

Abstract
Murine cytomegalovirus encodes numerous proteins that act on a variety of pathways to modulate the innate and adaptive immune responses. Here, we demonstrate that a chemokine-like protein encoded by murine cytomegalovirus activates the early innate immune response and delays adaptive immunity, thereby impairing viral clearance. The protein, m131/129 (also known as MCK-2), is not required to establish infection in the spleen; however, a mutant virus lacking m131/129 was cleared more rapidly from this organ. In the absence of m131/129 expression, there was enhanced activation of dendritic cells (DC), and virus-specific CD8+ T cells were recruited into the immune response earlier. Viral mutants lacking m131/129 elicited weaker production of alpha interferon (IFN-α) at 40 h postinfection, indicating that this protein exerts its effects during early rounds of viral replication in the spleen. Furthermore, while wild-type and mutant viruses activated plasmacytoid dendritic cells (pDC) equally at this time, as measured by the upregulation of costimulatory molecules, the presence of m131/129 stimulated more pDC to secrete IFN-α, accounting for the stronger IFN-α response than from the wild-type virus. These data provide evidence for a novel immunomodulatory function of a viral chemokine and expose the multifunctionality of immune evasion proteins. In addition, these results broaden our understanding of the interplay between innate and adaptive immunity.
INTRODUCTION
The study of viral virulence and immunomodulatory factors has provided us with a better understanding of innate and adaptive immune functions. We now recognize that viral products can act at multiple levels to sabotage the antiviral immune response. For example, Epstein-Barr virus, herpes simplex virus, and cytomegalovirus (CMV) encode proteins that interfere with major histocompatibility complex (MHC) class I expression, proteasome function, and/or peptide transport to prevent the processing and presentation of viral epitopes to activated CD8+ cytotoxic lymphocytes (CTL) (1). Viral products also target the innate immune response by inhibiting natural killer (NK) cell activation (2) or interfering with signaling by pattern recognition receptors, such as the toll-like receptors (TLR) (3).
CMV is a member of the herpesvirus family that is well known for its elaborate immune modulation and evasion strategies. Acute infection is typically asymptomatic in immunosufficient individuals, but despite an aggressive and ongoing immune response, the virus is able to persist in the host indefinitely, thanks to its sophisticated immune evasion strategies. A large proportion of the CMV genome is devoted to immune modulation and evasion, and many of these products have been characterized for both mouse CMV (MCMV) and human CMV (HCMV) (reviewed in reference 4), although our understanding is far from complete. CMV has been shown to target a variety of pathways, including cytokine and chemokine signaling, antibody Fc binding, apoptosis, complement activation, NK cell activation, and antigen processing and presentation. CMV has refined these strategies over millions of years of coevolution with its host, and as a consequence, the virus can provide us with valuable insights on how the immune response is regulated (5).
MCMV is attenuated by the deletion of the m131/129 open reading frame (also known as MCK-2) such that fewer inflammatory foci are observed in the liver and viral titers are lower in the salivary glands (6–8). Interestingly, the m131/129 protein exhibits some sequence homology with the CC-chemokines (9) and appears to act as an agonist for some chemokine receptors (8). Furthermore, m131/129 can recruit myeloid cells to the site of MCMV infection (10), leading to the suggestion that the main function of this protein is to facilitate dissemination of the virus. Interestingly, our earlier study of m131/129 mutant viruses also showed they are cleared from the spleens of susceptible BALB/c mice with faster kinetics than wild-type MCMV (6), indicating that m131/129 may influence the immune response during early acute infection. In this report, we establish that m131/129 inhibits the activation of CD8+ T cells in the spleen via an effect on the innate immune response to MCMV. This function is associated with enhanced levels of alpha interferon (IFN-α) production by plasmacytoid dendritic cells (pDC) during early infection, inhibition of interleukin-12 (IL-12) production and antigen presentation by conventional dendritic cells (cDC), and slower generation of virus-specific CD8+ cytotoxic lymphocytes (CTL). Thus, m131/129 modulates pathogen sensing and innate inflammation so as to delay activation of the adaptive immune response.
MATERIALS AND METHODS
Mice and viruses.
Inbred BALB/c mice were obtained from the Animal Resources Centre (Perth, WA, Australia), and congenic BALB.B6-CT6 mice (H-2d, I-Ad, NK1.1+, Ly49H−) (11) were bred in the Animal Services Facility of the University of Western Australia. All mice were housed under specific-pathogen-free conditions at the Animal Services Facility of the University of Western Australia. Experiments were performed with the approval of the Animal Ethics and Experimentation Committee of the University of Western Australia and according to the guidelines of the National Health and Medical Research Council of Australia. The viruses used in this study were as follows: MCMV-K181-Perth (K181) and the mutant virus lacking the m131/129 gene, Δm131-129-stop (6). The virus stocks used for in vivo studies were propagated in the salivary glands of 3-week-old BALB/c mice.
Treatment of mice.
Mice were infected intraperitoneally (i.p.) with salivary gland-propagated virus (SGV) diluted in phosphate-buffered saline (PBS)–0.05% fetal bovine serum. A total of 5 × 103 PFU of SGV was used for all experiments except for the experiment shown below in Fig. 2A, in which 1 × 104 PFU was used. To deplete CD4 and CD8 T cells, mice were inoculated i.p. with 100 μg of the GK1.5 (anti-CD4), YTS 169 (anti-CD8), or PKH 136 (anti-NK1.1) monoclonal antibodies diluted in phosphate-buffered saline–0.05% fetal bovine serum at days −2, 0, and +2 relative to MCMV infection. Depletion of relevant cell subsets was confirmed by flow cytometry.
Fig 2.

The architecture of the spleen is preserved in the absence of m131/129. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. (A) Spleens were collected on day 4 postinfection and prepared for tissue histology. Representative sections are shown for 3 mice/group. Magnification, ×10. (B) Spleens were harvested at the indicated times and homogenized for CCL2 detection by ELISA. CCL2 concentrations are plotted as the mean ± SEM (n = 3 to 5 mice). (C) Spleens were harvested at the indicated times and homogenized for CCL21 detection by ELISA. CCL21 concentrations were pooled for two independent experiments, normalized with respect to uninfected controls, and plotted as the mean ± SEM (n = 5 to 8 mice for all time points, except day 3, where n = 3).
