

Abstract
Hepatocellular carcinoma (HCC) is one of the most deadly cancers worldwide and has no effective treatment, yet the molecular basis of hepatocarcinogenesis remains largely unknown. Here we report findings from a whole-genome sequencing (WGS) study of 88 matched HCC tumor/normal pairs, 81 of which are Hepatitis B virus (HBV) positive, seeking to identify genetically altered genes and pathways implicated in HBV-associated HCC. We find beta-catenin to be the most frequently mutated oncogene (15.9%) and TP53 the most frequently mutated tumor suppressor (35.2%). The Wnt/beta-catenin and JAK/STAT pathways, altered in 62.5% and 45.5% of cases, respectively, are likely to act as two major oncogenic drivers in HCC. This study also identifies several prevalent and potentially actionable mutations, including activating mutations of Janus kinase 1 (JAK1), in 9.1% of patients and provides a path toward therapeutic intervention of the disease.
Liver cancer is the fifth most frequently diagnosed cancer and the third leading cause of cancer mortality worldwide (Jemal et al. 2011). Hepatocellular carcinoma (HCC), the most common primary liver malignancy, is refractory to nearly all currently available anti-cancer therapies. Hepatitis B virus (HBV) infection causes the majority of HCC cases worldwide and is the leading etiological agent in epidemic regions of China, South Korea, Southeast Asia, and sub-Saharan Africa (Parkin 2006). Genetic alterations in HBV-mediated HCC have been identified previously, including point mutations in TP53 and beta-catenin (CTNNB1), amplifications of MYC and FGF19, and HBV integrations into TERT and KMT2B (also known as MLL4) (Neuveut et al. 2010; Zender et al. 2010; Sawey et al. 2011). Recently a number of studies that published on genome or exome sequencing of HCC have begun to reveal the genetic landscape in HCC (Li and Mao 2012). However, the number of HCC samples sequenced is still limited, the list of genetic alterations is far from complete, and the key drivers of HBV-induced tumorigenesis remain poorly understood. Whole-genome sequencing (WGS) of a large number of matched tumor and normal samples provides a systematic and comprehensive approach for identifying recurrent somatic genetic alterations, including single nucleotide variations (SNVs), small insertions and deletions (indels), DNA copy number variations (CNVs), and structural variations (SVs) (Bass et al. 2011; Chapman et al. 2011). We applied WGS to survey a cohort of 88 well-annotated HCC tumors to identify somatically mutated genes and pathways potentially driving HCC. We report herein the commonly altered cancer pathways as well as recurrent and potentially actionable mutations, including novel and activating mutations in Janus kinase 1 (JAK1) that could lead to an opportunity to evaluate existing JAK inhibitors for the treatment of this deadly disease.
Results
Overview of genetic alterations
Eighty-eight primary HCC tumors and matched adjacent nontumor liver tissues were analyzed by whole-genome DNA sequencing to identify somatic mutations and HBV integration sites. The vast majority (92%, n = 81) of patients in this cohort were HBV carriers (i.e., HBsAg seropositive) suffering from chronic hepatitis B or cirrhosis. None of the patients were hepatitis C virus (HCV) positive (see Supplemental Table 1).
For each DNA sample except for 11 patients where either normal or tumor DNA is limited, we built two libraries with different insert sizes (170 bp and 800 bp) for sequencing because the larger insert library provides better physical coverage for capturing SVs. The average depth of base pair coverage was 36.1× except for three tumor/normal pairs sequenced at 100× coverage. The concordance of single nucleotide polymorphism (SNP) calls between WGS and genotype data generated from Illumina 550K arrays exceeded 98.8%.
We developed a method to detect somatic SNVs in WGS data (see Methods) and estimated its sensitivity to be 82.2% (88/107) when evaluated against a reference set of 107 somatic SNVs identified from whole-transcriptome sequencing and experimentally validated using Sequenom mass spectrometric genotyping (see Methods). To assess the false-positive rate, we randomly selected 497 somatic SNV calls, and 3.4% were invalidated by Sequenom method in both tumor and normal DNA samples (Supplemental Table 2). Moreover, our validation results showed that sensitivity was not much higher in 100× samples (87.5%, 42/48) than in 30× samples (78%, 46/59) while false-positive rates were similar in the two groups (2.2% vs. 4.1%). Hence, we selected 30× as sequencing coverage for the study because it is more cost-effective for surveying the landscape of somatic SNVs in a large cohort than 100×. In total, 823,835 somatic SNV mutations were detected in 88 tumors, of which 5015 (0.6%) are predicted to affect protein coding (4482 missense, 334 nonsense, and 199 splice site) (Supplemental Table 3). A total of 3739 genes were mutated including 809 genes with multiple protein-altering mutations (Supplemental Table 4). The average somatic mutation rate is 3.69 per Mb with a wide range across samples (0.07–39). The nonsynonymous to synonymous somatic SNV ratio of 2.99 and the mean protein-altering mutation rate of 1.8 per Mb are midrange among different cancer types (Greenman et al. 2007). The mutation prevalence and patterns observed are largely consistent with reports from a recent WGS study of a single, HCV-associated HCC genome (Supplemental Fig. 1; Totoki et al. 2011).
Using a somatic indel calling pipeline based on the SRiC method (Zhang et al. 2011) (see Methods), we also predicted 35,168 small (≤20-bp) deletions and 22,826 small insertions including 302 events affecting protein-coding (256 frame-shifted, 46 in-frame) in 285 genes (Supplemental Table 5). Eighty-eight of the 90 randomly selected 1- to ∼4-bp coding indels were validated (97.8%) by the Sequenom method in both tumor and normal DNA samples (Supplemental Table 6). In-frame deletions were found in oncogenes such as CTNNB1 (1.1%), MDM2 (1.1%), and IL6ST (1.1%) (Rebouissou et al. 2009), and frame-shifting deletions were found in tumor suppressors including ARID1A (2.3%), AXIN1 (2.3%), PTEN (1.1%), RB1 (1.1%), and TP53 (1.1%) (Supplemental Table 5).
We also detected 399 genomic HBV integration events affecting 115 coding genes and 4314 somatic structural variation (SV) events, including 260 gene fusions. Details of HBV integration and SV events have been described elsewhere (Sung et al. 2012; J Fernandez-Banet, NP Lee, KT Chan, H Gao, X Liu, WK Sung, W Tan, RT Poon, PA Rejto, M Mao, et al., in prep.).
