

Abstract
The NADPH oxidase (NOX), an oligomeric enzyme, plays a key role in polymorphonuclear neutrophil (PMN)-mediated host defense by producing cytotoxic superoxide anion (O2.−). Whereas in vitro and biochemical studies have examined the assembly and activation of this important host immune defense system, few studies have examined the function of NOX in human patients with primary immunodeficiencies other than chronic granulomatous disease. We studied the activation of NOX in PMN from patients with two distinct immunodeficiencies, interleukin-1 receptor associated kinase 4 (IRAK4) deficiency and nuclear factor kappa (NF-κB) essential modulator (NEMO or IKKγ) deficiency. We observed impaired O2.− generation by LPS-treated and fMLP-activated IRAK4-deficient PMN that correlated with decreased phosphorylation of p47phox and subnormal translocation of p47phox, p67phox, Rac2, and gp91phox/Nox2 to the membranes indicating that TLR4 signaling to the NOX activation pathway requires IRAK4. NEMO-deficient PMN generated significantly less O2.− in response to LPS-primed fMLP and translocated less p67phox than normal PMN although p47phox and Rac2 translocation were normal. Generally, responses of NEMO-deficient cells were intermediate between IRAK4-deficient cells and normal cells. Decreased LPS and fMLP induced phosphorylation of p38 MAPK in both IRAK4- and NEMO-deficient PMN implicates additional signal transduction pathways in regulating PMN activation by LPS and fMLP. Decreased activation of NOX may contribute to the increased risk of infection seen in patients with IRAK4- and NEMO-deficiency.
Keywords: Human, Neutrophils, Immunodeficiency Diseases, Cell Activation, Inflammation
INTRODUCTION
One of the mechanisms PMN use to kill phagocytosed microbes involves the generation of toxic oxygen metabolites derived from O2.− within the phagosome (1-3). Chronic granulomatous disease (CGD), resulting in increased rates of microbial infections, occurs in patients with mutations in NOX components that are unable to produce O2.− due to defective assembly or activation of the oligomeric holoenzyme from its cytosolic and membrane-associated components (1, 4-6). In nonstimulated PMN, NOX is dissociated and inactive: the cytosolic phox proteins (p47phox or Nox organizer, p67phox or Nox activator, and p40phox) are complexed together in a de-phosphorylated state, the membrane-integrated heterodimeric flavocytochrome b558 (Nox2 and p22phox subunits) mainly resides in the membranes of specific granules and secretory vesicles and Rac is maintained in a GDP-bound cytoplasmic complex (7-9). Aside from direct activation of NOX by certain stimuli, such as the bacterial chemoattractant fMLP or PMA, an additional state of activation, called the primed state, occurs upon stimulation of cell surface receptors such as TLR4, the signaling receptor for bacterial lipopolysaccharide (10, 11). The primed, adherent phenotype allows for PMN margination from the vasculature into the tissue and chemotaxis to the site of infection upon activation. Lipopolysaccharide (LPS), a major immunostimulatory component of the outer membrane of most Gram-negative bacteria, does not cause significant activation of NOX but strongly augments O2.− generation in response to a second stimulus such as fMLP. Primed PMN display altered organization of NOX, in that intracellular secondary granules and vesicles containing Nox2 fuse with phagosomes followed by recruitment to the membrane of phosphorylated cytosolic phox subunits and GTP-Rac (12, 13). Full activation of NOX requires additional signals (such as fMLP) and results in the transfer of electrons from cytoplasmic NADPH to molecular oxygen, culminating in the production of O2.−, a precursor of several potent antimicrobial compounds important for host innate immune response to infection (14). The pathways followed by signals from TLR4 to the NADPH oxidase have yet to be delineated.
A group of primary immunodeficiencies are caused by mutations in NF-κB essential modulator (NEMO, IKKγ), the regulatory subunit of a complex (with the kinases IKKα and IKKβ) that controls the degradation of IκB, the cytoplasmic inhibitor of NFκB. Mutations in NEMO are diverse and lead to a spectrum of symptoms that may not be present in individual patients including increased frequency of infections, decreased antibodies to carbohydrate antigens and ectodermal dysplasia (15). The known mutations occurring in the leucine zipper (LZ) and zinc finger (ZF) domains of the NEMO protein selectively impair CD40-triggered and NF-κB /c-Rel–mediated induction of IL-12 production by monocytes and monocyte-derived dendritic cells (16). Specific NFκB responses may be retained in some of these patients, for example RelA activation in response to LPS (17). NEMO also shuttles between the cytoplasm and nucleus where it binds and regulates activity of the transcriptional coactivator cAMP response element binding protein (CBP) thereby competing with NFκB and inhibiting transcription of NFκB responsive genes (18). NEMO may also interact with and regulate IL-1E-associated kinase (IRAK)1 in some settings (19) raising the question of a possible role in regulating signaling events at the TLR.
Another primary human immunodeficiency occurs in patients with mutations in IRAK4, a key early enzyme in the TLR/IL-1R/IL-18R signaling pathway. These patients suffer from recurrent, life-threatening pyogenic bacterial diseases, typically caused by Streptococcus pneumoniae (20, 21). IRAK4-deficiency, caused by homozygous or compound heterozygous IRAK4 mutations is rare (about 28 cases reported world-wide) (22) but the severe presentation may result in significant underreporting due to early death. With early recognition and appropriate clinical management, the susceptibility to infection of IRAK4-deficient patients typically decreases as they age suggesting that adaptive immunity progressively compensates for this innate immune defect (22).
Human donors with mutations in NEMO and IRAK4 resulting in primary immunodeficiencies present opportunities to dissect the relationship between specific immunological defects and human disease processes. To better understand the relevance of signals emanating from the TLR signal transduction pathway to the assembly and activation of the PMN NOX, we investigated PMN from these patients. Biochemical evidence for additional immune dysfunctions in NEMO- and IRAK4-deficient patients may contribute to better understanding of the diverse clinical phenotypes caused by these mutations.
