To determine whether each of these hepatic isoforms performs a unique function in the metabolism of fatty acid, we measured the incorporation of de novo synthesized fatty acid or exogenous fatty acid into complex lipids in primary mouse hepatocytes from control, Gpat1−/−, and Gpat4−/− mice. Although hepatocytes from each genotype incorporated a similar amount of exogenous fatty acid into triacylglycerol (TAG), only control and Gpat4−/− hepatocytes were able to incorporate de novo synthesized fatty acid into TAG. When compared with controls, Gpat1−/− hepatocytes oxidized twice as much exogenous fatty acid. To confirm these findings and to assess hepatic β-oxidation metabolites, we measured acylcarnitines in liver from mice after a 24-h fast and after a 24-h fast followed by 48 h of refeeding with a high sucrose diet to promote lipogenesis. Confirming the in vitro findings, the hepatic content of long-chain acylcarnitine in fasted Gpat1−/− mice was 3-fold higher than in controls. When compared with control and Gpat4−/− mice, after the fasting-refeeding protocol, Gpat1−/− hepatic TAG was depleted, and long-chain acylcarnitine content was 3.5-fold higher. Taken together, these data demonstrate that GPAT1, but not GPAT4, is required to incorporate de novo synthesized fatty acids into TAG and to divert them away from oxidation.
Introduction
Glycerol-3-phosphate acyltransferase (GPAT3; EC 2.3.1.15) initiates the pathway of TAG synthesis by esterifying a long-chain fatty acyl-CoA to a glycerol-3-phosphate backbone at the sn-1 position to form sn-1-acylglycerol-3-phosphate (lysophosphatidic acid) (1). A second esterification at the sn-2 position is catalyzed by acylglycerol-3-phosphate acyltransferase. The phosphate is then hydrolyzed by phosphatidic acid phosphohydrolase (lipin), and then a final acylation is catalyzed by diacylglycerol acyltransferase to form TAG. In addition to their roles in TAG synthesis, the phosphatidic acid and diacylglycerol intermediates in this pathway are precursors for all the glycerophospholipids, and they initiate signaling pathways.
The pathway of TAG biosynthesis is remarkable for the number of isoenzymes that catalyze each step. For example, four independent GPAT isoforms, each encoded by a separate gene, catalyze the formation of lysophosphatidic acid, but it is unclear why GPAT enzyme redundancy is required. Studies of mice with deleted GPATs show that the isoforms present in liver include primarily GPAT1 (2) and GPAT4 (3, 4), with lower expression of GPAT2 (5) and GPAT3 (6). Each of the GPAT isoforms is an intrinsic membrane protein whose active site faces the cytosol (7). GPAT1, located on the outer mitochondrial membrane and at contact sites with the endoplasmic reticulum (8), has a strong preference for saturated fatty acids (1). GPAT1 comprises 30–50% of total activity in liver (1), and Gpat1 mRNA expression is highly responsive to SREBP1c and is up-regulated when dietary carbohydrate intake and circulating insulin concentrations are high (9). Absent GPAT1 protects against the development of hepatic steatosis caused by a high fat diet (2) or by the up-regulation of SREBP1c in ob/ob mice (10). Conversely, GPAT1 overexpression causes hepatic steatosis (11) and insulin resistance (12). GPAT4, which is located on the endoplasmic reticulum and on lipid droplets (13), contributes 40–50% of total hepatic GPAT activity (3). Like GPAT1, GPAT4 also appears to be important for hepatic TAG synthesis because the TAG content in Gpat4−/− liver is 42% lower than in control mice (3).
Hepatic triacylglycerol synthesis is enhanced by the increased availability of long-chain fatty acids synthesized within hepatocytes from dietary glucose or from fatty acid that enters hepatocytes exogenously from dietary chylomicron remnants or as free fatty acids lipolyzed from adipose tissue. Because SREBP1c concomitantly up-regulates both GPAT1 and the enzymes that catalyze the de novo synthesis of fatty acids, including acetyl-CoA carboxylase and fatty acid synthase (14), we hypothesized that the function of GPAT1 and de novo lipogenesis might be intrinsically linked. To test this hypothesis, we compared the ability of GPAT1 and GPAT4 to use exogenous versus de novo synthesized fatty acids in control and knock-out mice and in their hepatocytes.
EXPERIMENTAL PROCEDURES
Materials
Type I collagenase was from Worthington Biochemical Corp. [1-14C]Acetic acid and [1-14C]palmitic acid were purchased from PerkinElmer Life Sciences. Silica G gel plates were from Whatman. Tissue culture plates were from BD Biosciences, and media were obtained from Invitrogen. Sigma was the source of 4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75) and all other chemicals, unless otherwise indicated.
Animal Care and Hepatocyte Isolation
Animal protocols were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee. Mice were housed in a pathogen-free barrier facility on a 12-h light/dark cycle with free access to water and food (Prolab 5P76 Isopro 3000; 5.4% fat by weight). Gpat1−/− (2) and Gpat4−/− (3, 15) mice had been backcrossed at least eight times onto a C57BL6/J background. At 8 weeks of age, littermate controls and Gpat1−/− and Gpat4−/− mice were either fasted for 24 h or fasted for 24 h and then refed a high sucrose diet for 48 h before sacrifice. The high sucrose diet was modified from Research Diets D12450B (10% kcal of fat, 20% kcal of protein, and 70% kcal of carbohydrate (525 g/kg of sucrose, 175 g/kg of corn starch)).