Significantly mutated genes
The statistical significance of the observed mutation prevalence for each gene was assessed in the context of the background mutation rate and gene sequence length (Youn and Simon 2011) (see Methods and Supplemental Table 3). The most significantly mutated genes include three genes that have been previously established to be mutated in HCC (TP53, CTNNB1, AXIN1), two genes known to be frequently mutated in other cancer types (JAK1, LRP1B), and six genes not previously reported to be mutated in HCC (EPS15, SLC10A1, CACNA2D4, ADCY2, FAM5C, and COL11A1) (Table 1). TP53 has the highest prevalence of protein-altering mutations identified in our HCC cohort (31/88 samples = 35.2%), consistent with earlier HCC studies (Neuveut et al. 2010). We found TP53 mutated tumors harbor higher levels of chromosome instability, are more likely to be poorly differentiated, and have poor survival (Supplemental Fig. 2). This is consistent with the hypothesis that large-scale chromosomal alteration is left unchecked in cells where TP53-mediated defenses against DNA damage were deactivated by TP53 mutation (Chow and Poon 2010). Interestingly, the R249S mutation in TP53, a hotspot linked to aflatoxin B exposure in China (Aguilar et al. 1994), was observed in nine cases, suggesting that aflatoxin B exposure could be a secondary etiological agent in this cohort dominated by HBV+ cases and the mutation could be as the result of selective advantage during mutagenesis induced by aflatoxins and HBV chronic infection (Gouas et al. 2009). CTNNB1 harbors protein-altering mutations in 15.9% of the tumors. Eleven of the 14 mutations are located in exon 3 and have been previously reported (Miyoshi et al. 1998). As exon 3 contains the phosphorylation site of GSK3B, a mediator of CTNNB1 degradation, prevalent mutations in this region may act to preserve CTNNB1 activities. We also detected a small deletion in exon 3 of CTNNB1 that removes the GSK3B phosphorylation sites (S33 and S37). In addition to CTNNB1, negative regulators of CTNNB1, including AXIN1 (4.5%), AXIN2 (2.3%), and APC (2.3%), are also mutated, implicating the canonical Wnt pathway as a major driver of hepatocarcinogenesis. Two known cancer genes not previously reported to be mutated in HCC are frequently mutated in our cohort—BRCA2 (5.7%) and IGF1R (5.7%). Furthermore, there are multiple protein-altering mutations in cancer genes such as FLT3 (3.4%), KDR (3.4%), PTEN (2.3%), MET (2.3%), ALK (2.3%), ROS1 (2.3%), NOTCH2 (2.3%), and FGFR2 (2.3%). Mutations in PIK3CA and HRAS were observed but were infrequent (1/88). We did not observe mutations in KRAS, which has been reported in vinyl chloride–associated HCC (Weihrauch et al. 2001).
Table 1.
Significantly mutated genes in primary HCC
Several of the frequently mutated genes identified in this study are not well-established cancer genes, but corroborating evidence from recent studies points to potential roles in cancer. The chromatin remodeling gene ARID2 was found mutated in 14.0% of HCV-associated HCC and 2.0% of HBV-associated HCC tumors recently (Li et al. 2011), and in our study, it was found mutated in three samples (3.4%) where all three mutations are nonsense. We also observed two mutations in ARID1A, a key component of the SWI-SNF chromatin remodeling complex reported to be frequently mutated in ovarian, colorectal, and gastric cancers (Wiegand et al. 2010; Jones et al. 2011; Wang et al. 2011). KMT2C (also known as MLL3), a member of the MLL gene family associated with aggressive acute leukemia (ALL) and a subunit of the histone H3K4 methyltransferase complex (Lee et al. 2009), is mutated in 8.0% of tumors. KMT2B, a paralog of KMT2C, is affected by HBV integration in 10.2% of tumors. Taken together, our findings suggest that chromatin remodeling may play a significant role in HCC carcinogenesis, echoing recent findings in medulloblastoma, renal carcinoma, gastric cancer, and ovarian cancer genome sequencing efforts (Wiegand et al. 2010; Varela et al. 2011).
In addition to well-known and emerging cancer genes, our study revealed several genes with poorly characterized functional roles in cancer but sufficient mutational evidence to warrant experimental investigation. One of the most significantly mutated genes is LRP1B with mutations in 10 tumors (11.4%) and LRP1B mutations have been previously reported in 9% of lung adenocarcinomas (Ding et al. 2008). EPS15, a phosphorylation substrate of EGFR and a translocation partner of MLL in acute leukemia, is significantly mutated with four mutations (4.5%). ADCY2 belongs to the adenylate cyclase family of enzymes responsible for the synthesis of cAMP and is mutated in five cases (5.7%). USH2A mutations in eight tumors (9.1%) including two nonsense mutations were identified in this study, indicating that USH2A might function as a tumor suppressor. Germline mutations in the USH2A gene are responsible for the most common subtype of Usher syndrome (USH), a genetic disorder that is a main cause of combined deaf-blindness (Reiners et al. 2006). Interestingly, three additional USH-associated genes, GPR98 (8.0%), PCDH15 (6.8%), and MYO7A (1.1%), are also mutated in this cohort, putatively linking the genetic causes of Usher syndrome with hepatocarcinogenesis.
JAK1 mutations and functional characterization
JAK1 (Janus kinase 1) is a member of the Janus kinase family that mediates the growth factor and cytokine-induced STAT signaling pathway and plays important roles in immunity, cell growth, and differentiation. Activating mutations in JAK1 and JAK2 have been described in patients with various hematologic malignancies, including acute leukemia and myeloproliferative neoplasm (MPN) (Quintas-Cardama et al. 2011). We identified seven distinct protein-altering mutations in JAK1 in eight HCC tumors (9.1%), with two of the mutations (S703I and S729C) recurring in two tumors. All mutations were validated by Sanger sequencing. Six of the mutations are located in the pseudo-kinase (JH2) and tyrosine kinase (JH1) domains (Fig. 1A). The tandem mutation Q644H/V645F was recently reported in one HCC tumor (Xie et al. 2009), and S703I has been independently shown to activate JAK1 in a mouse model system characterizing IL-9/JAK signaling (Supplemental Fig. 3; Hornakova et al. 2011). We therefore examined whether the seven JAK1 somatic mutations identified in our study constitutively activate the JAK/STAT pathway. Upon expression of the mutants in HEK293FT and a HCC cell line Hep3B, we observed increased autophosphorylation of JAK1 (S703I and S729C) and increased activation of STAT3 (Q644H/V645F, S703I, S729C, and L910P) compared with wild-type JAK1 (Fig. 1B). No changes in signaling were observed with the FERM domain mutant N242S or with the kinase domain mutant G902E. Cytokines such as IL6 activate STAT signaling in HCC cell lines, including Hep3B, and the presence of such receptor complexes has been shown to mediate mutant JAK1 signaling (Hornakova et al. 2009; Pilati et al. 2011); thus, we sought to examine whether IL6 regulates variant JAK1 signaling in Hep3B cells. Although JAK1 Q644H/V645F, S703I, S729C, and L910P constitutively increase JAK/STAT signaling compared with wild-type, IL6–dependent regulation of the JAK1 mutants is not enhanced beyond wild-type activity (Fig. 1B). We modeled the interaction between the JH1 and JH2 domains using a crystal structure of the JH1 domain (3EYG) (Williams et al. 2009) and a homology model of the JH2 domain oriented to have an interaction between the first N-terminal α-helices in each domain, patterned after previous models (Fig. 1C; Flex et al. 2008). Notably, three of the mutated residues located in the pseudo-kinase domain that constitutively activate the JAK/STAT pathway (Q644H/V645F, S703I) are predicted by the model to be at the JH1/JH2 interface. Mutation of these residues could alleviate the inhibition of JAK1 kinase activity by the pseudo-kinase domain; a negative regulatory function for the JH2 domain has been established in Janus kinases (Saharinen and Silvennoinen 2002), and the A634D mutation in the JAK1 JH2 domain has been reported to up-regulate kinase activity ligand-independently (Flex et al. 2008).
Figure 1.

