

Abstract
Glycans and glycan-binding proteins are central to a properly functioning immune system. Perhaps the best known example of this is the selectin family of surface proteins that are primarily found on leukocytes, and which bind to endothelial glycans near sites of infection or inflammation and enable extravasation into tissues. In the past decade, however, a number of other immune pathways that are dependent on or sensitive to changes in glycan-mediated mechanisms have been revealed. These include antibody function, apoptosis, Th1 versus Th2 skewing, T cell receptor signaling, and MHC class II antigen-presentation. Here, we highlight how regulated changes in protein glycosylation both at the cell surface and on secreted glycoproteins can positively and negatively modulate the immune response.
Keywords: Glycosylation, Glycans, Immune Regulation, Galectin, C-type Lectin, MHCII, Antibody
Host Glycans and their Ligands
Host glycans can take many forms, ranging from glycosaminoglycans (GAGs) such as heparin, chondroitin sulfate, and hyaluronan, to the canonical glycoprotein-associated glycans. Protein glycosylation is the enzymatic process of adding carbohydrates to specific sites on nascent proteins (Figure 1)1,2. Recent genomic studies reveal the presence of carbohydrate recognition domains (CRDs) throughout evolution, suggesting an important role for glycans and their recognition in physiology3. In addition, new techniques in mass spectrometric analysis allow definition of precise glycan compositions, including linkages and other features, providing a deeper understanding of glycan biology 4,5. With these advances, protein glycosylation (Figure 1; Box 1) is emerging as an important regulatory mechanism in the immune response. Given the overlapping epitopes found in N- and O-linked glycans, we will focus on these glycans and their binding partners, but O-GlcNAc (N-acetylglucosamine) and other glycans (e.g., GAGs and glycolipids) are also important in immunity as described elsewhere 6–11.
Figure 1. Protein Glycosylation and Important Glycan Epitopes.

Schematic representations of N-linked, O-linked (O-GalNAc), and O-GlcNAc protein glycosylation pathways. For the N-linked glycosylation pathway, the structures falling into ER-localized, high mannose, hybrid, and complex groups are indicated to illustrate the general steps of synthesis and structural diversification that occurs in the Golgi apparatus. Differences in branching patterns are also illustrated, together with details about the enzymes (Mgat1–5) responsible for the addition of GlcNAc residues that seed each branch. Branching is a modulated structural attribute of N-glycans during disease and inflammation, which can be regulated through differential expression of the Mgat genes. For the O-linked glycosylation pathway, each of the “Core” structures are shown to illustrate the structural relationships between each glycan type. All of the “Core” structures can serve as a platform for further diversification, as shown for Core 1 and Core 3. Within both N- and O-linked glycans, a number of key glycan epitopes and ligands that are relevant for the immune system are possible and indicated in red boxes. For example, sialyl-Lewisx is a ligand for selectin molecules, whereas LacNAc and poly-LacNAc represent minimal glycans for many of the galectins. We also show both Type-1 and Type-2 A/B/O(H) blood group antigens which can also be found in both N- and O-linked glycans as examples relevant to transplantation and blood transfusion.
Box 1. N- and O-Linked Protein Glycosylation.
Glycans attached to glycoproteins can be divided into three broad categories: asparagine (N)-linked, serine/threonine (O)-linked, and O-linked N-acetylglucosamine (O-GlcNAc). Many nuclear and cytoplasmic proteins are glycosylated by the addition of a single N-acetylglucosamine (GlcNAc) on serine or threonine residues by O-GlcNAc transferase (OGT), whereas essentially every secreted and integral membrane protein is glycosylated by N- or O-linked glycans within the ER and Golgi apparatus. Classical O-glycans are built de novo directly on the protein within the Golgi apparatus, which is initiated by the α-linked addition of a N-acetylgalactosamine (GalNAc) carbohydrate to a serine or threonine residue by the polypeptide GalNAc transferase (PP-GalNAcT) via the available hydroxyl group on the amino acid side chain, hence the name “O-linked”. If left unmodified, the single GalNAc is known as the Tn antigen; however, these structures are usually built up by the sequential action of Golgi-resident enzymes to form mature glycans, which are classified into eight “Cores” (Core 1–8) depending on the carbohydrate(s) proximal to and their linkage with the initiating GalNAc. For example, the addition of a galactose (Gal) residue by β1,3 linkage turns the Tn antigen into the Core 1 glycan (also known as the T antigen). Further addition of a GlcNAc residue to the Core 1 glycan by β1,6 linkage creates the Core 2 glycan. The other Core glycans are variations on this theme, with differing carbohydrates and linkages proximal to the initial GalNAc. In all cases, these glycans can be further elongated to highly complex structures carrying a variety of carbohydrates in multiple combinations and glycosidic linkages, including terminal sialic acids, Gal, GalNAc, GlcNAc, fucose (Fuc), and others.