Determination of viral titers.
Spleen, liver, lungs, and salivary glands were collected on the indicated days postinfection and processed to determine viral titers by standard plaque assay using M210B4 cells, as previously described (12).
Histochemistry.
Spleens were collected at the indicated days postinfection, formalin fixed, and processed for tissue histology by using standard methods. For detection of the immediate-early 1 (IE1) protein, spleen tissue was collected and processed for immunofluorescence according to the method of Hsu et al. (13). The monoclonal antibody 6/58/1 was used to detect IE1 (14).
Flow cytometry.
Single-cell suspensions prepared from the spleen were preincubated on ice for 30 min with PBS–2% fetal calf serum containing 10% normal goat serum and then stained with specific antibodies. Propidium iodide was incorporated in the final wash at 1 μg/ml, and then the labeled cells were analyzed on a FACSCanto apparatus (Becton, Dickinson, San Jose, CA). Antibodies used included anti-CD8α (53-6.7; Biolegend, San Diego, CA), anti-T cell receptor β (anti-TCR-β; H57-597; Biolegend), phycoerythrin-conjugated H-2Ld-168-YPHFMPTNL-176 MCMV IE1 tetramer (ImmunoID Tetramers, Melbourne, VIC, Australia), anti-CD11b (M1/70; Biolegend), anti-CD11c (N418; Biolegend), anti-F4/80 (BM8; eBioscience, San Diego, CA), anti-IA/IE (M5/114.15.2; Biolegend), anti-Ly6C (AL-21; BD Biosciences), anti-CD80 (1610A1; BD Biosciences), and anti-CD86 (GL1; BioLegend). Anti-IL-12 (C17.8; BioLegend) was used for intracellular staining after cells were fixed and permeabilized with cytofix/cytoperm solution (BD Biosciences, San Diego, CA). Type I IFN was detected ex vivo by using a mixture of two unconjugated rat monoclonal antibodies against IFN-α (F18 [Hycult Biotech, The Netherlands] and RMMA-1 [PBL Interferon Source, Piscataway, NJ]) and one unconjugated rat monoclonal antibody against IFN-β (RMMB-1; PBL Interferon Source), followed by detection with a biotinylated anti-rat (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and streptavidin (BD Biosciences) conjugates after cells were fixed and permeabilized with cytofix/cytoperm solution. After blocking with 10% normal rat serum, pDC were identified using 120G8 (prepared in-house) in conjunction with antibodies against IA/IE, CD11b, and CD11c. Appropriately stained controls were used to check compensation for all fluorochromes used. Analysis of the fluorescence-activated cell sorting data was performed with the FlowJo software (Ashland, OR).
In vivo CTL assay.
Virus-specific CTL-mediated cytotoxicity was assessed by measuring the elimination of targets pulsed with the IE1 viral peptide YPHFMPTNL. The in vivo CTL assay quantifies CTL activity by measuring the loss of specific peptide-pulsed targets in comparison to targets that have not been pulsed with peptide. Here, target cell lysis was measured using a modification of the previously described in vivo CTL assay (15). IE1 peptide-pulsed (0.02 μg/ml) splenocyte targets were labeled with a low concentration (final concentration, 0.025 μM) of 5,6-carboxyfluorescein diacetate, succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA), whereas control targets, not pulsed with peptide, were labeled with a high concentration (final, 0.25 μM) of CFSE. Labeled cells were washed to remove free CFSE, and differential labeling was confirmed by flow cytometry. CFSElow IE1-pulsed targets and CFSEhigh unpulsed control targets were resuspended in PBS, mixed in equal proportions, and a total of 4 × 107 cells per mouse were transferred intravenously into syngeneic mice that had been infected with MCMV for various periods of time. Mice were sacrificed 4 h later, and single-cell suspensions from the spleen were analyzed by flow cytometry. Transferred cells served as targets for peptide-specific CTL. The frequency of unpulsed targets served as an internal control for cell trafficking, recovery, and nonspecific cell loss. The loss of specific peptide-pulsed targets was a measure of CTL activity against targets pulsed with the IE1 viral peptide. Specific lysis was determined using the following formula: percent specific lysis of CFSE-labeled target cells in each mouse = [1 − (r for uninfected control mice)/(r for infected test mice)] × 100, where r = (frequency of unpulsed targets)/(frequency of peptide-pulsed targets). The assay is a very sensitive and specific method to measure MHC class I-restricted, CD8+ T cell-dependent cytotoxicity in vivo.
Quantitation of cytokines and chemokines by ELISA.
Serum and spleen cytokine levels were measured after infection with 5 × 103 PFU of MCMV. IFN-γ and IL-12 were measured by standard sandwich enzyme-linked immunosorbent assay (ELISA) with BD Biosciences antibodies. Detection was achieved with poly-horseradish peroxidase (poly-HRP) conjugated to streptavidin (CBL, Amsterdam, Netherlands) and K-Blue (Elisa Systems, Brisbane, Australia). IFN-α was quantitated with the Verikine mouse interferon alpha ELISA kit (PBL Interferon Source, Piscataway, NJ). CCL2 and CCL21 were also measured by ELISA, as previously described (16), using commercial antibodies (R&D Systems, Minneapolis, MN). Absorbance was measured at 450 nm with an automated ELISA reader (SpectraMAX 250; Molecular Devices, Sunnyvale, CA).
Statistical analysis.
For statistical analysis, the Mann-Whitney or Student's unpaired t test was performed using the statistical software package InStat (GraphPad Software, San Diego, CA). All data shown are means ± standard errors of the means (SEM) or means ± standard deviations, as indicated.
RESULTS
m131/129 contributes to the virulence of MCMV infection in the spleen.