Activating mutations in Janus kinase 1 (JAK1). (A) Seven distinct somatic mutations in JAK1 in the context of protein domains and active sites. Two mutations (S703I and S729C) are recurrent, found in two samples each. (B) HEK293FT and Hep3B cells were transiently transfected with either empty vector, Flag-tagged JAK1 WT, or Flag-tagged JAK1 variants for 48 h, serum-starved for 4 h, and in the case of Hep3B, treated with vehicle or 10 ng/mL IL6 for 15 min. Resultant JAK1 and STAT3 activation determined by immunoblotting lysates with anti-pJAK1 (Y1034/1035) and -pSTAT3 (Y705), respectively. Comparable expression of Flag-tagged JAK1 constructs and protein loading was confirmed using anti-M2 Flag and -beta actin. Representative result is shown in two to six independent experiments. (C) A model of the interaction between the JAK1 JH1 and JH2 domains was generated using the crystal structure of the JH1 domain (3EYG) and a homology model of the JH2 domain built using Discovery Studio (accelrys.com). The two residues reported to be in contact (Lys 924 and Glu 637) that were used to orient the domains are highlighted (gray CPK), as are the seven residues found to be mutated in this study (green CPK), and the small-molecule CP-690,550 bound in the JH1 ATP site (ball-and-stick). (D) Tumor samples are ranked by JAK1 activation scores based on independently derived JAK1 gene expression signature (Flex et al. 2008). Colors indicate JAK1 mutation statuses and whether the mutation is activating in vitro as experimentally determined. (E) Wild-type JAK1 or activating JAK1 mutants (S703I, S729C, and L910P) transduced Ba/F3 cells were cultured in the absence of IL3 in triplicate. Cell numbers were counted at indicated time points and data presented as mean ± SD. (F) JAK1 activating mutant (S703I and S729C)-transduced cells were treated with either ruxolitinib or BMS-911453 for 3 d. Cell viability was measured and data presented as mean ± SD.
To evaluate the endogenous functional consequences of JAK1 mutations, we performed gene expression profile analysis on the 88 HCC tumors using an independently derived gene signature from acute lymphoblastic leukemia patients with acquired JAK1 mutations that is enriched in genes known to be modulated by JAK/STAT signaling (Flex et al. 2008). All six HCC tumors harboring JAK1 activating mutations (Q644H/V645F, S703I, S729C, and L910P) exhibit a strong expression signature as quantified by principal component analysis (PCA), while the two tumors with nonactivating mutations (N242S and G902E) do not have the signature, suggesting a gain-of-function role for these activating JAK1 mutations in primary HCC tumors (Fig. 1D). To assess oncogenic consequences of the JAK1 activating mutations, we evaluated the ability of each to induce autonomous growth when expressed in cytokine-dependent Ba/F3 cells. Wild-type JAK1 or selected JAK1 mutants (S703I, S729C, and L910P) were transduced into Ba/F3 cells, and cell growth was assessed in the absence of the cytokine IL3. All three mutants conferred IL3–independent growth; in contrast, cells transduced with wild-type JAK1 remained dependent on the cytokine for survival, comparable to cells transduced with vector alone. Among the three activating mutations tested, it appeared that S729C exhibited the most transforming capability (Fig. 1E). JAK2 inhibitors have proven to be promising therapies for JAK2 mutation driven myeloproliferative neoplasms (Quintas-Cardama et al. 2011). We evaluated two mutant JAK1 transduced Ba/F3 cell lines for their sensitivity to the JAK1/2 dual inhibitor ruxolitinib (Quintás-Cardama et al. 2010), and the JAK2 inhibitor BMS-911453 (Purandare et al. 2012). Ruxolitinib displayed potent growth inhibition on both S703I and S729C transduced cells, whereas BMS-911453 only showed a partial inhibitory effect (Fig. 1F), and growth inhibition by ruxolitinib correlated with its complete inhibition of downstream STAT3 phosphorylation (Supplemental Fig. 4). The differing cellular activity observed with the two JAK compounds may be due to ruxolitinib being a 100-fold more potent inhibitor of the JAK1 enzyme (i.e., JAK1 IC50 of 3.3 nM for ruxolitinib vs. 356 nM for BM-911453). Taken together, these results suggest that the JAK1 mutations identified in this study are potentially oncogenic drivers in HCC through constitutive activation of downstream signaling.
Copy number changes
The landscape of copy number variations (CNVs) in HCC genomes is predominated by large-scale amplifications and deletions of chromosomal arms or entire chromosomes (amplified: 1q, 5p, 6p, 8q, 17q, 20q, and Xq; deleted: 4p-q, 8p, 13p-q, 16p-q, 17p, 21p-q, and 22q) (Fig. 2A; Supplemental Table 7; Supplemental Fig. 5). We performed a GISTIC analysis adapted to WGS data and revealed 1967 genes in 104 amplified regions and 2604 genes in 262 deleted regions (Supplemental Tables 8, 9). In total, 7.8% of the genome is amplified with an average gain of 0.65 copies, and 10.5% of the genome is deleted with an average loss of 0.77 copies. In significantly amplified or deleted regions, the G-scores of amplification for oncogenes as classified by Cancer Gene Census (Futreal et al. 2004) are threefold higher on average than tumor suppressors (0.079/0.025), whereas G-scores of deletion for tumor suppressors are 2.7-fold higher than oncogenes (0.244/0.095) (Supplemental Fig. 6). Outside of CNV regions, however, there is little difference overall in G-scores between oncogenes and tumor suppressors. The selective amplification of oncogenes and deletion of tumor suppressor genes suggest that these broad copy number changes in HCC genomes likely contribute to hepatocarcinogenesis.
Figure 2.

Landscape of genomic copy number alteration in HCC. (A) Shown is a genome-scale overview of copy number segments across 88 HCC samples rendered by IGV (Robinson et al. 2011). Each track in the upper panel represents one tumor sample, and the lower panels show the amplification and deletion G-score profiles across the genome. (B) Shown is a chromosomal overview of amplification and deletion peaks with the locations of putative cancer driver genes indicated. Arrows indicate focal peaks (<100 kb). Asterisks indicate genes also harboring nonsense mutations. (C) A log-scale plot of gene-level expression fold-change in CNV relative to non-CNV samples versus amplitude of copy gains. Genes with strong increase in expression driven by copy gains are labeled. Colors and shadings were used to indicate the CNV and cis-correlation statuses of genes. The size of the marker is proportional to the amplification G-score.
Applying a filter based on cis-correlation of DNA copy numbers and mRNA expression levels (see Methods), we focused on 685 candidate driver genes in amplified regions and 821 genes in deleted regions, including many known and putative oncogenes—MDM4 (1q), ELK4 (1q), PARP1 (1q), MYC (8q), PTK2 (8q), CCND1 (11q), FGF19 (11q), RPTOR (17q), and IRAK1 (Xq)—and tumor suppressors—ARID1A (1p), FBXW7 (4q), PTK2B (8p), CDKN2A/B (9p), RB1 (13q), AXIN1 (16p), TP53 (16q), and MAP2K4 (16q) (Fig. 2B). FGF19 along with CCND1 in the 11q13.3 amplicon were identified as two driver genes in HCC by a functional genomics screening (Sawey et al. 2011). We found that the CCND1/FGF19 region is focally amplified in four samples (4.5%) with the highest amplitude among all genes, and the amplification is concordant with a significant increase in gene expression (Fig. 2C). RSPO2, a member of the R-spondin family that activates beta-catenin signaling through LGR receptors (de Lau et al. 2011), had a similar pattern of overexpression driven by amplification. Conversely, well-known tumor suppressors such as TP53, RB1, CDKN2A, and CDKN2B reside in the most frequently deleted regions in HCC genomes. Other genes in regions that are frequently deleted include FBXW7, ARID1A, and MAP2K4, consistent with their putative roles as tumor suppressors.
