

Abstract
Severe acute respiratory syndrome (SARS) is an infectious and highly contagious disease that is caused by SARS coronavirus (SARS-CoV) and for which there are currently no approved treatments. We report the discovery and characterization of small-molecule inhibitors of SARS-CoV replication that block viral entry by three different mechanisms. The compounds were discovered by screening a chemical library of compounds for blocking of entry of HIV-1 pseudotyped with SARS-CoV surface glycoprotein S (SARS-S) but not that of HIV-1 pseudotyped with vesicular stomatitis virus surface glycoprotein G (VSV-G). Studies on their mechanisms of action revealed that the compounds act by three distinct mechanisms: (i) SSAA09E2 {N-[[4-(4-methylpiperazin-1-yl)phenyl]methyl]-1,2-oxazole-5-carboxamide} acts through a novel mechanism of action, by blocking early interactions of SARS-S with the receptor for SARS-CoV, angiotensin converting enzyme 2 (ACE2); (ii) SSAA09E1 {[(Z)-1-thiophen-2-ylethylideneamino]thiourea} acts later, by blocking cathepsin L, a host protease required for processing of SARS-S during viral entry; and (iii) SSAA09E3 [N-(9,10-dioxo-9,10-dihydroanthracen-2-yl)benzamide] also acts later and does not affect interactions of SARS-S with ACE2 or the enzymatic functions of cathepsin L but prevents fusion of the viral membrane with the host cellular membrane. Our work demonstrates that there are at least three independent strategies for blocking SARS-CoV entry, validates these mechanisms of inhibition, and introduces promising leads for the development of SARS therapeutics.
INTRODUCTION
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the causative agent of SARS, a life-threatening viral respiratory illness which emerged from Southern China in November 2002 and spread to other parts of the world, including North America, South America, and Europe (1, 2). The World Health Organization estimated the mortality rate of SARS to be up to 15% (3–5), and there is currently no approved therapeutic for the treatment of SARS infections. While SARS is currently not a public threat, the recent outbreak of a new human coronavirus (hCoV-EMC) (6) and the possibility of future outbreaks of infections with both SARS-CoV and other related viruses warrant continuous research for discovery of antiviral therapies.
Viral entry is an essential step of the virus life cycle that can be targeted for therapy (7). Compounds that can inhibit the entry of several viruses have been identified. RFI 641 and VP-14637 are small molecules that inhibit the entry of respiratory syncytial virus by binding at a hydrophobic pocket of the fusion (F) glycoprotein (8, 9). For human immunodeficiency virus (HIV), a number of inhibitors have been shown to block HIV entry by using diverse strategies. For example, maraviroc is a small-molecule anti-HIV drug that targets CCR5, a host protein used as a coreceptor during HIV entry (10–12). Enfuvirtide and SC29EK are peptides that are also used to block viral entry by binding to the viral transmembrane protein gp41 and blocking the final stage of fusion with the target cell (13–18). There are also several monoclonal antibodies that are currently in clinical trials, including KD-247 and PRO140, which block HIV entry by binding either the viral surface glycoprotein gp120 (19, 20) or the CCR5 coreceptor (21, 22).
The surface glycoprotein of SARS-CoV, SARS-S, comprises two components: S1, which contains the receptor binding domain (RBD); and S2, which contains the fusion peptide. SARS-CoV gains entry into permissive cells through interactions of the SARS-S RBD with the cell surface receptor angiotensin converting enzyme 2 (ACE2) (23, 24). These interactions are followed by endocytosis, and at the low pH in endosomes, SARS-S is cleaved by a cellular protease called cathepsin L, thereby exposing the S2 domain of the spike protein for membrane fusion (25–30). Previous studies have also shown that fusion of SARS-S-expressing cells with ACE2 receptor-expressing cells can also take place by a pH-independent mechanism at the cell surface (31, 32). SARS-S also regulates cell stress responses and apoptosis (33). Early studies have identified some SARS-CoV entry and replication inhibitors (34–40). Nonetheless, to date, there are no approved drugs for the treatment of SARS-CoV infection.
In this study, we used a cell-based assay to screen the Maybridge HitFinder small-molecule library of compounds to identify inhibitors of SARS-CoV entry. This chemical library contains ∼14,000 compounds that follow Lipinski's rule of five, an empirical rule that is used to evaluate “drug-likeness” and potential for oral bioavailability in humans (41, 42). We report the discovery of small molecules from that library that block SARS-CoV entry by three different strategies: (i) inhibition of early SARS-S–ACE2 interactions, (ii) inhibition of cathepsin L, and (iii) entry inhibition by blocking fusion.
MATERIALS AND METHODS
Cell lines and plasmids.
pCAGGS SARS-CoV Spike, pNL-4.3-luc, and pCDNA 3.1-ACE2 were provided by Paul Bates (University of Pennsylvania) (43). The pCDNA 3.1 soluble ACE2 plasmid was provided by S. Pohlmann (Institute of Virology, Hannover, Germany). A plasmid encoding the SARS-CoV spike RBD with the signal sequence of CD5 and the Fc domain of human IgG1, pCDM8-Spike RBD-Fc, was provided by Michael Farzan (New England Primate Research Center) (29, 44). pMDG-VSV-G was provided by Marc Johnson (University of Missouri, Columbia, MO) (45). pSV2tat72 was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Alan Frankel (46). TZM-bl cells were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc. (15, 47–50). 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 10 μg streptomycin (43).
Infection assay with pseudotyped virus.
293T cells were cotransfected with pCAGGS SARS-S DNA or pMDG-VSV-G DNA together with pNL-4.3-Luc-E−R− by using calcium phosphate transfection reagents (43). After 48 h of incubation at 37°C and 5% CO2, the supernatants, containing virions pseudotyped with SARS-S or the vesicular stomatitis virus glycoprotein (VSV-G), were harvested and stored at −80°C. For infection experiments, 293T cells transiently transfected with pCDNA 3.1-ACE2 were seeded into 96-well plates at a density of 1 × 104 cells/well. After 15 min, equal volumes of the viral supernatant were added to the wells with the cells, and the mixtures were incubated for another 48 h. Bright-Glo luciferase reagent (Promega, Madison, WI) was added to the cells, and the luciferase activity was determined using a Veritas microplate luminometer (Turner Veritas Biosystems). To determine the amount of pseudotyped virus to be used for screening and identification of SARS-CoV entry inhibitors, we titrated the viral supernatant (1 to 50 μl) and the ACE2-expressing 293T cells, and the amount of viral supernatant that gave ∼25,000 relative luciferase units was chosen for use in infection assays. The multiplicity of infection (MOI) in the pseudotype-based assays was estimated to be 10 U/cell by using an enzyme-linked immunosorbent assay (ELISA) for the detection of HIV p24. Experiments performed below this MOI gave inconsistent results with the pseudotype-based assay. To determine the MOI, six 5-fold serial dilutions of 50 μl of the pseudotyped viral stock were used to infect 1 × 104 ACE2/293T cells in a 96-well plate in quadruplicate, with noninfected ACE2/293T cells as a control. After 48 h, the supernatant in each well was replaced with a lysis buffer (137 mM NaCl, 1.94 mM K3PO4, 8.06 mM Na3PO4, and 2.7 mM KCl, pH 7.4, or phosphate-buffered saline [PBS] and 10% Triton X-100), and the p24 antigen in each sample was subsequently measured using an ELISA kit (Organon Teknika). The 50% tissue culture infective dose (TCID50) was calculated according to the Spearman-Karber formula, as follows: 1/TCID50 = Be, where e is the exponent for the reciprocal titer and B is the fold dilution used in the dilution series (http://www.europrise.org/documents/NEUTNET/SOPS/11_NHRBC_PBMC.pdf). After determination of the TCID50 of the viral stock (TCID50/ml), the TCID50 titer was then converted to the estimated number of infectious units per volume of virus material (U/ml) (similar to PFU/ml in a plaque assay) by multiplying the titer by 0.7 (51). To obtain the MOI in U/cell, the number of infectious particles was divided by the number of cells to be infected. For the purpose of screening to identify inhibitors of SARS-CoV entry, the compounds were incubated with ACE2-expressing 293T cells for 45 min, followed by addition of the appropriate amount of viral supernatant containing 100 TCID50 (MOI of 10 U/cell). The cells were further incubated for 48 h, followed by measurement of the luciferase activity using a Veritas microplate luminometer (Turner Veritas Biosystems).
