

Abstract
The B-cell lymphoma-2 (Bcl-2) family of proteins regulates the intrinsic, or mitochondrial pathway of apoptosis, the final common mechanism of cell death in response to a variety of physiologic and pharmacologic signals, and plays a central role in AML pathogenesis, prognosis and responsiveness to chemotherapy. Traditionally thought to be an important survival factor for multiple myeloma cells, the anti-apoptotic Bcl-2 family protein myeloid cell leukemia-1 (Mcl-1) has recently been shown in preclinical studies to be critical to the development and maintenance of AML, making it an attractive therapeutic target in this disease. Several characteristics, such as its very short half-life, distinguish Mcl-1 from other anti-apoptotic Bcl-2 family members. Additionally, Mcl-1 levels are regulated by a large number of pathways affecting its transcription, translation and degradation. A variety of approaches exploiting these features have been developed to inhibit directly or indirectly the anti-apoptotic function of Mcl-1. Many of these lend themselves well to combination therapies, leading to striking synergism, at least in preclinical models. In this brief review, we highlight some of the more promising strategies targeting Mcl-1 in AML, with a particular emphasis on rational combinations of novel agents.
Keywords: AML, Mcl-1, Obatoclax, Sorafenib, CDK inhibitors
1. Introduction
Multiple signal transduction pathways in normal and malignant cells converge on Bcl-2 family proteins, which stand at the crossroads between cell survival and apoptosis. The seminal event in the intrinsic pathway of apoptosis is mitochondrial outer membrane permeabilization (MOMP), induced by the “effector” Bcl-2 family proteins, Bax and Bak. The anti-apoptotic proteins are Bcl-2, Bcl-XL, Bcl-w, Mcl-1, Bfl-1/A1 and Bcl-B. The “multi-domain” pro-apoptotic proteins are Bax, Bak and Bok, which share homology in domains BH (Bcl-2 homology) 1–3. The other pro-apoptotic Bcl-2 family proteins share only the BH3 domain and are termed “BH3-only” proteins; these include Bim, Bid, Bad, Bik, Hrk, Bmf, Puma and Noxa. The BH3 domain enables binding to a hydrophobic groove on the surface of the anti-apoptotic Bcl-2 molecules. Under normal conditions, pro-apoptotic BH3-only proteins prevent the anti-apoptotic Bcl-2 proteins from engaging with the apoptosis effectors Bax and Bak, allowing the latter to trigger apoptosis upon receipt of a death signal. Alternatively, “direct activator” BH3-only proteins may be held in check by their anti-apoptotic counterparts. Fig. 1 illustrates some of these concepts.
Fig. 1.
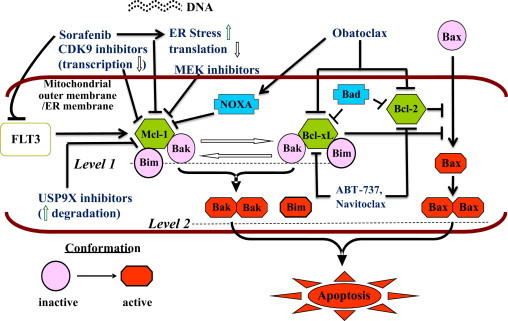
Schematic representation of interactions between pro- and anti-apoptotic members of the Bcl-2 family and various pharmacologic mechanisms of triggering apoptosis via the intrinsic, or mitochondrial pathway. Upon activation, the apoptosis effectors Bax and Bak induce permeabilization of the mitochondrial outer membrane, resulting in a cascade of events that trigger cell death. Under normal conditions, the anti-apoptotic proteins Bcl-2, Bcl-XL and Mcl-1 sequester apoptosis effectors (e.g., Bak) as well as the pro-apoptotic “BH3-only” protein Bim, thereby preventing apoptosis. The BH3-only proteins Bad and Noxa, as well as “BH3-mimetic” drugs such as navitoclax and obatoclax untether the apoptosis effectors and Bim from the anti-apoptotic proteins, leading to apoptosis. In addition, a variety of pharmacologic strategies may be used to down-regulate Mcl-1. These include inhibition of translation by sorafenib, transcriptional repression by CDK9 inhibitors such as flavopiridol (alvocidib) or roscovitine (seliciclib), MEK inhibitors, and inhibitors of deubiquitinase USP9X, which enhance its degradation. These agents have the potential to potentiate the effects of BH3-mimetics, and vice versa. As sorafenib potently inhibits FLT3, which up-regulates Mcl-1 and Bcl-XL and phosphorylates Bad (leading to its degradation) via several different downstream pathways (not shown), combinations involving this agent may be particularly effective in FLT3-mutated AML, as seen in preclinical studies of sorafenib and obatoclax.