MATERIALS & METHODS
Human samples
Blood samples were obtained under NIH Institutional Review Board-approved protocols 01-I-0202, 89-1-0158, and 99-CC-0168.
Among the studied three NEMO-deficient patients the mutations were: a nucleotide substitution at position 944 (A→C) resulting in E315A in the LZ domain of the NEMO (23), a C1207 → T point mutation in exon 10 causing Q403X truncation of the NEMO ZF domain (16, 24), and a T1249 → C point mutation in exon 10 resulting in C417R in the NEMO ZF domain (16). These mutations in NEMO of patients cause X-linked hyper-IgM syndrome with ectodermal dysplasia (XHM-ED), a clinical condition characterized by the abnormal development of ectoderm-derived structures leading to misshaped or absent teeth, lack of eccrine sweat glands, a paucity of hair follicles, susceptibility to mycobacterial disease, and immunodeficiency.
The patient with IRAK4 deficiency expresses a compound heterozygous genotype with two defective alleles, a point mutation (C21175T) and a two-nucleotide AC deletion (15978-15979), both of which encode truncated forms of the IRAK4 protein with disrupted kinase domains (21, 22, 25-27).
Purification of PMN
Human blood was anticoagulated with acid citrate dextrose (Vacutainer ACD Solution A; BD Biosciences, San Jose, CA) and PMN were purified as described previously (28). Microscopic evaluation of isolated cells indicated that greater than 97% were PMN and viability was greater than 95% by trypan blue exclusion.
PMN Oxidative burst assay
PMN from normal, NEMO-, or IRAK4-deficient subjects (40 μl per well at 2.5 × 106/ml Hanks’ Balanced Salt Solution with calcium and magnesium, HBSS+) were placed in a white polypropylene 96-well Uniplate (Whatman Inc., Sanford, Maine) and then incubated at 37°C under 5% CO2, with 5 μl of buffer or E.coli K12 LPS (InvivoGen, San Diego, CA) to achieve a final concentration of 100 ng LPS/ml. After 30 min incubation, the cells were quickly mixed with 50 μl of Diogenes chemiluminescent enhancer (National Diagnostics, Atlanta, GA) along with 5 μl of fMLP (Sigma-Aldrich, St. Louis, MO) for a final concentration of 0.1 μM. To measure maximal receptor-independent activation of NOX, 10 ng PMA/ml (Sigma-Aldrich, St. Louis, MO) was included in separate wells for comparison. Chemiluminescence was recorded for 1 sec per well every 2.5 min for 60 min in an Anthos Zenyth 3100 luminometer (Beckman Coulter, Inc., Fullerton, CA) at 37°C with intermittent shaking. For each group, the experiments were performed at least 3 times in duplicate. Data are presented kinetically and/or as the sum CLU (sum of all the measurements of chemiluminescence units over the first 60 min) to facilitate comparison. To confirm the chemiluminescence results, a cytochrome c reduction assay (25) was performed using LPS or fMLP as the priming agent for normal, NEMO-, or IRAK4-deficient cells.
Subcellular fractionation of PMN
PMN (1 × 107 cells/0.5ml HBSS) from normal, NEMO- or IRAK4-deficient subjects were incubated with buffer alone or treated with 100 ng LPS/ml for 30 min at 37°C under 5% CO2, and then stimulated with 0.1 μM fMLP for an additional 15 min in the absence or presence of LPS (chemiluminescence peaked at 15 minutes, Figure 1). Reactions were chilled on ice then cells were pelleted at 200 × g for 10 min at 4°C. The treated cells were resuspended in ice-cold relaxation buffer (100 mM KCl, 3.5 mM MgCl2, 3 mM NaCl, 10 mM HEPES pH 7.4, Complete Protease Inhibitor Cocktail [Roche Diagnostics Corp., Indianapolis, IN]) and disrupted by nitrogen cavitation in a cell disruption bomb (#4635, Parr Instrument Co., Moline, IL) (29, 30) after equilibration to 350 psi for 20 min at 4°C and decompression into a tube containing EDTA (final concentration 1.25 mM). Microscopy indicated that approximately 70-80% of the cells were broken. The suspension was then centrifuged at 1,000 × g for 10 min at 4°C to pellet unbroken cells and intact nuclei. The resulting post-nuclear supernatant was centrifuged at 12,000 × g for 30 min at 4°C to pellet cytoplasmic granules. The post-granular supernatant was further centrifuged at 20,000 × g for 40 min at 4°C to collect the pellet enriched for plasma membrane proteins (membrane fraction, MF). 50 μl NuPAGE LDS sample buffer with reducing agent (Invitrogen Corp., Carlsbad, CA) was added to MF enriched pellet and the samples were incubated at 95°C for 5 min prior to SDS-PAGE and western blotting.
Figure 1. O2.− generation following LPS and fMLP treatment of PMN.

PMN from normal, IRAK4-, or NEMO-deficient subjects were incubated with buffer or treated with 100 ng LPS/ml for 30 min at 37°C and then stimulated with 0.1 μM fMLP. PMN superoxide generation was measured using enhanced chemiluminescence as described in Methods. A) The kinetics of normal PMN chemiluminescence in response to the indicated treatments (mean±SE, n=6). B) To facilitate comparison of PMN chemiluminescent responses between groups, the sum of chemiluminescence over the first 60 minutes of the assay is presented. The experiments were performed on cells from 6 normal donors, 3 NEMO-deficient donors, and 1 IRAK-4 deficient donor tested on three different occasions. C) PMN chemiluminescence induced by 10 ng PMA/ml for donors described above. *=p<0.05.