Effects of inhibitors on cathepsin L and cathepsin B activity.
Purified recombinant cathepsin L (2 units) was incubated at 37°C with a 25 μM concentration of the fluorogenic substrate Z-Phe-Arg-7-amido-4-methylcoumarin and assay buffer (Calbiochem) in the presence or absence of various concentrations of the compounds (2 to 80 μM), in a total reaction volume of 200 μl. The amount of fluorescence produced as a result of the release of amido-4-methylcoumarin (AMC) from the substrate was measured using a fluorimeter at an excitation wavelength of 380 nm and an emission wavelength of 460 nm. The data obtained were analyzed using GraphPad Prism 5.0 software (GraphPad, Inc.). To monitor cathepsin B activity, purified recombinant cathepsin B (2 units) was incubated at 37°C with a 25 μM concentration of the fluorogenic substrate Z-Arg-Arg-7-amido-4-methylcoumarin and assay buffer (Calbiochem) in the presence or absence of CA074 (cathepsin B inhibitor) and SSAA09E1, in a total reaction volume of 200 μl. The amount of fluorescence produced as a result of the release of AMC from the substrate was measured using a fluorimeter with an excitation wavelength of 380 nm and an emission wavelength of 460 nm. The data obtained were analyzed using GraphPad Prism 5.0 software (GraphPad, Inc.).
Expression and purification of SARS-CoV spike receptor binding domain.
Expression and purification of the SARS-CoV spike receptor binding domain were carried out as described previously (29). Briefly, 293T cells were transfected with pCDM8-Spike RBD-Fc, a plasmid encoding the SARS-S RBD with the signal sequence of CD5 and the Fc domain of human IgG1. Twenty-four hours after transfection, cells were washed in PBS and then incubated in 293 serum-free medium II (Invitrogen). After 48 h, the medium was harvested and the protein was precipitated using protein A Sepharose beads at 4°C for 16 h. Beads were then washed in PBS with 0.5 M NaCl, and the bound protein was eluted with 50 mM sodium citrate-50 mM glycine, pH 2.0, and neutralized to pH 7.0 with 5 M sodium hydroxide. Purified proteins were concentrated with Millipore Amicon Ultra centrifugal filter units (nominal molecular weight limit [NMWL], 10,000) and then dialyzed against PBS.
Immunoprecipitation and Western blotting.
Soluble ACE2 protein was obtained from the supernatant of transiently transfected 293T cells, followed by dialysis against PBS and protein concentration using a Centricon Plus Ultrafilter with a 10-kDa molecular mass cutoff (Millipore, St. Louis, MO) (26). Soluble ACE2 protein and the purified receptor binding domain of the spike protein (RBD-Fc) were incubated in the presence of various concentrations of the inhibitors (0, 5, 10, and 20 μM) for 2 h at 4°C, followed by the addition of protein A Sepharose slurry to the mixture. The samples were incubated for an additional hour and then separated by 10% PAGE, followed by transfer to nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). ACE2 protein and SARS-S RBD-Fc were detected using a goat anti-human polyclonal ACE2 antibody (LifeSpan Biosciences, Inc.) with rabbit anti-goat–horseradish peroxidase (HRP) as the secondary antibody (Millipore, St. Louis, MO) and anti-human IgG–HRP (Millipore, St. Louis, MO), respectively.
Binding inhibition and flow cytometry.
Binding inhibition and flow cytometry were performed as described previously (29). Approximately 1 × 106 ACE2-transfected 293T cells were detached in PBS with 5 mM EDTA and washed with PBS with 0.5% bovine serum albumin (BSA). Various concentrations of the compounds (0, 5, 10, and 20 μM) were added to the cells and then allowed to incubate on ice for 30 min, followed by addition of the purified RBD-Fc, and then the mixture was incubated on ice for 1 h. The cells were washed 3 times with PBS and 0.5% BSA and then incubated with anti-human IgG–fluorescein isothiocyanate (FITC) conjugate (Sigma). Cells were washed again 3 times with PBS and 0.5% BSA. The cells were then fixed with 4% paraformaldehyde and analyzed by flow cytometry at the University of Missouri Cell and Immunobiology Core Facility to determine the percentage of cells bound to the spike receptor binding domain in the presence and absence of the inhibitors.
Quantitative cell-to-cell fusion assay.
TZM-bl cells that stably express luciferase under the control of the HIV-1 promoter when activated by Tat protein (15, 47–50) were transfected with pcDNA 3.1-ACE2 plasmid. Simultaneously, 293T cells were cotransfected with plasmids encoding wild-type SARS-S and Tat cDNA (46). All transfections were done with the Lipofectamine 2000 (Invitrogen) transfection reagent. Forty-eight hours after transfection, the TZM-bl cells were seeded into 96-well plates at a density of 1 × 104 cells/well. The ACE2-expressing TZM-bl cells were then overlaid with the same amount of 293T cells expressing SARS-S envelope and Tat protein (1 × 104 cells/well). Dimethyl sulfoxide (DMSO) or compounds were added to the TZM-bl cells either 45 min prior to the overlay with 293T cells or 2 h after the overlay with 293T cells. Three hours after the overlay, cells were treated with serum-free medium containing 2 μg/ml trypsin to induce fusion and replaced with complete DMEM after 30 min. Six hours after fusion induction, the cells were monitored or analyzed for the expression of luciferase by using the Bright-Glo luciferase reagent and a Veritas microplate luminometer.
SARS-CoV cytopathic effect assay.
The assay for the identification of potential inhibitors of SARS-CoV entry was performed essentially as described previously (52). Briefly, Vero E6 cells were dispensed into black, clear-bottom, 96-well plates at a density of 10,000 cells/well in 50 μl DMEM with phenol red, supplemented with 10% FBS and 1% l-glutamine, by use of a WellMate microplate dispenser (Matrix, Hudson, NH) and then were incubated for 24 h at 37°C and 5% CO2, with high humidity. For dose-response and compound 50% effective concentration (EC50) determinations, 25-μl aliquots of various concentrations of the compounds (0.052 μM to 120 μM) were added for final plate well concentrations ranging from 0.013 μM to 30 μM and a final DMSO concentration of 0.5%. Eight concentrations of each compound were added to 96-well plates in triplicate to measure the effective concentration at which each compound inhibited viral cytopathic effect (CPE) by 50% (EC50) or in duplicate, in the absence of virus, to measure the inhibitory concentration at which growth was inhibited by 50% (50% cytotoxic concentration [CC50]). The plates were transported to a biosafety level 3 (BSL3) facility (Southern Research Institute, AL), where they were infected with 25 μl of diluted Toronto-2 strain of SARS-CoV (a gift of Heinz Feldman) at a concentration of 100 TCID50 by use of a WellMate microplate dispenser (MOI of 0.1 PFU/cell). Internal controls consisted of wells containing cells only (cell control), cells infected with virus (virus control), and virus-infected cells treated with calpain inhibitor IV. Plates were then allowed to incubate at 37°C and 5% CO2 for 72 h. After incubation, 100 μl of Promega Cell Titer Glo (Promega, Madison, WI) was added to each well by use of a microplate dispenser. Plates were shaken for 2 min on a Lab-Line plate shaker. Luminescence was then measured using a PerkinElmer Envision plate reader.
The percent CPE inhibition was defined as follows: % CPE inhibition = [(test compound − virus control)/(cell control − virus control)] × 100, with “test compound” defined as the amount of luminescence obtained from wells with the compound, cells, and virus; “virus control” is defined as the amount of luminescence obtained from wells with the virus and cells only, and “cell control” is defined as the amount of luminescence obtained from wells with cells only. Percent cell viability was defined as follows: % cell viability = (test compound/cell control) × 100. An active compound, or “hit,” was defined as a compound that exhibited a % CPE inhibition of >50% without compromising cell viability. Eight concentrations of each compound were added to 96-well plates in triplicate to measure the EC50 or in duplicate in the absence of virus to measure the CC50 of the compound alone.
