

Abstract
Damaged and misfolded proteins that are no longer functional in the cell need to be eliminated. Failure to do so might lead to their accumulation and aggregation, a hallmark of many neurodegenerative diseases. Protein quality control pathways play a major role in the degradation of these proteins, which is mediated mainly by the ubiquitin proteasome system. Despite significant focus on identifying ubiquitin ligases involved in these pathways, along with their substrates, a systems-level understanding of these pathways has been lacking. For instance, as misfolded proteins are rapidly ubiquitylated, unconjugated ubiquitin is rapidly depleted from the cell upon misfolding stress; yet it is unknown whether certain targets compete more efficiently to be ubiquitylated. Using a system-wide approach, we applied statistical and computational methods to identify characteristics enriched among proteins that are further ubiquitylated after heat shock. We discovered that distinct populations of structured and, surprisingly, intrinsically disordered proteins are prone to ubiquitylation. Proteomic analysis revealed that abundant and highly structured proteins constitute the bulk of proteins in the low-solubility fraction after heat shock, but only a portion is ubiquitylated. In contrast, ubiquitylated, intrinsically disordered proteins are enriched in the low-solubility fraction after heat shock. These proteins have a very low abundance in the cell, are rarely encoded by essential genes, and are enriched in binding motifs. In additional experiments, we confirmed that several of the identified intrinsically disordered proteins were ubiquitylated after heat shock and demonstrated for two of them that their disordered regions are important for ubiquitylation after heat shock. We propose that intrinsically disordered regions may be recognized by the protein quality control machinery and thereby facilitate the ubiquitylation of proteins after heat shock.
Cells face the constant threat of protein misfolding and aggregation, and thus protein quality control pathways are important in selectively targeting damaged and misfolded proteins for degradation (1, 2). The ubiquitin proteasome system serves as a major mediator of this pathway by conjugating the small protein ubiquitin onto substrates through the E1-E2-E3 (ubiquitin-activating enzyme, ubiquitin-conjugating enzyme, and ubiquitin ligase, respectively) cascade for their recognition and degradation by the proteasome (3, 4). It is known that the activity of the ubiquitin-proteasome system is associated with many neurodegenerative diseases. For instance, ubiquitin is found enriched in protein inclusions associated with these diseases (5). Furthermore, proteasome activity has been shown to decrease with age in a large variety of organisms (6), leading to increased proteotoxicity in the cell.
Because of the importance of maintaining protein homeostasis, numerous ubiquitin ligases in different cellular compartments function in protein quality control pathways to target misfolded or damaged proteins for degradation via the proteasome. For instance, the conserved Hrd1 ubiquitin ligase is involved in the endoplasmic-reticulum-associated degradation pathway that targets endoplasmic reticulum proteins for retro-translocation to the cytoplasm and proteasome degradation (7). A major question is what features are recognized by ubiquitin ligases that allow them to selectively target terminally misfolded proteins for degradation, given that the folding rates and physicochemical properties vary largely from protein to protein. Several E3 ubiquitin ligases involved in cytosolic protein quality control target their substrates via their interactions with chaperone proteins. For instance, the CHIP ubiquitin ligase can directly bind to Hsp70 and Hsp90 proteins (8), which may hand over client proteins that are not successfully folded. Understanding which features are recognized by these degradation quality-control pathways might help us understand how certain misfolded proteins evade this system, leading to their accumulation and aggregation in the cell.
Many studies investigating degradation protein quality control have employed model substrates (e.g. mutated proteins that misfold) to reveal which components are involved in a given quality control machinery. However, these approaches do not typically reveal the whole spectrum of substrates for these pathways. Thus, alternative system-wide approaches are also needed to provide a bigger picture. Heat shock (HS)1 induces general misfolding at the proteome level by increasing thermal energy and was shown to cause an increase in ubiquitylation levels in the cell over 25 years ago (9, 10). However, the exact mechanism and pathways that target misfolded proteins have remained uncharacterized for a long time. We recently showed that the Hul5 ubiquitin ligase plays a major role in this heat stress response that mainly affects cytosolic proteins (11). Absence of Hul5 averts the ubiquitylation in the cytoplasm of several misfolded targets after HS, as well as low-solubility proteins in unstressed cells. Other E3 ubiquitin ligases are likely involved in this pathway (12). Interestingly, as ubiquitin constitutes about only 1% of the proteome, free unconjugated ubiquitin is rapidly depleted under stress conditions (13, 14). Given the limited amount of this protein, how does the cell triage ubiquitin among an excess of misfolded proteins? In order to gain systems-level insight, we sought to identify characteristics enriched among proteins ubiquitylated after HS using a combination of statistical and computational analysis, and we conducted additional proteomics and biochemical experiments to support our hypotheses. We discovered an unexpected susceptibility of intrinsically disordered proteins for ubiquitylation after misfolding stress.
EXPERIMENTAL PROCEDURES
Statistical Analysis and Protein Predictions
Saccharomyces cerevisiae protein sequences were obtained from the Saccharomyces Genome Database (released January 5, 2010) (15). Statistical tests and plotting were performed using the R programming language (16). Specifically, p values were obtained using a two-sided non-parametric Wilcoxon rank-sum test on continuous data (box plots and Amino Acid Compass) and Fisher's exact test on count data (bar plots). All data points were used for statistical testing, but outliers were removed for box plots to maintain clarity. Amino Acid Compass is an in-house program written in the R programming language. Unless indicated, all protein disorder predictions were done with DISOPRED2 (17) using the default settings. Secondary structure was predicted using NetSurfP, version 1.1 (18). Proteins were considered His-rich if they contained at least 6 histidine residues in a sliding 12-residue window. Ubiquitylation sites were mapped to disorder and secondary structure predictions using in-house scripts written in Perl. Further details are provided in the supplemental material.