Modified from Dai Y, Grant S. Cancer Research 2007;67:2908–11 (Fig. 1). ©2007. American Association for Cancer Research.
2. Mcl-1
Mcl-1 is distinguished from other anti-apoptotic Bcl-2 proteins in that it has a very short half-life (only 2–4 h in most cells), making it dependent upon active transcription and translation for its maintenance. Mcl-1 sequesters, in conjunction with Bcl-XL, the apoptotic effector Bak on the mitochondrial surface, protecting against Bak oligomerization and the induction of MOMP. Mcl-1 also interacts with and sequesters the pro-apoptotic proteins Bim and truncated Bid. Noxa antagonizes Mcl-1 and promotes its proteasomal degradation, freeing Bak and Bim (Fig. 1). Because of structural differences, Mcl-1 is not bound by ABT-737, a potent small-molecule “BH3-mimetic” antagonist of Bcl-2, Bcl-XL and Bcl-w, which thus mimics the actions of Bad (Fig. 1). Indeed, up-regulation of Mcl-1 represents an important mechanism underlying leukemia cell resistance to ABT-737.1
While up-regulation of Mcl-1 has been described at the time of AML relapse, in fms-like tyrosine kinase-internal tandem duplication (FLT3-ITD) AML stem cells, and in the regulation of stem cell self-renewal, the most compelling evidence to date in support of a critical role for Mcl-1 in the survival of human AML cells emanates from a recent study in which deletion of Mcl-1, but not loss or pharmacological blockade of Bcl-XL, Bcl-2, or Bcl-w, induced the death of transformed AML and cured disease in AML-afflicted mice.2
2.1. Simultaneously targeting multiple arms of the apoptotic regulatory machinery (Fig. 1)
As ABT-737 does not inhibit Mcl-1, combining such agents with drugs capable of down-regulating Mcl-1 is a particularly appealing therapeutic strategy in AML, analogous to Bad and Noxa, which synergistically induce apoptosis. In this context, cyclin dependent kinase (CDK) inhibitors that inhibit the cyclin T-CDK9 complex (positive transcription elongation factor b, P-TEFb), such as flavopiridol (alvocidib), roscovitine (seliciclib) or SCH727965 (dinaciclib), down-regulate Mcl-1 by acting as transcriptional repressors. Thus, roscovitine dramatically increases ABT-737 lethality in human leukemia cells.3 Coadministration of roscovitine and ABT-737 untethered Bak from Mcl-1 and Bcl-XL, respectively, triggering Bak activation and Bax translocation, with resultant induction of MOMP.
Another mechanism of down-regulating Mcl-1 involves inhibition of translation. It has been demonstrated that the multi-targeted tyrosine kinase inhibitor sorafenib mediates apoptotic cell death in human leukemia cells, at least in part, through down-regulation of Mcl-1 via inhibition of translation.4 Accordingly, sorafenib synergizes with ABT-737 to induce apoptosis in AML cell lines.5
ABT-737 may induce activation of extracellular signal regulated kinase and up-regulation of Mcl-1 in AML cells, arguing for a combination strategy involving inhibitors of mitogen activated protein kinase (MEK), which have been used successfully to suppress Mcl-1. Indeed, the MEK inhibitor PD0325901 potently synergizes with ABT-737 to promote killing of AML-derived cell lines, primary AML blasts, and CD34+38–123+ AML progenitor/stem cells, both in vitro and in vivo.6
Navitoclax (ABT-263) is an orally available analog of ABT-737. Based on the promising laboratory findings noted above, clinical trials of navitoclax in combination with sorafenib, transcriptional CDK inhibitors, or MEK inhibitors in patients with AML are under consideration.