Frequently altered cancer pathways
To further elucidate the functional impact of the complex alterations observed in HCC genomes, we performed an integrative analysis of somatic mutation, CNV, and HBV integration mapped to canonical cancer pathways from MSigDB (Liberzon et al. 2011), and identified frequent alterations across multiple pathways, including Wnt, JAK/STAT, G1/S cell cycle, and apoptosis (Supplemental Table 10; Supplemental Fig. 7). Pathway enrichment analysis (see Methods) further identified significant overlaps (FDR < 0.1) with cell adhesion, DNA repair, and chromatin modification pathways (Supplemental Table 10).
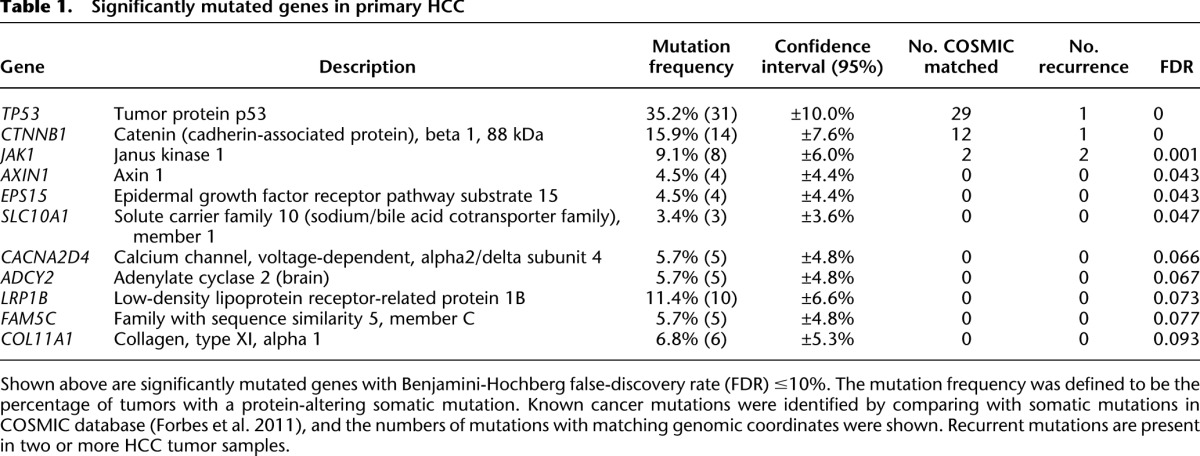
The Wnt pathway drives proliferation, maintains survival and promotes stemness in cancer cells (Klaus and Birchmeier 2008), and plays a well-known role in HCC tumorigenesis (Whittaker et al. 2010; Fatima et al. 2011). Overall, 62.5% of tumors in the cohort contain genomic alterations composed of SNV and CNV affecting core components of the Wnt pathway (Fig. 3A). Nonsynonymous somatic mutations occur in 23.9% of the tumors, including mutations in CTNNB1 (15.9%) and components of the destruction complex (APC and AXIN1/2) at a frequency of 8.0%. In addition, the tumor suppressor AXIN1 (Satoh et al. 2000) was deleted in 11.4% of tumors. Proximal to the destruction complex, amplifications were detected both in the pathway ligands (WNT9A, 25.0%; RSPO2, 21.6%) and in a pathway receptor (FZD6, 21.6%). It remains to be determined which genomic lesions translate into aberrant Wnt pathway activation in these tumors as measured by nuclear beta-catenin staining or the presence of a Wnt pathway signature. While the majority of the tumors in this study harbor genetic lesions of Wnt pathway components, activation of Wnt signaling can occur in additional or overlapping tumor cases through down-regulation of SFRP1 and DACT1 gene expression, known negative regulators of Wnt and Dvl family proteins, respectively (Kawano and Kypta 2003; Yau et al. 2005). Hepatocytes are capable of rapid regeneration and proliferation through activating the stem cell compartment in response to chronic liver damages inflicted by hepatitis, cirrhosis, and other hallmark conditions of HCC. An earlier study has shown that 28%–50% of HCCs express markers of liver progenitor cells, suggesting a link between HCC and these constantly proliferating stem cells (Mishra et al. 2009). Frequent mutations in the Wnt pathway revealed in this study, coupled with the observation that the recently identified R-Spondin receptor LGR5 (de Lau et al. 2011), an adult stem cell marker and beta-catenin target, is up-regulated in HCC further strengthens the hypothesis that HCC may have a significant stem cell lineage.
Figure 3.
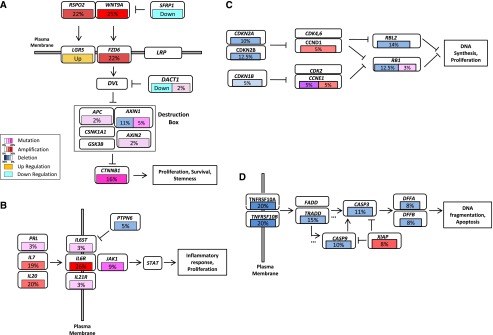
Frequently altered cancer pathways in HCC. Core pathway analysis identified frequent genomic alterations in multiple cancer pathways, including (A) Wnt, (B) JAK/STAT, (C) G1/S cell cycle, and (D) apoptosis pathways. Alterations include somatic mutation, DNA copy number changes correlated with gene expression, and HBV integration. Alteration frequency was represented as a percentage of all cases harboring a genomic alteration in one of the pathway genes shown. Gene expression up- or down-regulation in tumor relative to normal samples is shown but not included in the calculation of alteration frequencies. Alteration types and frequencies were represented by different colors and color gradients, respectively.
The pathogenesis of HCC is frequently linked to inflammatory responses triggered by chronic viral infection and cell necrosis. Cytokine-induced JAK/STAT pathway activation can lead to tumor-promoting inflammation in HCC, where the majority of cases were shown to harbor activated STAT3 (He and Karin 2011). Our findings of prevalent and activating JAK1 mutations shed light on a heretofore underappreciated role for JAK1 in promoting STAT3 signaling and hepatocarcinogenesis. Further, as much as 45.5% of the HCC tumors harbor genomic alterations in the JAK/STAT pathway, including frequent amplification and mutation of the cytokines PRL, IL7, and IL20, and the cytokine receptors IL6ST, IL6R, and IL21R (Fig. 3B), although it remains to be determined if these genetic alterations would lead to JAK/STAT pathway activation. It has been demonstrated experimentally that GNAS mutations activate STAT3 to induce inflammatory responses in a rare subtype of inflammatory liver cancer, suggesting cross-talk of the cAMP and JAK/STAT pathways (Nault et al. 2011). We noted that adenylate cyclase type 2 (ADCY2), an important member of the cAMP synthesis pathway activated by GNAS, is mutated in 5.7% of this HCC cohort. Furthermore, GNAS and four other members of the adenylate cyclase family (ADCY3, -8, -9, and -10) are also mutated in one case each. Taken together, our findings suggest that the JAK/STAT pathway, potentially acting in concert with the G-protein/cAMP pathway, may induce inflammatory responses that promote carcinogenesis in a substantial subset of HCC.