Cell Culture and Sample Preparation
All yeast strains are listed in supplemental Table S5. Yeast cells for mass spectrometry analysis were grown, lysed, and cell fractionated as described elsewhere (19); details are provided in the supplemental material. The PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling Technology, Danvers, MA) was used to immunoprecipitate GlyGly peptides following the manufacturer's protocol using 200 mg of protein lysate and two 3-ml HyperSep C18 columns (Thermo Fisher Scientific). For the purification of ubiquitylated proteins, about 1 μl of MagneHis (Promega, Madison, WI) was used per 100 μg of protein extract as described elsewhere (19, 20) (details are provided in the supplemental material). After trypsin digestion, peptides from lysates, soluble and pellet fractions, and GlyGly enriched peptides were purified with C18 stage tips without strong cation exchange fractionation (21). Ubiquitylated proteins in the soluble and pellet fractions were purified and fractionated on strong cation exchange stage tips. For the tandem affinity purification (TAP) experiments, 100 ml of cells carrying an H8-ubiquitin plasmid (BPM30 (19) or BPM512 in which the tagged ubiquitin was subcloned in pRS315) or an empty pRS316 vector were grown in SD-Ura or -Leu. For truncation analysis, the following fragments (numbered by their amino acids) were subcloned in a pRS313 plasmid containing a GAL1 promoter followed by a single N-terminal HA tag and PGK1 terminating sequencing at the 3′ UTR: Hal5 1–855 (full length; BPM441), Hal5 1–527 (N-terminal only; BPM442); Hal5 528–855 (C-terminal only; BPM443); Ppz1 1–692 (full length; BPM450): and Ppz1 339–692 (C-terminal only; BPM452). For galactose induction (2%, 60 min), cells carrying H8-ubiquitin and GAL1p-HA-fragment plasmids were first grown in a synthetic media (-Ura, -His) containing 2% raffinose and 0.5% dextrose and then switched to the same media without dextrose 60 min prior to induction. All cells were grown at 25 °C and collected at their exponential phase (A600 ∼ 0.8–1). When indicated, HS was performed at 45 °C for 20 min prior to glass bead lysis in a mixture of 8 m urea, 100 mm HEPES, 0.05% SDS, 10 mm chloroacetamide, 1 mm phenylmethylsulphonyl fluoride, 10 mm imidazole, and protease inhibitors. Immobilized metal affinity chromatography (IMAC) was performed as described elsewhere (11). Details of the reverse pull-down experiments are provided in the supplemental material. Immunoblots were performed with rabbit anti-TAP (PICAB1001, Fisher Scientific) and anti-Pgk1 (AP21371AF-N, Acris Antibodies, Herford, Germany) and mouse anti-HA (HA.C5 clone) antibodies. Fluorescent secondary antibodies (LI-COR Biosciences) were used for quantification analysis in a CLx Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Mass Spectrometry
Purified peptides were analyzed using a linear-trapping quadrupole Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) coupled to an Agilent 1100 Series Nanoflow HPLC. Samples were run with a 90-min gradient with a full-range scan at 60,000 resolution from 300 to 1600 Th and to fragment the top 15 peptide ions in each cycle. Parent ions were then excluded from tandem mass spectrometry for the next 30 s, as were singly charged ions.
Centroided fragment peak lists were processed to Mascot generic format using Proteome Discoverer (1.2). Fragment spectra were searched using the Mascot algorithm (2.3.0) against the Saccharomyces Genome Database (January 5, 2012, with 6147 protein sequences and 6147 randomized sequences) using the following parameters: peptide mass accuracy 10 ppm, fragment mass accuracy 0.6 Da, trypsin (one miscleavage, or three miscleavages for the GlyGly analysis, were allowed), 13C2 option, carbamidomethyl fixed modification (C), oxidation variable modification (M), electrospray ionization TRAP fragment characteristics. 15N/14N peak intensities were quantified by MSQuant (v1.5b7) using proteins with at least two peptides with an ion score of at least 15 (the averaged false discovery rate (FDR) of peptides based on Decoy analysis was <0.85%). We established the cut-off based on the 15N/14N ratio distribution in the control lysate sample, so that less than 5% of the quantified proteins were considered enriched in the control lysate (22).
Fragment spectra from GlyGly experiments were searched against Mascot using the same parameters, adding GlyGly as a variable modification. 14N/15N ratios were quantified by MSQuant for each peptide containing a GlyGly lysine (FDR of GlyGly peptides based on the Decoy database was <0.75%). An in-house Perl script corrected errors in finding the corresponding light or heavy labeled peptide. Similar to previous studies, we removed peptides with a C-terminus GlyGly and considered peptides with Log2 (ratios) ≥ 1 (23, 24).
RESULTS
We previously identified which proteins in Saccharomyces cerevisiae were ubiquitylated or further ubiquitylated upon HS using quantitative mass spectrometry (Fig. 1A) (11). Briefly, we compared two differentially labeled cell populations expressing octahistidine-tagged ubiquitin (H8-ubiquitin). After a transient 20-min 45 °C HS in one sample, the two populations were mixed together equally and lysed. Ubiquitylated proteins were purified via IMAC prior to quantitative mass spectrometry (supplemental Fig. S1A). In three independent experiments, we identified 419 proteins that were more ubiquitylated after HS (11). For this study, we generated a high-confidence dataset of 201 proteins that were further ubiquitylated after HS in at least two experiments (supplemental Table S1).
Fig. 1.

Heat-shock ubiquitylated proteins are enriched for intrinsically disordered proteins. A, schematic of experimental setup using mass spectrometry to identify proteins further ubiquitylated after HS using IMAC (a) or immunoprecipitation using antibodies against remnant GlyGly (GG) peptides (b). B, box plot of protein length comparing proteins ubiquitylated after HS and purified via IMAC to proteins in the yeast proteome and the lysate. Medians are indicated by thick horizontal lines. Boxes contain the inner 50% of data points. Whiskers show maximum and minimum values, excluding outliers. p values of all data points computed via Wilcoxon rank-sum test are indicated. C, Amino Acid Compass of ubiquitylated proteins after HS in orange, compared with proteins in the proteome in gray, showing fold ratios. Significant differences as determined by Wilcoxon rank-sum test after FDR correction are indicated by an orange dot. D, distribution of predicted percentage disorder by DISOPRED2 for proteins ubiquitylated after HS and purified via IMAC (orange) and in the whole cell lysate (gray). E, bar plot of data binned from D at 10% and 30% disorder using DISOPRED2 comparing ubiquitylated proteins enriched via IMAC (orange) with proteins in the lysate (gray). p values from Fisher's exact test comparing proportions in lysate versus IMAC.
