

Abstract
Noroviruses (NoVs) are the most important viral pathogens that cause epidemic acute gastroenteritis. NoVs recognize human histo-blood group antigens (HBGAs) as receptors or attachment factors. The elucidation of crystal structures of the HBGA-binding interfaces of a number of human NoVs representing different HBGA binding patterns opens a new strategy for the development of antiviral compounds against NoVs through rational drug design and computer-aided virtual screening methods. In this study, docking simulations and virtual screening were used to identify hit compounds targeting the A and B antigens binding sites on the surface of the capsid P protein of a GII.4 NoV (VA387). Following validation by re-docking of the A and B ligands, these structural models and AutoDock suite of programs were used to screen a large drug-like compound library (derived from ZINC library) for inhibitors blocking GII.4 binding to HBGAs. After screening >2 million compounds using multistage protocol, 160 hit compounds with best predicted binding affinities and representing a number of distinct chemical classes have been selected for subsequent experimental validation. Twenty of the 160 compounds were found to be able to block the VA387 P dimers binding to the A and/or B HBGAs at an IC50<40.0 µM, with top 5 compounds blocking the HBGA binding at an IC50<10.0 µM in both oligosaccharide- and saliva-based blocking assays. Interestingly, 4 of the top-5 compounds shared the basic structure of cyclopenta [a] dimethyl phenanthren, indicating a promising structural template for further improvement by rational design.
Introduction
Noroviruses (NoVs) are a group of single-stranded, positive sense RNA viruses constituting the Norovirus genus in the family Caliciviridae. NoVs have been recognized as the most important cause of viral epidemic acute gastroenteritis affecting people of all ages [1], [2]. In the United States NoVs cause 23 million infections each year and are responsible for more than 90% of the outbreaks of viral gastroenteritis. On a worldwide basis NoVs lead to 218,000 deaths in developing countries and 1.1 million episode of pediatric gastroenteritis in developed countries annually [3]. Thus, NoV associated diseases have been a heavy burden to public healthcare. NoVs are difficult to control owing to their widespread nature and the lack of effective vaccines and antivirals.
NoVs are non-enveloped viruses that are encapsulated by an icosahedral protein capsid comprising 180 copies of the single major structural protein, the capsid protein (VP1). Based on its structural features, the capsid protein is divided into two major domains, the shell (S) and the protruding (P) domains, each forming the interior shell and the arch-like protrusions of NoV capsid, respectively. The P domain can be further divided into P1 and P2 subdomains, constituting the leg and the head of the arch-shaped P dimer, respectively [4]. The P domain plays an important role in host immune response and receptor recognition. Heterologous expression of the P domain in E. coli forms P dimers [4] that is structurally and functionally indistinguishable from the authentic P dimers of viruses [5]–[11], providing a simple model for study of NoV-host interaction [12]–[17]. In addition, production of P domain with end modifications can also form 24 mer P particles [12], [14], [15] and 12 mer small P particle [13], which contain 12 and 6 copies of P dimers, respectively.
NoVs recognize human histo-blood group antigens (HBGAs) as receptors or attachment factors, which play an important role in host susceptibility to NoV infection [18]–[21]. HBGAs are complex carbohydrates that are presented abundantly on the surface of mucosal epithelia of gastrointestinal track, where they may function as anchors for NoVs to initiate an infection. Human HBGAs are highly polymorphic that contain three major families, the ABO, secretor and Lewis families. Human NoVs are also highly diverse and multiple receptor binding patterns with different ABO, secretor and Lewis antigens have been described. The HBGA interacting sites have been mapped to the P domain of NoV capsid [4], [12]–[14], [22]. Further X-ray crystallography of the recombinant P dimers of a number of human NoVs representing different HBGA binding patterns in complex with different HBGA oligosaccharides has been resolved which provided valuable structural basis of the HBGA-NoV interactions [5]–[10].
The HBGA binding interfaces are located at the top of the P dimer, corresponding to the outermost surface of the capsid. The carbohydrate binding pockets involve several scattered amino acid residues in the P domain that form extensive hydrogen bond network with individual saccharides, and thus stabilizing the binding of HBGAs to the capsid protein. Structure-based mutagenesis followed by functional analyses further confirmed the observed HBGA binding sites [7], [15], [16]. This detailed structural information of NoV-HBGA interactions opens a way to a new strategy for antiviral development through Computer-Aided Drug Design (CADD), while the established biological assays of NoV P dimer-HBGA interaction provide a convenient approach for validation of hit compounds identified by CADD.