In contrast, N-glycans begin as a relatively large core structure which is initially synthesized as a lipid precursor on the cytoplasmic face of the endoplasmic reticulum (ER) membrane, moved into the ER lumen during synthesis by the ER-localized Flipase, and then added to nascent polypeptides at the available primary amine on asparagine residue side chains within the “N-x-S/T” consensus sequence, hence the name “N-linked” 101. Although not all N-xS/T sites are glycosylated, this transfer is catalyzed in the ER by the oligosaccharyltransferase (OST) enzyme complex, which is the target of the often used drug tunicamycin which prevents the addition of N-glycans and causes significant ER stress and the initiation of the unfolded protein response. Within the ER, N-glycans play a role in the quality control of protein folding by mediating interactions with ER-resident chaperones such as calreticulin and calnexin; however, once out of the ER and into the Golgi, the N-glycans on nascent proteins are trimmed to “high mannose” structures dominated by terminal mannoses before being rebuilt into the complex N-glycans common to mammalian glycoproteins. The transition between a high mannose N-glycan and a complex-type N-glycan is the addition of a GlcNAc on one arm of the mannose core by GlcNAcT1 (Mgat1). This transitional glycan structure with a single GlcNAc (which can be elongated with the other carbohydrates) and terminal mannose residues on the other arms is called a “hybrid” N-glycan. Upon subsequent addition of GlcNAc residues, the N-glycans fall into the “complex N-glycan” category, with each additional GlcNAc representing a new glycan “branch”. As these GlcNAc branches are added, it is common to term the resulting N-glycans bi-, tri-, and tetra-antennary, based on how many branches are present. N-glycans typically develop into structures with at least two branches (bi-antennary) and are built up through the sequential action of Golgi enzymes in much the same fashion as O-glycans.
N- and O-linked glycans are found on nearly all membrane and secreted proteins produced by mammalian cells and can carry key immunologic epitopes, such as the canonical blood group antigens H, A, and B as well as the Lewis blood antigens (e.g., sialyl-LewisX) (Figure 1)1,2. Another epitope found within N- and O-linked glycans is N-acetyllactosamine (LacNAc), which is a disaccharide of galactose (Gal) and GlcNAc that can be present as a single disaccharide unit or in repeated fashion (poly-LacNAc) (Figure 1). LacNAc or poly-LacNAc structures represent the minimal glycan structure required for many of the mammalian β-D-galactoside-binding lectins, galectins 12, which are integral to the regulation of immunity. In addition to epitopes found within the N- and O-linked glycans themselves, modifications such as sialylation and fucosylation (Box 1) can alter the biology of the glycan substantially and are highly regulated (Figure 1). For example, glycans with the LacNAc motif are ligands for galectin-1 (gal-1), but addition of a terminal sialic acid can “cap” the epitope and prevent gal-1 binding 13. Likewise, lymphocyte development is controlled through fucosylation of the NOTCH pathway (reviewed in [14,15]).