Upon infection with MCMV, high viral titers were detected in the spleens of BALB/c mice from day 2 postinfection, and viral replication peaked around day 4 and then declined from day 6 onwards (Fig. 1A) (17). When BALB/c mice were infected with recombinant MCMV lacking m131/129 (here referred to as Δm131), similar viral titers were seen in the spleen over the first 5 days; however, by day 6 postinfection, viral titers were significantly lower (P = 0.028) (Fig. 1A). We subsequently examinined spleen tissue sections for expression of the IE1 protein and found that at day 2 postinfection, expression of IE1 in the spleens of mice infected with Δm131 was comparable to that observed in mice infected with wild-type (WT) virus (Fig. 1B and C). However, by day 5 postinfection, when IE1 was readily detectable in mice infected with the wild-type virus, fewer infected cells were found in mice inoculated with Δm131 (Fig. 1B and C).
Fig 1.

Recombinant MCMV lacking m131/129 is controlled in the spleen earlier than wild-type virus. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. (A) Spleens were harvested at the indicated times and homogenized for plaque assays. Viral titers are plotted as the means ± SEM (n = 3 to 9 mice). (B) On days 2 and 5 postinfection, spleens were collected and frozen for immunohistochemistry to detect IE1 expression. Representative sections are shown for 3 mice per group. Magnification, ×20. (C) The number of fluorescent IE1+ cells was quantified from six fields for each group on days 2 and 5 postinfection by using ImageJ. Values are expressed per mm2.
The improved clearance of Δm131 was associated with reduced tissue pathology. On day 4 postinfection, a time when there was little difference in viral titers (Fig. 1A), mice infected with Δm131 exhibited far less disorganization of the spleen and very little disruption of the lymphoid follicles compared to mice infected with wild-type virus (Fig. 2A). This observation prompted us to assay local production of two chemokines, CCL2 and CCL21, which are known to play a role in tissue inflammation and organization of the lymphoid tissues. CCL2 was produced in the spleen in response to infection, and levels were similar for both viruses on days 2 and 3; however, significantly higher levels were detected on days 4 and 5 (P = 0.0018 and P = 0.0049, respectively) in mice infected with the wild-type virus than in mice infected with Δm131 (Fig. 2B). In contrast, production of CCL21 declined upon infection, and the decline was faster after infection with wild-type virus than with Δm131 (Fig. 2C). Consistent with the ELISA results, by day 5 postinfection, reverse transcription-PCR revealed that the wild-type virus had induced an ∼30-fold reduction in CCL21 mRNA expression, compared to only an ∼3-fold reduction for Δm131 (data not shown). Therefore, changes in spleen architecture induced by MCMV infection correlate with a steady decline in CCL21 production and a late increase in CCL2 production, both of which are moderated in the absence of m131/129 expression.
Activation of antiviral CTL is improved in the absence of m131/129 expression.
BALB/c mice limit MCMV infection by generating an effective antiviral CTL response. The immunodominant epitope recognized by CTL in these mice is derived from the IE1 protein (18). By using MHC class I (H-2Ld) tetramers containing this epitope, we enumerated and characterized the antigen-specific CD8+ T cell response to MCMV, as described in our previous studies (19, 20). IE1-specific CD8+ T cells were detectable in the spleen 4 days after MCMV infection, with the numbers being higher (∼3-fold) in mice infected with Δm131 than in mice infected with wild-type virus (Fig. 3A). By day 6, however, there were ∼2.5-fold more IE1-specific CD8+ T cells in mice infected with wild-type virus, though the total number had increased in both groups of mice (Fig. 3A). IE1-specific CD8+ T cells displayed an activated/effector phenotype in response to both viruses, where the majority of cells expressed CD69 and PD1, upregulated CD44, and lost CD62L expression in mice infected with either virus on day 4 (see Fig. S1 in the supplemental material). Interestingly, IE1-specific CD8+ T cells that were CD127− KLRG1+ were readily detectable on day 4 in mice infected with Δm131 but not those infected with wild-type virus, although by day 6 both groups exhibited similar numbers (see Fig. S1).
Fig 3.

Early control of recombinant MCMV lacking m131/129 is mediated by an improved CD8+ T cell response in the spleen. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. (A) Spleens were collected at the indicated times postinfection and prepared for flow cytometry to determine the frequency of CD8+ T cells that could bind IE1 tetramer. The absolute number of IE1-specific CD8+ T cells was calculated using the absolute cell count and is plotted as the mean ± SEM (n = 6 mice per group), with results from one of three representative independent experiments shown. (B) On the indicated days, mice were administered CFSE-labeled target cells loaded with IE1 peptide, and the proportion of specific killing was determined for the spleen 4 h later using flow cytometry. Values are plotted as the mean ± SEM (n = 6 mice per group). (C to E) BALB/c mice were either left undepleted (C), treated with three doses of anti-CD8 (D), or three doses of anti-CD4 (E), and then infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. Viral titers were determined in the spleens 6 days postinfection and plotted for each mouse per group. Results of one representative experiment of two independent experiments are shown. (F) BALB/c.CT6 congenic mice were either left undepleted or treated with three doses of anti-NK1.1 to deplete NK cells, then infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. Viral titers were determined in the spleen 6 days postinfection and are plotted for each mouse per group.
Next, we measured CTL activity against IE1 peptide-pulsed targets in an in vivo killing assay. CTL activity was higher in mice infected with Δm131 on day 4 postinfection (73.82% ± 1.044% versus 49.80% ± 1.792% [mean ± SEM]) (P < 0.0001) (Fig. 3B). Taken together, these data demonstrate that CD8+ T cell activation was faster and more efficient in mice infected with Δm131 than in mice infected with wild-type virus.
CD8+ T cells are responsible for early clearance of MCMV lacking m131/129.