HCC genomes suffer extensive damage in the form of large-scale copy number alterations and viral integrations, which if left unchecked, would be expected to trigger TP53-mediated apoptosis and cell cycle arrests. Frequent mutations and deletions of TP53 appear to have disabled this important line of cellular defense in an aggregate of 43.2% of HCC tumors, which are more chromosomal instable and have poor survival. We found that up to 42.0% of HCC tumors harbor genomic alteration in the genes operating the G1/S check point of the cell cycle (Fig. 3C). RB1, a key inhibitor of cell cycle progression, harbors nonsense mutations in three cases (3.4%) and genomic deletion in 11 cases (12.5%). CDKN2A and CDKN2B, which negatively regulate CDK activities, are deleted in 10.2% and 12.5% of the cases, respectively. On the other hand, promoters of cell cycle progression are activated by DNA amplification (CCND1, 4.5%) and HBV integration (CCNE1, 4.5%), resulting in markedly higher gene expression. The apoptosis pathway, an executor of cell death, could be deactivated by DNA copy number changes in as much as 45.5% of HCC tumors (Fig. 3D). Death receptors such as TNFRSF10A/B are deleted in 22.7% of cases, and downstream signaling is further disrupted by genomic deletions of TRADD, CASP3, CASP9, DFFA, and DFFB. In contrast, XIAP, an inhibitor of caspases and apoptosis is amplified in 8.0% of tumors. Hence, the functions of TP53-related pathways such as the G1/S check point and apoptosis also appear to be genetically altered through selective activation and deactivation of key regulators.
HCC molecular classification
Three HCC subclasses (S1, S2, and S3) have been identified based on a meta-analysis of nine gene expression profiling studies (Hoshida et al. 2009). While gene expression signatures and some clinical phenotypes for each subclass have been described, the underlying genetic alterations remain largely unknown. We also clustered 88 HCC tumors in our study into three subclasses based on gene expression array data and applying supervised methods using previously defined gene signatures (Fig. 4A; Hoshida et al. 2009). To delineate the genetic basis of the HCC subclasses, SNV, CNV, and HBV integration data for significantly altered genes and pathways were mapped to three subclasses, revealing distinct genetic profiles for each subclass (Fig. 4B,D; Supplemental Fig. 8, Supplemental Tables 12, 13). S1 and S2 express high level of genes involved in cell cycle control and cell proliferation. Most S1 and S2 tumors are poorly or moderately differentiated with a high rate of recurrence. A subset of S1 and S2 tumors harbors HBV integration into the KMT2B gene locus. S1 tumors also express a high level of genes in immune response and angiogenesis. S2 has the highest frequency of TP53 mutation and the highest serum AFP level. S3 tumors are well or moderately differentiated with a gene expression profile reflecting normal liver function. In addition, S3 has relatively high frequency of CTNNB1 and JAK1 mutations and has HBV integration into the TERT gene locus. Integrative analysis of gene expression profiles, genetic alterations, and clinical characteristics appears to at least partially explain the observed difference in progression-free survival of these subclasses (Fig. 4C).
Figure 4.
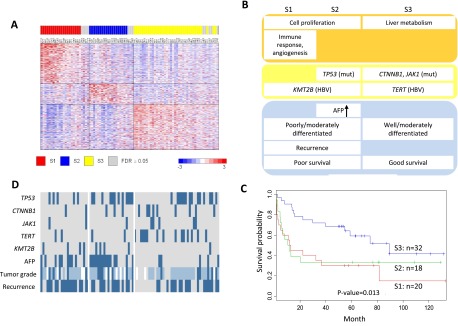
Molecular subclassification of HCC. (A) Gene expression profile of previously defined signatures (Hoshida et al. 2009) in three HCC subclasses. High and low expressions are represented by red and blue color, respectively. Seventy-six of the 88 tumors (86%) are assigned to subclasses with high confidence (FDR < 0.05). (B) Genetic and clinical profiles of the three HCC subclasses. Tumors with somatic SNVs in CTNNB1, TP53, and JAK1, HBV integrations into TERT and MLL4 (also known as KMT2B), AFP level >400 ng/mL, or tumor recurrence are represented by dark blue color. For tumor grade, poor, moderate, and well differentiation are represented by dark blue, light blue, and gray color, respectively. Tumors with missing tumor grade information are shown in white. (C) Kaplan-Meier survival plot for the three HCC subclasses. (D) Schematic summary of gene expression, genetic, and clinical profiles for each HCC subclass.
Potentially actionable genetic alterations
Our findings of several mutations and gene amplifications in HCC have clinical implications (Table 2). Inhibitors of these enzymes or key components of the pathways are already in clinical development for non-HCC indications. The prevalence of these genetic alternations in HCC discovered from our study warrants testing some of these existing inhibitors in HCC preclinical models and patients.
Table 2.
Potentially actionable mutations and matched clinical stage inhibitors
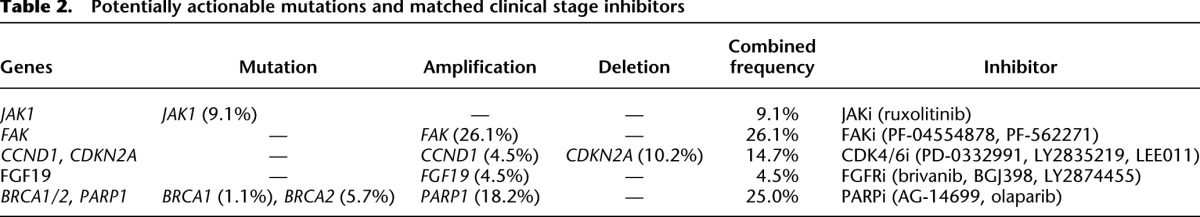
Recurrent JAK1 mutations in HCC have not been reported previously. Nine percent of HCC tumors in this cohort have JAK1 mutations. The majority of mutations reside in the pseudo-kinase and kinase domains and are functionally activating, and mutant-induced cell growth can be suppressed by the JAK1/2 dual inhibitor ruxolitinib. Our data suggest ruxolitinib, recently approved by the FDA to treat myelofibrosis (Mesa et al. 2012), should be considered for testing in HCC patients with activating JAK1 mutations.
Preclinical studies have shown that cell lines with elevated cyclin D1 and decreased CDKN2A (also known as p16) expression are most sensitive to cyclin D kinase 4/6 inhibitors (Finn et al. 2009). PD-0332991, a selective CDK4/6 inhibitor, is being tested in advanced breast cancer with CCND1 amplification and/or loss of CDKN2A (p16). We observed 4.5% HCC tumors with CCND1 focal amplification and 10.2% with CDKN2A deletion. Our data suggest this subset of HCC tumors could be candidates of CDK4/6 inhibitor trials.
Two focal adhesion kinase (FAK, also known as PTK2) inhibitors are in phase 1 clinical trials and have demonstrated modest clinical benefits in a small unselected population. FAK amplification has been associated with poor survival in breast and gastric cancers (Park et al. 2011; Yom et al. 2011). With 26.1% of HCC tumors harboring FAK amplifications, our data suggest testing FAK inhibitors in the FAK amplified HCC models.
PARP inhibitors such as olaparib have been showing efficacy in BRCA-deficient ovarian and breast cancer trials (Audeh et al. 2010; Tutt et al. 2010). We find the DNA repair pathway extensively altered in HCC with PARP1 amplified in 18.2% of cases and mutations in BRCA1 (1.1%) and BRCA2 (5.7%). Though these alterations remain to be functionally tested, our findings provide empirical evidence for the inclusion of HCC in the PARP inhibitor trials.