2.2. Combining down-regulation of Mcl-1 with antagonism of Mcl-1 function
Obatoclax (GX15-070) is another small-molecule BH3-mimetic that inhibits Mcl-1 in addition to Bcl-2, Bcl-xL and Bcl-w through a Noxa-dependent mechanism. Obatoclax potently interferes with the sequestration of Bak by Mcl-1, untethering Bak and overcoming resistance to apoptosis conferred by Mcl-1.7 In leukemia cell lines and primary AML cells, obatoclax both triggers apoptosis and induces autophagy.8 Apoptosis in this setting was preceded by the release of Bak from Mcl-1, liberation of Bim from both Bcl-2 and Mcl-1, and the formation of an active Bak/Bax complex. A recent study has demonstrated marked potentiation by obatoclax of sorafenib-induced apoptosis in multiple AML cell lines and primary AML cells but not normal CD34+ cells.9 Indeed, exposure to comparable or significantly higher concentrations of sorafenib and obatoclax alone or in combination exhibited minimal lethality toward normal CD34+ cells. Up-regulation of Bcl-2 family anti-apoptotic proteins represents an important mechanism of therapeutic resistance to sorafenib, and can be abolished by the concomitant administration of obatoclax. In line with the known FLT3-inhibitory action of sorafenib, synergism occurred at much lower sorafenib concentrations in FLT3-mutated cells. In FLT3-ITD AML, up-regulation of Mcl-1 is dependent on FLT3 signaling and an essential effector of FLT3-ITD-mediated drug resistance, and down-regulation of Mcl-1 sensitizes FLT3-ITD hematopoietic cells to cytotoxic and targeted therapies.10 In light of these observations, a National Cancer Institute-sponsored phase I/Ib trial (with a limited expansion cohort in FLT3-mutated patients) of sorafenib and obatoclax in patients with relapsed/refractory and selected poor prognosis types of AML or MDS is planned by the Southeast Phase II consortium. This strategy of simultaneously down-regulating Mcl-1 with sorafenib and inhibiting its function (and that of Bcl-2 and Bcl-XL) with obatoclax to trigger apoptosis in AML cells is depicted schematically in Fig. 1.
2.3. Combinations of agents capable of down-regulating Mcl-1 with cytotoxic chemotherapy
Preclinical studies have shown that in addition to triggering apoptosis, flavopiridol may recruit surviving leukemia cells to a proliferative state, thereby priming such cells for the S-phase-related cytotoxicity of cytarabine. Pretreatment with flavopiridol enhanced the pro-apoptotic and cytotoxic effects of cytarabine on primary bone marrow blasts from patients with refractory acute leukemias.11 Based on this concept, several clinical trials of timed sequential therapy with flavopiridol, cytarabine and mitoxantrone (FLAM) have been conducted. In a phase II study in 45 newly diagnosed patients with poor-risk AML, 67% achieved complete remission (CR).12 Accordingly, a randomized phase II study comparing FLAM to cytarabine and daunorubicin in newly diagnosed patients with AML (NCT01413880) is currently ongoing.
Several groups have combined sorafenib with chemotherapy in AML. In a phase II study, 51 adults with previously untreated AML received sorafenib, cytarabine and idarubicin. While the overall CR rate was 75%, it was significantly higher (p=0.033) in FLT3-mutated patients (93%) than in FLT3-wild type (FLT3-WT) patients (66%).13 While responses to the combination of sorafenib and low dose cytarabine in unselected older patients with AML or high risk MDS were disappointing,14 encouraging activity was observed in both FLT3-mutated and FLT3-WT pediatric patients with relapsed/refractory AML receiving sorafenib in combination with clofarabine and cytarabine on a phase I trial.15 Whether Mcl-1 down-regulation by sorafenib (or flavopiridol) contributes to the anti-leukemic actions of genotoxic agents remains to be determined.