To isolate hepatocytes, after mice were anesthetized with 250 mg/kg of Avertin, a 22-gauge catheter was inserted into the vena cava, and the liver was perfused with wash buffer (10 mm Hepes, 132 mm NaCl, 6.7 mm KCl, 20 mm glucose, 0.5 m adenosine, 100 mm EGTA, 140 nm insulin, pH 7.4) and then with digestion buffer (100 mm HEPES, 57 mm NaCl, 6.7 mm KCl, 5 mm CaCl2, 20 mm glucose, 0.45 mg/ml type 1 collagenase, pH 7.4). The digested liver was removed and placed in a 100-mm dish on ice with Hanks'/BSA buffer (10 mm HEPES, 137 mm NaCl, 5.4 mm KCl, 1.26 mm CaCl2, 0.8 mm MgSO4, 4.2 mm NaHCO3, 0.34 mm Na2HPO4, 0.44 mm KH2PO4, 5 mm glucose, pH 7.4). The hepatocytes were dissociated and filtered through a cell strainer, centrifuged at 4 °C at 50 × g for 3 min, and then washed once in Williams' medium E. The cells were resuspended in Williams' medium E:Percoll (16 ml:14 ml), centrifuged at 4 °C at 400 × g for 8 min, and washed with Williams' medium E. Cells were plated onto 60-mm collagen-coated dishes and incubated for 90 min in Williams' medium E + 10% FBS. After the cells attached, the medium was changed to Williams' medium E, 100 units of penicillin/streptomycin, 50 nm insulin, 0.1 μm dexamethasone, and 0.1% BSA.
GPAT Activity
Hepatocytes were cultured overnight in 60-mm plates and then scraped and homogenized in cold Medium I (250 mm sucrose, 10 mm Tris, pH 7.4, 1 mm EDTA, 1 mm dithiothreitol) with 10 up-and-down strokes with a Teflon glass motor-driven homogenizer. Liver was minced and then homogenized similarly. Total membranes were isolated by centrifuging the homogenate at 100,000 × g for 1 h. GPAT specific activity was assayed for 10 min at room temperature in a 200- μl reaction mixture containing 75 mm Tris-HCl, pH 7.5, 4 mm MgCl2, 1 mg/ml BSA (essentially fatty acid-free), 1 mm dithiothreitol, 8 mm NaF, 800 μm [3H]glycerol 3-phosphate, and 80 μm palmitoyl-CoA (20). The reaction was initiated by adding 10–30 μg of membrane protein after incubating the membrane protein on ice for 15 min in the presence or absence of 2 mm N-ethylmaleimide (NEM), which inactivates GPAT isoforms 2, 3, and 4. The reaction products were extracted into CHCl3, dried under N2, resuspended in 4 ml of Cytoscint, and counted on a scintillation counter. NEM-resistant activity (GPAT1) was calculated by subtracting NEM-sensitive activity from total activity.
RNA Extraction and RT-PCR
Total RNA was extracted from overnight cultures of hepatocytes with TRIzol (Invitrogen). cDNA synthesis and real time RT-PCR were performed as described (3). Primer sequences for each GPAT isoform and data normalization used were described (3, 32).
Labeling, Oxidation, and Lipid Extraction
Sixteen to 20 h after plating, hepatocytes were labeled with 0.5 ml of labeling buffer (Krebs Ringer buffer, 10 mm HEPES, 5.5 mm glucose, 0.25% fatty acid-free bovine serum albumin (BSA), and 1 mm carnitine) containing 500 μm [1-14C]acetic acid or [1-14C]palmitic acid for 2 h. Media were removed, and CO2 was liberated by adding 100 μl of 70% perchloric acid and trapped in 1 m NaOH. An aliquot of the NaOH was counted to measure radiolabeled CO2. To quantify intracellular oxidation metabolites, fresh Krebs Ringer buffer and 100 μl of 70% perchloric acid were added to each plate, and acidified supernatants were collected. The acidified media and cell supernatants were incubated overnight with BSA and then centrifuged at 14,000 × g for 20 min. Aliquots of the supernatant were counted to measure radiolabeled acid-soluble metabolites (ASM), a measure of incomplete fatty acid oxidation. Cells were washed with 1% BSA and then with phosphate-buffered saline before lipids were extracted (16). Lipid extracts and standards were separated by thin layer chromatography on Partisil LK5D silica gel plates in a two-phase system: chloroform:methanol:ammonium hydroxide (65:25:4, v/v/v) run to 50% of the plate and then dried and followed by heptane:isopropyl ether:acetic acid (60:40:4, v/v/v) run to the top of the plate. Radiolabeled lipids were quantified with a Bioscan AR-2000 imaging scanner. Lipid standards were visualized by iodine staining.
Blood Chemistries and Lipids
Before blood collection, mice were either fasted for 24 h or fasted 24 h and then fed a high sucrose diet for 48 h. Plasma TAG (Stanbio, Boerne, TX), free fatty acid (Wako Diagnostics, Richmond, VA), glucose (glucose CII reagent, Wako Diagnostics), and β-hydroxybutyrate (Wako Diagnostics) were determined by enzymatic colorimetric methods.
Lipid Extraction and TAG Measurement in Liver
Liver lipids were extracted (12), dried under N2 gas, resuspended in 200 μl of tert-butanol:methanol:Triton X-100 (3:1:1, v/v), and analyzed colorimetrically as described above.
Liver Acylcarnitine Measurement
Livers were snap-frozen in liquid nitrogen, pulverized, and weighed. The pulverized liver (50–100 mg) was homogenized in acetonitrile:water:formic acid (50:49.7:0.3; v/v) at a final concentration of 50 mg/ml. Acylcarnitines were analyzed by flow injection tandem mass spectrometry (17). Samples were stored at −80 °C before analysis.
Statistics
Data represent means ± S.E. of at least three independent experiments performed in triplicate unless indicated otherwise. In vitro models were analyzed by one-way analysis of variance comparing each genotype versus controls and post hoc comparisons of diet conditions within each genotype. Data were considered significant at p < 0.05.