The fibroblast growth factor ligand (FGF19) is amplified in 4.5% of HCC tumors. A neutralizing anti-FGF19 antibody blocks clonogenicity and tumorigenicity of HCC models harboring FGF19 amplification (Sawey et al. 2011). As a proof-of-concept study, we tested the FGFR small-molecule inhibitor PD173074 in a panel of 14 HCC cell lines for inhibition of cell proliferation. Indeed those HCC cell lines harboring FGF4/FGF19 amplification and overexpressing FGFR3/FGFR4 display increased sensitivity to PD173074 (Supplemental Table 14). Based on our data and the emerging preclinical data, we propose to test FGFR inhibitors including brivanib, a dual VEGFR/FGFR inhibitor, which is being evaluated in phase 3 HCC trials, in HCC patients with FGF amplifications and FGFR overexpressions.
Discussion
The results of this study highlight important differences between HCC and other solid tumors such as lung, breast, prostate, and colon cancers. While EGFR, PI3K, and MAPK pathway alterations are common in other cancer types, Wnt/beta-catenin and JAK/STAT are the two major oncogenic pathways in HCC. These findings may explain why some targeted therapies that are effective in other tumor types have not demonstrated significant improvement on HCC patient survival. Cataloging major mutations and pathways by cancer genome sequencing can lead to a better understanding of tumor biology and the discovery and prioritization of drug targets. The time line of drug development may be shortened when compounds already in late-stage preclinical or clinical development for other indications are available for testing in HCC (Chin et al. 2011). Activating JAK1 mutations discovered from this study fit into the class of so-called actionable mutations, as do amplifications of FAK, CCND1, FGF19, and PARP, and inactivating mutations in BRCA1 and 2. Most importantly, this study demonstrates that morbicentric genome sequencing is not only a powerful tool to study disease biology and identify new drug targets, but also provides opportunities for promptly translating scientific discoveries into improvements in patient care.
Methods
Samples and genomic analyses
Sample collection, gene expression array analysis, SNP array analysis, and whole-genome sequencing have been described in previous publications (Lamb et al. 2011; Sung et al. 2012). Microarray data have been deposited in the GEO database under accession numbers GSE28127 (for SNP arrays) and GSE25097 (for expression arrays). Sequence data have been deposited in the European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena/) under accession number ERP001196 and in GigaDB (http://dx.doi.org/10.5524/100034).
Somatic mutation detection
Sequencing reads were aligned to the hg19 reference genome using SOAP2 (Li et al. 2009) followed by removal of PCR duplicates, low-quality (Q < 20), and nonuniquely mapped reads. Mutations in tumor were first predicted by SOAPsnv (http://soap.genomics.org.cn/SOAPsnv.html) using a sensitive score threshold, and a P-value was calculated using Fisher's exact test for all putative mutation sites based on the distribution of read support for different alleles in tumor and matched normal samples (Supplemental Fig. 9). A somatic mutation is called if the following criteria were met: (1) read depth ≥10 in both tumor and normal samples; (2) read support of mutant allele in tumor tissue not a result of sequencing error (binomial test, f = 0.1, P > 0.01); (3) quality score not significantly lower than other alleles (Wilcoxon rank sum test, P > 0.01); (4) mutant allele frequency change between tumor and adjacent normal ≥20% and Fisher's exact test P < 0.01; (5) mutant allele not significantly enriched in repeatedly aligned reads; and (6) mutant allele not significantly enriched within 10 bp of 5′ or 3′ ends of reads (Fisher's exact test, P > 0.01).
Indels in tumor samples were predicted by the split reads (SRiC) method (Zhang et al. 2011). Gapped alignment was performed using BWA aligner (Li and Durbin 2009), allowing only one gap located >5 bp to either read ends for each mapped read. To identify somatic indels, all reads from normal samples were aligned to sequence templates representing reference and variation genotypes for each detected tumor indel. Tumor indels with six or more matched normal reads as well as those matching dbSNP indel records were filtered as germline indels.
Somatic SNV and indel validation
Genotyping assays were performed via iPLEX MassARRAY system (Sequenom). Both PCR and MassEXTEND primers for each mutation were in silico designed by MassARRAY Assay design 4.0 software. Multiplex PCR were carried out by GeneAmp PCR System 9700 Dual 384-Well Sample Block Module (Applied Biosystems), followed by dephosphorylation, single base extention reaction, and desalting. MassARRAY Nanodispenser RS1000 was used to automatically spot reactions to 384 SpectroCHIP, which was further placed into the MALDI-TOF mass spectrometer. All genotype calls by MassARRAY Typer 4.0 were manually confirmed by examining the spectra for each assay and sample.
Significantly mutated genes analysis
We used the method described previously by Youn and Simon (2011) to compute the significance of observed mutations on each gene. The statistical model takes both mutation prevalence and functional impact into consideration. Functional impact was evaluated as mutation score assigned in the following order: missense < inframe indel < mutation in splice sites < frameshift indel = nonsense. Different types of missense mutations were also assigned different scores based on BLOSUM80 matrix. This method assumes that passenger mutations, including silent and nonsilent mutations, were generated from the same background mutation process. Incorporating different background mutation rates of each sample, background distribution of mutation score for each gene was computed. The P-value for each gene was calculated from this background distribution and the test statistics from the observed mutation scores across samples. The P-value was adjusted using the Benjamini-Hochberg method to estimate the false-discovery rate (FDR). Significantly mutated genes were selected if FDR ≤10%.
Validation of JAK1 mutations
We designed 19 pairs of primers that uniquely amplify 24 exons of JAK1 gene using Primer Premier 5.00 (Premier Biosoft) and Oligo 6.71 (Molecular Biology Insights). PCR was performed on Dual 96-well GeneAmp PCR System 9700 (Applied Biosystems). The 50-μL reaction contains 1× PCR buffer, 0.2 mM dNTP mixture, 200 nM forward and reverse primers, 2.5 units Taq (Takara Bio), and 20 ng template DNA. Cycling conditions were 1 min at 94°C for initial denaturation of the DNA and polymerase activation, followed by 35 cycles of 30 sec denaturation at 95°C, 30 sec annealing at 62°C∼67°C, and 30 sec ∼90 sec extension at 72°C. The products were sequenced by 3730xl DNA Analyzer (Applied Biosystems). All sequences were analyzed by the Sequencing Analysis Software version 5.2 (Applied Biosystems).
Experimental characterization of JAK1 mutations
Flag-tagged human JAK1 ORFs for wild-type and mutant variants (N242S, Q644H/V645F, S703I, S729C, G902E, and L910P) were cloned into pcDNA3.1(+) (Invitrogen). Anti-M2 Flag HRP and -beta actin (Sigma), anti-pJAK1 (pY1034/1035), -pSTAT3 (pY705), and -STAT3 (Cell Signaling) were used for Western blotting. All cell culture reagents were from Hyclone.
All cells were cultured at 37°C in 5% CO2 humidified air. HEK293FT cells were maintained in DMEM (high glucose) supplemented with 10% fetal bovine serum, 4 mM L-glutamine, 1 mM sodium pyruvate. Hep3B cells were maintained in MEM/EBSS supplemented with 10% fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate. For Western blotting experiments, HEK293FT and Hep3B cell cultures were seeded the day before at 800,000 and 400,000 cells/well, respectively, in a six-well dish and transfected with a total of 1.5 μg plasmid DNA using Fugene 6 (Roche) or FuGENE HD (Promega) at a ratio of 3:1 (Fugene:DNA).