3. Conclusion
Long known to be a key mediator of resistance to treatment in multiple myeloma, Mcl-1 has recently emerged as a promising and rational therapeutic target in AML. A variety of approaches are potentially available through which Mcl-1 levels may theoretically be lowered, e.g., by interfering with its transcription or translation, or by promoting its degradation with deubiquitinase (e.g., USP9X) inhibitors. Alternatively, its function may be disrupted, e.g., by agents such as obatoclax or gossypol/AT-101. Particularly promising are combination approaches that simultaneously disrupt multiple cooperative pro-survival pathways and/or activate their pro-apoptotic counterparts. The results of translational efforts attempting to recapitulate some of these exciting preclinical findings in AML patients are eagerly awaited.
Contributions
P.B. drafted the article. S.G. revised the article critically for important intellectual content.
Acknowledgments
This work was supported by the following awards to S.G. from the National Institutes of Health: Nos. CA93738, CA100866, CA148431, CA137823, CA142509, and CA130805, and an award from the Leukemia and Lymphoma Society of America.
References
- 1.Konopleva M., Contractor R., Tsao T., Samudio I., Ruvolo P.P., Kitada S. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Glaser S.P., Lee E.F., Trounson E., Bouillet P., Wei A., Fairlie W.D. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes and Development. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S., Dai Y., Harada H., Dent P., Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Research. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 4.Rahmani M., Davis E.M., Bauer C., Dent P., Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. Journal of Biological Chemistry. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 5.Zhang W., Konopleva M., Ruvolo V.R., McQueen T., Evans R.L., Bornmann W.G. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
- 6.Konopleva M., Milella M., Ruvolo P., Watts J.C., Ricciardi M.R., Korchin B. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–787. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Nguyen M., Marcellus R.C., Roulston A., Watson M., Serfass L., Murthy Madiraju S.R. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konopleva M., Watt J., Contractor R., Tsao T., Harris D., Estrov Z. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax) Cancer Research. 2008;68:3413–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahmani M., Aust M.M., Attkisson E., Williams D.C., Jr., Ferreira-Gonzalez A., Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089–6098. doi: 10.1182/blood-2011-09-378141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasper S., Breitenbuecher F., Heidel F., Hoffarth S., Markova B., Schuler M. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer Journal. 2012;2:e60. doi: 10.1038/bcj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karp J.E., Ross D.D., Yang W., Tidwell M.L., Wei Y., Greer J. Timed Sequential Therapy of Acute Leukemia with Flavopiridol. Clinical Cancer Research. 2003;9:307–315. [PubMed] [Google Scholar]
- 12.Karp J.E., Blackford A., Smith B.D., Alino K., Seung A.H., Bolanos-Meade J. Clinical activity of sequential flavopiridol, cytosine arabinoside, and mitoxantrone for adults with newly diagnosed, poor-risk acute myelogenous leukemia. Leukemia Research. 2010;34:877–882. doi: 10.1016/j.leukres.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravandi F., Cortes J.E., Jones D., Faderl S., Garcia-Manero G., Konopleva M.Y. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. Journal of Clinical Oncology. 2010;28:1856–1862. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macdonald D.A., Assouline S.E., Brandwein J., Kamel-Reid S., Eisenhauer E.A., Couban S. Phase I/II study of sorafenib (Bay 43-9006) in combination with low dose cytarabine (LDAC) in elderly patients with AML or high-risk MDS from The NCIC Clinical Trials Group: Trial IND.186. Leukemia and Lymphoma. 2012, Nov. 15 doi: 10.3109/10428194.2012.737917. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Inaba H., Rubnitz J.E., Coustan-Smith E., Li L., Furmanski B.D., Mascara G.P. Phase I pharmacokinetic and pharmacodynamic study of the mssultikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. Journal of Clinical Oncology. 2011;29:3293–3300. doi: 10.1200/JCO.2011.34.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]