The Z factor values were calculated as follows: Z = [1 − (3σc + 3σv)/(μc − μv)], where σc is the standard deviation of the cell control, σv is the standard deviation of the virus control, μc is the mean cell control signal, and μv is the mean virus control signal (53).
Cytotoxicity studies on 293T cells were also performed by assessing the effects of the inhibitors on cellular viability, using a commercially available XTT cytotoxicity assay kit (Roche Diagnostics, Indianapolis, IN) that measures metabolism of XTT {2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide)}. This assay was conducted as previously described (54), and the results were in agreement with those obtained for Vero cells by cytotoxicity tests using Promega Cell Titer Glo (Promega, Madison, WI). The latter kit quantitates the amount of ATP present, which signals the presence of metabolically active cells.
SARS-CoV replicon assay with RNA detection by RT-qPCR.
The SARS-CoV replicon and mutants were generated as previously described (41, 55). Briefly, 293T cells were grown to 95% confluence on 35-mm-diameter plates and transfected with 4 μg of SARS-CoV replicon, a SARS-CoV nonreplicative construct (NRC) (Rep1b deletion mutant), or mock plasmid by using Lipofectamine reagent (Invitrogen) as directed by the manufacturer. Compounds (20 μM) were added to the replicon-transfected cells and NRC-transfected cells. At 48 h posttransfection (hpt), the total intracellular RNA was extracted using TRIzol (Invitrogen), followed by treatment with DNase I to digest remaining DNA. The extracted RNA was used as a template for subsequent reverse transcription–quantitative real-time PCR (RT-qPCR) analysis of N gene mRNA synthesis (NC). The reverse primer URB-28630RS (5′-TGCTTCCCTCTGCGTAGAAGCC-3′), complementary to nucleotides 511 to 532 of the N gene, and the forward primer URB-29VS (5′-GCCAACCAACCTCGATCTCTTG-3′), containing nucleotides 29 to 50 of the Urbani leader sequence, were used for amplification using a SuperScript One-Step RT-qPCR system with Platinum Taq DNA polymerase (Invitrogen) as suggested by the manufacturer. The SuperScript system is a real-time qPCR system that uses Sybr green for detection and quantitation of amplified DNA. The sequences of the forward and reverse primers used for the amplification of U6 mRNA as an endogenous control were as follows: U6 forward primer, 5′-CTCGCTTCGGCAGCACA-3′; and U6 reverse primer, 5′-AACGCTTCACGAATTTGCGT-3′. Primer pair amplification efficiencies were determined using 1:10 cDNA dilutions; test and housekeeping gene primer pairs with similar efficiencies were used for the qPCRs. Samples were normalized internally using the cycle threshold (CT) of the U6 housekeeping gene, as follows: ΔCT = (CT NC) − (CT U6). This was followed by determination of the mean for each sample, since the reactions were performed in triplicate. The mean value for each sample was normalized to the mean value for the NRC cells by using the following equation: ΔΔCT = ΔCT(sample) − ΔCT(NRC). The relative quantity (RQ) values were calculated as follows: RQ = (2−ΔΔCT). The RQ value for each sample was then normalized to the RQ value for the NRC (which is 1) in order to obtain percent relative RQ values. The data were plotted as percentages of relative replicon activity against inhibitor concentrations, in μM, using GraphPad Prism 5.0 (GraphPad Inc.). Data presented represent assays performed in triplicate in 3 independent experiments.
RESULTS
Primary screening for SARS-CoV entry inhibitors.
To identify compounds that can inhibit SARS-CoV entry into susceptible cells, screening of a library of pharmacologically active small molecules was carried out using the SARS/HIV-luc pseudotyped virus infection assay. Compounds that reduced luciferase activity by 50% or less were selected as potential leads. Approximately 3,000 compounds were screened from the Maybridge Hitfinder chemical library, and 44 compounds were found to cause ≤50% reduction in luciferase activity at 10 μM.
Validation of initial hits with VSV/HIV-luc pseudotyped virus.
To exclude compounds that reduced luciferase activity due to cytotoxicity, luciferase inhibition, or HIV replication inhibition from the SARS/HIV pseudotype-based assay, we tested these compounds with a VSV/HIV pseudotyped virus infection assay. Of the 44 compounds tested, we selected for further characterization 3 compounds that did not reduce luminescence below 80% in the VSV/HIV pseudotyped virus infection assay, indicating that these 3 compounds are specific inhibitors of SARS-CoV entry (Fig. 1 and 2). The compounds are (i) SSAA09E1 {[(Z)-1-thiophen-2-ylethylideneamino]thiourea}, (ii) SSAA09E2 {N-[[4-(4-methylpiperazin-1-yl)phenyl]methyl]-1,2-oxazole-5-carboxamide}, and (iii) SSAA09E3 [N-(9,10-dioxo-9,10-dihydroanthracen-2-yl)benzamide]. The structures of these compounds are shown in Fig. 3. Additional compounds that reduced luciferase activity by 20% to 50% in the VSV/HIV infection assay were considered to be less specific SARS/HIV pseudotyped virus inhibitors and are the subject of a separate investigation.
Fig 1.

Specificity of initial hits selected from the initial screen. Forty-four compounds identified as inhibitors during the initial screening of the Maybridge Hitfinder chemical library were tested again using either SARS/HIV pseudotyped virions (A) or control VSV/HIV pseudotyped virions (B) to determine whether the inhibitors specifically blocked SARS-CoV entry. Compounds A (SSAA09E1), B (SSAA09E2), and C (SSAA09E3) were selected for further characterization, as they were efficient inhibitors of SARS/HIV but not VSV/HIV entry (<20% decrease in luminescence). Additional compounds that decreased luminescence by ∼20 to 50% in VSV/HIV entry experiments were considered to be less specific SARS/HIV pseudotype inhibitors and will be characterized in future studies. Experiments were performed three independent times, and error bars represent standard deviations for the three measurements. RLU, relative light units.
Fig 3.

Chemical names and structures of the selected entry inhibitors.
Dose dependence assay.
To further validate the efficiencies of these compounds for SARS/HIV pseudotyped virus entry inhibition, we performed dose-response experiments with these 3 compounds, using SARS/HIV and VSV/HIV pseudotype-based infection assays. The results indicated that these 3 compounds decreased luciferase activity with increasing concentrations of the compounds (Fig. 2). The calculated EC50s are shown in Table 1. It should be noted that inhibition was significantly less efficient (higher EC50 values) at higher MOIs (more virus).
Fig 2.

Dose-response assays of inhibition of SARS/HIV and VSV/HIV pseudotype entry. ACE2-expressing 293T cells were infected with SARS/HIV (■) or control VSV/HIV (▲) pseudovirions in the presence of various concentrations of inhibitors. Panels A, B, and C present data for compounds SSAA09E2, SSAA09E1, and SSAA09E3, respectively. Data shown are mean values with standard deviations derived from three independent experiments.
Table 1.
Concentration parameters for entry inhibitors in pseudotyped-based and antiviral-based assays
| Compound | Pseudotype-based assay in 293T cellsa,c |
Infectious virus assay in Vero cellsb,c |
||||
|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | Selectivity index | EC50 (μM) | CC50 (μM) | Selectivity index | |
| SSAA09E3 | 9.7 ± 0.8 | 20 | 2 | 0.15 | 17 | 113 |
| SSAA09E1 | 6.7 ± 0.4 | >100 | >16 | ND | ND | ND |
| SSAA09E2 | 3.1 ± 0.2 | >100 | >33 | ND | ND | ND |
The MOI for the pseudotype-based experiments with 293T cells was 10. The CC50 in 293T cells was determined by the XTT assay as described in the text.
The MOI for the infectious virus experiments with Vero cells was 0.1. The CC50 in Vero cells was determined by the Cell Titer Glo assay (Promega) as described in the text.