RESULTS
Isolated and Cultured Primary Hepatocytes Retained GPAT Specific Activity
To compare the contributions of GPAT1 and GPAT4 to the total amount of GPAT activity, we isolated and cultured primary hepatocytes from control, Gpat1−/−, and Gpat4−/− mice and measured GPAT specific activity in the presence and absence of NEM, which inhibits GPAT2, 3, and 4 (Fig. 1A). When compared with wild type hepatocytes, total GPAT specific activity in Gpat1−/− hepatocytes was 35% lower and minimal residual NEM-resistant GPAT activity remained, whereas NEM-sensitive GPAT activity was unchanged. Total GPAT specific activity in Gpat4−/− hepatocytes was 42% lower than in control hepatocytes; NEM-resistant GPAT specific activity was unchanged, but NEM-sensitive GPAT activity was 60% lower. The residual NEM-sensitive GPAT activity is likely due to GPAT2 and -3. These results show that isolated and cultured primary hepatocytes retained GPAT activity and that the specific activity was similar in Gpat1−/− and Gpat4−/− hepatocytes.
FIGURE 1.
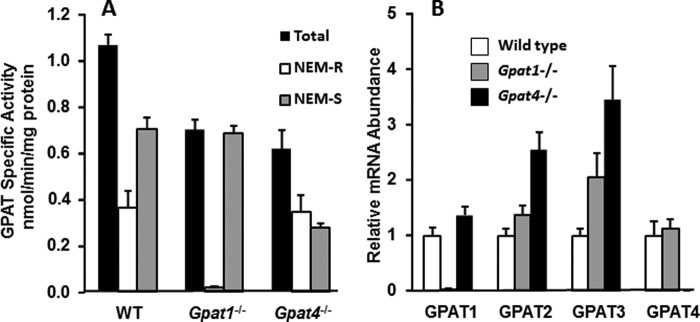
GPAT specific activity and mRNA expression in primary hepatocytes from wild type, Gpat1−/−, and Gpat4−/− mice. A, GPAT specific activity was measured in total particulate preparations of primary hepatocytes cultured for 16 h; results show triplicates from two independent experiments. Total, N-ethylmaleimide-resistant (NEM-R), and N-ethylmaleimide-sensitive (NEM-S) GPAT specific activities are shown. B, mRNA expression of each GPAT isoform was measured by RT-PCR. Total RNA was obtained from primary hepatocytes in triplicate from two experiments. Data are presented as means ± S.E.
To determine whether other GPAT isoforms were affected by deletion of either GPAT1 or GPAT4, we measured mRNA expression of each GPAT isoform (Fig. 1B). In Gpat1−/− hepatocytes, Gpat3 mRNA abundance was 2-fold higher than in wild type hepatocytes, but NEM-sensitive GPAT activity did not change, indicating a lack of compensation. In the absence of GPAT4, the mRNA expression of Gpat2 and -3 increased 2.5- and 3.5-fold, respectively. GPAT2 activity is extremely low in liver (5), and even when the amount of Gpat2 mRNA doubles, it probably does not contribute substantially to total NEM-sensitive GPAT activity. However, because we cannot differentiate between GPAT3 and GPAT4 protein or activity, the 42% lower NEM-sensitive GPAT activity (Fig. 1B) could reflect partial compensation by GPAT3.
The Incorporation of de Novo Synthesized Fatty Acid into TAG Required GPAT1
Because GPAT1 is up-regulated by SREBP1c, we hypothesized that it might be functionally and specifically linked to de novo lipogenesis, such that newly synthesized fatty acids would be incorporated into TAG. Using [1-14C]acetate, we measured the ability of control, Gpat1−/−, and Gpat4−/− hepatocytes to incorporate de novo synthesized fatty acid into TAG and PL. The incorporation of acetate into TAG and PL was equal in hepatocytes from wild type and Gpat4−/− mice; however, in Gpat1−/− hepatocytes, the incorporation of [14C]acetate into PL was 78% lower than in controls, and the incorporation into TAG was virtually absent (Fig. 2, A and B). When wild type and Gpat4−/− hepatocytes were cultured in the presence of the fatty acid synthase inhibitor C75, [14C]acetate incorporation into TAG was inhibited 80–85% (Fig. 2A). However, in Gpat1−/− hepatocytes, C75 had no additional effect, indicating that the absence of GPAT1 in hepatocytes had abolished the incorporation of the de novo synthesized fatty acid. In wild type and Gpat4−/− hepatocytes, C75 decreased acetate incorporation into PL by 60%, but only by 27% in Gpat1−/− hepatocytes (Fig. 2B). In each group of hepatocytes, the addition of C75 decreased the incorporation of acetate into free fatty acids by more than 72% (data not shown). These results show that GPAT1, but not GPAT4, is required to incorporate de novo synthesized fatty acid into complex lipids.
FIGURE 2.
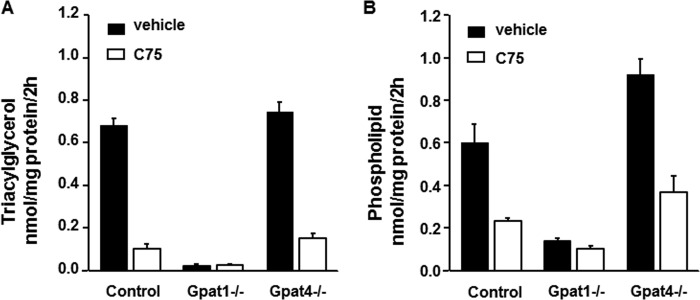
Gpat1−/− hepatocytes incorporated less [14C]acetate into glycerolipids. Hepatocytes were labeled with 500 μm [1-14C]acetate in the presence or absence of 20 μg/ml C75 for 2 h. A and B, [1-14C]acetate incorporation into triacylglycerol (A) or phospholipids (B) in control, Gpat1−/−, or Gpat4−/− hepatocytes. Data show means ± S.E. of two experiments performed in triplicate.