In our previous study, we found that the improved control of Δm131 in the spleen was prevented by pretreatment with anti-asialo GM1 or a combination of antibodies depleting both CD4+ and CD8+ T cells (6). In order to better define the cell populations required for improved control of the Δm131 virus in the spleen, CD8+ or CD4+ T cells were individually depleted prior to infection with Δm131 or wild-type virus, and viral titers were determined at day 6 postinfection. As expected, Δm131 titers were over 10-fold lower than wild-type virus in undepleted mice (Fig. 3C). CD8 depletion completely abrogated the difference between Δm131 and wild-type titers (Fig. 3D). CD4 depetion did not alter Δm131 viral titers (Fig. 3E). The potential role of NK cells was examined using BALB/c.CT6 congenic mice, in whom NK cells can be specifically depleted by using an antibody against the NK1.1 determinant. In these mice, Δm131 titers were also over 10-fold lower than wild-type virus in the spleen on day 6, and NK cell depletion did not alter this phenotype (Fig. 3F). Thus, early control of Δm131 in the spleen can be attributed solely to CD8+ T cells.
Cytokine production is influenced by m131/129.
Cytokines that are produced during MCMV infection can have a profound effect on the course of the disease (17, 21–23). Since the course of infection was altered in the spleen by the absence of m131/129 expression, we compared the local expression of IFN-γ, IL-12, and IFN-α following infection with wild-type and Δm131 viruses.
BALB/c mice infected with wild-type virus exhibited a sharp peak of IFN-γ production on day 2 and a trough on day 3, followed by a second peak on day 5 (Fig. 4A). The IFN-γ response to Δm131 was identical to that to wild-type virus until day 5, when production plateaued rather than rising to a second peak (Fig. 4A). There was a single peak of IL-12 production on day 2 postinfection that was ∼2-fold higher for Δm131 than for wild-type virus and was sustained at later times postinfection (Fig. 4B). A peak of IFN-α production was observed at 40 h postinfection and was largely lost by 50 h (Fig. 4C). Both viruses elicited similar kinetics; however, IFN-α peak levels were ∼2-fold lower in response to infection with Δm131 (Fig. 4C). Therefore, wild-type MCMV expressing m131/129 induced high levels of IFN-α and moderate levels of IL-12 production during early infection, followed by rising levels of IFN-γ; in contrast, a stronger IL-12 response and weaker production of IFN-α and IFN-γ were elicited in the absence of m131/129 expression.
Fig 4.
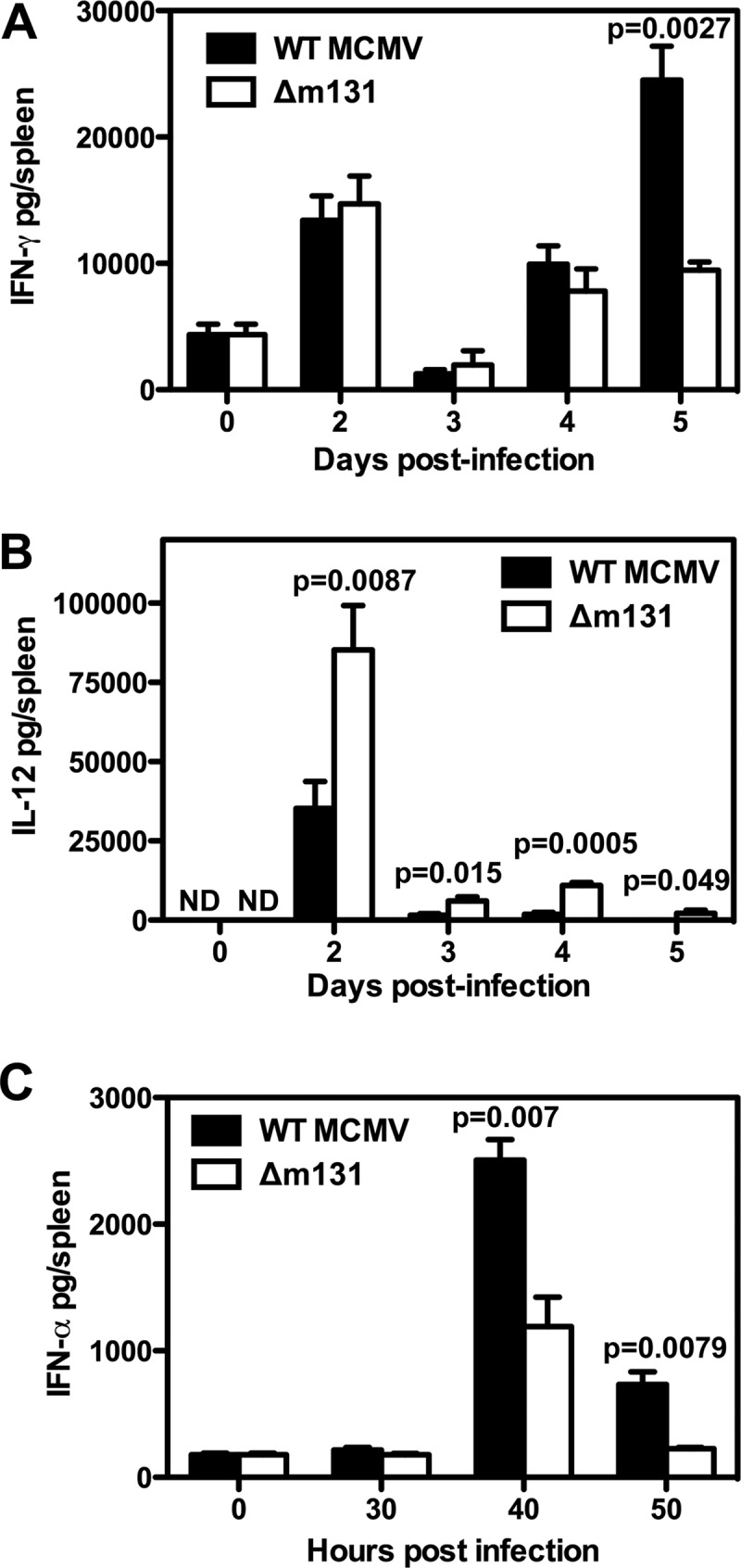
m131/129 modulates cytokine production in the spleen. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. Spleens were harvested at the indicated times and homogenized for cytokine detection by ELISA. (A) IFN-γ concentrations are plotted as the mean ± SEM (n = 3 to 6 mice). (B) IL-12 concentrations are plotted as the mean ± SEM (n = 3 to 6 mice). (C) IFN-α concentrations are plotted as the mean ± SEM (n = 5 to 7 mice).