HS Ubiquitylated Proteins Are Enriched for Intrinsically Disordered Proteins
In order to understand how the ubiquitin-proteasome system rations unconjugated ubiquitin among different misfolded proteins, we aimed to identify characteristics that are enriched among proteins that are ubiquitylated after misfolding stress. We hypothesized that these characteristics could explain why certain proteins are more prone to misfolding or more likely to be recognized by the ubiquitylation machinery.
We first examined the size of these proteins based on the number of amino acids in each protein. Because the distribution of protein length in the proteome does not follow a normal distribution (supplemental Fig. S1B), we used the non-parametric Wilcoxon rank-sum test to assess statistical significance (details are provided in the supplemental material). We found that on average, the 201 proteins ubiquitylated after HS are significantly larger relative to proteins in the yeast proteome, with a median increase of 186 residues (Fig. 1B). To ensure that the difference in size was not due to bias introduced by the mass spectrometer, we assembled a list of 481 reference proteins that were identified from the whole cell lysate samples in two out of the three experiments (supplemental Table S1). These correspond to relatively abundant proteins in the cell and represent proteins normally sampled in our proteomic experiments. Similar to the whole-proteome comparison, we observed a significant increase in size when comparing ubiquitylated proteins to the reference lysate sample (Fig. 1B), indicating that the increased length was not caused by instrument sampling. This trend, along with the main findings that follow, also was confirmed by using the Kolmogorov–Smirnov test as an independent statistical test (supplemental Fig. S7).
We next asked whether the amino acid compositions of these proteins were enriched or depleted toward certain residues. We used the Amino Acid Compass visualization tool that we developed to compare the fold-change of the percentage of each amino acid in the sequences of proteins ubiquitylated after HS relative to the whole proteome. We found that several hydrophobic (leucine, isoleucine, and methionine) and aromatic residues (phenylalanine, tryptophan, and tyrosine) and cysteine were significantly depleted (Fig. 1C). These differences were found to be statistically significant after using the Benjamini and Hochberg FDR correction, which adjusts p values to have an overall expected 5% incorrect rejection rate among statistically significant tests (25) (supplemental Fig. S1C).
Hydrophobic residues are typically embedded within folded domains; thus we hypothesized that an underrepresentation of these amino acid residues could be explained by an overrepresentation of disordered regions that are depleted in hydrophobic residues. Disordered regions lack unique secondary and tertiary structures in solution because of their reduced hydrophobic content. Proteins that contain many disordered regions are called “intrinsically disordered” and are estimated to constitute over 30% of eukaryotic proteomes (17). Their unique lack of structure enables higher flexibility and increased surface area, which have been suggested to enhance interaction with other macromolecules, but which also require tighter control in order to maintain proteostasis (26), as their overexpression often leads to cytotoxicity (27). Disordered regions possess bias for distinct amino acids (e.g. they contain less cysteine and tryptophan and more proline residues) (28), which is consistent with the amino acid distributions of proteins ubiquitylated after HS (Fig. 1C). When comparing the predicted percentage of disorder of each protein based on DISOPRED2 (17), we observed a bimodal distribution of the proteins further ubiquitylated after HS (with proteins predicted to be highly structured or with disordered regions; Fig. 1D). The bimodal distribution was supported by the observation that the distribution of certain individual amino acid residues such as serine, asparagine, and glycine were also bimodal (supplemental Fig. S1D). To assess statistical significance, we partitioned the reference lysate by categorizing proteins into three classes—structured (≤10% disorder), intermediate (10% to 30% disorder), and disordered (>30% disorder)—as done previously (26). The proportion of these three classes of proteins was significantly different (Fisher's exact test; Fig. 1E). This trend was confirmed (i) by using IUPRED (29) as an independent disorder prediction program (supplemental Fig. S1E), (ii) by comparison with the proteome (supplemental Figs. S1F and S1G), and (iii) when we assessed all 419 proteins that were more ubiquitylated after HS in any one of the three replicate experiments (supplemental Fig. S1H). Taken together, these data indicate a significant enrichment of proteins that contain large regions of predicted disorder.
We noticed that several disordered proteins ubiquitylated upon HS contained short stretches of histidine residues that could increase their enrichment (supplemental Table S1); nevertheless, disordered proteins remained enriched after the exclusion of these proteins (supplemental Fig. S1I). To fully exclude any bias from the enrichment method, we used an independent antibody-based approach to identify which proteins were ubiquitylated after HS (Fig. 1A). In this method, ubiquitylated peptides were enriched using an antibody that specifically recognizes the two remnant glycine residues of ubiquitin (GlyGly) that are left on conjugated lysines after trypsin digestion (23). We compared two cell populations differentially labeled that were either heat-shocked (14N) or not (15N) prior to lysis and GlyGly-peptide enrichment. Overall, we identified 1321 GlyGly peptides via mass spectrometry. Excluding ubiquitin, we quantified 461 unique GlyGly peptides (Fig. 2A) from 240 proteins (supplemental Table S2), of which 143 proteins were further ubiquitylated upon HS (proteins with a peptide log2 ratio ≥ 1). We also analyzed an aliquot of the whole cell lysate to ensure equal loading of the two labeled cell populations and quantified 304 proteins. We found that these proteins were significantly longer on average (Fig. 2B) and were enriched for proteins with predicted disordered regions (Fig. 2C). We repeated this experiment and confirmed that there was significant enrichment for longer and more disordered proteins (Figs. S2A–S2C). Overall, these results indicate that proteins ubiquitylated after HS are significantly enriched for longer proteins and proteins with more intrinsic disorder.
Fig. 2.