Both GPAT1 and GPAT4 Can Incorporate Exogenous Fatty Acids into TAG and Phospholipid
In addition to de novo synthesis, liver fatty acids are also derived from exogenous sources. To distinguish between the functions of GPAT1 and GPAT4 in the use of exogenous fatty acids, we measured the incorporation of [1-14C]palmitate into glycerolipids in primary hepatocytes. In contrast to fatty acids derived from de novo synthesis, the incorporation of [1-14C]palmitate into TAG and PL was not altered by the absence of either GPAT1 or GPAT4 (Fig. 3, A and B), indicating that neither isoform was essential for the use of exogenous fatty acid. Thus, in both Gpat4−/− and Gpat1−/− hepatocytes, the remaining GPAT isoforms can fully incorporate exogenous fatty acid into glycerolipids.
FIGURE 3.
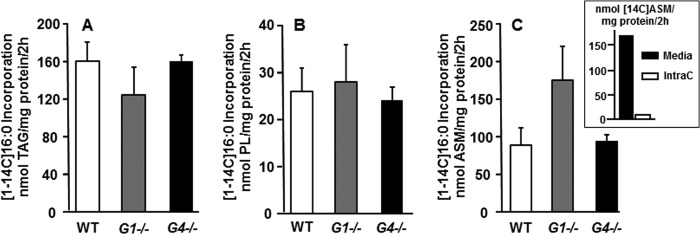
GPAT1 was not required for the incorporation of exogenous fatty acid, and β-oxidation was higher in Gpat1−/− hepatocytes. Hepatocytes from wild type, Gpat1−/− (G1−/−), or Gpat4−/− (G4−/−) mice were labeled with 500 μm [1-14C]palmitic acid for 2 h. A–C, [1-14C]palmitic acid incorporation into triacylglycerol (A), phospholipids (B), and ASM (C). Data show means ± S.E. of two experiments performed in triplicate. Inset, comparison of cell media and intracellular (IntraC) ASM. Data show means of one experiment performed in quadruplicate.
In Hepatocytes, the Absence of GPAT1, but Not GPAT4, Increased Fatty Acid Oxidation
Previous studies have strongly suggested that GPAT1 competes with CPT1 for acyl-CoAs at the outer mitochondrial membrane (12). To confirm this competition for acyl-CoAs and to determine whether GPAT4 located on the endoplasmic reticulum competes similarly, we measured the incorporation of [1-14C]palmitate into ASM, a measure of incomplete fatty acid oxidation. Gpat1−/− hepatocytes oxidized [14C]palmitate at a rate 200% higher than control hepatocytes, but the incorporation of [14C]palmitate into ASM by Gpat4−/− hepatocytes was unchanged (Fig. 3C). Of the total ASM extracted from the media and cells, intracellular ASM comprises only 5% of the total ASM (Fig. 3C, inset); thus, media ASM reflect 95% of the total ASM produced. These data show that only GPAT1 competes with CPT1 for acyl-CoAs, thereby diminishing the flux of fatty acid into the mitochondria for oxidation.
Hepatic GPAT1 Diminished Fatty Acid Transport into Mitochondria in Vivo
To confirm the disparate effects of each GPAT isoform on the β-oxidation of exogenous versus de novo synthesized fatty acid in vivo, we measured hepatic acylcarnitine content in wild type, Gpat1−/−, and Gpat4−/− mice after food was removed for 24 h. Because fasting increases the flux of adipose-derived fatty acids into the liver, more fatty acids are transported to the mitochondria via CPT1, thereby elevating the content of hepatic long-chain acylcarnitines. These metabolites are subsequently reconverted to acyl-CoAs in the mitochondrial matrix, where they enter the β-oxidation pathway. In fasting mice of each genotype, the content of free carnitine (C0), acetylcarnitine (C2), and short-chain (C3-C6) and medium-chain (C8-C12) acylcarnitine species was similar (Fig. 4, A and C). Consistent with the hypothesized ability of GPAT1 to compete with CPT1 for acyl-CoAs, total long-chain (C14-C22) acylcarnitine content in fasted Gpat1−/− liver was 3-fold higher than in control liver (Fig. 4C). The most abundant long-chain acylcarnitine species, 14:0, 16:1, 16:0, 18:2, 18:1, and 18:0, were 2–6-fold higher than in control liver (Fig. 4E). In contrast, and consistent with the in vitro data, the acylcarnitine content in livers of fasted Gpat4−/− mice was similar to that of control mice.
FIGURE 4.
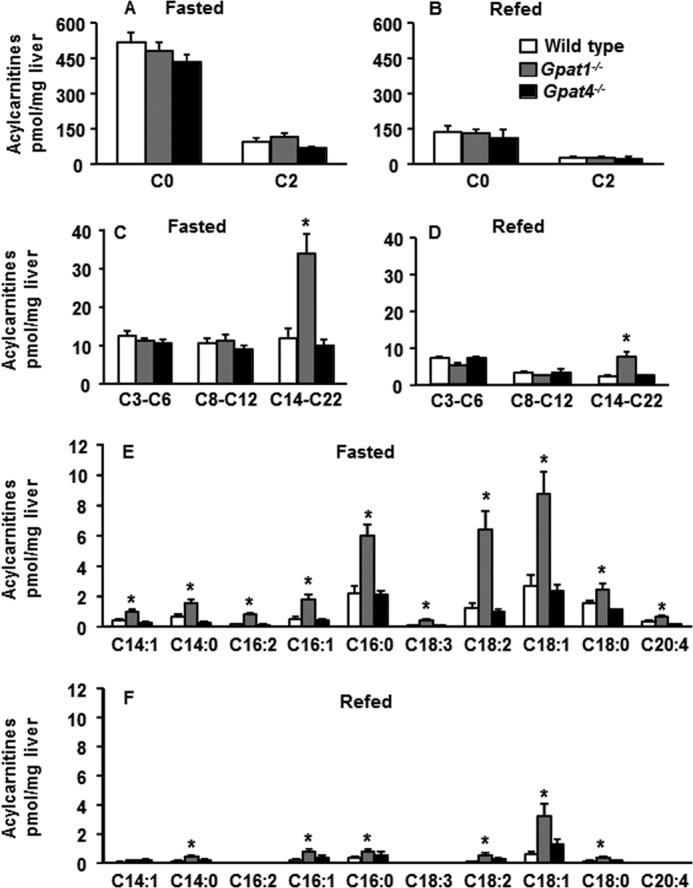
Long-chain acylcarnitine content was higher in Gpat1−/− liver than controls after a 24-h fast and after 48 h of refeeding. Wild type, Gpat1−/−, and Gpat4−/− mice were fasted for 24 h or fasted for 24 h and then refed with a high sucrose diet for 48 h. A and B, hepatic content of free carnitine (C0) and acetylcarnitine (C2). C and D, short-, medium-, and long-chain acylcarnitine content of mouse liver. E and F, major hepatic long-chain acylcarnitine species from mice. Data are shown as means ± S.E. Significant differences are indicated by * for genotype. (p < 0.05, analysis of variance) (n = 6 diet, genotype).