The number of antigen-presenting cells is not influenced by m131/129.
In order to learn more about the effects of Δm131 on the spleen, we enumerated a range of cell types 2 to 4 days after infection. The number of conventional dendritic cells (CD11c+ MHC-IIhi) is not affected by infection with wild-type MCMV until day 3, when a 50% reduction in DC numbers was noted (Fig. 5A). pDC (CD11clo SiglecH+ CD11b−) were lost from the spleen within 2 days of infection with wild-type virus (Fig. 5B), as were red pulp macrophages (CD11clo CD11blo F4/80hi) and monocytes (CD11bhi CD115+ Ly6Glo) (Fig. 5C and D, respectively). Infection with Δm131 depleted antigen-presenting cell populations with the same kinetics and to the same extent as infection with the wild-type virus. We also found that both viruses had similar effects on NK cell and granulocyte numbers (data not shown).
Fig 5.

m131/129 does not influence the number of antigen-presenting cells in the spleen. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. Spleens were collected at the indicated time points and prepared for flow cytometry. (A) The number of cDC was calculated using the absolute cell count and the frequency of cells that were CD11chi MHC-IIhi. (B) The number of pDC was calculated using the absolute cell count and the frequency of cells that were CD11b− CD11cint SiglecH+. (C) The number of red pulp macrophages was calculated using the absolute cell count and the frequency of cells that were CD11blo CD11clo F4/80hi. (D) The number of monocytes was calculated using the absolute cell count and the frequency of cells that were CD11bhi CD115+ Ly6G−/lo.
m131/129 influences activation of both pDC and cDC.
Since infection with a virus lacking m131/129 led to significant differences in early cytokine production, particularly IFN-α and IL-12, along with enhanced recruitment of antiviral CD8+ T cells, we investigated activation and cytokine production in pDC and cDC. Both viruses stimulated upregulation of CD86 on the surface of pDC and cDC to a similar extent at 40 h postinfection (Fig. 6A). Similarly, we observed no differences in the upregulation of CD40 (data not shown). We then examined the production of type I interferons by using a combination of three monoclonal antibodies against IFN-α and IFN-β and intracellular cytokine detection. At 40 h postinfection, IFN-α/β was only detected in pDC (data not shown), and the number of pDC producing IFN-α/β was significantly higher (P = 0.0003) in spleens of mice infected with wild-type MCMV compared to Δm131 (Fig. 6B), as was the frequency of pDC producing IFN-α/β (WT, 16.57 ± 1.396% [n = 6]; Δm131, 8.695 ± 0.6365% [n = 4]). We also found that the frequency (WT, 19.23 ± 2.290% [n = 6]; Δm131, 10.47 ± 1.439% [n = 4]) and number of pDC producing IL-12 were similarly elevated in response to wild-type MCMV (Fig. 6C). In contrast, there was no difference in the number of cDC producing IL-12 (Fig. 6D), although the frequency of IL-12-producing cDC was mildly elevated at this time point in mice infected with Δm131 (WT, 5.352 ± 0.3170% [n = 6]; Δm131, 7.538 ± 0.8932% [n = 4]; P = 0.0271). These results demonstrate that expression of m131/129 directly enhances both IFN-I and IL-12 production by pDC during acute infection. In addition, enhanced IFN-I production coincided with significantly lower titers of the wild-type virus in the spleen 40 h postinfection (Fig. 6E). Taken together, these data indicated m131/129 directly enhances the antiviral response of pDC in the spleen, leading to better control of early replication of MCMV.
Fig 6.

m131/129 influences the activation of pDC and cDC. BALB/c mice were infected with 5 × 103 PFU wild-type MCMV or Δm131 i.p. (A to D) Spleens were collected 40 h postinfection, and the expression of CD86 (as measured by mean fluorescence) was determined for pDC, CD8α+ cDC, and CD11b+ cDC in the spleen by using flow cytometry (A). At the same time, the frequencies of pDC producing IFN-α and IL-12 were determined using intracellular cytokine staining to calculate the number of IFN-α+ pDC (B) and IL-12+ pDC (C) based on the absolute cell count. (D) The number of cDC producing IL-12 was also calculated. (E) Spleens were harvested at 40 h postinfection and homogenized for use in a plaque assay. Viral titers are plotted as the mean ± SEM (n = 7 mice). (F and G) Spleens were collected 50 h postinfection, and the expression levels of CD80 and CD86 (as measured by mean fluorescence intensity [MFI]) were determined for CD11b+ and CD8α+ cDC in the spleen (F). At the same time, the frequencies of CD11b+ and CD8α+ cDC producing IL-12 were determined using intracellular cytokine staining. The numbers of CD11b+ and CD8α+ cDC that were IL-12+ were calculated from the absolute cell count. All values are plotted as the mean ± SEM (n = 3 to 5 mice per group), and results from one representative experiment of at least two independent experiments are shown.
We then went on to examine cDC activation after the peak of IFN-α production (50 h postinfection) and found higher levels of CD80 and CD86 were expressed by CD8α+ cDC, but not CD11b+ cDC, in mice infected with Δm131 compared to those infected with wild-type virus (Fig. 6F). We also detected a higher number of IL-12+ cells among both CD8α+ and CD11b+ cDC in mice infected with Δm131 (Fig. 6G). Therefore, cDC, particularly CD8α+ cDC, appear to be activated more efficiently by Δm131 than wild-type virus during acute infection, potentially accounting for the improved antiviral CD8+ T cell response observed in the absence of m131/129.
DISCUSSION
The chemokine-like protein m131/129 was originally identified for its effects on inflammatory cell recruitment and dissemination of MCMV (6–8). In this report, we describe a novel effect of this viral protein on CD8+ T cell activation via interplay with the innate response to the virus. A recombinant MCMV encoding a mutation within the m131/129 open reading frame productively infects the spleen and replicates as efficiently as wild-type virus, but infection is controlled more rapidly. We demonstrated that the early control of Δm131 infection of the spleen can be attributed to the faster generation of virus-specific CD8+ T cell responses. Improved T cell responses were associated with increased activation of cDC in the context of a weaker IFN-α response by pDC. This is the first time the m131/129 viral protein has been shown to enhance IFN-I production by pDC and influence DC and CD8+ T cell activation, thereby accounting for its effect on the efficacy of the adaptive immune response to MCMV.