CADD is a common approach in drug discovery that typically involves following major steps: 1) construction and validation of computational models of the target protein based on its crystal structures with known functional (HBGA binding) sites; 2) virtual high throughput screening (VHTS) of a large number of chemical compounds to identify candidate inhibitors (hit compounds) that are predicted to bind to the functional site of the protein with sufficiently high affinity; and 3) validation of the candidate inhibitors through biological and biochemical assays. Further development of promising candidate inhibitors includes assessment of their toxicity, pharmacokinetic and rational re-design based on structures of individual candidates, with the goals of improving efficacy while lowering toxicity and other undesired properties.
VHTS has been widely used for candidate compound discovery due to an advantage in elimination of undesired molecules from compound libraries, so that the cost and labor can be greatly reduced in a drug discovery project. A number of public compound libraries are currently available for VHTS, including Zinc (http://zinc.docking.org/) [23], NCI [http://cactus.nci.nih.gov/download/nci/], UC Irwine Chem BD [24], and Ligand-Depot [25]. VHTS of large databases of chemical compounds has been repeatedly shown to successfully identify hit compounds that can effectively inhibit the function of a given protein [26]–[29].
In this study we report the first attempt to identify inhibitors of HBGA binding to NoVs as potential antivirals against NoVs through a CADD procedure. The crystal structures of the VA387 (a member of the predominant GII.4 NoVs) P dimer interacting with HBGA were employed to construct computer models for VHTS. After validations of the models by virtual docking simulation using the type A- and B-trisaccharides as ligands, VTHS of a large drug-like compound library was performed, resulting in 255 hit compounds. A total of 160 compounds of the compounds were further tested by biological assays and five revealed strong blocking activity on P dimer-HBGA interaction with an IC50<10 µM. Our results suggest that the CADD approach can facilitate the development of antivirals against human NoVs and the five highly active compounds could become a basis for developing promising drug candidates against NoVs.
Materials and Methods
Computer Models of P Dimers and Oligosaccharides
The two PDB files revealing the crystal structures of the P dimers of VA387 (a predominant GII.4 NoVs) in complex with A (2OBS) and B (2OBT) HBGA oligosaccharides, respectively [6], were downloaded from Protein Data Bank (PDB, http://www.pdb.org). The protein structures with removal of the HBGA ligands were used as model of P dimer, while the extracted HBGA structures were used as models of A and B trisaccharides. Preparation of the HBGA model included specifying rotatable bonds, assigning partial charges, and preparing grid boxes for docking simulations, which was performed using AutoDock package. Preparation of the P dimer model included adding polar hydrogens, checking missing atoms, assigning charges and solvation parameters. Information of HBGA oligosaccharides interacting with the amino acids that constitute the HBGA binding sites were used to set parameters for molecular docking.
Docking Simulation
Re-docking simulations were used for validation of the structural models and docking protocols. The results obtained by docking the native ligands (A and B trisaccharides) to the P dimer were compared with the structural data on P dimer in complex with HBGAs [6]. Using AutoDock 3 package [30] we performed a series of rigid (P dimer) body simulations, followed by additional assessment of the results from “flexible ligand-flexible key binding residues of the P dimer” docking simulations. We found that rigid body docking was qualitatively consistent with flexible docking simulations, and able to reproduce experimentally observed conformations of the P dimer - HBGAs complexes. In particular, majority of over 200 different poses generated in repeated rigid body simulations were found to be in good agreement with experimental data. Grid boxes and grid densities for rigid body docking were optimized to provide sufficient accuracy and to cover the binding site(s) that might occur over the whole P dimer molecule (blind docking). Docked conformations of ligands were generated by AutoDock’s Lamarckian genetic algorithm (GA) [31]. Docking parameters used for the simulations are listed in Table 1. Docking simulations and the VTHS (see below) were conducted on the Cincinnati Children’s Hospital Medical Center’s BMI computational cluster with over 200 processing cores (at least 2.4 GHz) running 64-bit SuSE Linux operating system.