Because GPAT1 was essential for the incorporation of de novo synthesized fatty acids into complex lipids in vitro, we predicted that the absence of hepatic GPAT1 in vivo would also increase the transport of de novo synthesized fatty acids into mitochondria. To test this hypothesis, we fasted mice for 24 h and then refed them with a high sucrose diet for 48 h to stimulate de novo lipogenesis. The total long-chain (C14-C22) acylcarnitine content of Gpat1−/− liver remained 3.5-fold higher than that of controls, and the most abundant long-chain acylcarnitine species, 14:0, 16:1, 16:0, 18:2, 18:1 and 18:0, ranged from 2- to 5-fold higher (Fig. 4, D and F). In contrast, no differences were observed in the hepatic acylcarnitine content of Gpat4−/− mice that had been fasted and then refed (Fig. 4, B, D, and F). Thus, GPAT1, but not GPAT4, was required in vivo to diminish CPT1-mediated transport of both de novo synthesized and exogenous fatty acid.
Hepatic GPAT1 Was Essential for the Incorporation of de Novo Synthesized Fatty Acid into TAG
Because the absence of GPAT1 increased hepatic β-oxidation of de novo synthesized fatty acids in vivo, we predicted that the liver TAG content would be low. After a 24-h fast, hepatic TAG content in control, Gpat4−/−, and Gpat1−/− liver was similar. After refeeding a high sucrose diet to enhance de novo lipogenesis, the TAG content in control and Gpat4−/− liver increased 2–2.5-fold (Fig. 5A); however, the hepatic TAG content of Gpat1−/− liver did not increase. These data support the conclusion that GPAT1 is essential for the incorporation of de novo synthesized fatty acid into TAG and for diverting those fatty acids away from mitochondrial oxidation.
FIGURE 5.
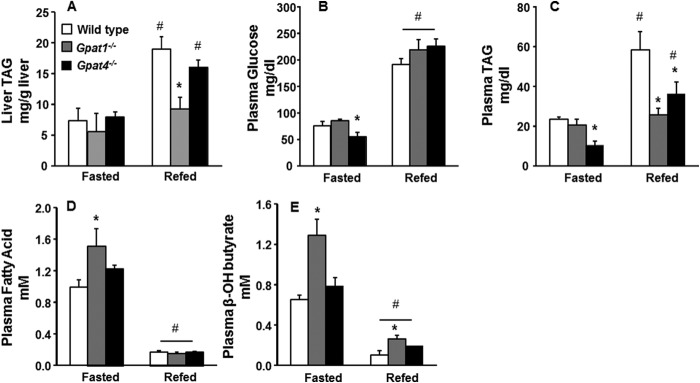
Liver and plasma metabolites. A, liver TAG content of wild type, Gpat1−/−, and Gpat4−/− mice fasted for 24 h or fasted for 24 h and then refed with a high sucrose diet for 48 h. B, plasma glucose; C, plasma TAG; D, plasma fatty acid; E, plasma β-hydroxybutyrate. Data are shown as means ± S.E. Significant differences are indicated by * for genotype and by # for diet challenge. (p < 0.05, analysis of variance) (n = 6 diet, genotype).
The Absence of GPAT1, but Not GPAT4, Increased Ketogenesis during Fasting
When the concentration of plasma glucose decreases during an extended fast, adipocyte-derived fatty acids flood into the liver, their oxidation rate increases, and ketone bodies are synthesized and secreted into the blood. The accumulation of long-chain acylcarnitines in the liver together with an elevated plasma concentration of ketone bodies suggests an increased rate of hepatic fatty acid oxidation. Because GPAT1 diminishes the oxidation of exogenous fatty acid in vitro and the absence of GPAT1 in liver results in the accumulation acylcarnitines, we predicted that fasting would result in a higher rate of hepatic β-oxidation in Gpat1−/− mice than in controls. To confirm this hypothesis, we measured plasma ketone bodies in control, Gpat1−/−, and Gpat4−/− mice. After a 24-h fast, the concentrations of plasma glucose and TAG in control and Gpat1−/− mice were similar (Fig. 5, B and C), but the plasma fatty acid concentration in Gpat1−/− mice was 1.5-fold higher than controls (Fig. 5D). Consistent with elevated fatty acid oxidation in the Gpat1−/− mice, the plasma concentration of β-hydroxybutyrate was 2-fold higher than controls (Fig. 5E).
Ketogenesis Remained Elevated in Gpat1−/− Mice during Refeeding
After a 24-h fast, refeeding with high sucrose drives hepatic de novo lipogenesis, promotes TAG storage, and blocks fatty acid oxidation (18). Because our data suggested that GPAT1 esterifies de novo synthesized fatty acids, we asked whether the rate of hepatic fatty acid oxidation would remain elevated during short-term high sucrose refeeding. Although a high carbohydrate diet diminishes PPARα gene targets related to β-oxidation and ketone body formation, in the absence of GPAT1, the plasma β-hydroxybutyrate concentration in Gpat1−/− mice was 3-fold higher than in control or Gpat4−/− mice (Fig. 5D), consistent with a continued higher rate of hepatic fatty acid oxidation. Consistent with diminished hepatic TAG synthesis in both Gpat1 and Gpat4 null mice, when compared with controls, the plasma TAG concentration was 48% lower in Gpat1−/− mice and 25% lower in Gpat4−/− mice (Fig. 5C). Plasma glucose and fatty acid concentrations in each genotype were similar (Fig. 5, B and E).