EC50 and CC50 values were obtained from dose-response curves, using Graphpad Prism 5.0. Experiments were performed at least three independent times at least in duplicate. Data are means ± standard deviations for three independent experiments. ND, not determined.
Inhibition of cathepsin L activity.
To understand the mechanism by which each of these compounds inhibits SARS-CoV entry, we tested their effects on cathepsin L proteolytic activity, since this enzyme has been implicated in the cleavage of SARS-S to render the S2 domain of S accessible for cell fusion during SARS-CoV entry (27). In this experiment, we did a dose dependence fluorescence inhibition assay of cathepsin L activity, and the results indicated that only SSAA09E1 inhibited cathepsin L activity, with a 50% inhibitory concentration (IC50) of 5.33 ± 0.61 μM (Fig. 4A), which is similar to the potency with which the compound blocks SARS/HIV entry. However, several studies have implicated a related enzyme, cathepsin B, in the entry of some coronaviruses, such as feline enteric coronavirus (56) and, to a lesser extent, SARS-CoV (27, 57). Notably, cathepsin B has a similar but distinct cleavage specificity from that of cathepsin L. Hence, to determine the specificity of SSAA09E1 as a cathepsin L activity inhibitor, we performed cathepsin B activity assays in the presence and absence of SSAA09E1 as described in Materials and Methods. A known specific cathepsin B inhibitor, CA074, was used as a positive control for this assay. The results show that although SSAA09E1 is the only one of the three compounds that can inhibit cathepsin L activity, it does not block cathepsin B activity, further suggesting that SSAA09E1 may inhibit SARS-CoV entry by specifically suppressing cathepsin L activity (Fig. 4B).
Fig 4.
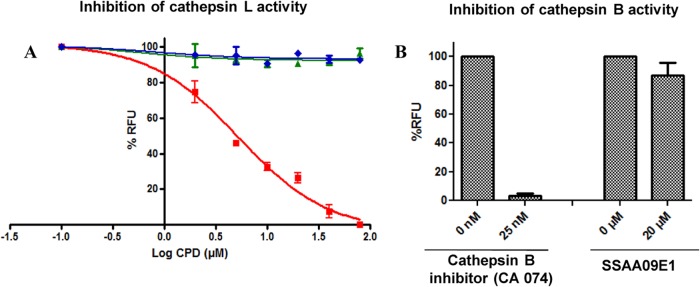
(A) Effects of compounds on cathepsin L activity. Purified recombinant cathepsin L (2 units/assay mixture) was incubated with a 25 μM concentration of a fluorogenic substrate for cathepsin L (Z-Phe-Arg-7-amido-4-methylcoumarin) in the presence or absence of various concentrations of SSAA09E1 (■), SSAA09E2 (◆), and SSAA09E3 (▲), as described in the text. Data shown are mean values with standard deviations derived from three independent experiments. CPD, compound. (B) Effect of SSAA09E1 on cathepsin B activity. The specificity of SSAA09E1 was evaluated by testing its ability to block cathepsin B activity. Purified recombinant cathepsin B (2 units/assay mixture) was incubated with a 25 μM concentration of a fluorogenic substrate for cathepsin B (Z-Arg-Arg-7-amido-4-methylcoumarin) (with the first Arg being the difference from the cathepsin L substrate) in the presence or absence of a known specific cathepsin B inhibitor (CA074) or SSAA09E1. Data shown are mean values with standard deviations derived from three independent experiments. RFU, relative fluorescence units.
Effects of compounds on interactions of SARS-S RBD with soluble ACE2.
To further investigate the inhibition mechanism of SARS-CoV entry by these compounds, we used an immunoprecipitation and immunoblot assay, as described in Materials and Methods, to examine their effects on the interaction of the SARS-S RBD with the soluble ACE2 receptor. The results indicate that only SSAA09E2 interferes with the interaction of the RBD with ACE2 (Fig. 5). To ensure that SSAA09E2 does not interfere with surface expression of ACE2, we incubated ACE2-expressing cells with DMSO or SSAA09E2 for 48 h, followed by surface labeling of the cells with anti-ACE2 antibody–FITC conjugate. The results (not shown) establish that SSAA09E2 does not prevent surface expression of ACE2 and likely affects initial recognition of the virus by directly interfering with SARS-S RBD–ACE2 interactions.
Fig 5.
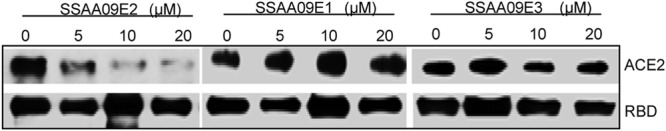
Effects of inhibitors on interactions of SARS-S RBD with soluble ACE2. The purified SARS-S RBD was incubated with purified soluble ACE2 in the presence and absence of increasing concentrations (0 to 20 μM) of the three SARS-CoV entry inhibitors. Immunoprecipitation and immunoblot analyses were carried out as described in the text. Experiments were independently confirmed three times.
Effects of compounds on fusion of the SARS-S envelope with the host cellular membrane.
Following endocytosis and cathepsin L processing of the SARS-S envelope, the S2 domain of the spike fuses with the host cellular membrane by using the class I fusion mechanism. Fusion is followed by the release of the viral genome for viral replication (27, 30, 58, 59). To investigate whether any of the three compounds inhibited entry by interfering with fusion of the viral membrane with the host membrane, we performed a fusion assay as described in Materials and Methods. As shown in Fig. 6A and B, SSAA09E2 appeared to inhibit fusion only when added prior to overlay of SARS-S 293T cells with ACE2-expressing TZM-bl cells (Fig. 6B), consistent with the above data suggesting that it interferes directly with SARS-S RBD–ACE2 interactions. SSAA09E1 did not inhibit fusion when it was added prior to or after the overlay, consistent with the above data that predict that in an assay of this type, where fusion occurs at the surface of the cells and not at an endosomal compartment requiring function of cathepsin L, this inhibitor that blocks cathepsin L would not be likely to inhibit fusion. This is consistent with a previous report that showed that SARS-S-driven cell-cell fusion was independent of cathepsin L (60). Finally, SSAA09E3 inhibited fusion of the viral membrane with the host cellular membrane when it was added before or after overlay of the two cell types, consistent with a scenario where it blocks the process of membrane fusion regardless of the initial recognition between the SARS-S RBD and ACE2.
Fig 6.
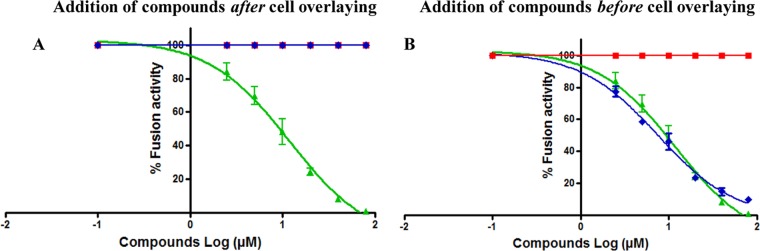
Effects of compounds on SARS-S-mediated cell-to-cell fusion. The fusion assay was performed by overlaying TZM-bl cells transfected with a plasmid encoding ACE2 on 293T cells transfected with plasmids encoding SARS-S and Tat. After 3 h of incubation, 2 μg/ml trypsin was added to induce fusion. Cell fusion was determined at 6 h postinduction, by measuring luciferase activity as described in the text. To investigate the effects of the inhibitors on fusion of the SARS-S envelope with the host cellular membrane, compounds SSAA09E1 (■), SSAA09E2 (◆), and SSAA09E3 (▲) were added either after the cell overlay but before fusion induction (A) or before cell overlay and fusion induction (B). Experiments were performed three times, and error bars represent standard deviations from the means.
Time-of-addition experiments to establish whether the compounds target “early” or “late” steps of the entry process.