Table 1. Parameters used for docking simulation.
| Iteration Parameters | |
| Translation step | 2 Å |
| Quaternion step | 50° |
| Torsion step | 50° |
| Lamarckian Genetic Algorithm Parameters | |
| Number of Genetic Algorithm runs | 255 |
| Initial population size | 300 |
| Maximum number of energy evaluations | 2.5 million |
| Maximum number of generations | 35,000 |
| No. of top individuals that automatically survive | 1 |
| Rate of gene mutation | 0.02 |
| Rate of crossover | 0.8 |
| Number of generations for picking worst individual | 10 |
| Number of iterations of pseudo Solis and Wets local search | 300 |
| Number of consecutive successes before changing | 4 |
| Number of consecutive failures before changing | 4 |
| Probability of performing local search on an individual | 0.06 |
| Grid Parameters | |
| Grid Spacing | 0.375 Å |
| Number of grid points in x, y, and z directions | 78, 50, 45 |
VHTS of the Compound Library
A subset of the public compound library ZINC, consisting of 2,066,906 drug-like compounds was downloaded from the ZINC database (http://zinc.docking.org/). In the initial stage of VTHS, these compounds were subjected to the automatic molecular docking using the “rigid P dimer-flexible ligand” approach and AutoDock ver. 3. The primary screening was done using coarse-level docking with limited sampling [26], [32]. Compounds predicted to have <0.1 µM inhibition constant (Ki<10−7) were subsequently subjected to a secondary round of screening with improved sampling. The protocol used for secondary screening (see Table 2) involved an increased number of overall simulation runs, increased number of energy evaluation, increased size of the GA population [33], and a finer grid resolution (decreased from 0.6 to 0.375 Ang). The identified top 255 hit compounds that were predicted to bind the HBGA binding site with better affinity than that of A and B oligosaccharides were selected for experimental validation. The bound P dimer structures of VA387 (2OBS) [6] with removal of the A-trisaccharides were used as model for the screening. The docking parameters for the primary VHTS were similar to those used in molecular docking (Table 2). To reduce the docking time, trivial parallelism of VHTS was exploited by performing docking simulations for subsets of compounds on individual computing nodes using a pipeline described in Biesiada et al [32].
Table 2. Parameters used in the primary and secondary screening of the VHTS.
| Iteration Parameters | Primary | Secondary |
| Translation step | 2 Å | 2 Å |
| Quaternion step | 50° | 50° |
| Torsion step | 50° | 50° |
| Lamarckian Genetic Algorithm Parameters | ||
| Number of Genetic Algorithm runs | 10 | 100 |
| Initial population size | 50 | 500 |
| Maximum number of energy evaluations | 150,000 | 500,000×torsion |
| Maximum number of generations | 27,000 | 27,000 |
| No. of top individuals that automatically survive | 1 | 1 |
| Rate of gene mutation | 0.02 | 0.02 |
| Rate of crossover | 0.8 | 0.8 |
| Number of generations for picking worst individual | 10 | 10 |
| Number of iterations of pseudo Solis and Wets local search | 300 | 300 |
| Number of consecutive successes before changing | 4 | 4 |
| Number of consecutive failures before changing | 4 | 4 |
| Probability of performing local search on an individual | 0.06 | 0.06 |
| Grid Parameters | ||
| Grid Spacing | 0.6 Å | 0.375 Å |
| Grid points in x, y, and z directions | 49, 32, 29 | Split into two grids |
Purchases of the Hit Compounds
160 top hits (which were actually selected based on their availability from a somewhat larger initial set of 255 hit compounds obtained in virtual screening) were purchased from Molport (http://www.molport. com) supplied by Maybridg, TimTec, ChemBridge, Pharmeks, Specs, Otava, ChemDiv, InterBioScreen Ltd, Vitas-M Laboratory, Princeton Biomolecular Research and Enamine Ltd. The information including ZINC numbers and structures of the 160 chemicals were listed in Table S1 and Figure S1. All compounds were dissolved at 100 µg/ml in PBS (pH 7.4) containing 1% DMSO as stock solutions.
Validation of Hit Compounds by NoV/HBGA Blocking Assays
Saliva-based NoV-HBGA binding assays were performed as described previously using P dimers of VA387 (GII.4) as NoV surrogates and saliva samples and/or synthetic oligosaccharides as HBGA sources [4], [34], [35]. Briefly, synthetic oligosaccharides and/or boiled saliva samples with defined HBGAs phenotype were coated on 96-well microtiter plates, after blocking with nonfat milk, P dimer of VA387 were added. The bound P dimer was detected using a guinea pig antiserum against NoVs VLPs, followed by the addition of horseradish peroxidase (HRP)-conjugated goat anti-guinea pig IgG. The bound HRP conjugates were colorized by the TMB kit (Kirkegaard & Perry Laboratories), which was read an EIA spectrum reader (Tecan). The synthetic oligosaccharide-PAA conjugates (2 µg/ml, GlycoTech Corporation, Rockville, MD) were captured to a microtiter plate through coated streptavidin (5 µg/ml) [36].