DISCUSSION
With the discovery of four independent GPAT isoforms that differ in tissue distribution, subcellular location, acyl-CoA preference, and transcriptional regulation, questions have arisen as to the specific function of each isoform (1). Gpat2 mRNA is most strongly expressed in testes and probably plays a minor role in liver, but the mRNA expression and activities in liver of the remaining GPAT isoforms are high (1, 5, 6, 19). Although studies suggest that GPAT3 is regulated by PPARγ and PPARδ in keratinocytes (20) and by insulin-mediated phosphorylation in 3T3-L1 adipocytes (21), the regulation and specific contributions of hepatic GPAT3 to glycerolipid synthesis are unknown. The availability of Gpat1 and Gpat4 knock-out mice, however, has allowed us to directly compare them and to delineate the specific roles of each isoform.
The function of GPAT1 in the esterification of fatty acids has been best studied. GPAT1 esterification rates for 16:0-CoA are 3–10 times higher than those observed with 18:0-, 18:1-, 18:2n6-, and 18:3n3-CoAs (21–24). Examination of the glycerolipid species in Gpat1−/− liver have confirmed the preference of GPAT1 for 16:0-CoA because the absence of GPAT1 results in decreases in the amounts of 16:0 esterified to phosphatidylcholine and phosphatidylethanolamine in membranes from total liver (2) and from liver mitochondria (25). The 16:0 deficiencies at the sn-1 position of phosphatidylcholine and phosphatidylethanolamine are compensated by increases in the content of 18:1 at the sn-1 position and of 20:4ω6 at the sn-2 position.
A second function of GPAT1 is related to de novo lipogenesis. This role is implied by the fact that the specific activity of GPAT1 is low in liver during fasting, in part because of inhibition by AMP-activated kinase (26), and is up-regulated transcriptionally when lipogenesis from carbohydrate precursors is enhanced. Similar to acyl-CoA carboxylase and fatty acid synthase, Gpat1 mRNA, protein, and specific activity increase in response to SREBP1c up-regulation, which, in turn, is stimulated by insulin and depressed via liver X receptor by polyunsaturated fatty acids (9). Thus, GPAT1 is markedly elevated in ob/ob mice, and a deficiency in ob/ob mice of either Gpat1 itself (10) or of SREBP1c (14) protects against the development of a fatty liver. In addition to promoting the synthesis of TAG, indirect evidence suggests that GPAT1 diverts acyl-CoAs away from CPT1 and β-oxidation because long-chain acylcarnitines and β-hydroxybutyrate increase in Gpat1−/− mice fed an obesogenic diet that is high in sucrose and palm oil (12). Thus, under conditions when the synthesis of fatty acid is elevated, the presence of GPAT1 would ensure that the resulting fatty acids are esterified rather than oxidized. Our current data directly support this idea.
In isolated hepatocytes, incubation with [14C]acetate showed that only the deficiency of GPAT1 blocked the incorporation of de novo synthesized fatty acids into glycerolipids. The incorporation was equivalent in control and Gpat4−/− hepatocytes and was diminished equally by the fatty acid synthase inhibitor C75. These data suggest that fatty acids synthesized de novo from glucose require GPAT1 to be incorporated into TAG. Perhaps because of the preference of GPAT1 for 16:0-CoA, the predominant species synthesized de novo by fatty acid synthase, GPAT1 was also responsible for most of the acetate-derived fatty acid that was incorporated into PL. Inhibition of fatty acid synthesis by C75 had a weaker effect on PL synthesis than it did on TAG synthesis, suggesting that de novo synthesized fatty acids may be preferentially directed to TAG storage, consistent with the fact that lipogenesis is up-regulated when high carbohydrate intake and insulin concentrations activate SREBP1c. The final step in the synthesis of TAG is catalyzed by DGAT1 and DGAT2. Stable isotope studies show that only DGAT2 incorporates fatty acids synthesized de novo (27).
Because Gpat1−/− hepatocytes failed to incorporate label from [14C]acetate into TAG, we concluded that GPAT1 is required for the synthesis of TAG from de novo lipogenesis. Confirming this interpretation, after the fasting-refeeding protocol to promote hepatic de novo lipogenesis, Gpat1−/− liver contained 53% less TAG than did controls. When compared with fasted mice, the hepatic TAG content in both refed control and refed Gpat4−/− mice increased 2- and 3-fold, respectively, but liver TAG content in refed Gpat1−/− mice did not change, indicating that Gpat1−/− failed to incorporate de novo-derived fatty acid into hepatic TAG. A direct comparison of hepatic β-oxidation revealed that when compared with controls, long-chain acylcarnitines and β-hydroxybutyrate in Gpat1−/− mice were 3.5- and 2-fold higher, respectively. Thus, when hepatic GPAT1 was absent, the de novo synthesized fatty acids were oxidized rather than incorporated into TAG. These data are consistent with our in vitro findings and confirm that GPAT1 is required to incorporate de novo synthesized fatty acids into TAG and diminish fatty acid oxidation.
In humans, VLDL secretion is stimulated by dietary sugars, and after a high carbohydrate load, de novo synthesized fatty acids comprise roughly 37% of plasma VLDL-TAG (28, 29). Consistent with a role for GPAT1 in incorporating de novo synthesized fatty acids into VLDL, Gpat1−/− mice fed a diet containing 60% of calories from sucrose were protected from hypertriglyceridemia and showed diminished VLDL secretion rates (30). After the fasting-refeeding protocol, plasma TAG concentrations in Gpat1−/− mice did not change, suggesting that GPAT1 is essential for the postprandial lipemia that is stimulated by carbohydrate feeding. In contrast, plasma TAG in refed control and Gpat4−/− mice increased 2- and 3.5-fold, respectively, consistent with the continued presence and function of GPAT1 in using de novo synthesized TAG in VLDL.