Translocation of NOX components to MF
Proteins (MF or cell lysates; 10 μl equivalent to 1 × 106 cells per lane) were resolved by electrophoresis using NuPAGE 4-12% Bis-Tris Gels (Invitrogen Corp., Carlsbad, CA) and transferred to nitrocellulose membranes using submerged electroblotting. After transfer, the membranes were washed at room temp. 1 × 5 min in 15 ml Tris Buffered Saline (TBS), followed by incubation for 1 hour at room temp. in 15 ml of blocking buffer (5% Blotting Grade Blocker; Bio-Rad Laboratories, Hercules, CA) in TBS with 0.1% Tween-20 (TBS-T; Sigma-Aldrich, St. Louis, MO). The membranes were then washed 3 × 5 min with 15 ml TBS-T. For detection of NOX subunits, the membranes were probed with antibodies against p47phox (610354 monoclonal; BD Biosciences, San Jose, CA), p67phox (610912 monoclonal; BD Biosciences, San Jose, CA), p40phox (sc-18253 polyclonal; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), or Nox2 (anti gp91phox goat serum; provided by Dr. Thomas L. Leto, LHD, NIAID/NIH, Bethesda, MD) at 1:1000 dilution in 10 ml of blocking buffer with gentle agitation overnight at 4°C. The membranes were then washed 3 × 5 min with 15 ml TBS-T and incubated with HRP-conjugated anti-mouse (7076; Cell Signaling Technology, Inc., Danvers, MA) or anti-goat (sc-2741, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) (1:2000) IgG secondary antibody in 10 ml of blocking buffer with gentle agitation for 60 min at room temp. Rac2 was detected with a rabbit polyclonal (sc-96, Santa Cruz Biotechnology, Inc., Santa Cruz, CA)) antibody (1:500) and HRP-conjugated anti-rabbit IgG [7074; Cell Signaling Technology, Inc., Danvers, MA] as secondary antibody (1:2000). The membranes were subsequently washed 3 × 5 min with 15 ml TBS-T. Bound antibodies were detected by enhanced chemiluminescence (SuperSignal West Dura Extended Duration Substrate; Pierce Biotechnology, Inc., Rockford, IL).
Cell fractionation studies were normalized using cell equivalents. Validation of the protein quantitation method was performed using a dose curve of cells and found to be linear over the ranges tested. Specifically, after washing blots, chemiluminescence enhancer was added and the band intensity of MF or cell lysate proteins was quantified using the Molecular Imager ChemiDoc XRS System (Bio-Rad; Hercules, CA). For each blot, the software determined optimum exposure and boxes of equal area were drawn around specific bands and the resulting luminescence measured in units of pixel intensity/mm2 using Quantity One (4.5.2) densitometry quantitation analysis software.
Membrane translocation of Nox2 was also measured using FACS. 1 × 106 PMN/ml FACS buffer (PBS with 1% fetal bovine serum (Thermo Fisher Scientific Inc., Waltham, MA)) were treated with 100 ng/ml LPS for 30 min or 0.1 μM fMLP for 15 min at 37°C, washed with ice-cold FACS buffer, and incubated with FITC labeled anti-flavocytochrome b558 mAb 7D5 (D162-4; MB International, Woburn, MA) in the dark for 30 minutes at 4°C. Cells were then washed twice with ice-cold FACS buffer, and resuspended in PBS at 2 × 106 PMN/ml for analysis on a FACSCalibur cytometer with CellQuest software (BD Biosciences, San Jose, CA). Cells stained with an isotype control (IgG1) were used for comparison. A total of 10,000 gated cells each were analyzed from two separate experiments. Flow cytometry data were analyzed by Flow JO software (Tree Star, San Carlos, CA).
Phosphorylation of p47phox in PMN
PMN (5 × 106) from normal, NEMO-, or IRAK4-deficient subjects were incubated with buffer alone or treated for 30 min at 37°C with 100 ng LPS/ml and then stimulated for an additional 15 min at 37°C with or without 0.1 μM fMLP. The treated cells were centrifuged at 1000 × g for 10 min at 4°C and the pellets were lysed in Cell Lysis Buffer (Cell Signaling Technology, Inc., Danvers, MA) containing 20 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1mM EGTA, 1 mM Na2EDTA, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1% Triton X-100, 1 μg/ml leupeptin, 1 mM β-glycerophosphate and Complete Protease Inhibitor Cocktail (Roche Diagnostics Corp., Indianapolis, IN). Following incubation for 30 min at 4°C, the lysed cells were centrifuged at 1000 × g for 10 min at 4°C. Immunoprecipitation was performed by incubating cleared supernatant with 5 μg mouse anti-p47phox antibody for 16 hrs at 4°C with constant rotation. The immune complexes were collected by incubation with 20 μl agarose-immobilized Protein A/G (sc-2003; Santa Cruz Biotechnology, Santa Cruz, CA) for 4 hrs at 4°C with constant rotation. The immunoprecipitate was washed 2 × 5 min with cell lysis buffer, solubilized in 50 μl LDS sample buffer with reducing agent and then incubated at 95°C for 5 min. The samples were sonicated (10 sec × 3) in an ice bath to shear DNA prior to electrophoresis and immunoblotting. Phosphorylation of p47phox was determined using 1:1000 dilution of rabbit polyclonal phospho-(Ser) PKC substrate antibody (2261 polyclonal; Cell Signaling Technology, Inc., Danvers, MA) that is specific to cPKC substrates including p47phox containing phospho-serine, followed by HRP-conjugated anti-rabbit IgG secondary antibody (1:2000) and developed as above. The blots were then stripped using Restore Western Blot stripping buffer (Pierce Biotechnology, Inc., Rockford, IL) and reprobed with antibody against p47phox to allow determination of the ratio of phospho p47phox to total p47phox.