Blocking effects of the hit compounds were measured by a pre-incubation of the P dimers with the compounds at given concentrations for 30 min before the P dimers were added to the coated saliva samples or HBGA oligosaccharides in a binding assay [37]. The blocking activity of a compound was defined as its concentration yielding 50% inhibition (IC50) in the binding assay. IC50 calculation was performed using Probit regression analysis and correlation analysis between IC50 and Ki values was performed with nonparametric Spearman’s r (two-tailed) by using SPSS statistical software version 13.0 (SPSS, Chicago, IL).
MTS Cytotoxicity Assay
This assay was performed using CellTiter 96 aqueous nonradioactive cell proliferation kits (Promega, Madison, WI) as described elsewhere [36]. Briefly, HeLa and LLC-MK2 cells were seeded at 5×104 cells/ml onto a 96-well plate overnight. After an incubation with each compound at various concentrations for 3 days (the compound was added one time at the beginning of the incubation), the culture medium was replaced with fresh one with 100 µl of MTS-phenazine methosulfate/well. After a further incubation at 37°C for 2 h, the color products of MTS were measured with a plate reader at 490 nm. The cytotoxicities of individual compounds were indicated by the decrease in cellular reduction of MTS into the colored product. The 50% cytotoxic concentrations (CC50 s) were determined as the concentrations of the compounds that caused 50% inhibition of cell growth compared with that of control cells without a compound.
Results
Validation of the NoV Model and the Molecular Docking Protocol
The crystal structures of NoV P dimers of VA387 (GII.4) with HBGA oligosaccharides (2OBS and 2OBT) [6] were used to build target structures for molecular docking and VTHS of the ZINC compound library. The models were first employed for validation by re-docking simulations using a type A trisaccharide as a ligand through software AutoDock 3 [30]. As shown in Figure 1, the vast majority of multiple poses of the A trisaccharide obtained in repeated simulations docked well to the experimentally mapped HBGA binding site of VA387 that was formed by seven amino acids: Ser343, Arg345, His347, Asp374, Gln376, Ser441 and Gly442. It was noted that some conformations of the A trisaccharide docked to an undefined nearby site. The predicted inhibition constants (Ki) were relatively low (with the best predicted Ki of about 1.6 µM), reflecting the nature of trisaccharide-protein interactions. Similar results were obtained when docking simulations were performed using the type B trisaccharide as a ligand (data not shown). Thus, these data are consistent with the crystal structures of the VA387 P dimer and provide validation of our computational models and molecular docking protocols.
Figure 1. Predicted docking poses for the A trisaccharides docked to the VA387 P dimer clusters at the proved HBGA binding site.

The majority of the predicted poses (each represented by a magenta ball) docked to the experimentally resolved HBGA binding site that was formed by Ser343, Arg345, His347, Asp374, Gln376, Ser441 and Gly442 (circled by a yellow dashed line). Some of the poses docked to a nearby site that was previously suggested as an alternative binding site for HBGA (Tan et al., 2003). The structure of the P dimer of VA387 (2OBS) is shown using ribbon model, each monomer in green and blue, respectively. The amino acids that constitute the experimentally mapped HBGA binding site were shown using stick model and circled by a yellow dashed line, while amino acids forming the nearby site were also shown in stick model. The squared region containing the HBGA binding site is enlarged in the up-left panel.
VTHS and Laboratory Validation of Drug-like Compounds Against NoV Binding to HBGAs
Multistage screening of ∼ 2.07 million compounds from the ZINC drug-like library has resulted in identification of 255 hit compounds with predicted inhibition constants Ki values less than 100 µM against VA387 binding to the A and/or B trisaccharides (Figure 2). A total of 160 compounds from the 255 compounds were purchased from several different companies based on their availability (see Materials and Methods) and tested by saliva-based blocking assays using VA387 P dimers as NoV surrogates and type A and B saliva samples as HBGA sources. Twenty compounds (12.5%) exhibited >50% inhibitory effects on the P dimer-saliva interactions at a concentration <40 µM (Figure 3). The specificities of the top 20 compounds were further studied by both saliva- and synthesized HBGA oligosaccharide-based blocking assays. Five of the 20 compounds showed strong inhibitions with IC50 s <10 µM; five others revealed good inhibitions with IC50 s ranging from 10 to 20 µM, while the remaining ten compounds exhibited moderate inhibitions with IC50 s ranging from 20–40 µM (Table 3). All of the top five strongest inhibitors (ZINC04041115, ZINC05260830, ZINC05223451, ZINC04831336 and ZINC04026813) revealed similar levels of inhibition in a dose-dependent manner against the VA387 P dimer binding to the synthetic A and B oligosaccharides (Figure 4). A marginal correlation between the IC50 values from the block assays and the Ki values from the docking simulation of the 20 top-list compounds were observed (Spearman r = 0.561, p = 0.01, data not shown).