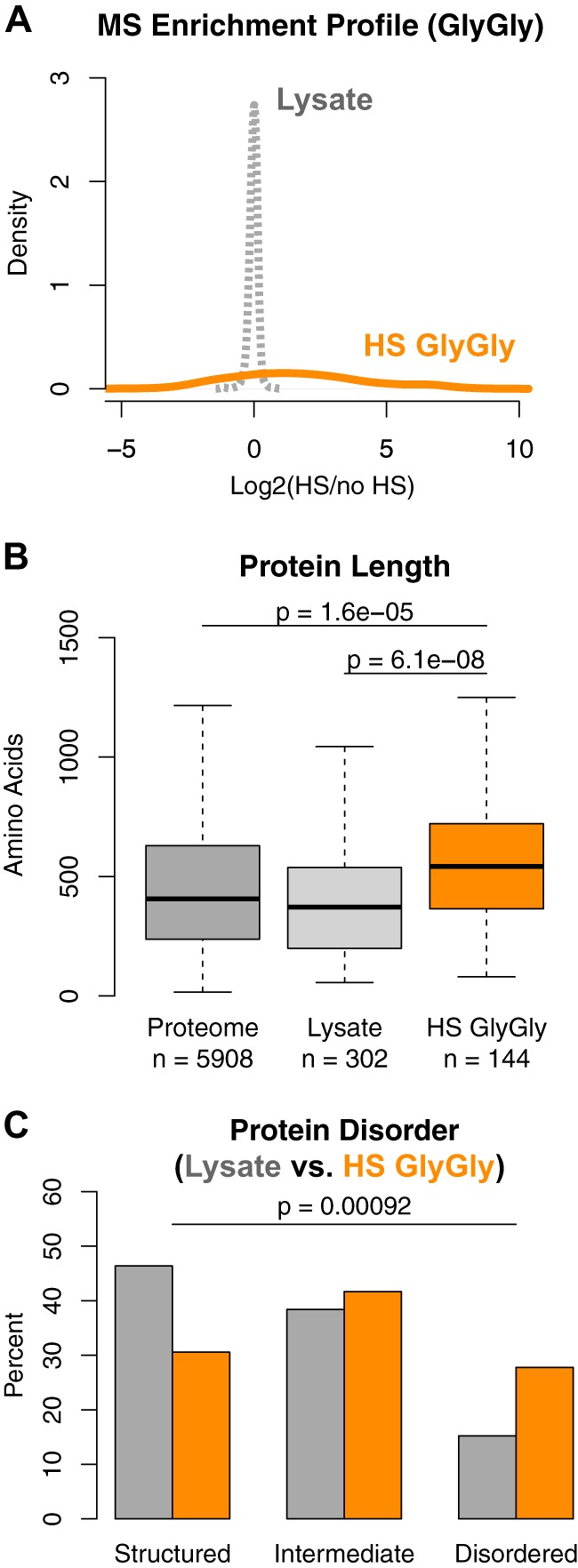
GlyGly analysis confirms enrichment for disordered proteins among proteins ubiquitylated after heat shock. A, distribution of 14N-HS/15N-noHS peptide ratios on a base two logarithmic scale. Ratios from the corresponding lysate in gray and ubiquitylated peptides enriched via immunoprecipitation using anti-GlyGly antibodies in orange. Ratios of peptides found only after heat shock were set to 100 (log2(100) = 6.64). B, bar plot of data binned at 10% and 30% disorder using DISOPRED2 comparing proteins ubiquitylated after HS that were enriched by the GlyGly antibody (orange) versus proteins in the whole cell lysate (gray). p values from Fisher's exact test comparing proportions in lysate versus GlyGly. C, bar plot of lysines ubiquitylated after HS predicted to be in regions disordered.
Although the GlyGly approach does not allow one to distinguish between mono- and poly-ubiquitylation, we observed a greater than 2-fold increase of ubiquitin peptides modified on K48 and K63 after HS, and a more modest enrichment for K11, K27, and K29 (supplemental Fig. S2D). This result indicates that there was an overall increase in poly-ubiquitylation levels (with primarily K48 and K63 linkages) and suggests that a portion of proteins modified after HS were poly-ubiquitylated.
We also found that lysines ubiquitylated after HS are not specifically enriched in regions that were predicted to be disordered (supplemental Fig. S2F). One possibility is that the GlyGly antibody approach might allow better detection of proteins that are prominently ubiquitylated on fewer lysine residues. In contrast, peptides derived from proteins conjugated at multiple sites might not be efficiently identified, as each ion, in comparison, would be of lower intensity. This is important when considering misfolded proteins that presumably adopt many possible configurations, and for which the selection of conjugated lysines is likely stochastic. This might be specifically exacerbated on disordered domains that are more flexible. In support of the latter explanation, we found that proteins ubiquitylated after HS detected using the GlyGly antibody approach had a lower lysine composition than those detected via the IMAC approach (supplemental Fig. S2G). This suggests that the antibody approach might be less sensitive toward detecting proteins that are ubiquitylated stochastically at different lysines.
Distinct Subpopulations of Proteins Are Ubiquitylated after HS
Because structured and disordered protein regions have fundamentally different biophysical properties, they likely have different susceptibilities for being targeted by the ubiquitin-proteasome system. Therefore, we considered these two classes separately for further analysis and confirmed that they constituted two distinct subpopulations of proteins. For instance, the amino acid compositions of highly structured and disordered proteins ubiquitylated after HS were, as expected, different (Fig. 3A). Furthermore, when considering secondary structure in proteins ubiquitylated after HS, there were fewer predicted alpha-helices and more predicted beta-sheets in the disordered proteins (Fig. 3B). The trend was similar for the disordered proteins identified with the GlyGly approach, although it was not significant, presumably because of the lower sample size (supplemental Fig. S3A). In contrast, we observed no significant difference when comparing the secondary structures of highly structured proteins (supplemental Fig. S3B), and we found no significant enrichment for a particular SCOP/Pfam domain in structured proteins ubiquitylated after HS (data not shown). As well, the difference in protein length was more pronounced when considering only disordered proteins ubiquitylated after HS (supplemental Fig. S3C). Importantly, disordered proteins ubiquitylated after HS were also on average longer than all disordered proteins in the proteome and disordered proteins in the reference lysate sample (supplemental Fig. S3D; disordered proteins are on average longer than other proteins).
Fig. 3.