To further confirm the stages of SARS entry at which the compounds exhibit inhibitory activity, we performed time-of-addition experiments using the infection assay with the SARS/HIV pseudotyped virions and added the compounds at various time points after infection, as shown in Fig. 7. The results showed that entry inhibition by SSAA09E2 was effective only if the compound was added for up to 1 h postinfection. In contrast, the other two compounds, SSAA09E1 and SSAA09E3, were effective until at least 3 h postinfection (Fig. 7). As a control, we used the known cathepsin L inhibitor Z-FY(t-Bu)-DMK (Calbiochem), which also showed effective inhibition until 3 h postinfection and, to a lesser extent, at 6 h postinfection. Taken together, these data strongly suggest that SSAA09E2 targets an “early” step of SARS/HIV entry, such as initial recognition of the SARS-S RBD by ACE2, whereas SSAA09E1 and SSAA09E3 inhibit later stages of viral entry.
Fig 7.

Time-of-addition experiment to validate inhibition mechanism. ACE2-expressing 293T cells infected with SARS/HIV pseudovirions in the presence of the inhibitors (10 μM) were tested for suppression of luciferase activity. Compounds were added at 0, 0.2, 0.5, 1, 3, 6, 9, 12, and 24 h postinfection (hpi), and luciferase activity was measured at 48 hpi as described in the text. A cathepsin L inhibitor [Z-FY(t-Bu)-DMK] (50 nM) was used as a control. Experiments were performed twice in triplicate, and error bars represent standard deviations from the means.
Inhibition of infectious SARS-CoV.
The antiviral activity of SSAA09E3 was determined by using a SARS-CoV cytopathic effect assay as described in Materials and Methods. Independent experiments were carried out at the Southern Research Institute and the University of Louisville. The results showed that SSAA09E3 inhibits SARS-CoV infection of Vero cells with a submicromolar EC50 (Table 1), resulting in a promising selectivity index (>100).
Effects of compounds on postentry steps of SARS-CoV replication.
Infectious SARS-CoV was significantly more sensitive to the inhibitors than the SARS/HIV pseudotyped virions. This could have been the result of differences in conditions, such as the cell type or the 100-fold difference in MOI, but it was also possible that fully infectious virus was more sensitive to inhibitors because other steps of the viral life cycle were also blocked. To determine whether the 3 compounds also affected steps that are downstream from viral entry, we performed a SARS-CoV replicon assay as described in Materials and Methods. This assay was described in detail in a previous publication (41). We performed these experiments with the SARS-CoV replicon in the presence and absence of SSAA09E1, SSAA09E2, and SSAA09E3. As a control, we used SSYA10-001, a compound that we discovered previously in a search for SARS-CoV helicase (nsp13) inhibitors (41). As shown in Fig. 8, none of the entry inhibitors interfered with replication of the SARS-CoV replicon, whereas, as expected, our nsp13 helicase inhibitor, SSYA10-001 (41), suppressed SARS-CoV replicon function. These data suggest that the three compounds presented here are specific for inhibiting the entry process of the virus.
Fig 8.
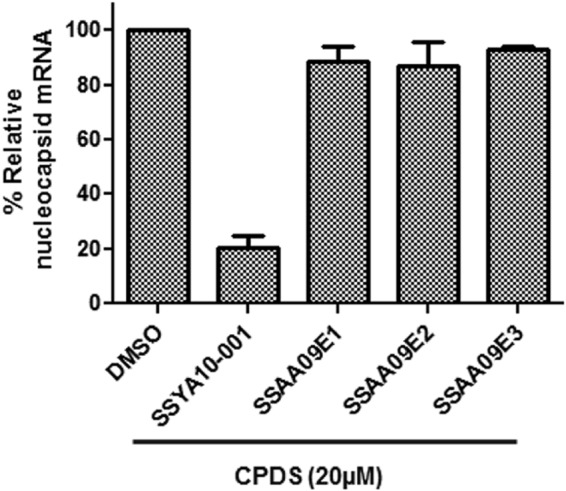
Effects of compounds on postentry steps of SARS-CoV life cycle. Compounds were tested for the ability to block SARS-CoV replication in a replicon-based system. HEK 293T cells were transfected with a plasmid encoding a SARS-CoV replicon in the presence of 20 μM SSAA09E1, SSAA09E2, or SSAA09E3 as described in the text. SSYA10-001 is a SARS-CoV replication inhibitor that targets the nsp13 helicase. Total RNA was isolated at 48 h posttransfection and analyzed by RT-qPCR as described in the text and as we have previously published (41, 55). Experiments were repeated three times, each time in triplicate, and error bars represent standard deviations for three independent experiments.
DISCUSSION
Interference with viral entry can be an effective therapeutic strategy for preventing viral infection. Proof of principle was provided by the discovery and use of enfuvirtide, a peptide-based drug that blocks HIV entry by targeting the gp41 component of the HIV surface glycoprotein (equivalent to S2 in SARS-CoV) (13, 15, 16). However, enfuvirtide is a peptide with low oral bioavailability and must be administered by injection, unlike small-molecule drugs, which are typically taken orally, avoiding the problems associated with injections. For the treatment of HIV infection, there is also a single approved small molecule that blocks HIV entry, maraviroc, which binds to the CCR5 coreceptor and prevents interactions with the gp120 component of the HIV surface glycoprotein (equivalent to S1 in SARS-CoV). Here we describe the discovery and initial characterization of three small molecules that can inhibit SARS-CoV entry by three different mechanisms.
In order to identify specific inhibitors of SARS-CoV, we searched for compounds that block entry of SARS/HIV but not VSV/HIV pseudotyped virions (43, 61). This strategy enabled elimination of compounds that are cytotoxic (since successful VSV/HIV infection requires unimpeded cell growth) and of false-positive hits from compounds that inhibit proteins involved in HIV replication, as well as compounds that block translation or inhibit luciferase. Some potential inhibitors of SARS-CoV entry were not characterized further because they also inhibited entry of VSV/HIV pseudotyped virions. Such compounds are being examined in ongoing studies. Notably, the current screening strategy could be used to identify compounds that specifically block VSV entry by focusing on inhibitors of VSV/HIV but not SARS/HIV entry. The present study focused on three compounds that are specific inhibitors of SARS-CoV entry, namely, SSAA09E1, SSAA09E2, and SSAA09E3. SSAA09E3 was more cytotoxic than the other compounds, possibly because of interactions with unknown host factors. Nonetheless, the compound was not cytotoxic at the concentrations where it exhibited activity against infectious SARS-CoV at a low MOI (0.1) in Vero cells, as well as against SARS/HIV pseudotypes at a high MOI (21) in 293T cells. These compounds were identified by screening ∼3,000 compounds from the Maybridge Hitfinder chemical library, which contains compounds that follow Lipinski's rule of five, an empirical rule that is used to evaluate “drug-likeness” and potential for oral bioavailability in humans. SSAA09E1 is a thiourea derivative. Several thiourea derivatives have previously been described to have potent antiviral activities against HIV and hepatitis C virus (HCV) (62, 63). These compounds were shown to have an elimination half-life of 1 to 3 h after parenteral administration, with moderate systemic clearance. Some were also shown to exhibit rapid distribution to tissues, with particularly high levels of accumulation in the lungs, adipose tissue, skin, urinary bladder, adrenal glands, uterus, and ovaries (62, 63). These compounds were shown to be absorbed rapidly following oral and parenteral administration, with higher bioavailability for intraperitoneal administration than for the oral route (34, 64). SSAA09E2 is an oxazole-carboxamide derivative. Some oxazole-carboxamide derivatives have been described for their inhibitory activities against some host enzymes which are targets for the potential treatment of obesity and diabetes or some treatments of neurodegenerative diseases, such as Alzheimer's disease, and neurological diseases, such as bipolar disorder (65, 66). These compounds were shown to have good oral bioavailability and high potencies against their targets, with low half-lives and volumes of distribution and moderate clearance (65, 66). The third compound, SSAA09E3, is a benzamide derivative, and related compounds have been shown to have antifibrotic and antimetastatic effects, with good oral bioavailability, ranging from 8.95% (mice) to 85% (dogs), and low cytotoxicity. They were also shown to exhibit rapid systemic distribution to the liver, kidneys, and lungs (67). All these properties of closely related compounds of these novel inhibitors suggest that these compounds may be promising druggable leads that can be effective antiviral agents which could potentially exhibit good pharmacokinetics.