Phosphorylation of p38 MAPK
PMN (5 × 106) from normal, NEMO-, or IRAK4-deficient subjects were incubated with buffer alone or treated for 30 min at 37°C with 100 ng LPS/ml and then stimulated for an additional 15 min at 37°C with or without 0.1 μM fMLP. The treated cells were centrifuged at 500 × g for 10 min at 4°C, lysed by adding 50 μl LDS sample buffer with reducing agent and then incubated at 95°C for 5 min. The phosphorylation of p38 MAPK was assessed in PMN lysates using 1:1000 phospho-p38 MAPK (Thr180/Tyr182) [9211 polyclonal; Cell Signaling Technology, Inc., Danvers, MA] antibody, respectively. The blots were stripped as above and reprobed with p38 MAP Kinase Antibody [9212 polyclonal; Cell Signaling Technology, Inc., Danvers, MA] to determine the ratio between phosphor- and total-protein kinase.
Data Analysis
The number of experiments analyzed is indicated in each figure. Data were collected using a minimum of three experiments and used to calculate means ± SE. Statistical significance between normal and patients was calculated using the Student’s t test, paired t test among the multiple groups of a subject and unpaired t test between the corresponding groups of different subjects (Prism, Graphpad software, San Diego, CA) and was considered significant at p ≤ 0.05.
RESULTS
LPS priming for O2.− generation in PMN
LPS pretreatment increases PMN responsiveness to secondary stimulation by fMLP. To determine whether PMN from NEMO- or IRAK4-deficient subjects demonstrated an altered priming response compared to those from normal donors, cells were treated for 30 min at 37°C with LPS and then stimulated with the chemo attractant fMLP and respiratory burst activity monitored by chemiluminescence as described in Methods. Normal PMN treated with LPS alone (Figure 1A and 1B) generated little O2.− and fMLP alone induced a transient short-lived burst. LPS significantly augmented the O2.− generation upon subsequent fMLP treatment compared to unprimed cells treated with fMLP alone (Figure 1A and 1B). PMN from NEMO-deficient patients also demonstrated LPS priming of fMLP-induced O2.− generation, however they produced ~50% less chemiluminescence (p<0.05) than normal PMN (Figure 1B). In contrast to the effect of LPS pretreatment on normal and, to a lesser extent on NEMO-deficient PMN, LPS failed to prime IRAK4-deficient PMN (Figure 1B, ~10% of normal chemiluminescence). These results were confirmed using the cytochrome c reduction assay to measure extracellular superoxide release (25) (data not shown). PMA, a receptor-independent trigger of superoxide production, induced similar levels of superoxide in normal, NEMO-deficient and IRAK4-deficient PMN indicating that the latent stores of NOX activity were equivalent (Figure 1C). Immunoblots probed with antibodies against NOX proteins and other components of the NADPH oxidase activation pathway demonstrated that normal, NEMO-, or IRAK4-deficient PMN express similar amounts of Nox2, p47phox, p67phox, p40phox, Rac2, TLR4 and p38 proteins (Table I).
Table I.
NOX essential and regulatory proteins expression
| Normal | NEMO | IRAK4 | |
|---|---|---|---|
| Nox2 | 3378±392 | 3571±731 | 3078±356 |
| p47phox | 5944±688 | 6140±893 | 5735±996 |
| p67phox | 5094±746 | 5474±1286 | 5011±808 |
| p40phox | 1474±92 | 1682±155 | 1363±252 |
| Rac2 | 3039±545 | 3241±410 | 2125±444 |
| TLR4 | 3857±138 | 3611±93 | 4227±266 |
| p38 | 2631±73 | 2726±56 | 2604±91 |
To confirm that protein content was similar between different patient populations, total cell lysates were prepared from 6 different normal donors, 3 different NEMO-deficient patients, and 3 PMN preparations from a single IRAK4-deficient patient (purified on 3 different days over a 24 month period). Lysates from 1 × 106 cell equivalents were subjected to western blotting and the resulting bands were measured as described in methods. To minimize day-to-day variation in western blotting, western blots were run on three different days with different combinations of samples. Numbers presented in Table 1 are mean quantitative chemiluminescence values (pixels intensity/mm2) ±SE. Statistical analysis failed to detect significant differences between any of the patient groups.
Membrane translocation of Nox2 in PMN
The integral membrane proteins Nox2 and p22phox form the catalytic center of the NOX. In resting PMN, 15% of the Nox2 is located in the plasma membrane while the remaining 85% associates with membranes of the specific granules and secretory vesicles (8). Upon activation, either by the binding of opsonized microorganisms or by binding of chemoattractants to their cognate surface receptors, the vesicles and granules fuse with the phagosomal membrane to deliver Nox2 to the site of O2.− production (8). With non-particulate stimuli, NOX assembly can occur at the plasma membrane (7).
To determine whether the observed impairment in O2.− production in the patients with NEMO- or IRAK4-deficiency was due to decreased association of Nox2 with the PMN plasma membrane, PMN from normal, NEMO- or IRAK4-deficient donors were incubated for 30 min at 37°C with or without LPS and then with or without fMLP for an additional 15 min at 37°C. A membrane fraction (MF) was generated (see Methods) and analyzed by immunoblotting for Nox2. Treatment of normal PMN with LPS alone resulted in a small increase (p<0.05) in Nox2 in the MF (Figure 2A). Treatment of normal PMN with fMLP alone did not increase Nox2 in the MF (Figure 2A). Treatment of normal PMN with LPS alone (p<0.05) or LPS followed by fMLP (p<0.01) resulted in significantly enhanced Nox2 association with MF consistent with previous reports (5, 8, 11, 22). Although NEMO-deficient cells responded normally to these stimuli, LPS did not activate IRAK4-deficient PMN and Nox2 association with MF in response to either fMLP alone or fMLP plus LPS. Interestingly, IRAK-4 deficient cells recruited significantly less Nox2 to the MF in response to PMA than normal or NEMO-deficient PMN possibly contributing to the somewhat decreased chemiluminescence response of these cells (Figure 1B).