Figure 2. Distribution of the lowest Ki values of the top 255 compounds docked at the HBGA binding sites of the VA387 P dimer.
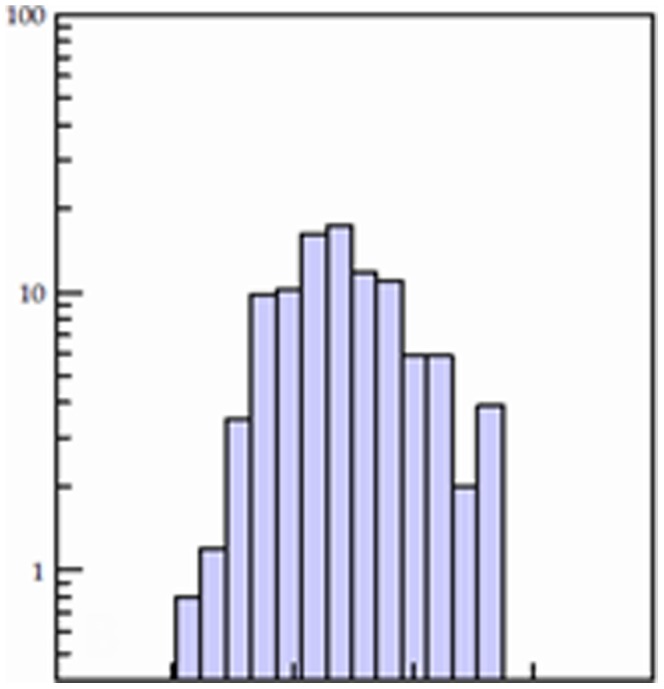
Figure 3. Structures of the top 20 hit compounds against binding of VA387 to the A and B saliva.

Table 3. The basic features of the 20 most inhibitory lead-like compounds.
| ZINC-codes | MW(Da) | MolecularFormula | Ki value(µM)a | IC50 (µM)b | |
| Saliva A | Saliva B | ||||
| ZINC04041115 | 344.49 | C21H32N2O2 | 0.64 | 2.38±0.15 | 2.54±0.21 |
| ZINC05260830 | 318.49 | C21H34O2 | 1.25 | 2.90±0.33 | 2.93±0.18 |
| ZINC05223451 | 306.48 | C20H34O2 | 1.14 | 3.37±0.13 | 3.39±0.24 |
| ZINC04831336 | 374.56 | C24H38O3 | 0.36 | 7.63±0.27 | 7.65±0.33 |
| ZINC04026813 | 345.55 | C20H31N3S | 0.16 | 8.70±1.03 | 8.97±0.63 |
| ZINC04095376 | 307.35 | C18H17N3O2 | 3.25 | 12.6±0.87 | 12.8±0.64 |
| ZINC04725822 | 296.36 | C18H20N2O2 | 1.65 | 13.2±1.04 | 13.1±0.87 |
| ZINC00128665 | 277.36 | C19H19NO | 1.58 | 14.1±0.53 | 14.9±0.71 |
| ZINC06166484 | 460.55 | C25H24N4O3S | 1.16 | 17.0±1.06 | 16.8±0.87 |
| ZINC04084183 | 418.53 | C23H34N2O5 | 0.42 | 18.7±0.35 | 18.6±0.62 |
| ZINC04081424 | 344.49 | C22H32O3 | 0.35 | 22.7±1.36 | 23.7±1.28 |
| ZINC04450155 | 323.35 | C18H17N3O3 | 7.77 | 24.2±0.74 | 24.3±0.87 |
| ZINC00124088 | 318.33 | C19H14N2O3 | 13.2 | 24.5±0.98 | 26.3±1.24 |
| ZINC00968234 | 314.46 | C21H30O2 | 0.92 | 24.8±2.02 | 25.1±1.32 |
| ZINC04062835 | 274.4 | C18H26O2 | 2.49 | 28.5±1.54 | 29.4±1.24 |
| ZINC00652738 | 374.51 | C23H34O4 | 1.67 | 30.9±1.04 | 30.1±1.35 |
| ZINC00181174 | 332.35 | C20H16N2O3 | 3.85 | 33.5±0.68 | 33.7±1.27 |
| ZINC04014899 | 327.46 | C21H29NO2 | 1.56 | 33.9±1.67 | 34.5±1.93 |
| ZINC03814360 | 290.44 | C19H30O2 | 2.20 | 36.9±1.47 | 35.7±1.06 |
| ZINC04298453 | 309.33 | C19H16FNO2 | 22.0 | 38.0±2.01 | 39.4±1.53 |
determined by docking;
determined by blocking assays; the data were indicated by mean ± standard deviation.