The process of viral entry involves interactions between the receptor binding domain of SARS-S and the receptor ACE2 (Fig. 9). SARS-S is hydrolyzed by cathepsin L to S1 and S2 domains (25–30). In addition to cathepsin L, other cellular factors may also be important for completion of viral entry (57, 68–70). The viral and cellular membranes eventually fuse through a type I fusion mechanism (64). We hypothesized that the SARS-CoV entry inhibitors could block viral entry by interfering with any of these stages of entry.
Fig 9.
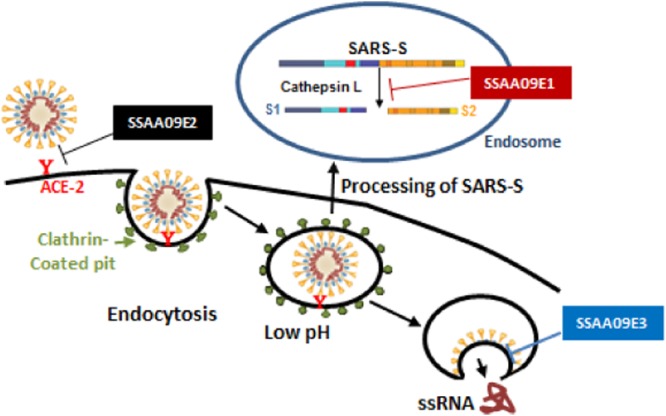
Stages of SARS-CoV entry inhibited by novel SARS-CoV small-molecule inhibitors. Following interaction of SARS-S with the ACE2 receptor on the permissive cell surface, the virus is endocytosed. After endocytosis of the virus, cathepsin L cleaves SARS-S to S1 and S2, allowing subsequent fusion of the viral membrane with the endosomal membrane. Our data suggest that SSAA09E2 prevents viral entry by blocking the interaction of SARS-S with the ACE2 receptor, SSAA09E1 impedes viral entry by inhibiting cathepsin L processing of the SARS-S envelope in the endosome, and SSAA09E3 inhibits viral entry by preventing fusion of the viral membrane with the host cellular membrane.
To test this hypothesis, we carried out immunoprecipitation and immunoblot assays to monitor the interaction of the SARS-S RBD with soluble ACE2 in the presence and absence of the inhibitors. Interestingly, only SSAA09E2 prevented SARS-S binding to ACE2 (Fig. 5 and 9). This result was confirmed by a flow cytometry binding assay, which was conducted as described in Materials and Methods, using ACE2-expressing 293T cells and a recombinant RBD (data not shown). To determine whether SSAA09E2 binds to ACE2-expressing cells, we incubated the compound with the cells for 45 min and then washed the supernatant from the cells, followed by the addition of SARS/HIV pseudotyped virions to the cells. Comparable inhibition was observed, consistent with binding of SSAA09E2 directly to the ACE2 receptor or to other cellular factors that may facilitate the SARS-S–ACE2 interaction (data not shown). We do not expect SSAA09E2 to bind at the ACE2 protease active site, because it did not inhibit ACE2's enzymatic activity (data not shown). Further studies are in progress to test this hypothesis. Our data are consistent with previous work reporting that ACE2 inhibitors do not block SARS-CoV entry, and also with crystal structures of the SARS-CoV RBD–ACE2 complex that show that the protein interactions do not directly involve residues of the ACE2 active site (71, 72).
We determined that only SSAA09E1 can inhibit cathepsin L (Fig. 4 and 9). Cathepsin L inhibitors would also be useful for the treatment of neoplastic diseases in which cathepsin L has been implicated (73–75). It was previously shown that cathepsin L inhibitors suppress SARS-CoV replication (27). Published cathepsin L inhibitors, including dipeptide epoxyketones, calpain inhibitor III, oxocarbazate, and MDL28170, appear to be peptidomimetic in nature (37, 38, 40, 43). SSAA09E1 is a small molecule that is likely to have improved bioavailability. Despite the close resemblance of the amino acid sequences at the cleavage sites of both cathepsins L and B, SSAA09E1 did not inhibit cathepsin B activity. This strongly suggests that SSAA09E1 may bind directly to cathepsin L to inhibit its enzymatic function.
The cell-to-cell fusion assay allowed us to distinguish between those inhibitors that block fusion by preventing direct interactions of SARS-S with ACE2 and those that target events after the initial interactions. Because SSAA09E1 inhibits cathepsin L activity, which is not required for fusion in this assay (fusion takes place at the surface), it did not affect fusion in this assay. SSAA09E2 lost its inhibition ability when it was added after ACE2 and SARS-S were allowed to interact (preincubation with ACE2-TZM-bl cells before the overlay with SARS-S-expressing 293T cells), suggesting that it blocks entry by interfering with the initial recognition of SARS-S and ACE2 rather than with the fusion process. In contrast, SSAA09E3 prevented fusion even after the SARS-S–ACE2 interactions were allowed to occur (no difference in potency when the compound was added before or after ACE2-TZM-bl cells were overlaid with the SARS-S-expressing 293T cells) (Fig. 6). While these data show that SSAA09E3 blocks the fusion step of viral entry, it remains unclear whether the inhibitor binds directly to the spike protein and interferes with downstream conformational changes required for efficient fusion or if it interferes with the involvement of other required host factors.
To further establish the stages of entry inhibited by these compounds, we performed time-of-addition experiments. The results clearly showed that none of the compounds exhibited any significant inhibition when added later than 3 h postinfection, suggesting that the SARS coronavirus entry process is completed within this time frame. SSAA09E2 did not show any significant inhibition after 1 h, indicating that either SSAA09E2 is specific for preventing SARS-S interaction with the ACE2 receptor or it inhibits other, unknown early entry processes apart from receptor binding. If SSAA09E2 only inhibits the SARS-S–ACE2 interaction, then it can be inferred from this result that the complete binding process of the virus to the host cell happens within 1 h. The results for SSAA09E1 and SSAA09E3 also suggest that trafficking of the virus-endosome complex to a low-pH region after endocytosis does not take more than 3 h, indicating that it takes ≤3 h for cathepsin L processing of the SARS-S envelope and fusion of the viral envelope with the host cellular membrane to take place postinfection.
SSAA09E3 was shown to have promising submicromolar antiviral activity, with a selectivity index of >100, which is considered the cutoff for subsequent development studies. SSAA09E3 appeared to be more potent in the assays with fully infectious virus than in the pseudotype-based assays (submicromolar EC50 versus EC50 of 9.7 μM). This difference was likely due to the large differences in MOI. For example, the MOI needed for a measurable signal in the less sensitive pseudotype-based assay was up to 2 orders of magnitude higher than the MOI used in the SARS-CoV CPE assay (Table 1).
In conclusion, we have identified small molecules that inhibit SARS-CoV entry by at least three different mechanisms. SSAA09E2 is the first known small molecule to block SARS-CoV by interfering with the ACE2–SARS-S RBD interaction (Fig. 5 and 9), SSAA09E1 is a nonpeptidomimetic small molecule that inhibits cathepsin L (Fig. 4 and 9), and SSAA09E3 blocks SARS-CoV entry by preventing fusion of the viral membrane with the host cellular membrane (Fig. 6 and 9).
Recently, an outbreak of a new human coronavirus (hCoV-EMC) closely related to SARS-CoV has resulted in public health concerns and warnings by the WHO and CDC. Despite the strong homology between the two coronaviruses, many of the differences are at the surface glycoprotein region that is expected to interact with the receptor. In addition, recent reports identified dipeptidyl peptidase 4 (DPP4; also known as CD26) as a functional receptor for hCoV-EMC (6, 76). Therefore, since SARS-CoV and hCoV-EMC use different receptors and also differ in their receptor binding proteins, it is unlikely that inhibitors that directly block spike-receptor interactions, such as SSAA09E2, would affect hCoV-EMC entry. Since it is not clear whether SSAA09E3 blocks fusion by directly binding to the spike protein or by interfering with the use of other required host factors, it is not possible to predict whether the inhibitor would also inhibit hCoV-EMC entry. However, as Gierer and colleagues have recently shown that hCoV-EMC is activated by cathepsins B and L for viral entry (76), we expect that SSAA09E1 will be able to block hCoV-EMC entry and may have anti-hCoV-EMC activity.