HEK293FT and Hep3B cells were serum starved 48 h post-transfection in 0% serum for 4 h. Cells were lysed on ice in cold RIPA buffer (150 mM NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris at pH 8.0; Sigma) plus protease and phosphatase inhibitors (HALT protease and phosphatase inhibitor cocktail; Pierce). Cell lysates were sonicated in an ice water bath for 5 min and then centrifuged at 16,500g for 20 min at 4°C. A portion of the supernatant was removed, mixed 1:1 with 6× Laemmli sample buffer, and boiled for 5 min. Samples were subjected to SDS-PAGE and transferred to nitrocellulose using the iBlot dry blotting system (Invitrogen). Membranes were blocked in 5% nonfat milk/TBS-T for 1 h at room temperature. Western blotting was performed using the aforementioned primary antibodies in 3% BSA/TBS-T, secondary anti-mouse or -rabbit IgG antibody-HRP conjugates in 1% BSA/TBS-T (GE Healthcare), and enhanced chemiluminescence (SuperSignal West Pico or SuperSignal West Femto Pierce).
Human JAK1 ORFs for wild-type and mutant variants (N242S, S703I, S729C, G902E, and L910P) were constructed in the bicistronic retrviral vector pMX-IRES-GFP (Cell Biolabs). Retroviral supernatants were used to infect Ba/F3 cultured in RPMI 1640 medium containing 10% FBS and 10% WEHI-3B cell condition media. GFP-positive cells were isolated by flow-cytometric sorting and subsequently expanded. Equal GFP expression levels of transduced cells were confirmed by FACS analysis. For assaying cytokine independence, transduced Ba/F3 cells were cultured in the absence of IL3. Cell viability was determined by Trypan blue exclusion.
For compound treatment, mutant JAK1 transduced Ba/F3 cells were plated on 96-well plates (2000 cells/well) in RPMI 1640 medium containing 2% FBS. They were treated with compounds at indicated concentrations for 3 d. Cell viability was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the manufacturer's instructions.
For Western blot analysis, JAK1 (S703I)-transduced Ba/F3 cells were treated with 1 μM compounds for 2 h in serum-free media. Approximately 30 μg of lysate were resolved by electrophoresis through a polyacrylamide gel (BioRad Laboratories). Following transfer to nitrocellulose membrane, the membrane was subjected to immunoblot analysis using pSTAT3 (pY705) and STAT3 antibodies (Cell Signaling), and mouse α-actin antibody (Sigma-Aldrich). Secondary antibodies were purchased from Licor Biosciences. The images were analyzed using Odyssey Imager (Licor Biosciences).
JAK1 pseudo-kinase and tyrosine kinase domain interaction modeling
We modeled the interaction between the pseudo-kinase and tyrosine kinase domains using a crystal structure of the tyrosine kinase domain (3EYG) (Williams et al. 2009) and a homology model of the pseudo-kinase domain oriented to have an interaction between the first N-terminal α-helices in each domain, patterned after previous models (Flex et al. 2008) using Discovery Studio (accelrys.com).
JAK1 gene signature analysis
The JAK1 mutation gene signature was derived from acute lymphoblastic leukemia patients (Flex et al. 2008). The 112 differentially expressed genes were mapped to expression profiles of our cohort. Using principal component analysis (PCA), we then computed the first principal component value for each sample as the JAK1 activation score.
CNV analysis
We used SegSeq (Chiang et al. 2009) to infer somatic copy number variation (CNV) in HCC genomes using WGS reads. The resulting copy number segments were mapped to individual genes to determine gene-level copy numbers and copy gain/loss statuses using thresholds of ≥3 copies for gain and ≤1.25 for loss. To infer recurrently amplified or deleted genomic regions, we reimplemented the GISTIC algorithm (Beroukhim et al. 2007) using copy numbers in 1-kb windows instead of SNP array probes as markers. G-scores were calculated for genomic and gene-coding regions based on the frequency and amplitude of amplification and deletion affecting the gene respectively. A significant CNV region is defined as having amplification G-score >0.08 or deletion G-score <0.09, corresponding to a P-value threshold of 0.05 from permutation-derived null distribution (Beroukhim et al. 2007). For each gene, we calculated the Pearson correlation coefficient between gene-level copy numbers and mRNA gene expression levels previously published (Lamb et al. 2011). A gene was defined to be “cis-correlated” if its Pearson correlation coefficient is ≥0.3 and its gene expression fold-change in CNV versus non-CNV tumor samples is consistent with the direction of CNV (≥1.25 for amplified and ≤0.8 for deleted genes).
Canonical pathway analysis
Thirty-three canonical cancer pathways were selected from MSigDB (Liberzon et al. 2011). For each gene and tumor sample, also called an event, a binary status (1, altered; 0, no change) is determined for somatic mutation, cis-correlated CNV, and HBV integration. The integrated alteration status is simply a sum of three alteration statuses. Pathway alteration prevalence is represented at the sample-level as the proportion of tumors harboring alteration in any pathway gene. In addition, prevalence is calculated at the event level as the percentage of events altered and gene level as the percentage of pathway genes harboring one or more alterations.
Pathway enrichment analysis
Pathway enrichment analyses of genes harboring somatic SNV or CNV were performed using the KEGG or Gene Ontology canonical pathways. P-values were calculated based on hypergeometric distribution with FDR correction using the Benjamini and Hochberg method (Hochberg and Benjamini 1990). GO term enrichment analysis was performed using only the FAT terms and DAVID (Dennis et al. 2003).
HCC molecular classification
A previously defined 619-gene signature for three HCC subclasses and binary weights for each gene were used to define cluster centroid. Each of the 88 HCC samples in the current study was assigned to the closest centroid based on expression of the 619-gene signature. A permutation test was used to assess the statistical significance, and an FDR of 0.05 was used to define high-confidence class assignment. To identify genes overexpressed in each subclass, t-tests were carried out to compare gene expressions in each subclass with those in the rest of HCC samples. A P-value <0.05 and fold change >1.5 were used to define statistical significance. Significantly overexpressed genes in each subclass were subsequently mapped to gene ontology, and significantly overrepresented gene ontology terms were identified based on hypergeomatric distribution with a false discovery rate <0.1.
Data access
Annotated somatic variants and interactive variant analysis results are available online at www.ingenuity.com/acrg2012.
Competing interest statement
Z.K., J.X., R.H., P.A.R., J.F., K.W., H.E., S.D., P.W., M.Q., and M.M. are employees of Pfizer Inc. S.L., T.D.B., M.D.W., J.W., I.H.W., J.L., M.E.E., A.A., and C.R. are employees of Eli Lilly and Company. K.H., R.C., W.Z., C.Z., A.L., J.H., and H.D. are employees of Merck and Company.
Acknowledgments
This work was funded by Asian Cancer Research Group, Inc. (ACRG), a not-for-profit organization formed by Eli Lilly, Merck, and Pfizer. We thank Stephen Friend and Gang Jin for initiating the establishment of ACRG. We are grateful to all the past and present members of ACRG, especially Kerry Blanchard, Yaron Turpaz, Jonathon Sedgwick, Greg Tucker-Kellogg, Xiang S. Ye, Gary Gilliland, Carolyn Buser, Peter Shaw, Neil Gibson, and Steven Adams. We also thank Ingenuity Systems for providing the Ingenuity Variant Analysis system to enable readers to analyze annotated somatic variants interactively online.
Author contributions: The study was initiated and designed by M.M., H.D., C.R., and J.L. WGS sequencing and SNV validation was done by X.L., G.W., Q.Z., and B.W. Data analysis was conducted by H.Z., Z.K., S.L., R.C., K.H., Y.L., and H.G. M.D.W., P.W., M.Q., I.H.W., and T.D.B. did the JAK1 functional studies. P.A.R. analyzed the JAK1 protein structure. Z.G. was the project manager. N.P.L., R.T.P., and S.T.F. coordinated the collection of specimens and clinical data. M.M., Z.K., and S.L. wrote the manuscript. Z.K., S.L., H.Z., P.A.R., T.D.B., M.D.W., P.W., M.M., and N.P.L. produced tables and figures. All the authors contributed to the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.154492.113.