Figure 4. Validation and titration of inhibitory activities of the top 5 hit compounds to HBGA oligosaccharide-PAA conjugates.

All compounds revealed significantly blocking activities against VA387 binding to the oligosaccharide-PAA conjugated A and B. The concentrations of the compounds used in the assays were adjusted according to their blocking activities in the type A and type B saliva screening. The IC50 concentrations of these 5 compounds were lower than 10 µM. Triplicate tests for each compound were performed, and the mean reduction of binding activity was presented.
Structure Comparisons of the Top 20 Hit Compounds
The chemical structure of the 20 top-list compounds are shown in Figure 3 according to their IC50 s in blocking the binding of NoV P dimer to HBGAs. Interestingly, over a half of them share the basic structure of cyclopenta [a] dimethyl phenanthren. Particularly, four of the five most potent inhibitors share this common structure, suggesting that the cyclopenta [a] phenanthren with dimethyl may represent a promising class of compounds for further refinement.
The Cytotoxicity of Top 5 Hit Compounds
This was tested in human cervical carcinoma cells (HeLa) and rhesus monkey kidney epithelial cells (LLC-MK2). The resulting CC50 s were 203.2 (ZINC04041115), 266.8 (ZINC05260830), 170.6 (ZINC05223451), 225 (ZINC04831336) and 212.4 (ZINC04026813) µM, respectively.
Discussion
Currently there is no effective intervention available against NoV gastroenteritis. We described in this study a search for small compounds to inhibit NoV binding to HBGAs as potential antivirals against NoVs. The computer-aided drug development (CADD) approach was used. Computational models of NoV P dimers and their ligands (A and B trisaccharides) were constructed based on available crystal structures [6]. After validation of the models and docking protocol, over two million compounds in the ZINC library were screened by docking simulations and virtual screening. A total of 255 hit candidates have been identified, among which 160 were further tested by HBGA blocking assays. Twenty compounds (12.5%) exhibited >50% inhibitory effects (IC50) at concentrations below 40 µM, in which five have an IC50<10 µM. This study suggested the CADD is a useful approach for antiviral development against NoVs.
Virtual screening based on validated computer models allows elimination of undesired molecules from large compound libraries, thus greatly reducing the cost and time of drug discovery process. In our case, experimentally screening of a library with more than two million compounds is impossible due to the huge workloads. However, the VHTS approach resulted in only 255 hit candidates with theoretical binding affinity higher than the native HBGA ligands within a few weeks of computation time, which greatly facilitated our study to the next step of biological assays for validation. Nevertheless, computer-aided virtual screening also has shortcomings. For example, some active compounds or structures may be screened off by virtual screening and thus may not be tested. This limitation should be kept in mind when the CADD is used.
We identified five compounds with high affinities (IC50 s <10 µM) to the HBGA binding sites of GII.4 NoVs. Further development of effective antivirals against NoVs based on these top five compounds is possible. However, we also are aware that the assays used in our study were based on monovalent interaction which may not represent the natural interactions between NoVs and human HBGAs which are multivalent. Thus, future studies to develop polyvalent inhibitors by conjugation of the hit compounds with large molecular back-bones for higher efficient blocking against NoVs are necessary. In addition, in the measurement of the cytotoxicity (CC50) of the top 5 hit compounds, the compounds were added to the cell media at the beginning of the three-day incubation without further monitoring their stability. This experiment needs to be improved in our future studies. Furthermore, we noticed that all the five top candidates have the common structures of steroid pregnanolone. This needs to be considered in our future studies to avoid potential side effects of hormone activities of the candidates as well as their derivatives.
In our docking simulations using the A or B trisaccharide as ligands, both ligands could be docked to an undefined nearby site around Arg 289 (Figure 1). This type of extra docking site is understandable owing to the protein/carbohydrate interaction nature that is involved in conformational network interactions. It is known that a single amino acid difference can significantly change the outcome of HBGA interaction in a mutagenesis studies of the HBGA binding interface of NoVs [4], [15], [16], [38] [7], [8], [16], [39]. The receptor binding sites of human NoVs are known highly conserved and no extra binding site was found in any known crystal structures of NoVs. Thus, the extra docking site was ignored in our study.