These three novel inhibitors provide useful leads for the discovery of antivirals that could prevent SARS and SARS-related infections and may also become useful tools for studying fundamental mechanisms of coronavirus replication.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants AI076119, AI094715, AI079801, and AI087489. We also acknowledge support by a grant from the Ministry of Knowledge and Economy, Bilateral International Collaborative R&D Program, Republic of Korea.
We are grateful to Paul Bates for providing the pCAGGS SARS-CoV Spike, pNL-4.3-luc, and pCDNA 3.1-ACE2 plasmids and to Michael Farzan for the pCDM8-Spike RBD-Fc plasmid. We thank Stefan Pohlmann and Marc Johnson for providing the pCDNA 3.1 soluble ACE2 and pMDG-VSV-G plasmids, respectively. We are thankful for the pSV2tat72 plasmid and TZM-bl cells, which were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pSV2tat72 from Alan Frankel and TZM-bl from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, Chu CM, Hui PK, Mak KL, Lim W, Yan KW, Chan KH, Tsang NC, Guan Y, Yuen KY, Peiris JS. 2003. Lung pathology of fatal severe acute respiratory syndrome. Lancet 361:1773–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poutanen SM, Low DE, Henry B, Finkelstein S, Rose D, Green K, Tellier R, Draker R, Adachi D, Ayers M, Chan AK, Skowronski DM, Salit I, Simor AE, Slutsky AS, Doyle PW, Krajden M, Petric M, Brunham RC, McGeer AJ. 2003. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 348:1995–2005 [DOI] [PubMed] [Google Scholar]
- 3. Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA, Walmsley SL, Mazzulli T, Avendano M, Derkach P, Ephtimios IE, Kitai I, Mederski BD, Shadowitz SB, Gold WL, Hawryluck LA, Rea E, Chenkin JS, Cescon DW, Poutanen SM, Detsky AS. 2003. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 289:2801–2809 [DOI] [PubMed] [Google Scholar]
- 4. Frieman M, Basu D, Matthews K, Taylor J, Jones G, Pickles R, Baric R, Engel DA. 2011. Yeast based small molecule screen for inhibitors of SARS-CoV. PLoS One 6:e28479. 10.1371/journal.pone.0028479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsu LY, Lee CC, Green JA, Ang B, Paton NI, Lee L, Villacian JS, Lim PL, Earnest A, Leo YS. 2003. Severe acute respiratory syndrome (SARS) in Singapore: clinical features of index patient and initial contacts. Emerg. Infect. Dis. 9:713–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bupp K, Roth MJ. 2005. Alteration and analyses of viral entry with library-derived peptides. Adv. Virus Res. 65:147–172 [DOI] [PubMed] [Google Scholar]
- 8. Douglas JL, Panis ML, Ho E, Lin KY, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Cihlar T. 2003. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J. Virol. 77:5054–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Razinkov V, Gazumyan A, Nikitenko A, Ellestad G, Krishnamurthy G. 2001. RFI-641 inhibits entry of respiratory syncytial virus via interactions with fusion protein. Chem. Biol. 8:645–659 [DOI] [PubMed] [Google Scholar]
- 10. Lieberman-Blum SS, Fung HB, Bandres JC. 2008. Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin. Ther. 30:1228–1250 [DOI] [PubMed] [Google Scholar]
- 11. MacArthur RD, Novak RM. 2008. Reviews of anti-infective agents. Maraviroc: the first of a new class of antiretroviral agents. Clin. Infect. Dis. 47:236–241 [DOI] [PubMed] [Google Scholar]
- 12. Wood A, Armour D. 2005. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog. Med. Chem. 43:239–271 [DOI] [PubMed] [Google Scholar]
- 13. Anonymous 1999. Pentafuside. DP 178, T 20. Drugs R D 2:347–349 [DOI] [PubMed] [Google Scholar]
- 14. Berkhout B, Eggink D, Sanders RW. 2012. Is there a future for antiviral fusion inhibitors? Curr. Opin. Virol. 2:50–59 [DOI] [PubMed] [Google Scholar]
- 15. Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74:8358–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartt JK, Liang T, Sahagun-Ruiz A, Wang JM, Gao JL, Murphy PM. 2000. The HIV-1 cell entry inhibitor T-20 potently chemoattracts neutrophils by specifically activating the N-formylpeptide receptor. Biochem. Biophys. Res. Commun. 272:699–704 [DOI] [PubMed] [Google Scholar]
- 17. Miller MD, Hazuda DJ. 2004. HIV resistance to the fusion inhibitor enfuvirtide: mechanisms and clinical implications. Drug Resist. Updat. 7:89–95 [DOI] [PubMed] [Google Scholar]
- 18. Naito T, Izumi K, Kodama E, Sakagami Y, Kajiwara K, Nishikawa H, Watanabe K, Sarafianos SG, Oishi S, Fujii N, Matsuoka M. 2009. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob. Agents Chemother. 53:1013–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eda Y, Murakami T, Ami Y, Nakasone T, Takizawa M, Someya K, Kaizu M, Izumi Y, Yoshino N, Matsushita S, Higuchi H, Matsui H, Shinohara K, Takeuchi H, Koyanagi Y, Yamamoto N, Honda M. 2006. Anti-V3 humanized antibody KD-247 effectively suppresses ex vivo generation of human immunodeficiency virus type 1 and affords sterile protection of monkeys against a heterologous simian/human immunodeficiency virus infection. J. Virol. 80:5563–5570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsushita S, Takahama S, Shibata J, Kimura T, Shiozaki K, Eda Y, Koito A, Murakami T, Yoshimura K. 2005. Ex vivo neutralization of HIV-1 quasi-species by a broadly reactive humanized monoclonal antibody KD-247. Hum. Antibodies 14:81–88 [PubMed] [Google Scholar]
- 21. Cilliers T, Willey S, Sullivan WM, Patience T, Pugach P, Coetzer M, Papathanasopoulos M, Moore JP, Trkola A, Clapham P, Morris L. 2005. Use of alternate coreceptors on primary cells by two HIV-1 isolates. Virology 339:136–144 [DOI] [PubMed] [Google Scholar]
- 22. Dau B, Holodniy M. 2009. Novel targets for antiretroviral therapy: clinical progress to date. Drugs 69:31–50 [DOI] [PubMed] [Google Scholar]
- 23. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767 [DOI] [PubMed] [Google Scholar]
- 24. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, Gramberg T, Pohlmann S. 2004. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 319:1216–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 102:11876–11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tripet B, Howard MW, Jobling M, Holmes RK, Holmes KV, Hodges RS. 2004. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 279:20836–20849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong SK, Li W, Moore MJ, Choe H, Farzan M. 2004. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 279:3197–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, Schwartz O, Subbarao K, Nabel GJ. 2004. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78:5642–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dimitrov DS. 2003. The secret life of ACE2 as a receptor for the SARS virus. Cell 115:652–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. 2003. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 312:1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, Regla-Nava JA, Alvarez E, Oliveros JC, Zhao J, Fett C, Perlman S, Enjuanes L. 2011. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 7:e1002315. 10.1371/journal.ppat.1002315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. 2003. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 361:2045–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haga S, Nagata N, Okamura T, Yamamoto N, Sata T, Sasazuki T, Ishizaka Y. 2010. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antiviral Res. 85:551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knowles SR, Phillips EJ, Dresser L, Matukas L. 2003. Common adverse events associated with the use of ribavirin for severe acute respiratory syndrome in Canada. Clin. Infect. Dis. 37:1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah PP, Wang T, Kaletsky RL, Myers MC, Purvis JE, Jing H, Huryn DM, Greenbaum DC, Smith AB, 3rd, Bates P, Diamond SL. 2010. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 78:319–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Struck AW, Axmann M, Pfefferle S, Drosten C, Meyer B. 2012. A hexapeptide of the receptor-binding domain of SARS corona virus spike protein blocks viral entry into host cells via the human receptor ACE2. Antiviral Res. 94:288–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Vonderen MG, Bos JC, Prins JM, Wertheim-van Dillen P, Speelman P. 2003. Ribavirin in the treatment of severe acute respiratory syndrome (SARS). Neth. J. Med. 61:238–241 [PubMed] [Google Scholar]