Figure 2. Membrane recruitment of Nox2.

Normal, IRAK4-, or NEMO-deficient PMN were incubated with buffer or treated for 30 min at 37°C with 100 ng LPS/ml and then stimulated for an additional 15 min at 37°C with 0.1 μM fMLP prior to fractionation. Translocation of Nox2 (91kDa) to MF was probed by A) immunoblotting and quantitated by densitometry (means ±SE, n=6 normal donors, 3 NEMO-deficient donors, or 1 IRAK4-deficient subject tested on three different days, *=p<0.05) with the inset depicting a representative immunoblot. B) FACS analysis of plasma membrane Nox2 in normal, NEMO-, or IRAK4-deficient PMN. A representative experiment (with matched normal control) is shown for each NEMO-, and IRAK4-deficient donor. The dashed histograms show resting Nox2 levels and the solid lines show Nox2 expression after stimulation with either LPS or fMLP.
To confirm these results, an antibody against Nox2 (mAb 7D5) was used to stain PMN for FACS analysis. Compared to isotype control (data not shown), mAb 7D5 stained PMN from normal, NEMO-, or IRAK4-deficient patients to a similar level (Figure 2B). Furthermore, normal, NEMO-, or IRAK4-deficient cells treated with fMLP increased staining with Nox2 antibody. Normal or NEMO-deficient cells stimulated with LPS also significantly increased Nox2 display on the plasma membrane but IRAK4-deficient PMN did not. Both immunodetection of Nox2 in membrane fractions and FACS analysis using a Nox2 specific antibody demonstrate that Nox2 membrane translocation in response to LPS requires IRAK4 but not NEMO.
Translocation of cytosolic NOX components to MF of normal and immunodeficient PMN
Although Nox2 contains the critical redox components of NOX, it requires several cytosolic cofactors (i.e., p47phox, p67phox and Rac2) for maximum enzymatic activity (8). While the failure of IRAK4-deficient PMN to recruit Nox2 to membranes could explain the observed defect in chemiluminescence, NEMO-deficient PMN recruited Nox2 normally but still failed to produce normal levels of chemiluminescence. We therefore measured recruitment of the known cytosolic regulators of NOX activity to the membrane fraction to attempt to elucidate the molecular basis of this defect.
As shown in Figure 3, treatment of normal PMN with LPS alone resulted in a significant (p<0.05) increase in the translocation of p47phox (Figure 3A) and Rac2 (Figure 3C), but not p67phox (Figure 3B) to the MF. Subsequent stimulation with fMLP significantly augmented the translocation of p47phox, p67phox and Rac2 in normal PMN compared to fMLP alone (Figure 3). LPS treated NEMO-deficient cells failed to translocate p47phox to the membrane (Figure 3A). Under LPS-primed fMLP-stimulated conditions, NEMO-deficient PMN displayed normal translocation of p47phox (Figure 3A) and Rac2 (Figure 3C), but significantly less (p<0.05) translocation of p67phox (Figure 3B). IRAK4- (Figure 3A-C) deficient PMN did not translocate p47phox, p67phox or Rac2 to MF in response to LPS treatment and subsequent fMLP stimulation. Compared to IRAK4-deficient PMN, LPS-primed fMLP-stimulated cells of NEMO-deficient patients were more responsive (p<0.05) for the translocation of cytosolic NOX components. PMA induced translocation (Figure 3) and total cell levels (Table I) of these components were indistinguishable between the patient groups indicating that these results are due to differential activation of the pathway leading to O2.− production rather than cellular differences in the components of these pathways.
Figure 3. Translocation of p47phox, p67phox, and Rac2 to membranes.

Cells were treated as described in the legend of Figure 1. Translocation to MF of A) p47phox (47kDa), B) p67phox (67kDa) and C) Rac2 (21kDa) was determined by densitometry. Representative blots are shown in the insets and the quantitative results are expressed as means ±SE, n=6 normal donors, 3 NEMO-deficient donors, or 1 IRAK4-deficient subject tested on three different days, *=p<0.05.
Phosphorylation of p47phox
In the resting state, p47phox is folded in a conformation involving intramolecular interactions between an internal SH3 domain and auto-inhibitory domain (5, 31). Phosphorylation of p47phox relieves the inhibitory intramolecular interaction exposing the SH3 domain and allowing downstream signaling (31). Biochemical and structural studies have demonstrated that phosphorylation of p47phox is a prerequisite for translocation to the membrane and is required for its function as an adapter protein for assembly of NOX (31-34). At least eleven different serines on the C-terminal portion of p47phox are phosphorylated during activation and are reported to play a role in NOX activation (34, 35). We therefore measured phosphorylation of p47phox in normal, NEMO- and IRAK4-deficient PMN to assess its possible role in the translocation of cytosolic phox proteins to MF in these cells.
Stimulation of normal PMN with either LPS or fMLP significantly up-regulated the phosphorylation of p47phox (Figure 4). As in normal cells, LPS or fMLP stimulation of NEMO-deficient PMN caused the phosphorylation of p47phox (p<0.05; NEMO-deficient PMN resting vs LPS or fMLP stimulated) (Figure 4). Although IRAK4-deficient PMN phosphorylated p47phox normally in response to fMLP or PMA, LPS induced phosphorylation of p47phox was not significantly different compared to control (Figure 4). As fMLP itself appeared to induce near maximal receptor-dependent phosphorylation of p47phox, PMN of NEMO- and IRAK4-deficient patients all achieved a similar level of p47phox phosphorylation under LPS primed fMLP stimulated conditions. In contrast, LPS-induced translocation of p47phox to the membrane appears to require an additional NEMO and IRAK-4 dependent step.