The shared structures of cyclopenta [a] dimethyl phenanthren among most hit compounds suggest an important lead class of compounds for future development, although such structures also shared with the steroid pregnanolone. The low toxicity of all the top five compounds to HeLa and LLC-MK2 cells further suggested this basic structure as a promising lead class. Thus, future studies to define the major functional domains for the blocking activities against NoVs as well as to avoid or reduce the potential hormone activities of pregnanolone of these compounds are critical for our rational design for maximal blocking activities with least toxicity or side effects.
Human NoVs are diverse in recognizing different HBGAs. However, the receptor binding interfaces of different NoVs are structurally conserved among strains within the two major genogroups of human NoVs. This suggests that the CADD approach described in this study may be extended to other NoVs recognizing different HBGAs because of a potential common mechanism of virus/HBGA interaction for all human NoVs. In addition to the GII.4 NoVs that were explored in this study, the crystal structures of several other NoVs with different genetic backgrounds and HBGA binding profiles have also been determined, including the GI.1 Norwalk virus [5], [8], GI.2 FUV258 [11], GII.9 VA207 [7], GII.10 Vietnam 026 [10] and GII.12 Hiro [10]. Future studies to develop antivirals for other NoVs using the same CADD procedures described in this study may be fruitful, because a good correlation between the IC50 s in the blocking assays and the Ki values by the docking simulations has been found which support the VHTS as a useful approach for antiviral development against NoVs.
Supporting Information
The structures of the 160 hit compounds that were purchased for validation by blocking assay.
(DOC)
(DOCX)
Acknowledgments
The work on the virtual screening described in this study was performed by Monica Chhabra as a master degree dissertation, entitled “Modeling and Analysis of Ligand Docking to Norovirus Capsid Protein for the Computer Aided Drug Design ”, under directions by Drs. Jarek Meller and Yizong Cheng at University of Cincinnati.
Funding Statement
This study was partially funded by Guangdong Province ‘211 Project’ and the National Natural Science Foundation of China (30901992) to Xu-Fu Zhang, and National Institutes of Health R01 AI055649, R01 AI089634, and P01 HD13021 to Xi Jiang. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Glass RI, Parashar UD, Estes MK (2009) Norovirus gastroenteritis. N Engl J Med 361: 1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lopman B, Gastanaduy P, Park GW, Hall AJ, Parashar UD, et al. (2012) Environmental transmission of norovirus gastroenteritis. Curr Opin Virol 2: 96–102. [DOI] [PubMed] [Google Scholar]
- 3. Patel MM, Widdowson MA, Glass RI, Akazawa K, Vinje J, et al. (2008) Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg Infect Dis 14: 1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tan M, Hegde RS, Jiang X (2004) The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol 78: 6233–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bu W, Mamedova A, Tan M, Xia M, Jiang X, et al. (2008) Structural basis for the receptor binding specificity of Norwalk virus. J Virol 82: 5340–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao S, Lou Z, Tan M, Chen Y, Liu Y, et al. (2007) Structural basis for the recognition of blood group trisaccharides by norovirus. J Virol 81: 5949–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Tan M, Xia M, Hao N, Zhang XC, et al. (2011) Crystallography of a lewis-binding norovirus, elucidation of strain-specificity to the polymorphic human histo-blood group antigens. PLoS pathogens 7: e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi JM, Hutson AM, Estes MK, Prasad BV (2008) Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci U S A 105: 9175–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shanker S, Choi JM, Sankaran B, Atmar RL, Estes MK, et al. (2011) Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: implications for epochal evolution. J Virol 85: 8635–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansman GS, Biertumpfel C, Georgiev I, McLellan JS, Chen L, et al. (2011) Crystal Structures of GII.10 and GII.12 Norovirus Protruding Domains in Complex with Histo-Blood Group Antigens Reveal Details for a Potential Site of Vulnerability. J Virol 85: 6687–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kubota T, Kumagai A, Ito H, Furukawa S, Someya Y, et al. (2012) Structural basis for the recognition of Lewis antigens by genogroup I norovirus. J Virol 86: 11138–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan M, Fang P, Chachiyo T, Xia M, Huang P, et al. (2008) Noroviral P particle: Structure, function and applications in virus-host interaction. Virology 382: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan M, Fang PA, Xia M, Chachiyo T, Jiang W, et al. (2011) Terminal modifications of norovirus P domain resulted in a new type of subviral particles, the small P particles. Virology 410: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan M, Jiang X (2005) The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. Journal of Virology 79: 14017–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan M, Xia M, Cao S, Huang P, Farkas T, et al. (2008) Elucidation of strain-specific interaction of a GII-4 norovirus with HBGA receptors by site-directed mutagenesis study. Virology 379: 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan M, Xia M, Chen Y, Bu W, Hegde RS, et al. (2009) Conservation of carbohydrate binding interfaces: evidence of human HBGA selection in norovirus evolution. PLoS ONE 4: e5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng DP, Widdowson MA, Glass RI, Vinje J (2010) Molecular epidemiology of genogroup II-genotype 4 noroviruses in the United States between 1994 and 2006. J Clin Microbiol 48: 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan M, Jiang X (2005) Norovirus and its histo-blood group antigen receptors: an answer to a historical puzzle. Trends Microbiol 13: 285–293. [DOI] [PubMed] [Google Scholar]
- 19. Tan M, Jiang X (2007) Norovirus-host interaction: implications for disease control and prevention. Expert Rev Mol Med 9: 1–22. [DOI] [PubMed] [Google Scholar]
- 20. Tan M, Jiang X (2010) Norovirus gastroenteritis, carbohydrate receptors, and animal models. PLoS pathogens 6: e1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan M, Jiang X (2011) Norovirus-host interaction: Multi-selections by human histo-blood group antigens. Trends in microbiology 19: 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan M, Meller J, Jiang X (2006) C-terminal arginine cluster is essential for receptor binding of norovirus capsid protein. Journal of Virology 80: 7322–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Irwin JJ, Shoichet BK (2005) ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model 45: 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen JH, Linstead E, Swamidass SJ, Wang D, Baldi P (2007) ChemDB update - full-text search and virtual chemical space. Bioinformatics 23: 2348–2351. [DOI] [PubMed] [Google Scholar]
- 25. Feng ZK, Chen L, Maddula H, Akcan O, Oughtred R, et al. (2004) Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics 20: 2153–2155. [DOI] [PubMed] [Google Scholar]
- 26. Biesiada J, Porollo A, Velayutham P, Kouril M, Meller J (2011) Survey of public domain software for docking simulations and virtual screening. Human genomics 5: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cavasotto CN, Orry AJW (2007) Ligand docking and structure-based virtual screening in drug discovery. Current topics in medicinal chemistry 7: 1006–1014. [DOI] [PubMed] [Google Scholar]
- 28. Villoutreix BO, Eudes R, Miteva MA (2009) Structure-based virtual ligand screening: recent success stories. Combinatorial chemistry & high throughput screening 12: 1000–1016. [DOI] [PubMed] [Google Scholar]
- 29. Wolf A, Shahid M, Kasam V, Ziegler W, Hofmann-Apitius M (2010) In silico drug discovery approaches on grid computing infrastructures. Current clinical pharmacology 5: 37–46. [DOI] [PubMed] [Google Scholar]
- 30. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry 19: 1639–1662. [Google Scholar]
- 31. Chang DT, Oyang YJ, Lin JH (2005) MEDock: a web server for efficient prediction of ligand binding sites based on a novel optimization algorithm. Nucleic Acids Res 33: W233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Biesiada J, Porollo A, Meller J (2012) On setting up and assessing docking simulations for virtual screening. Methods in molecular biology (Clifton, N J ) 928: 1–16. [DOI] [PubMed] [Google Scholar]
- 33. Hetenyi C, van der Spoel D (2002) Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein science : a publication of the Protein Society 11: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang P, Farkas T, Zhong W, Tan M, Thornton S, et al. (2005) Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J Virol 79: 6714–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang XF, Dai YC, Zhong W, Tan M, Lv ZP, et al. (2012) Tannic acid inhibited norovirus binding to HBGA receptors, a study of 50 Chinese medicinal herbs. Bioorg Med Chem 20: 1616–1623. [DOI] [PubMed] [Google Scholar]
- 36. Taube S, Perry JW, McGreevy E, Yetming K, Perkins C, et al. (2012) Murine noroviruses bind glycolipid and glycoprotein attachment receptors in a strain-dependent manner. J Virol 86: 5584–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng X, Jiang X (2007) Library screen for inhibitors targeting norovirus binding to histo-blood group antigen receptors. Antimicrob Agents Chemother 51: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tan M, Huang P, Meller J, Zhong W, Farkas T, et al. (2003) Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: evidence for a binding pocket. J Virol 77: 12562–12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Rougemont A, Ruvoen-Clouet N, Simon B, Estienney M, Elie-Caille C, et al. (2011) Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J Virol 85: 4057–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The structures of the 160 hit compounds that were purchased for validation by blocking assay.
(DOC)
(DOCX)