An accelerated CD8+ T cell response against MCMV has been reported in Cmv1r congenic BALB/c mice that express the Ly49H NK cell receptor derived from C57BL/6 mice (23). Ly49H facilitates recognition and killing of MCMV-infected cells by NK cells and dramatically improves control of the virus in the spleen and lung (11, 24–26). Under these conditions, where NK cells mediate a reduction in early viral load, Cmv1r congenic BALB/c mice were found to produce significantly lower levels of both IFN-α and IL-12 than wild-type BALB/c mice after MCMV infection. Cmv1r congenic BALB/c mice also exhibited faster activation of CD8+ T cells, which was associated with the conservation of DC numbers in the spleen (23). The authors of that study concluded that NK cells promoted the CD8+ T cell response to MCMV by limiting the early viral load, which in turn limited the production of IFN-α, thereby ensuring DC populations were preserved (23). Our data for Δm131 infection of wild-type BALB/c (i.e., lacking Ly49H) demonstrated a reduction in IFN-α production despite a high viral load, a finding that has been confirmed using an independent mutant virus lacking m131 expression generated by the Mocarski laboratory (L. Daley-Bauer and E. Mocarski, personal communication). Furthermore, we found that a qualitative, rather than a quantitative, difference in the DC compartment of the spleen was associated with an accelerated CD8+ T cell response against MCMV.
Mocarski and colleagues recently reported that inflammatory monocytes recruited by m131/129 suppress the CD8+ T cell response to MCMV 7 days after infection (27). We have now shown that m131/129 also influences early CD8+ T cell activation (from day 4 postinfection), and this occurs in the absence of monocyte recruitment to the spleen. It should be noted that our results are not directly comparable with those of Daley-Bauer et al. (27), since we employed a different route of infection (intraperitoneal versus subcutaneous). Nevertheless, when taken together, these results indicate that MCMV is capable of modulating both the initiation and the duration of the antiviral CD8+ T cell response.
Type I IFNs are crucial for host survival, with the lethal dose for MCMV infection being around 100-fold lower in mice lacking IFN-α/β receptors (28). Three phases of IFN-I production have been identified following MCMV infection; the first phase is characterized by an IFN-I peak at 6 to 8 h, the second phase peaks at around 36 h postinfection, and a third, smaller peak is detected at 48 h (29–33). Stromal cells are largely responsible for IFN-I production in the spleen during the first phase (33), pDC are the major source of IFN-I at 36 h (21, 34, 35), and the third phase is attributed to cDC (30, 31, 36). In addition, IFN-α production at this time point relies on Toll-like receptor signals (30, 31). Our results support this notion, since IFN-I was only detected in pDC at 40 h postinfection. Importantly, expression of m131/129 directly influenced the number of pDC producing IFN-I during the second phase, and therefore the magnitude of the IFN-I response. Further investigation of the interaction of MCMV with pDC and the role of m131/129 is likely to provide further insight into the mechanisms that regulate IFN-I production in response to viral infection.
Recent work by Benedict and colleagues has provided more detail on the induction of the first phase of IFN-I production in the spleens of C5BL/6 mice during MCMV infection and has demonstrated that early production of IFN-I does not require Toll-like receptor signals, which is in contrast to the second phase. Furthermore, early IFN-I production depends on lymphotoxin-α/β production by B cells, while the late phase is unaffected. Finally, stromal cells are responsible for the bulk of early IFN-I production (33, 37, 38). These findings have led to the suggestion that MCMV may target the lymphotoxin pathway; however, it is unclear how the virus exerts its effect on lymphotoxin production by B cells. When we examined serum and spleen samples from BALB/c mice 8 h postinfection, we could not detect IFN-α by using a commercial ELISA (data not shown), suggesting that the early phase is either much weaker or lacking in MCMV-susceptible mouse strains. However, we did find evidence of IFN-I production, since CD69 was upregulated on NK and T cells in the spleen, a phenotype that can be attributed to the nonspecific effects of IFN-α (39), and there was no difference between Δm131 and wild-type virus at 30 h postinfection (data not shown). Therefore, m131/129 appears to influence only the Toll-receptor-dependent phase of IFN-I production.
Once produced, IFN-α acts quickly to promote NK cell cytotoxicity, an activity required for early control of MCMV in mouse strains that express Ly49H (e.g., C57BL/6) (29, 31, 32, 40). IFN-I also activates NK cells in MCMV-susceptible mouse strains (e.g., BALB/c) (reference 31 and our unpublished results). Interestingly, the type I interferons can contribute to early control of MCMV in BALB/c mice, since administration of low oral doses of IFN-α/β prior to infection reduced early viral titers in the spleen and liver (41), probably by directly inhibiting viral replication (42). Thus, depending on the dose and timing of secretion, IFN-I can have a profound effect on the course of the host response to MCMV. We observed that activation of cDC and the antiviral CD8+ T cell response was improved in the context of a weaker IFN-I response, although it is not clear how these events are related. Δm131 replicates better than wild-type virus in the spleen 40 h after infection, which is likely to be a consequence of the weaker IFN-I response. Higher titers of Δm131 could provide greater stimulation for local cDC, accounting for the increase in costimulatory molecule expression and IL-12 production, leading to earlier activation of CD8+ T cells. In this scenario, m131/129 appears to protect the virus, because it induces more IFN-I during the early stages of infection.