Freely available online through the Genome Research Open Access option.
References
- Aguilar F, Harris CC, Sun T, Hollstein M, Cerutti P 1994. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 264: 1317–1319 [DOI] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 376: 245–251 [DOI] [PubMed] [Google Scholar]
- Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A, et al. 2011. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet 43: 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, et al. 2007. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci 104: 20007–20012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. 2011. Initial genome sequencing and analysis of multiple myeloma. Nature 471: 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES 2009. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 6: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Andersen JN, Futreal PA 2011. Cancer genomics: From discovery science to personalized medicine. Nat Med 17: 297–303 [DOI] [PubMed] [Google Scholar]
- Chow J, Poon RY 2010. DNA damage and polyploidization. Adv Exp Med Biol 676: 57–71 [DOI] [PubMed] [Google Scholar]
- de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, et al. 2011. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476: 293–297 [DOI] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3. [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. 2008. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima S, Lee NP, Luk JM 2011. Dickkopfs and Wnt/β-catenin signalling in liver cancer. World J Clin Oncol 2: 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. 2009. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, Ariola C, Fodale V, Clappier E, Paoloni F, et al. 2008. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 205: 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouas D, Shi H, Hainaut P 2009. The aflatoxin-induced TP53 mutation at codon 249 (R249S): Biomarker of exposure, early detection and target for therapy. Cancer Lett 286: 29–37 [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. 2007. Patterns of somatic mutation in human cancer genomes. Nature 446: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Karin M 2011. NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res 21: 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg Y, Benjamini Y 1990. More powerful procedures for multiple significance testing. Stat Med 9: 811–818 [DOI] [PubMed] [Google Scholar]
- Hornakova T, Staerk J, Royer Y, Flex E, Tartaglia M, Constantinescu SN, Knoops L, Renauld JC 2009. Acute lymphoblastic leukemia-associated JAK1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor α homodimers. J Biol Chem 284: 6773–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornakova T, Springuel L, Devreux J, Dusa A, Constantinescu SN, Knoops L, Renauld JC 2011. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica 96: 845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. 2009. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69: 7385–7392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D 2011. Global cancer statistics. CA Cancer J Clin 61: 69–90 [DOI] [PubMed] [Google Scholar]
- Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D, et al. 2011. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat 33: 100–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y, Kypta R 2003. Secreted antagonists of the Wnt signalling pathway. J Cell Sci 116: 2627–2634 [DOI] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W 2008. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 8: 387–398 [DOI] [PubMed] [Google Scholar]
- Lamb JR, Zhang C, Xie T, Wang K, Zhang B, Hao K, Chudin E, Fraser HB, Millstein J, Ferguson M, et al. 2011. Predictive genes in adjacent normal tissue are preferentially altered by sCNV during tumorigenesis in liver cancer and may rate limiting. PLoS ONE 6: e20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW 2009. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci 106: 8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mao M 2012. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett doi: 10.1016/j.canlet.2012.09.027 [DOI] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J 2009. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967 [DOI] [PubMed] [Google Scholar]
- Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, et al. 2011. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet 43: 828–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27: 1739–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa RA, Yasothan U, Kirkpatrick P 2012. Ruxolitinib. Nat Rev Drug Discov 11: 103–104 [DOI] [PubMed] [Google Scholar]
- Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, Shetty K, Johnson L, Reddy EP 2009. Liver stem cells and hepatocellular carcinoma. Hepatology 49: 318–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y 1998. Activation of the β-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res 58: 2524–2527 [PubMed] [Google Scholar]
- Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, et al. 2011. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol 58: 184–191 [DOI] [PubMed] [Google Scholar]
- Neuveut C, Wei Y, Buendia MA 2010. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol 52: 594–604 [DOI] [PubMed] [Google Scholar]
- Park JH, Lee BL, Yoon J, Kim J, Kim MA, Yang HK, Kim WH 2011. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum Pathol 41: 1664–1673 [DOI] [PubMed] [Google Scholar]
- Parkin DM 2006. The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118: 3030–3044 [DOI] [PubMed] [Google Scholar]
- Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JT, Paradis V, Nault JC, Izard T, Bioulac-Sage P, Couchy G, et al. 2011. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med 208: 1359–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purandare AVMT, Wan H, You D, Penhallow B, Han X, Vuppugalla R, Zhang Y, Ruepp SU, Trainor GL, Lombardo L, et al. 2012. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia 26: 280–288 [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama AVK, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, et al. 2010. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115: 3109–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S 2011. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov 10: 127–140 [DOI] [PubMed] [Google Scholar]
- Rebouissou SAM, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P, Zucman-Rossi J 2009. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 457: 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U 2006. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res 83: 97–119 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P, Silvennoinen O 2002. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem 277: 47954–47963 [DOI] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. 2000. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 24: 245–250 [DOI] [PubMed] [Google Scholar]
- Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L, et al. 2011. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell 19: 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Zheng H, Li S, Chen R, Liu X, Li Y, Lee N, Lee W, Ariyaratne P, Tennakoon C, et al. 2012. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44: 765–769 [DOI] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T, et al. 2011. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet 43: 464–469 [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 376: 235–244 [DOI] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. 2011. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469: 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al. 2011. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet 43: 1219–1223 [DOI] [PubMed] [Google Scholar]
- Weihrauch M, Benick M, Lehner G, Wittekind M, Bader M, Wrbitzk R, Tannapfel A 2001. High prevalence of K-ras-2 mutations in hepatocellular carcinomas in workers exposed to vinyl chloride. Int Arch Occup Environ Health 74: 405–410 [DOI] [PubMed] [Google Scholar]
- Whittaker S, Marais R, Zhu AX 2010. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 29: 4989–5005 [DOI] [PubMed] [Google Scholar]
- Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. 2010. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363: 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NK, Bamert RS, Patel O, Wang C, Walden PM, Wilks AF, Fantino E, Rossjohn J, Lucet IS 2009. Dissecting specificity in the Janus kinases: The structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol 387: 219–232 [DOI] [PubMed] [Google Scholar]
- Xie HJ, Bae HJ, Noh JH, Eun JW, Kim JK, Jung KH, Ryu JC, Ahn YM, Kim SY, Lee SH, et al. 2009. Mutational analysis of JAK1 gene in human hepatocellular carcinoma. Neoplasma 56: 136–140 [DOI] [PubMed] [Google Scholar]
- Yau TO, Chan CY, Chan KL, Lee MF, Wong CM, Fan ST, Ng IO 2005. HDPR1, a novel inhibitor of the WNT/beta-catenin signaling, is frequently downregulated in hepatocellular carcinoma: Involvement of methylation-mediated gene silencing. Oncogene 24: 1607–1614 [DOI] [PubMed] [Google Scholar]
- Yom CK, Noh DY, Kim WH, Kim HS 2011. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res Treat 128: 647–655 [DOI] [PubMed] [Google Scholar]
- Youn A, Simon R 2011. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics 27: 175–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM 2010. Cancer gene discovery in hepatocellular carcinoma. J Hepatol 52: 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZD, Du J, Lam H, Abyzov A, Urban AE, Snyder M, Gerstein M 2011. Identification of genomic indels and structural variations using split reads. BMC Genomics 12: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]