Figure 4. Phosphorylation of p47phox.

Cells were treated as described and p47phox was immunoprecipitated from cell lysates then detected using phospho-(Ser) PKC substrate antibody. The blots were then stripped and reprobed with antibody against p47phox (47kDa). The mean ratio of densitometric intensity between the phospho p47phox and total p47phox is depicted as means ±SE, n=4 normal donors, 2 NEMO-deficient donors, or 1 IRAK4-deficient subject tested on three different days, *=p<0.05. Representative phospho and total p47phox blots are shown.
p38 MAPK Phosphorylation in PMN
LPS triggers PI3K signaling along at least two divergent pathways: PI3K-Rac-PAK and PI3K-Akt-p38 MAPK (36). Moreover, phosphorylation of p47phox in vitro by p38 MAPK is reported to modulate NOX priming and activation (37). Some groups have found that p38 MAPK plays a critical role in priming (38, 39), whereas others claim the involvement of p38 MAPK only in NOX activation (40). We measured p38 MAPK phosphorylation in an attempt to localize the defects contributing to decreased chemiluminescence in NEMO- and IRAK4-deficiency. In normal and NEMO-deficient PMN, stimulation by LPS, fMLP or PMA alone resulted in significant levels of p38 phosphorylation (Figure 5). Under LPS-primed fMLP-stimulated conditions, NEMO-deficient PMN displayed significantly less (p<0.05) phosphorylation of p38 MAPK compared with normal PMN. In IRAK4-deficient PMN, fMLP induced p38 phosphorylation was completely normal, however, there was no response to LPS alone and the response in LPS+fMLP treated cells was no greater than fMLP alone. Compared with normal and NEMO-deficient PMN, PMA-induced p38 phosphorylation was significantly lower in IRAK4-deficient PMN.
Figure 5. p38 MAPK Phosphorylation.

Phosphorylation of p38 MAPK was determined in cell lysates prepared as described in Methods using phospho-p38 MAPK (Thr180/Tyr182) antibody. The blots were then stripped and reprobed with antibody against total p38 (43kDa). The data presented are the means of the ratios of the intensities of phospho-to the total-p38 ±SE, n=6 normal donors, 3 NEMO-deficient donors, or 1 IRAK4-deficient subject tested on three different days,*=p<0.05.
DISCUSSION
In vivo, it is unlikely that PMN ever encounter individual stimuli such as purified LPS or fMLP. Rather, the PMN must integrate many signals emanating from different pathways (e.g., TLRs, NODs, Dectins, fMLP-receptors, complement receptors) to orchestrate its responses to microbes. In complex systems, such as the NOX that involve assembly and activation of large numbers of subunits, a reductionist approach is essential to attempt to dissociate the different steps of activation but must be balanced by in vivo studies. The patient populations utilized in this study present a unique opportunity to study the effect of deficiencies in LPS signaling pathways on the assembly and activation of NOX. The results presented here (summarized in Table II) indicate that in human PMN, NEMO and IRAK4 are required for normal LPS-induced priming of O2.− production.
Table II.
Summary of results
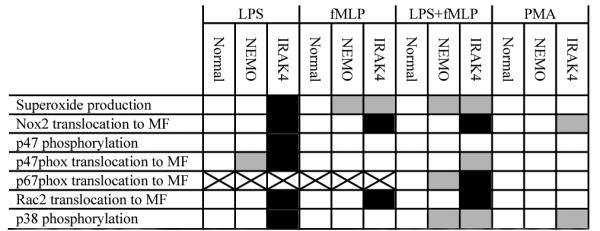
|
 = comparable response compared to increase in normal;
= comparable response compared to increase in normal;  = reduced response compared to increase in normal;
= reduced response compared to increase in normal;  = no response compared to increase in normal. No increase in p67phox translocation was observed in LPS- and FMLP-treated normal PMN
= no response compared to increase in normal. No increase in p67phox translocation was observed in LPS- and FMLP-treated normal PMN
NEMO mutations cause several overlapping phenotypes including ectodermal dysplasia, hyper-IgM, hypo-IgA, hypo-IgG and immune dysfunction predisposing to infection by some pyogenic bacteria, atypical mycobacteria, and some viruses (15). Our findings reveal an additional host defense defect in these patients that may contribute to increased susceptibility to microbial infection. Despite being able to respond normally to PMA, NEMO-deficient PMN failed to produce normal levels of O2.− in response to fMLP alone and more strikingly fMLP after pre-treatment with LPS (Figure 1). Although recruitment of Nox2 to membranes during LPS and fMLP stimulation of NEMO-deficient cells was normal, the recruitment of p67phox to membranes was markedly decreased. Interestingly, phosphorylation of p47phox was also normal in NEMO-deficient cells suggesting that additional regulatory signals, such as p67phox translocation, play a role in regulating NOX activity. The observed subnormal phosphorylation of p38 MAPK in NEMO-deficient PMN treated with LPS and fMLP implicates NEMO or a NEMO-dependent gene product in the pathway between fMLP receptor, TLR4 and NOX. It has been shown that IRAK1, another component of the TLR4 signaling complex, forms a complex with NEMO upon IL-1β treatment (19) raising the question whether NEMO plays a role in the membrane proximal TLR signaling complex. Further complexity arises from the diversity of genotypes that make up NEMO-deficiency. It would be interesting to see if the various reported mutants behave differently in their ability to support TLR signaling to NOX. Indeed, one of the patients studied carries a NEMO mutation (C417R) that is not ubiquitinated in response to CD40L stimulation and consequently does not activate the NFkB family member C-Rel but retains normal activation of RelA in response to LPS (17).