Although glycan function is highly specific to the particular glycoprotein on which the glycan is present, it often relies upon glycan-binding proteins, such as galectins, sialic acid binding Ig-like lectins (siglecs), and C-type lectin receptors (CLRs). The regulation of glycan structures and glycan-binding protein expression, and their complex interplay, are increasingly recognized as crucial features of a properly functioning immune system (Figure 2). Glycan function in the immune system is perhaps best known for its role in selectin binding (reviewed in [16–18]). Selectins are surface-localized members of the CLR family that bind to glycans and mediate leukocyte rolling and ultimately lead to extravasation into inflamed or infected tissues with the assistance of the integrins. As such, the regulation of glycan structures on endothelial and leukocyte surfaces during an inflammatory response is a key factor in the recruitment and homing of immune cells to the affected area.
Figure 2. Pathways for immune regulation by glycans and glycan-binding proteins.

Glycan binding proteins, such as the galectin and C-type lectin families, play both positive and negative roles in the induction of an immune response. For example, galectin-1 (Gal-1) and galectin-3 (Gal-3) hold CD45 at the cell surface, such that T cell receptor signaling is muted by preventing Lck phosphorylation, while Gal-1 can also impact the differentiation of the FoxP3− Tr1 subset of inducible T regulatory cells through an unknown mechanism. The C-type lectin DC-SIGN functions not only to recognize foreign carbohydrates from H. pylori and M. tuberculosis, but it also binds to sialylated IgG molecules (sIgG) as part of an anti-inflammatory cascade. Galactosylated IgG (gIgG) crosslinks the inhibitory FcγRIIB receptor with Dectin-1, which leads to inhibition of C5aR signaling, whereas galectin-9 associates with glycans on Tim-3 leading to apoptosis. The glycans themselves can regulate these interactions through programmed changes in glycan structure and diversification, which could be mediated by changes in expression of glycosylation-specific Golgi enzymes or their carbohydrate substrate concentrations.
Here, we highlight how regulated changes in protein glycosylation both at the cell surface and on secreted glycoproteins can positively and negatively modulate the immune response. This is well documented for the glycan-binding proteins selectins and siglecs that control leukocyte homing and self versus non-self discrimination, respectively and has been reviewed elsewhere 18–24. New work shows that glycans and their binding partners regulate antibody function, antigen presentation (e.g., MHCII), skewing of T cell responses, immune signaling cascades and innate recognition of microbial products. Like regulation of transcription and protein phosphorylation, the regulation of glycan synthesis and glycan-binding lectin expression is central to the immune response.
Glycosylation acts as a switch between pro- and anti-inflammatory IgG functionality
Antibodies are tetrameric glycoproteins that play a crucial role in directing immune responses. IgG is the major antibody isotype in the circulation and IgA is the dominant form in the mucosa. Each antibody consists of two copies of the heavy chain and two copies of the light chain, which together contain variable antigen specificity-determining (Fab) domains positioned away from a conserved functional (Fc) domain that modulates the cellular response in a way that is dependent on the type of cell-surface Fc receptor proteins engaged by the antibody (e.g. FcγRs for IgG). All IgG Fc domains contain a single, highly conserved, glycosylation site (Asn297) that carries complex N-glycans (Box 1; Figure 1) that are required for Fc receptor binding, probably due to the structural role they play in stabilizing the overall conformation of the region 25. Over 30 glycan variations have been detected, but they are typically limited to biantennary structures (Figure 1) with and without terminal sialic acids 26. Recent evidence suggests that Fc-localized N-linked glycans directly impact IgG antibody function.
Glycans modulate IgG Fc function, in part, by altering the binding affinity for different FcγRs. IgG containing glycans that lack core fucose have increased affinity for FcγRIIIA 27, a receptor involved in antibody-dependent cell-mediated cytotoxicity (ADCC) 28. IgG containing a bisecting GlcNAc (Figure 1) have increased affinity for FcγRIII 29, a receptor that mediates neutrophil recruitment 30 and which is required for the development of collagen-induced arthritis 31. Terminal sialylation seems in general to decrease affinity to FcγRs 32.