Distinct subpopulations of structured and disordered proteins are ubiquitylated after heat shock. A, Amino Acid Compass of highly structured proteins ubiquitylated after HS (green) and intrinsically disordered proteins ubiquitylated after HS (orange) compared with the proteome (gray). Significantly different amino acids at an FDR-corrected p < 0.05 using Wilcoxon rank-sum test compared with the proteome are indicated by a dot of the respective color. B, box plot showing secondary structure in structured regions of disordered proteins ubiquitylated after HS versus all disordered proteins in the proteome. p values computed using Wilcoxon rank-sum test. C, box plot showing the differences in abundance between highly structured proteins ubiquitylated after HS and all structured proteins or the proteome, and between intrinsically disordered proteins ubiquitylated after HS and all disordered proteins or the proteome.
We also observed that these two structural classes of ubiquitylated proteins displayed different abundances in the cell. Using protein abundance measurements based on epitope TAP-tagging (30), we observed that highly structured proteins ubiquitylated after HS were significantly more abundant relative to all highly structured proteins in the lysate (Fig. 3C). In contrast, disordered proteins ubiquitylated after HS were less abundant relative to all disordered proteins in the lysate (Fig. 3C). This trend was also noted in comparisons with the same categories in the proteome (supplemental Fig. S3E) and when repeating the analysis using proteins from the GlyGly approach (supplemental Fig. S3F). These results indicate that disordered proteins ubiquitylated after HS correspond to less abundant proteins in the cell that are not readily identified with our instrument setup, unless we perform an enrichment for ubiquitylated polypeptides. Overall, differences in amino acid composition, secondary structure, and protein abundance suggest that distinct subpopulations of proteins are ubiquitylated after HS.
Length Affects the Solubility of Abundant Proteins after HS
Our observations were driven by comparisons with the whole proteome or with proteins readily identified in whole cell lysates. However, these analyses provide no comparison with the portion of the proteome that is actually misfolded upon HS. Furthermore, it is not clear whether proteins with disordered regions actually aggregate after HS. We previously observed that after HS a large portion of ubiquitylated proteins display low solubility following a 16,000g centrifugation (11), presumably as a result of aggregation. One possibility is that some proteins might be more likely to misfold upon denaturing stress.
To determine which proteins aggregate after HS and to compare them to the proteins that are ubiquitylated, we performed a series of quantitative mass spectrometry experiments to compare the solubility of proteins and ubiquitylated proteins in untreated 15N-labeled cells to that in heat-shocked (20 min, 45 °C) 14N-labeled cells (Fig. 4A). To enrich for low-solubility proteins, cells were lysed in a native buffer, and then lysates were centrifuged at 16,000g. Ubiquitylated proteins were further purified via IMAC from both soluble and insoluble fractions (this approach was preferred to the GlyGly enrichment because it allows the identification of more proteins and the quantification of more peptides per protein). We quantified 266 proteins in the whole lysate, 733 proteins in the soluble fraction, and 695 proteins in the insoluble fraction (supplemental Table S3). When comparing the distributions of the 14N/15N ratios, we found that there were overall fewer proteins in the soluble fraction of the HS-treated cells than in unstressed cells (Fig. 4A). The median ratio value was 0.64, indicating that about 36% of the assessed portion of the proteome lost solubility. We separated proteins in three different pools based on their ratios and found that proteins further depleted in the soluble fraction of heat-shocked cells (i.e. less soluble) were significantly longer (Fig. 4B), whereas proteins in the lysate showed no significant difference in protein length (supplemental Fig. S4A). Of the insoluble proteins, 638 were enriched after HS (Fig. 4A). Again, proteins with higher ratios (i.e. presumably less soluble) were longer on average (supplemental Fig. S4B). We confirmed these results with an independent experiment (Figs. S4C–S4E). All together, these data suggest that the solubility of the assessed proteins (which consisted mainly of abundant and highly structured proteins) after HS is associated with protein length.
Fig. 4.

Length affects the solubility of proteins after heat shock. A, schematic diagram of fractionation and purification workflow with the number of proteins quantified for each fraction indicated (left panel) and the distribution of log2(HS/no HS) in indicated fractions (right panel). “Ubi” refers to the IMAC sample enriched for ubiquitylated proteins, and L, S, and P denote lysate, soluble, and pellet, respectively. B, box plot of length of proteins remaining soluble after HS binned by their log2 ratios (<−0.71; −0.71 to −0.51; >−0.51). C, distribution of predicted percentage disorder by DISOPRED2 for insoluble proteins ubiquitylated after HS and purified via IMAC (orange) and insoluble proteins after HS (black). p values from Fisher's exact test comparing insoluble proportions in lysate versus IMAC.
We then assessed the distribution of ubiquitylated proteins. We identified 261 ubiquitylated proteins in the soluble fraction and 721 ubiquitylated proteins in the insoluble fraction (Fig. 4A; supplemental Table S3). The median ratio of ubiquitylated proteins in the soluble fraction was 0.67, indicating that these proteins in general were depleted in HS-treated cells. These ubiquitylated proteins had most likely become insoluble or less ubiquitylated, and were therefore not further analyzed. Of the insoluble ubiquitylated proteins, 661 were enriched after HS, including 241 proteins predicted to be highly structured (i.e. with less than 10% predicted disorder). When we compared these structured proteins to structured proteins in the insoluble fraction, we could find only minor changes in the amino acid composition (supplemental Fig. S4F), and no enrichment for a particular secondary structure (supplemental Fig. S4G) or protein fold (data not shown). All together, these results indicate that highly structured proteins ubiquitylated after HS are simply sampled from the most abundant proteins in the low-solubility fraction, in which there is an enrichment for longer proteins.