Previous experiments demonstrated that MCMV induces a breakdown in the architecture of the spleen during acute infection involving the downregulation of CCL21 expression (43). This phenomenon occurred in both MCMV-susceptible (BALB/c) and -resistant (C57BL/6) mouse strains, and the extent of tissue disruption increased with viral load (43). Subsequent experiments by Mueller et al. demonstrated that the organization of lymphoid organs breaks down in a variety of infections, including those caused by vaccinia virus, influenza virus, and Listeria monocytogenes, and determined that these changes are caused by IFN-γ-dependent inhibition of CCL21 production (44). In agreement with these data, we found that an increase in IFN-γ production on day 5 postinfection was associated with a marked reduction in CCL21 and obvious disorganization of the spleen in mice infected with wild-type MCMV. Similarly, the initial decline in CCL21 production observed on day 2 postinfection also coincided with a peak of IFN-γ production in the spleen. At this point (days 2 to 4 postinfection), though, there was little difference in IFN-γ production in mice infected with either virus, yet downregulation of CCL21 was at least 30% greater for wild-type virus than Δm131. Thus, an additional mechanism may contribute to the inhibition of CCL21 production and the breakdown in splenic architecture observed after infection with wild-type virus (i.e., in the presence of m131/129 expression).
Our understanding of the function of m131/129 is far from complete. Previous studies have established that a homologue of m131/129 is present in rat CMV (45) but absent from guinea pig CMV (GPCMV) (46) and HCMV. Despite the absence of sequence homology, the genes located in this region of the CMV genome may encode functional homologues and, in this regard, there are some striking similarities between MCMV, GPCMV, and HCMV. For example, the GPCMV genes that correspond to m131/129 are critical for viral dissemination and high titers in the salivary glands (46). In addition, one of the GPCMV genes in this region shares weak homology with UL130, a chemokine-like protein crucial for HCMV entry into epithelial and endothelial cells and subsequent transmission to leukocytes (47–49), which is thought to assist dissemination of the virus. Interestingly, this coding region is highly conserved in MCMV, GPCMV, and HCMV passaged in vivo, yet it is lost when viruses are propagated in vitro (46, 50–52). Such similarities raise the possibility that m131/129 plays a role in cell entry and/or influence cell tropism for MCMV, either on its own or in combination with other proteins encoded by neighboring genes, as has been observed for UL130 (49). To this end, it is worth noting that early titers of Δm131 were higher than wild-type virus in the spleen, and thus m131/129 is not required by MCMV to establish infection in the spleen. However, m131/129 may be important for spread of the virus to other cell types, and this may account for its effect of IFN-α production during the second phase of production.
Our appreciation for the role of chemokines in immune function has grown over the last decade, and with it has come a greater understanding of viral infection and immune evasion strategies (53, 54). The data presented in this paper highlight the success of this strategy for viruses. Importantly, our data demonstrate that the effects of IFN-α on CD8+ T cell activation and CTL function can be mediated by a direct effect on the activation status of DC. These results also deepen our understanding of the potential effects of type I interferons on adaptive immunity, and thus they will be relevant to the design of therapeutic strategies for both CMV and other viral infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by fellowships and grants from the National Health and Medical Research Council of Australia (Canberra).
We thank Hyacinth Tabarias for technical assistance and Simone Ross, Helen Molder, and Kelly Hunt for assistance with animal care.
Footnotes
Published ahead of print 8 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00187-13.
REFERENCES
- 1. Horst D, Verweij MC, Davison AJ, Ressing ME, Wiertz EJ. 2011. Viral evasion of T cell immunity: ancient mechanisms offering new applications. Curr. Opin. Immunol. 23:96–103 [DOI] [PubMed] [Google Scholar]
- 2. Lanier LL. 2008. Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 8:259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8:911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller-Kittrell M, Sparer TE. 2009. Feeling manipulated: cytomegalovirus immune manipulation. Virol. J. 6:4. 10.1186/1743-422X-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beutler B, Georgel P, Rutschmann S, Jiang Z, Croker B, Crozat K. 2005. Genetic analysis of innate resistance to mouse cytomegalovirus (MCMV). Brief. Funct. Genomic. Proteomic. 4:203–213 [DOI] [PubMed] [Google Scholar]
- 6. Fleming P, Davis-Poynter N, Degli-Esposti M, Densley E, Papadimitriou J, Shellam G, Farrell H. 1999. The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J. Virol. 73:6800–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES. 2001. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J. Virol. 75:9966–9976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES. 1999. Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc. Natl. Acad. Sci. U. S. A. 96:10881–10886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. MacDonald MR, Burney MW, Resnick SB, Virgin HI. 1999. Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J. Virol. 73:3682–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, Mocarski ES. 2006. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood 107:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scalzo AA, Brown MG, Chu DT, Heusel JW, Yokoyama WM, Forbes CA. 1999. Development of intra-natural killer complex (NKC) recombinant and congenic mouse strains for mapping and functional analysis of NK cell regulatory loci. Immunogenetics 49:238–241 [DOI] [PubMed] [Google Scholar]
- 12. Allan JE, Shellam GR. 1984. Genetic control of murine cytomegalovirus infection: virus titres in resistant and susceptible strains of mice. Arch. Virol. 81:139–150 [DOI] [PubMed] [Google Scholar]
- 13. Hsu KM, Pratt JR, Akers WJ, Achilefu SI, Yokoyama WM. 2009. Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J. Gen. Virol. 90:33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keil GM, Fibi MR, Koszinowski UH. 1985. Characterization of the major immediate-early polypeptides encoded by murine cytomegalovirus. J. Virol. 54:422–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barber DL, Wherry EJ, Ahmed R. 2003. Cutting edge: rapid in vivo killing by memory CD8 T cells. J. Immunol. 171:27–31 [DOI] [PubMed] [Google Scholar]
- 16. Kohler RE, Caon AC, Willenborg DO, Clark-Lewis I, McColl SR. 2003. A role for macrophage inflammatory protein-3 alpha/CC chemokine ligand 20 in immune priming during T cell-mediated inflammation of the central nervous system. J. Immunol. 170:6298–6306 [DOI] [PubMed] [Google Scholar]