IRAK4 deficiency, which results in pyogenic infections in children, also results in defective LPS- or IL-1β-induced PMN priming but conserved responses to platelet activating factor or TNFα (25, 26). IRAK4 mutant forms appear to bind with higher affinity than wild type to MyD88, another important adaptor in TLR signaling, and consequently decrease availability of MyD88 for IRAK1 dependent signaling (41). IRAK4 has also been shown to bind and directly phosphorylate p47phox in neutrophils upon LPS stimulation (42). Consistent with this finding, we did not detect p47 phosphorylation in response to LPS alone in IRAK4 deficient PMN but did detect phosphorylation in response to fMLP and PMA. We are unable to discern whether the absence of p47phox and p38 phosphorylation is due to the absence of IRAK4 kinase activity itself or the failure of other kinases to be activated in an IRAK4-dependent manner. Another possible pathway from TLR signaling to NOX activation arises from studies showing that, at least for TLR2, Rac1 and PI3K interacted directly with the intracellular signaling domain of TLR2 and were required for NFkB signaling (43). Although we have not directly examined activation of PI3K, inhibitors of this enzyme do indeed decrease activation of NOX in normal, NEMO-deficient, and IRAK4-deficient PMN in response to LPS and fMLP (data not shown) suggesting that at least part of the NOX priming and activating signal moves through PI3K. We also found that Rac2 (the neutrophil-specific homologue of Rac1) was not recruited to PMN membranes in LPS/fMLP stimulated IRAK4-deficient cells. While further biochemical evidence is required, this observation suggests that IRAK4 is upstream of Rac activation.
It will be interesting to establish whether more natural stimuli, (e.g., intact microbes) are also less able to activate NOX in cells from patients with NEMO or IRAK4 deficiency. More importantly, does defective NOX activation in NEMO- or IRAK4-deficiency play a role during the innate immune response to infection in vivo? Although the defect in NOX activation in NEMO-deficiency is less dramatic than IRAK4-deficiency in vitro, the consequences may be more severe in the background of altered acquired immunity in NEMO-deficiency (e.g., defective immunoglobulin class-switching). Interestingly, survival of IRAK4-deficient patients past childhood is associated with a significant decrease in the risk of further infection (22) suggesting that the acquired immune system (when able to develop) can compensate for the innate signaling defect. Mapping alternative pathways leading to protective activation of the immune system should provide pharmacologic targets to aid in the treatment of these patients.
ACKNOWLEDGEMENTS
The authors thank Dr. Thomas L. Leto for helpful discussion and Drs. Harry L. Malech, Steven M. Holland, and Ashish K. Jain for arranging access to patient materials. We are grateful to the patients for their essential contributions to this study.
Footnotes
This research was supported by the Intramural Research Program of the NIH, the NIAID and the NIH Clinical Center.
REFERENCES
- 1.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. 2000;343:1703–1714. doi: 10.1056/NEJM200012073432307. [DOI] [PubMed] [Google Scholar]
- 3.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 4.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 5.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 6.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohno Y, Seligmann BE, Gallin JI. Cytochrome b translocation to human neutrophil plasma membranes and superoxide release. Differential effects of N-formylmethionylleucylphenylalanine, phorbol myristate acetate, and A23187. J Biol Chem. 1985;260:2409–2414. [PubMed] [Google Scholar]
- 8.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapouge K, Smith SJ, Groemping Y, Rittinger K. Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem. 2002;277:10121–10128. doi: 10.1074/jbc.M112065200. [DOI] [PubMed] [Google Scholar]
- 10.Qureshi ST, Medzhitov R. Toll-like receptors and their role in experimental models of microbial infection. Genes Immun. 2003;4:87–94. doi: 10.1038/sj.gene.6363937. [DOI] [PubMed] [Google Scholar]
- 11.DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101:455–463. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8:1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- 13.Zhao T, Benard V, Bohl BP, Bokoch GM. The molecular basis for adhesion-mediated suppression of reactive oxygen species generation by human neutrophils. J Clin Invest. 2003;112:1732–1740. doi: 10.1172/JCI19108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamori T, Inanami O, Nagahata H, Cui Y, Kuwabara M. Roles of p38 MAPK, PKC and PI3-K in the signaling pathways of NADPH oxidase activation and phagocytosis in bovine polymorphonuclear leukocytes. FEBS Lett. 2000;467:253–258. doi: 10.1016/s0014-5793(00)01167-4. [DOI] [PubMed] [Google Scholar]
- 15.Uzel G. The range of defects associated with nuclear factor kappaB essential modulator. Curr Opin Allergy Clin Immunol. 2005;5:513–518. doi: 10.1097/01.all.0000191241.66373.74. [DOI] [PubMed] [Google Scholar]
- 16.Jain A, Ma CA, Lopez-Granados E, Means G, Brady W, Orange JS, Liu S, Holland S, Derry JM. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J Clin Invest. 2004;114:1593–1602. doi: 10.1172/JCI21345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Temmerman ST, Ma CA, Borges L, Kubin M, Liu S, Derry JM, Jain A. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood. 2006;108:2324–2331. doi: 10.1182/blood-2006-04-017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma UN, Yamamoto Y, Prajapati S, Gaynor RB. Nuclear role of I kappa B Kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J Biol Chem. 2004;279:3509–3515. doi: 10.1074/jbc.M309300200. [DOI] [PubMed] [Google Scholar]