Specific Features Are Associated with Disordered Proteins Prone to Ubiquitylation after HS
Although a major portion of proteins in the pellet was highly structured, proteins predicted to be disordered were significantly enriched among insoluble proteins that were ubiquitylated (Fig. 4C). These data show that disordered proteins ubiquitylated after HS also displayed low solubility after HS. We indeed found that prion-like proteins enriched with glutamine or asparagine (supplemental Fig. S5A) (31–33) were overrepresented among disordered proteins ubiquitylated upon HS (Fig. 5A). This included the poly-Q protein Pin3 that we and others previously found to be further ubiquitylated after HS (11, 34). However, these proteins were not readily identified via mass spectrometry in the low-solubility fraction as a whole, presumably because highly abundant proteins, which also aggregate upon acute HS, precluded their identification. Their specific enrichment with ubiquitin indicates that a larger pool of these proteins is ubiquitylated. In contrast, a smaller pool of a given highly structured and abundant protein is ubiquitylated. Thus, we reasoned that these disordered proteins must contain specific elements or characteristics that favor their targeting by the ubiquitylation machinery.
Fig. 5.

Ubiquitylated disordered proteins are aggregation prone; fewer are essential, and they contain more regions involved in interactions. A, bar plot of percentage of disordered proteins that are aggregation prone in the indicated protein groups. B, bar plot of percentage of essential proteins for groups indicated. C, box plot of Scansite motifs in disordered regions of the indicated protein groups. D, box plot of the percentage of the indicated disordered regions with ANCHOR binding sites.
Cells might have adapted to become less dependent on proteins that are more susceptible to misfolding or protein quality control, and thus genes encoding these proteins might not be essential for viability under standard laboratory growth conditions. Significantly fewer genes encoding for disordered proteins that are ubiquitylated after HS are essential relative to all encoded disordered proteins in the proteome and all proteins in the proteome (Fig. 5B). A similar trend was also observed among disordered proteins identified via the GlyGly approach, although the difference was not statistically significant, presumably because of the smaller sample size (supplemental Fig. S5B). Interestingly, proteins encoded by essential genes are significantly longer (supplemental Fig. S5C), but disordered proteins ubiquitylated after HS are both less essential and longer.
Ubiquitylated Disordered Proteins Have Exposed Protein Binding Sites
Given that ubiquitylated disordered proteins were enriched in the low-soluble fraction, we wondered whether specific features on these disordered regions might make them prone to ubiquitylation (e.g. many disordered domains are involved in protein–protein interactions) (35–37). To test this possibility, we first used Scansite to look for sequence motifs on disordered proteins that could be readily recognized by other proteins (38). We discovered that on average, disordered proteins ubiquitylated after HS had more motifs in a given length relative to all disordered proteins (Fig. 5C) and to disordered proteins in the lysate (supplemental Fig. S5D). Then we confirmed these results using the Eukaryotic Linear Motif repository (39) (supplemental Fig. S5E). A prominent motif enriched corresponded to basophilic serine/threonine kinase sites (see supplemental Table S4 for a complete list). Corroborating these results, we observed the same trend using the independent method ANCHOR to find regions with favorable energetics for protein binding interactions (40) (Fig. 5D, supplemental Fig. S5F) and when using the data set derived from the insoluble fraction (supplemental Figs. S5G–S5I), and there was a similar magnitude change in the GlyGly experiment, although it was not significant, possibly because of the small sample size (supplemental Fig. S5J). These results show a significant enrichment of protein binding sites among disordered proteins ubiquitylated after HS that is consistent when compared with three groups (ubiquitylated proteins compared with the proteome, lysate, and insoluble fraction) using three prediction methods.
Disordered Regions Are Implicated in Mediating Ubiquitylation
Our computational analyses indicated that proteins predicted to have large disordered domains are prevalent among proteins ubiquitylated after HS and suggested that these intrinsically disordered regions could facilitate substrate recognition. We next sought to experimentally confirm that some of these disordered proteins are ubiquitylated after HS. From among the proteins that were identified in our mass spectrometry analysis, we selected seven proteins with various regions that were predicted to be disordered (Rvs167, Rtk1, Ppz1, Hal5, Sip1, Pbp1, and Osh3; supplemental Fig. S6A). We obtained cells in which each tested disordered protein, expressed from its endogenous promoter, was C-terminally TAP tagged (30). We expressed H8-ubiquitin in these cells, which were either subjected to HS or not. After cell lysis, ubiquitylated proteins were enriched via IMAC in denaturating conditions, followed by immunoblotting against the TAP tag. In each case, we could detect a stronger high molecular weight signal in HS-treated cells that was not detected in cells not expressing H8-ubiquitin (Fig. 6A, supplemental Fig. S9A). For comparison, a similar increase in poly-ubiquitylation was observed for a highly structured protein that was also assessed (Cpr1; Fig. 6B, supplemental Fig. S9B). Cpr1 was also found mono-ubiquitylated, but this modification was not affected by HS. In contrast, several disordered proteins (e.g. Osh3) also displayed a stronger band after HS that was consistent with the attachment of a single ubiquitin moiety. We previously observed a similar pattern for several Hul5 substrates (11); Hul5 is presumably an E4 enzyme that elongates ubiquitin chains initiated by other E3 ubiquitin ligases (41). To further verify that these proteins were ubiquitylated, we selected two candidates (Rvs167 and Ppz1) and performed a reverse pull-down in which the TAP-tagged proteins underwent immunoprecipitation followed by immunoblotting in order to assess the presence of HA-tagged ubiquitin. In both cases, there was an increase in the ubiquitylation signal following HS (supplemental Fig. S6B). These data provide additional evidence that these disordered proteins are further ubiquitylated after HS.
Fig. 6.

Disordered proteins are ubiquitylated after heat shock, and disordered regions mediate their ubiquitylation. A, purification of proteins conjugated to ubiquitin. Cells expressing (+) or not expressing (−) the indicated proteins fused to the TAP tag and His8-ubiquitin (H8-Ubi) were grown exponentially at 25 °C and were subjected (+) or not (−) to heat shock (HS) at 45 °C for 20 min. After lysis, equal amounts of whole cell lysates (WCL) were subjected to IMAC. The top panels correspond to immunoblotting with α-TAP antibodies of the IMAC fractions. The middle and bottom panels correspond to immunoblotting of the corresponding WCL with α-TAP and α-Pgk1 antibodies. Molecular weights are indicated on the left. Bars and asterisks denote regions with increased ubiquitylation after HS and a signal not specific to ubiquitylation, respectively. B, same as A, but for a structured protein. C, D, cells expressing His8-ubiquitin that carried a plasmid with the indicated HA tagged fragments under the control of the GAL1 promoter were incubated (+) with 2% galactose or not (−) prior to HS at 45 °C for 20 min. Immunoblottings were performed as described in A. FL, N, and C denote full-length, N-terminal, and C-terminal fragments, respectively. Quantification of the ubiquitylation signal was done by measuring the total signal intensity in the indicated section of the gel (denoted by a line with dots) after subtracting the background in the non-induced control lane, and the resultant value was then compared with the signal intensity of the same fragment in the WCL (normalized to 1 in FL). The asterisk denotes a signal not specific to ubiquitylation and the pound sign designates a region that was not used for the background subtraction. All untruncated images are provided in supplemental Fig. S9.