- 17. Sumaria N, van Dommelen SL, Andoniou CE, Smyth MJ, Scalzo AA, Degli-Esposti MA. 2009. The roles of interferon-gamma and perforin in antiviral immunity in mice that differ in genetically determined NK-cell-mediated antiviral activity. Immunol. Cell Biol. 87:559–566 [DOI] [PubMed] [Google Scholar]
- 18. Reddehase MJ, Rothbard JB, Koszinowski UH. 1989. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 337:651–653 [DOI] [PubMed] [Google Scholar]
- 19. Andrews DM, Andoniou CE, Fleming P, Smyth MJ, Degli-Esposti MA. 2008. The early kinetics of cytomegalovirus-specific CD8+ T-cell responses are not affected by antigen load or the absence of perforin or gamma interferon. J. Virol. 82:4931–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, Fleming P, Tabarias H, Hill GR, van der Most RG, Scalzo AA, Smyth MJ, Degli-Esposti MA. 2010. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J. Exp. Med. 207:1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. 2003. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 197:885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heise MT, Virgin HW, IV 1995. The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J. Virol. 69:904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, Sacher T, Tomasello E, Vivier E, Koszinowski UH, Dalod M. 2007. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 3:e123. 10.1371/journal.ppat.0030123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM. 2001. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292:934–937 [DOI] [PubMed] [Google Scholar]
- 25. Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. 2001. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2:951–956 [DOI] [PubMed] [Google Scholar]
- 26. Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. 1990. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J. Exp. Med. 171:1469–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daley-Bauer LP, Wynn GM, Mocarski ES. 2012. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity 37:122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Presti RM, Pollock JL, Dal Canto AJ, O'Guin AK, Virgin HW, IV 1998. Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J. Exp. Med. 188:577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 175:6723–6732 [DOI] [PubMed] [Google Scholar]
- 31. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21:107–119 [DOI] [PubMed] [Google Scholar]
- 32. Orange JS, Biron CA. 1996. Characterization of early IL-12, IFN-αβ, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 156:4746–4756 [PubMed] [Google Scholar]
- 33. Schneider K, Loewendorf A, De Trez C, Fulton J, Rhode A, Shumway H, Ha S, Patterson G, Pfeffer K, Nedospasov SA, Ware CF, Benedict CA. 2008. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe 3:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. 2010. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33:955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zucchini N, Bessou G, Robbins SH, Chasson L, Raper A, Crocker PR, Dalod M. 2008. Individual plasmacytoid dendritic cells are major contributors to the production of multiple innate cytokines in an organ-specific manner during viral infection. Int. Immunol. 20:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. 2005. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 6:1011–1019 [DOI] [PubMed] [Google Scholar]
- 37. Banks TA, Rickert S, Benedict CA, Ma L, Ko M, Meier J, Ha W, Schneider K, Granger SW, Turovskaya O, Elewaut D, Otero D, French AR, Henry SC, Hamilton JD, Scheu S, Pfeffer K, Ware CF. 2005. A lymphotoxin-IFN-beta axis essential for lymphocyte survival revealed during cytomegalovirus infection. J. Immunol. 174:7217–7225 [DOI] [PubMed] [Google Scholar]
- 38. Benedict CA, Banks TA, Senderowicz L, Ko M, Britt WJ, Angulo A, Ghazal P, Ware CF. 2001. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus detente. Immunity 15:617–626 [DOI] [PubMed] [Google Scholar]
- 39. Kranzer K, Bauer M, Lipford GB, Heeg K, Wagner H, Lang R. 2000. CpG-oligodeoxynucleotides enhance T-cell receptor-triggered interferon-gamma production and up-regulation of CD69 via induction of antigen-presenting cell-derived interferon type I and interleukin-12. Immunology 99:170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA. 2002. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169:4279–4287 [DOI] [PubMed] [Google Scholar]
- 41. Bosio E, Beilharz MW, Watson MW, Lawson CM. 1999. Efficacy of low-dose oral use of type I interferon in cytomegalovirus infections in vivo. J. Interferon Cytokine Res. 19:869–876 [DOI] [PubMed] [Google Scholar]
- 42. Sainz B, Jr, LaMarca HL, Garry RF, Morris CA. 2005. Synergistic inhibition of human cytomegalovirus replication by interferon-alpha/beta and interferon-gamma. Virol. J. 2:14. 10.1186/1743-422X-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benedict CA, De Trez C, Schneider K, Ha S, Patterson G, Ware CF. 2006. Specific remodeling of splenic architecture by cytomegalovirus. PLoS Pathog. 2:e16. 10.1371/journal.ppat.0020016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, Ahmed R, Matloubian M. 2007. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science 317:670–674 [DOI] [PubMed] [Google Scholar]
- 45. Kaptein SJ, van Cleef KW, Gruijthuijsen YK, Beuken EV, van Buggenhout L, Beisser PS, Stassen FR, Bruggeman CA, Vink C. 2004. The r131 gene of rat cytomegalovirus encodes a proinflammatory CC chemokine homolog which is essential for the production of infectious virus in the salivary glands. Virus Genes 29:43–61 [DOI] [PubMed] [Google Scholar]
- 46. Nozawa N, Yamamoto Y, Fukui Y, Katano H, Tsutsui Y, Sato Y, Yamada S, Inami Y, Nakamura K, Yokoi M, Kurane I, Inoue N. 2008. Identification of a 1.6 kb genome locus of guinea pig cytomegalovirus required for efficient viral growth in animals but not in cell culture. Virology 379:45–54 [DOI] [PubMed] [Google Scholar]
- 47. Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78:10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 70:78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jordan S, Krause J, Prager A, Mitrovic M, Jonjic S, Koszinowski UH, Adler B. 2011. Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr shows reduced growth in salivary glands due to a fixed mutation of MCK-2. J. Virol. 85:10346–10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith LM, Shellam GR, Redwood AJ. 2006. Genes of murine cytomegalovirus exist as a number of distinct genotypes. Virology 352:450–465 [DOI] [PubMed] [Google Scholar]
- 53. Alcami A, Lira SA. 2010. Modulation of chemokine activity by viruses. Curr. Opin. Immunol. 22:482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seet BT, McFadden G. 2002. Viral chemokine-binding proteins. J. Leukoc. Biol. 72:24–34 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.