- 19.Cooke EL, Uings IJ, Xia CL, Woo P, Ray KP. Functional analysis of the interleukin-1-receptor-associated kinase (IRAK-1) in interleukin-1 beta-stimulated nuclear factor kappa B (NF-kappa B) pathway activation: IRAK-1 associates with the NF-kappa B essential modulator (NEMO) upon receptor stimulation. Biochem J. 2001;359:403–410. doi: 10.1042/0264-6021:3590403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapel H, Puel A, von Bernuth H, Picard C, Casanova JL. Shigella sonnei meningitis due to interleukin-1 receptor-associated kinase-4 deficiency: first association with a primary immune deficiency. Clin Infect Dis. 2005;40:1227–1231. doi: 10.1086/428733. [DOI] [PubMed] [Google Scholar]
- 21.Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, Arditi M, Gallin JI, Vogel SN. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med. 2003;198:521–531. doi: 10.1084/jem.20030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–2422. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, Frucht DM, Christel K, von Bernuth H, Jouanguy E, Feinberg J, Durandy A, Senechal B, Chapgier A, Vogt G, de Beaucoudrey L, Fieschi C, Picard C, Garfa M, Chemli J, Bejaoui M, Tsolia MN, Kutukculer N, Plebani A, Notarangelo L, Bodemer C, Geissmann F, Israel A, Veron M, Knackstedt M, Barbouche R, Abel L, Magdorf K, Gendrel D, Agou F, Holland SM, Casanova JL. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–1759. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, Nurko S, Rasmussen WL, Kohler JR, Gellis SE, Ferguson BM, Strominger JL, Zonana J, Ramesh N, Ballas ZK, Geha RS. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest. 2002;109:1501–1509. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhns DB, Long Priel DA, Gallin JI. Endotoxin and IL-1 hyporesponsiveness in a patient with recurrent bacterial infections. J Immunol. 1997;158:3959–3964. [PubMed] [Google Scholar]
- 26.Picard C, Puel A, Bustamante J, Ku CL, Casanova JL. Primary immunodeficiencies associated with pneumococcal disease. Curr Opin Allergy Clin Immunol. 2003;3:451–459. doi: 10.1097/00130832-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Takada H, Yoshikawa H, Imaizumi M, Kitamura T, Takeyama J, Kumaki S, Nomura A, Hara T. Delayed separation of the umbilical cord in two siblings with Interleukin-1 receptor-associated kinase 4 deficiency: rapid screening by flow cytometer. J Pediatr. 2006;148:546–548. doi: 10.1016/j.jpeds.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 28.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol. 2007;178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 29.Dorseuil O, Quinn MT, Bokoch GM. Dissociation of Rac translocation from p47phox/p67phox movements in human neutrophils by tyrosine kinase inhibitors. J Leukoc Biol. 1995;58:108–113. doi: 10.1002/jlb.58.1.108. [DOI] [PubMed] [Google Scholar]
- 30.Kjeldsen L, Sengelov H, Borregaard N. Subcellular fractionation of human neutrophils on Percoll density gradients. J Immunol Methods. 1999;232:131–143. doi: 10.1016/s0022-1759(99)00171-4. [DOI] [PubMed] [Google Scholar]
- 31.Leto TL, Adams AG, de Mendez I. Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc Natl Acad Sci U S A. 1994;91:10650–10654. doi: 10.1073/pnas.91.22.10650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ago T, Nunoi H, Ito T, Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J Biol Chem. 1999;274:33644–33653. doi: 10.1074/jbc.274.47.33644. [DOI] [PubMed] [Google Scholar]
- 33.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 34.Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/s0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- 35.Martyn KD, Kim MJ, Quinn MT, Dinauer MC, Knaus UG. p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood. 2005;106:3962–3969. doi: 10.1182/blood-2005-03-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280:4279–4288. doi: 10.1074/jbc.M412820200. [DOI] [PubMed] [Google Scholar]
- 37.El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, Babior BM. Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys. 1996;334:395–400. doi: 10.1006/abbi.1996.0470. [DOI] [PubMed] [Google Scholar]
- 38.Brown GE, Stewart MQ, Bissonnette SA, Elia AE, Wilker E, Yaffe MB. Distinct ligand-dependent roles for p38 MAPK in priming and activation of the neutrophil NADPH oxidase. J Biol Chem. 2004;279:27059–27068. doi: 10.1074/jbc.M314258200. [DOI] [PubMed] [Google Scholar]
- 39.Partrick DA, Moore EE, Offner PJ, Meldrum DR, Tamura DY, Johnson JL, Silliman CC. Maximal human neutrophil priming for superoxide production and elastase release requires p38 mitogen-activated protein kinase activation. Arch Surg. 2000;135:219–225. doi: 10.1001/archsurg.135.2.219. [DOI] [PubMed] [Google Scholar]
- 40.Lal AS, Clifton AD, Rouse J, Segal AW, Cohen P. Activation of the neutrophil NADPH oxidase is inhibited by SB 203580, a specific inhibitor of SAPK2/p38. Biochem Biophys Res Commun. 1999;259:465–470. doi: 10.1006/bbrc.1999.0759. [DOI] [PubMed] [Google Scholar]
- 41.Medvedev AE, Thomas K, Awomoyi A, Kuhns DB, Gallin JI, Li X, Vogel SN. Cutting edge: expression of IL-1 receptor-associated kinase-4 (IRAK-4) proteins with mutations identified in a patient with recurrent bacterial infections alters normal IRAK-4 interaction with components of the IL-1 receptor complex. J Immunol. 2005;174:6587–6591. doi: 10.4049/jimmunol.174.11.6587. [DOI] [PubMed] [Google Scholar]
- 42.Pacquelet S, Johnson JL, Ellis BA, Brzezinska AA, Lane WS, Munafo DB, Catz SD. Cross-talk between IRAK-4 and the NADPH oxidase. Biochem J. 2007;403:451–461. doi: 10.1042/BJ20061184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]