To address whether intrinsically disordered regions are required to mediate ubiquitylation after HS, we selected two proteins for truncation analysis: Hal5, a putative kinase, and Ppz1, a phosphatase. In both proteins, the N-terminal regions are predicted to be disordered and rich in motifs that bind protein partners, whereas the C-terminal regions are predicted to form ordered structures (supplemental Fig. S6A). The HA-tagged fragments were expressed after induction with galactose, alongside H8-ubiquitin, and the cells were subjected to HS. After IMAC and HA immunoblotting, we compared the ubiquitylation levels (i.e. after IMAC) with expression levels (i.e. in whole cell lysate). For Hal5, both N-terminal and C-terminal regions were expressed at similar levels, and the relative ubiquitylation levels were higher for the disordered region (Fig. 6C, supplemental Fig. S9C). For Ppz1, the disordered regions alone could not be adequately expressed, presumably because of instability (data not shown). Nevertheless, the relative ubiquitylation level of the full-length protein was higher than that with the truncation missing the disordered region (Fig. 6D, supplemental Fig. S9D). Altogether, these data indicate that disordered regions of these two assessed proteins are important for mediating ubiquitylation after HS.
DISCUSSION
In this study, we discovered characteristics of proteins that are ubiquitylated after misfolding stress using three proteomic approaches: (i) purifying proteins conjugated to H8-ubiquitin, (ii) using an antibody against a diglycine remnant of ubiquitylation, and (iii) isolating ubiquitylated proteins in the insoluble fraction. A major finding is the presence of two classes of proteins that are ubiquitylated after HS: highly structured proteins, and proteins with a high percentage of intrinsically disordered residues. We experimentally verified that there was indeed an increase in ubiquitylation for a subset of proteins with disordered regions. Furthermore, we showed for two proteins that these disordered regions are important for mediating ubiquitylation.
The highly structured proteins ubiquitylated after HS were among the most abundant proteins in the proteome (our findings are summarized in supplemental Fig. S8). Because the most abundant ions are selected for fragmentation and sequencing by the mass spectrometer, these proteins likely reflect a selective sampling of the proteome by the instrument. In line with this view, we found no significant differences in secondary structure or protein folds between these structured proteins and all structured proteins in general. The only prominent characteristic was a length-dependent propensity to aggregate after HS.
In contrast, intrinsically disordered proteins that are ubiquitylated after HS were on average less abundant than other proteins in the proteome and relative to all intrinsically disordered proteins. Furthermore, these disordered proteins are significantly longer, and fewer are encoded by essential genes than other disordered proteins. Interestingly, these disordered regions also have more motifs that are recognized by protein domains. Taken together, this indicates that a distinct subset of disordered proteins is readily ubiquitylated after HS. The observation that these proteins have prominent interaction regions suggests that intrinsically disordered regions likely recruit the ubiquitylation machinery to facilitate the conjugation of ubiquitin onto structured regions. Alternatively, secondary structures present in disordered proteins might also influence ubiquitylation, as beta-sheets were found enriched.
A priori, the ubiquitylation of intrinsically disordered proteins after misfolding stress was unexpected (42). At least two explanations might account for this: their ubiquitylation (i) is needed for regulatory reasons or (ii) is a result of protein quality control pathways. Regarding the former explanation, the fact that intrinsically disordered proteins are often involved in regulatory and signaling processes (28, 43) might suggest that their ubiquitylation either inhibits or activates proteins necessary for a response to HS. In this case, the increased ubiquitylation of low-abundant, intrinsically disordered proteins after HS might be testimony of a targeted control of these proteins under stress conditions. It would be interesting to determine whether other post-translational modifications like phosphorylation also are involved in this process. Regarding the latter explanation, it has been shown that the availability of many intrinsically disordered proteins is tightly controlled by various cellular mechanisms (26), and it has been proposed that these mechanisms reduce the risk of unwanted promiscuous interactions by motifs in intrinsically disordered protein segments (44). Promiscuous interactions are also averted by binding of the motifs to specific partners in unstressed cells, but upon stress they could act as a mechanism by which disordered proteins could recruit the protein quality control machinery.
Supplementary Material
Acknowledgments
We thank Dr. N. Stoynov for help with mass spectrometry analyses, Dr. E. Conibear for the TAP-tagged strains, Dr. L. Hicke for the HA-ubiquitin plasmid, Drs. A. Chruscicki and L. Howe for the cross-linked IgG magnetic beads, and other members of T.M. lab for their encouragement and discussion.
Footnotes
* T.M. is supported by a grant (MOP-89838) and a New Investigator Career Award from the Canada Institutes of Health Research and a Career Investigator Award from the Michael Smith Foundation for Health Research. J.G. is supported by PrioNet Canada and a Career Investigator Award from the Michael Smith Foundation for Health Research.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are::
- HS
- Heat shock
- FDR
- false discovery rate
- GlyGly
- two remnant glycine residues of ubiquitin
- HS
- heat shock
- IMAC
- immobilized metal affinity chromatography.