- 40. Zhou Y, Agudelo J, Lu K, Goetz DH, Hansell E, Chen YT, Roush WR, McKerrow J, Craik CS, Amberg SM, Simmons G. 2011. Inhibitors of SARS-CoV entry—identification using an internally-controlled dual envelope pseudovirion assay. Antiviral Res. 92:187–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adedeji AO, Singh K, Calcaterra NE, Dediego ML, Enjuanes L, Weiss S, Sarafianos SG. 2012. Severe acute respiratory virus syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 56:4718–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26 [DOI] [PubMed] [Google Scholar]
- 43. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. 2004. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U. S. A. 101:4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H. 1999. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96:667–676 [DOI] [PubMed] [Google Scholar]
- 45. Mangor JT, Monsma SA, Johnson MC, Blissard GW. 2001. A GP64-null baculovirus pseudotyped with vesicular stomatitis virus G protein. J. Virol. 75:2544–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frankel AD, Pabo CO. 1988. Cellular uptake of the Tat protein from human immunodeficiency virus. Cell 55:1189–1193 [DOI] [PubMed] [Google Scholar]
- 47. Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J. Virol. 83:8289–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72:2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takeuchi Y, McClure MO, Pizzato M. 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J. Virol. 82:12585–12588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang H, Blair CD, Olson KE, Clem RJ. 2008. Effects of inducing or inhibiting apoptosis on Sindbis virus replication in mosquito cells. J. Gen. Virol. 89:2651–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Severson WE, Shindo N, Sosa M, Fletcher T, 3rd, White EL, Ananthan S, Jonsson CB. 2007. Development and validation of a high-throughput screen for inhibitors of SARS CoV and its application in screening of a 100,000-compound library. J. Biomol. Screen. 12:33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73 [DOI] [PubMed] [Google Scholar]
- 54. Durk RC, Singh K, Cornelison CA, Rai DK, Matzek KB, Leslie MD, Schafer E, Marchand B, Adedeji A, Michailidis E, Dorst CA, Moran J, Pautler C, Rodriguez LL, McIntosh MA, Rieder E, Sarafianos SG. 2010. Inhibitors of foot and mouth disease virus targeting a novel pocket of the RNA-dependent RNA polymerase. PLoS One 5:e15049. 10.1371/journal.pone.0015049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Almazan F, Dediego ML, Galan C, Escors D, Alvarez E, Ortego J, Sola I, Zuniga S, Alonso S, Moreno JL, Nogales A, Capiscol C, Enjuanes L. 2006. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J. Virol. 80:10900–10906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Regan AD, Shraybman R, Cohen RD, Whittaker GR. 2008. Differential role for low pH and cathepsin-mediated cleavage of the viral spike protein during entry of serotype II feline coronaviruses. Vet. Microbiol. 132:235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. 2011. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85:4122–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M, Rottier PJ, Choe H. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 281:3198–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569 [DOI] [PubMed] [Google Scholar]
- 60. Simmons G, Bertram S, Glowacka I, Steffen I, Chaipan C, Agudelo J, Lu K, Rennekamp AJ, Hofmann H, Bates P, Pohlmann S. 2011. Different host cell proteases activate the SARS-coronavirus spike-protein for cell-cell and virus-cell fusion. Virology 413:265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, Simmons G, Hofmann H, Kuri T, Weber F, Eichler J, Drosten C, Pohlmann S. 2010. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 84:1198–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen CL, Venkatachalam TK, Waurzyniak B, Chelstrom L, Uckun FM. 2001. In vivo toxicity, pharmacokinetic features and tissue distribution of N-[2-(2,5-dimethoxyphenylethyl)]-N′-[2-(5-bromopyridyl)]-thiourea (HI-236), a potent non-nucleoside inhibitor of HIV-1 reverse transcriptase. Arzneimittelforschung 51:574–581 [DOI] [PubMed] [Google Scholar]
- 63. Kang IJ, Wang LW, Yeh TK, Lee CC, Lee YC, Hsu SJ, Wu YS, Wang JC, Chao YS, Yueh A, Chern JH. 2010. Synthesis, activity, and pharmacokinetic properties of a series of conformationally-restricted thiourea analogs as novel hepatitis C virus inhibitors. Bioorg. Med. Chem. 18:6414–6421 [DOI] [PubMed] [Google Scholar]
- 64. Jardetzky TS, Lamb RA. 2004. Virology: a class act. Nature 427:307–308 [DOI] [PubMed] [Google Scholar]
- 65. Gentile G, Merlo G, Pozzan A, Bernasconi G, Bax B, Bamborough P, Bridges A, Carter P, Neu M, Yao G, Brough C, Cutler G, Coffin A, Belyanskaya S. 2012. 5-Aryl-4-carboxamide-1,3-oxazoles: potent and selective GSK-3 inhibitors. Bioorg. Med. Chem. Lett. 22:1989–1994 [DOI] [PubMed] [Google Scholar]
- 66. Qian Y, Wertheimer SJ, Ahmad M, Cheung AW, Firooznia F, Hamilton MM, Hayden S, Li S, Marcopulos N, McDermott L, Tan J, Yun W, Guo L, Pamidimukkala A, Chen Y, Huang KS, Ramsey GB, Whittard T, Conde-Knape K, Taub R, Rondinone CM, Tilley J, Bolin D. 2011. Discovery of orally active carboxylic acid derivatives of 2-phenyl-5-trifluoromethyloxazole-4-carboxamide as potent diacylglycerol acyltransferase-1 inhibitors for the potential treatment of obesity and diabetes. J. Med. Chem. 54:2433–2446 [DOI] [PubMed] [Google Scholar]
- 67. Kim YW, Kim YK, Lee JY, Chang KT, Lee HJ, Kim DK, Sheen YY. 2008. Pharmacokinetics and tissue distribution of 3-((5-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)-1H-imidazol-2-yl)methyl)benzamide; a novel ALK5 inhibitor and a potential anti-fibrosis drug. Xenobiotica 38:325–339 [DOI] [PubMed] [Google Scholar]
- 68. Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7:e1001258. 10.1371/journal.ppat.1001258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 85:873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang N, Ma P, Lang J, Zhang Y, Deng J, Ju X, Zhang G, Jiang C. 2012. Phosphatidylinositol 4-kinase IIIbeta is required for severe acute respiratory syndrome coronavirus spike-mediated cell entry. J. Biol. Chem. 287:8457–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li F, Li W, Farzan M, Harrison SC. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868 [DOI] [PubMed] [Google Scholar]
- 72. Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, Wong SK, Huang IC, Xu K, Vasilieva N, Murakami A, He Y, Marasco WA, Guan Y, Choe H, Farzan M. 2005. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24:1634–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lankelma JM, Voorend DM, Barwari T, Koetsveld J, Van der Spek AH, De Porto AP, Van Rooijen G, Van Noorden CJ. 2010. Cathepsin L, target in cancer treatment? Life Sci. 86:225–233 [DOI] [PubMed] [Google Scholar]
- 74. Leto G, Sepporta MV, Crescimanno M, Flandina C, Tumminello FM. 2010. Cathepsin L in metastatic bone disease: therapeutic implications. Biol. Chem. 391:655–664 [DOI] [PubMed] [Google Scholar]
- 75. Xu X, Yuan G, Liu W, Zhang Y, Chen W. 2009. Expression of cathepsin L in nasopharyngeal carcinoma and its clinical significance. Exp. Oncol. 31:102–105 [PubMed] [Google Scholar]
- 76. Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pohlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 87:5502–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]