The absence of either GPAT1 or GPAT4 did not block the incorporation of exogenous fatty acid into TAG or PL. Normal incorporation seems surprising because TAG content in both Gpat1−/− and Gpat4−/− liver is 40–50% lower than controls (2, 31). However, our hepatocyte studies measured TAG synthesis from [14C]palmitate in the absence of insulin. Normally, after a 24-h fast, when insulin levels are low, fatty acids released from adipocytes constitute the majority of the fatty acid entering the liver to be stored or incorporated into VLDL-TAG, a process that is not sensitive to plasma insulin levels (28). Under these conditions, fatty acids entering the liver might be preferentially oxidized for energy, and the remaining low GPAT activity would suffice for a minimal demand for TAG synthesis for storage or for VLDL secretion. Consistent with this idea and with our in vitro results, liver TAG content was similar in fasting control, Gpat1−/−, and Gpat4−/− mice. After feeding in humans, concomitant with a rise in plasma insulin, both dietary fatty acids and those derived from de novo synthesis are incorporated into TAG (28). Because the TAG content in Gpat1−/− liver was lower than in controls after high sucrose refeeding, it is likely that insulin-induced SREBP1c activity drove fatty acid flux toward TAG storage; however, in the absence of GPAT1, the remaining GPAT activity was unable to meet the demand for higher TAG synthesis.
Because Gpat1−/− mice fed low or high fat diets for 4 months had a higher content of plasma and liver acylcarnitines and higher concentrations of plasma β-hydroxybutyrate, we had speculated that GPAT1 might normally divert fatty acids away from CPT1 and β-oxidation (12). The present results directly support this hypothesis by showing that when compared with controls, Gpat1−/− hepatocytes produced twice as much ASM from [14C]palmitate and that plasma β-hydroxybutyrate and hepatic acylcarnitines were markedly elevated in Gpat1−/− mice under both fasted and refed conditions. In contrast, ASM production was similar in Gpat4−/− and control hepatocytes under both conditions, indicating that lack of GPAT4 did not increase the use of excess acyl-CoAs for β-oxidation.
To directly compare β-oxidation metabolites from the two Gpat null genotypes, we investigated acylcarnitines. After a 24-h fast, the hepatic content of long-chain acylcarnitines was equivalent in control and Gpat4−/− mice. Confirming the hypothesis that GPAT1 competes with CPT1 for acyl-CoAs and diverts them away from mitochondria, the content of long-chain acylcarnitines in Gpat1−/− livers was 3-fold higher than controls. Thus, despite the observation that exogenous fatty acid incorporation into TAG was unaffected by a deficiency of either GPAT1 or GPAT4, these results further support the unique function of GPAT1 in diverting fatty acids away from β-oxidation.
These studies clearly differentiate the roles of the major hepatic isoforms, GPAT1 and GPAT4. GPAT1, but not GPAT4, reciprocally regulates hepatic de novo glycerolipid synthesis and fatty acid oxidation. In this role, the absence of GPAT1 protects mice from high fat diet- and genetic-induced hepatic steatosis and hepatic insulin resistance (10). Conversely, the increased expression of GPAT1 may contribute to insulin resistance, both by diverting fatty acids away from oxidation and toward glycerolipid synthesis and by increasing lipid intermediates such as diacylglycerol and phosphatidic acid, which impair hepatic insulin signaling (11, 32).
This work was supported, in whole or in part, by National Institutes of Health Grants DK56598 (to R. A. C.), DK089312 (to D. M. M.), DK083157 (to A. A. W.), and T32HD057824 (to D. E. C.).
- GPAT
- glycerol-3-phosphate acyltransferase
- ASM
- acid-soluble metabolite(s)
- C75
- 4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid
- CPT
- carnitine palmitoyltransferase-1
- DGAT
- diacylglycerol acyltransferase
- NEM
- N-ethylmaleimide
- PL
- phospholipid
- TAG
- triacylglycerol
- PPAR
- peroxisome proliferator-activated receptor.
REFERENCES
- 1. Coleman R. A., Mashek D. G. (2011) Mammalian triacylglycerol metabolism: Synthesis, lipolysis, and signaling. Chem. Rev. 111, 6359–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hammond L. E., Gallagher P. A., Wang S., Hiller S., Kluckman K. D., Posey-Marcos E. L., Maeda N., Coleman R. A. (2002) Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol. Cell. Biol. 22, 8204–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagle C. A., Vergnes L., Dejong H., Wang S., Lewin T. M., Reue K., Coleman R. A. (2008) Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4 as the enzyme deficient in Agpat6−/− mice. J. Lipid Res. 49, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen Y. Q., Kuo M. S., Li S., Bui H. H., Peake D. A., Sanders P. E., Thibodeaux S. J., Chu S., Qian Y. W., Zhao Y., Bredt D. S., Moller D. E., Konrad R. J., Beigneux A. P., Young S. G., Cao G. (2008) AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase (GPAT). J. Biol. Chem. 283, 10048–10057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang S., Lee D. P., Gong N., Schwerbrock N. M., Mashek D. G., Gonzalez-Baró M. R., Stapleton C., Li L. O., Lewin T. M., Coleman R. A. (2007) Cloning and functional characterization of a novel mitochondrial N-ethylmaleimide-sensitive glycerol-3-phosphate acyltransferase (GPAT2). Arch. Biochem. Biophys. 465, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao J., Li J. L., Li D., Tobin J. F., Gimeno R. E. (2006) Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19695–19700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coleman R. A., Lee D. P. (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 43, 134–176 [DOI] [PubMed] [Google Scholar]