REFERENCES
- 1. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 2. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 3. Pickart C. M., Eddins M. J. (2004) Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695, 55–72 [DOI] [PubMed] [Google Scholar]
- 4. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Ann. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berke S. J., Paulson H. L. (2003) Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Curr. Opin. Genet. Dev. 13, 253–261 [DOI] [PubMed] [Google Scholar]
- 6. Carrard G., Bulteau A.-L., Petropoulos I., Friguet B. (2002) Impairment of proteasome structure and function in aging. Int. J. Biochem. Cell Biol. 34, 1461–1474 [DOI] [PubMed] [Google Scholar]
- 7. Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Höhfeld J., Patterson C. (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96 [DOI] [PubMed] [Google Scholar]
- 9. Hough R., Pratt G., Rechsteiner M. (1986) Ubiquitin-lysozyme conjugates. J. Biol. Chem. 261, 2400–2408 [PubMed] [Google Scholar]
- 10. Parag H. A., Raboy B., Kulka R. G. (1987) Effect of heat shock on protein degradation in mammalian cells: involvement of the ubiquitin system. EMBO J. 6, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fang N. N., Ng A. H. M., Measday V., Mayor T. (2011) Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 13, 1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang N. N., Mayor T. (2012) Hul5 ubiquitin ligase: good riddance to bad proteins. Prion 6, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carlson N., Rogers S., Rechsteiner M. (1987) Microinjection of ubiquitin: changes in protein degradation in HeLa cells subjected to heat-shock. J. Cell Biol. 104, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dantuma N. P., Groothuis T. A., Salomons F. A., Neefjes J. (2006) A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J. Cell Biol. 173, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cherry J. M., Hong E. L., Amundsen C., Balakrishnan R., Binkley G., Chan E. T., Christie K. R., Costanzo M. C., Dwight S. S., Engel S. R., Fisk D. G., Hirschman J. E., Hitz B. C., Karra K., Krieger C. J., Miyasato S. R., Nash R. S., Park J., Skrzypek M. S., Simison M., Weng S., Wong E. D. (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–D705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ihaka R., Gentleman R. (1996) R: a language for data analysis and graphics. Journal of Computational and Graphical Statistics 5, 299–314 [Google Scholar]
- 17. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 [DOI] [PubMed] [Google Scholar]
- 18. Petersen B., Petersen T. N., Andersen P., Nielsen M., Lundegaard C. (2009) A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayor T., Graumann J., Bryan J., MacCoss M. J., Deshaies R. J. (2007) Quantitative profiling of ubiquitylated proteins reveals proteasome substrates and the substrate repertoire influenced by the Rpn10 receptor pathway. Mol. Cell. Proteomics 6, 1885–1895 [DOI] [PubMed] [Google Scholar]
- 20. Mayor T., Lipford J. R., Graumann J., Smith G. T., Deshaies R. J. (2005) Analysis of polyubiquitin conjugates reveals that the Rpn10 substrate receptor contributes to the turnover of multiple proteasome targets. Mol. Cell. Proteomics 4, 741–751 [DOI] [PubMed] [Google Scholar]
- 21. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 22. Wilde I. B., Brack M., Winget J. M., Mayor T. (2011) Proteomic characterization of aggregating proteins after the inhibition of the ubiquitin proteasome system. J. Proteome Res. 10, 1062–1072 [DOI] [PubMed] [Google Scholar]
- 23. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat Soc. Series B Stat. Methodol. 57, 289–300 [Google Scholar]
- 26. Gsponer J., Futschik M. E., Teichmann S. A., Babu M. M. (2008) Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vavouri T., Semple J. I., Garcia-Verdugo R., Lehner B. (2009) Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138, 198–208 [DOI] [PubMed] [Google Scholar]
- 28. Radivojac P., Iakoucheva L. M., Oldfield C. J., Obradovic Z., Uversky V. N., Dunker A. K. (2007) Intrinsic disorder and functional proteomics. Biophys. J. 92, 1439–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dosztanyi Z., Csizmok V., Tompa P., Simon I. (2005) The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 347, 827–839 [DOI] [PubMed] [Google Scholar]
- 30. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 31. Alberti S., Halfmann R., King O., Kapila A., Lindquist S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harrison P. M., Gerstein M. (2003) A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol. 4, R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michelitsch M. D., Weissman J. S. (2000) A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U.S.A. 97, 11910–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chernova T. A., Romanyuk A. V., Karpova T. S., Shanks J. R., Ali M., Moffatt N., Howie R. L., O'Dell A., McNally J. G., Liebman S. W., Chernoff Y. O., Wilkinson K. D. (2011) Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol. Cell 43, 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim P. M., Sboner A., Xia Y., Gerstein M. (2008) The role of disorder in interaction networks: a structural analysis. Mol. Syst. Biol. 4, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buljan M., Chalancon G., Eustermann S., Wagner G. P., Fuxreiter M., Bateman A., Babu M. M. (2012) Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Babu M. M., van der Lee R., de Groot N. S., Gsponer J. (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440 [DOI] [PubMed] [Google Scholar]
- 38. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dinkel H., Michael S., Weatheritt R. J., Davey N. E., Van Roey K., Altenberg B., Toedt G., Uyar B., Seiler M., Budd A., Jödicke L., Dammert M. A., Schroeter C., Hammer M., Schmidt T., Jehl P., McGuigan C., Dymecka M., Chica C., Luck K., Via A., Chatr-Aryamontri A., Haslam N., Grebnev G., Edwards R. J., Steinmetz M. O., Meiselbach H., Diella F., Gibson T. J. (2012) ELM—the database of eukaryotic linear motifs. Nucleic Acids Res. 40, D242–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mészáros B., Simon I., Dosztányi Z. (2009) Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 5:e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crosas B., Hanna J., Kirkpatrick D. S., Zhang D. P., Tone Y., Hathaway N. A., Buecker C., Leggett D. S., Schmidt M., King R. W., Gygi S. P., Finley D. (2006) Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127, 1401–1413 [DOI] [PubMed] [Google Scholar]
- 42. Galea C. A., High A. A., Obenauer J. C., Mishra A., Park C. G., Punta M., Schlessinger A., Ma J., Rost B., Slaughter C. A., Kriwacki R. W. (2009) Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J. Proteome Res. 8, 211–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iakoucheva L. M., Brown C. J., Lawson J. D., Obradović Z., Dunker A. K. (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584 [DOI] [PubMed] [Google Scholar]
- 44. Tsvetkov P., Reuven N., Shaul Y. (2009) The nanny model for IDPs. Nat. Chem. Biol. 5, 778–781 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.