- 8. Pellon-Maison M., Montanaro M. A., Coleman R. A., Gonzalez-Baró M. R. (2007) Mitochondrial glycerol-3-P acyltransferase 1 is most active in outer mitochondrial membrane but not in mitochondrial associated vesicles (MAV). Biochim. Biophys. Acta 1771, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wendel A. A., Li L. O., Li Y., Cline G. W., Shulman G. I., Coleman R. A. (2010) Glycerol-3-phosphate acyltransferase 1 deficiency in ob/ob mice diminishes hepatic steatosis but does not protect against insulin resistance or obesity. Diabetes 59, 1321–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagle C. A., An J., Shiota M., Torres T. P., Cline G. W., Liu Z.-X., Wang S., Catlin R. L., Shulman G. I., Newgard C. B., Coleman R. A. (2007) Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J. Biol. Chem. 282, 14807–14815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hammond L. E., Neschen S., Romanelli A. J., Cline G. W., Ilkayeva O. R., Shulman G. I., Muoio D. M., Coleman R. A. (2005) Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J. Biol. Chem. 280, 25629–25636 [DOI] [PubMed] [Google Scholar]
- 13. Wilfling F., Wang H., Haas J. T., Krahmer N., Gould T. J., Uchida A., Cheng J. X., Graham M., Christiano R., Fröhlich F., Liu X., Buhman K. K., Coleman R. A., Bewersdorf J., Farese R. V., Jr., Walther T. C. (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moon Y. A., Liang G., Xie X., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Brown M. S., Goldstein J. L., Horton J. D. (2012) The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 15, 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beigneux A. P., Vergnes L., Qiao X., Quatela S., Davis R., Watkins S. M., Coleman R. A., Walzem R. L., Philips M., Reue K., Young S. G. (2006) Agpat6—a novel lipid biosynthetic gene required for triacylglycerol production in mammary epithelium. J. Lipid Res. 47, 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 17. An J., Muoio D. M., Shiota M., Fujimoto Y., Cline G. W., Shulman G. I., Koves T. R., Stevens R., Millington D., Newgard C. B. (2004) Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver, and whole-animal insulin resistance. Nat. Med. 10, 268–274 [DOI] [PubMed] [Google Scholar]
- 18. McDevitt R. M., Bott S. J., Harding M., Coward W. A., Bluck L. J., Prentice A. M. (2001) De novo lipogenesis during controlled overfeeding with sucrose or glucose in lean and obese women. Am. J. Clin. Nutr. 74, 737–746 [DOI] [PubMed] [Google Scholar]
- 19. Harada N., Hara S., Yoshida M., Zenitani T., Mawatari K., Nakano M., Takahashi A., Hosaka T., Yoshimoto K., Nakaya Y. (2007) Molecular cloning of a murine glycerol-3-phosphate acyltransferase-like protein 1 (xGPAT1). Mol. Cell Biochem. 297, 41–51 [DOI] [PubMed] [Google Scholar]
- 20. Lu B., Jiang Y. J., Kim P., Moser A., Elias P. M., Grunfeld C., Feingold K. R. (2010) Expression and regulation of GPAT isoforms in cultured human keratinocytes and rodent epidermis. J. Lipid Res. 51, 3207–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bremer J., Bjerve K. S., Borrebaek B., Christiansen R. (1976) The glycerolphosphate acyltransferases and their function in the metabolism of fatty acids. Mol. Cell Biochem. 12, 113–125 [DOI] [PubMed] [Google Scholar]
- 22. Kelker H. C., Pullman M. E. (1979) Phospholipid requirement of acyl coenzyme A:sn-glycerol-3-phosphate acyltransferase from rat liver mitochondria. J. Biol. Chem. 254, 5364–5371 [PubMed] [Google Scholar]
- 23. Monroy G., Kelker H. C., Pullman M. E. (1973) Partial purification and properties of an acyl coenzyme A:sn-glycerol 3-phosphate acyltransferase from rat liver mitochondria. J. Biol. Chem. 248, 2845–2852 [PubMed] [Google Scholar]
- 24. Yet S.-F., Moon Y. K., Sul H. S. (1995) Purification and reconstitution of murine mitochondrial glycerol-3-phosphate acyltransferase. Functional expression in baculovirus-infected insect cells. Biochemistry 34, 7303–7310 [DOI] [PubMed] [Google Scholar]
- 25. Hammond L. E., Albright C. D., He L., Rusyn I., Watkins S. M., Doughman S. D., Lemasters J. J., Coleman R. A. (2007) Increased oxidative stress is associated with balanced increases in hepatocyte apoptosis and proliferation in glycerol-3-phosphate acyltransferase-1 deficient mice. Exp. Mol. Pathol. 82, 210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muoio D. M., Seefeld K., Witters L. A., Coleman R. A. (1999) AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 338, 783–791 [PMC free article] [PubMed] [Google Scholar]
- 27. Qi J., Lang W., Geisler J. G., Wang P., Petrounia I., Mai S., Smith C., Askari H., Struble G. T., Williams R., Bhanot S., Monia B. P., Bayoumy S., Grant E., Caldwell G. W., Todd M. J., Liang Y., Gaul M. D., Demarest K. T., Connelly M. A. (2012) The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J. Lipid Res. 53, 1106–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Timlin M. T., Barrows B. R., Parks E. J. (2005) Increased dietary substrate delivery alters hepatic fatty acid recycling in healthy men. Diabetes 54, 2694–2701 [DOI] [PubMed] [Google Scholar]
- 29. Parks E. J., Skokan L. E., Timlin M. T., Dingfelder C. S. (2008) Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 138, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lewin T. M., de Jong H., Schwerbrock N. J., Hammond L. E., Watkins S. M., Combs T. P., Coleman R. A. (2008) Mice deficient in mitochondrial glycerol-3-phosphate acyltransferase-1 have diminished myocardial triacylglycerol accumulation during lipogenic diet and altered phospholipid fatty acid composition. Biochim. Biophys. Acta 1781, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vergnes L., Beigneux A. P., Davis R., Watkins S. M., Young S. G., Reue K. (2006) Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 47, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang C., Wendel A. A., Keogh M. R., Harris T. E., Chen J., Coleman R. A. (2012) Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1667–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]