One effective treatment for patients with autoimmune disease is the use of high-dose intravenous immunoglobulin (IVIg), which robustly suppresses the inflammatory response. In 2006, it was discovered that IgG antibodies with Fc domains carrying α2,6-linked sialylated N-glycans represent the anti-inflammatory portion of the IVIg 26. Although sialylated IgG (sIgG) comprises only about 10 % of total IVIg, when enriched, sIgG shows a 100-fold increase in anti-inflammatory activity over IVIg by weight in a mouse model of arthritis 26. In parallel experiments, in vitro α2,3-linked sialylation of IgG Fc did not result in anti-inflammatory activity, demonstrating that this effect is specific for the α2,6 linkage 33.
Although sIgG shows reduced binding affinity (decreased Ka values) to both activating and inhibitory FcγRs 26, the anti-inflammatory mechanism of the sIgG fraction of IVIg appears to be mediated through binding to the CLR DC-SIGN (SIGN-R1 in mice) (Figure 2) 34. Murine marginal zone macrophages, or human DCs, bind to sIgG and produce IL-33 leading to expansion of IL-4-secreting basophils. Effector macrophages then respond by increasing expression of the inhibitory ITIM-containing Fc receptor FcγRIIB, which shifts the response away from the damaging pro-inflammatory cascade (Figure 2). This mechanism was reported to suppress arthritis in one murine model of the disease 34, but more recent data suggests other mechanisms through which N-glycans may enable IgG-mediated suppression of the immune response 35.
Most FcγRs send activating signals that can synergize with the complement protein C5a and its receptor C5aR to elicit a pro-inflammatory cascade that is relevant to several immune-mediated diseases 36. Recent findings reveal that IgG1 immune complexes can suppress C5aR to inhibit inflammation through the coordinated action of both FcγRIIB and the CLR dectin-1 (Figure 2) 35. More specifically, IgG1 complexes induce FcγRIIB and dectin-1 association, leading to a blockade in ERK1/2 phosphorylation downstream of C5aR signaling. In contrast to earlier work 26,33,34, this study revealed that the inhibitory IgG1 requires high amounts of galactosylation (gIgG), while the presence of terminal sialic acids appears irrelevant for activity, further demonstrating the power of IgG glycosylation in determining biological functions.
Given these data, it is not surprising that changes in concentration of IgG Fc glycans are reported in patients with autoimmunity and inflammatory disease. For example, rheumatoid arthritis (RA) and Wegener’s granulomatosis patients produce lower levels of sIgG compared to normal controls 37,38. During remission, these patients show increased amounts of α2,6-sialylated IgG in the circulation, suggesting that sialylation is dynamically regulated 38. Loss of terminal galactose residues (agalactosylation) on IgG1, and subsequent loss of inhibitory gIgG, is also seen in patients suffering from RA and can appear in circulation preceding disease onset 39,40, however, the regulatory mechanisms leading to the generation of these IgG glycoforms remains unclear. One study showed that IL-21 is capable of modulating IgG glycosylation from B cells in vitro 41. Another group demonstrated the need for an intact promoter region of ST6Gal-1, the enzyme responsible for creating α2,6 sialyl-linkages, to produce anti-inflammatory IgG during inflammatory progression 42. Work from a third group suggests that antigen-specific regulation of sIgG forms under tolerogenic T cell-dependent signaling during allergic inflammation 43. Much work remains to fully understand the complex regulatory program required to dynamically manipulate IgG glycosylation.
MHC class II glycosylation modulates antigen presentation
The central event for initiation of T cell responses is αβ T cell receptor (αβTCR) recognition of an antigenic epitope presented in the context of a class I or class II MHC (MHCI/MHCII) molecule. Most textbooks and articles diagram this interaction as that of a heterodimeric αβTCR protein associating with a heterodimer MHC molecule with a bound peptide antigen, which is based on numerous crystal structures of such ternary complexes (e.g., [44]). However, these structures are derived from recombinant molecules that lack their mammalian patterns of glycosylation. Although glycosylation has been thought of as inconsequential for both MHCI and MHCII, recent studies suggest that the glycans on both the αβTCR and MHCII can impact T cell activation.
αβTCRs are heavily glycosylated, with each subunit containing multiple acceptor sites for N-linked glycosylation. Glycan occupancy of one of these sites is needed for cell surface localization 45, suggesting a role in early ER quality control events. Due to the spatial orientation of αβTCR N-glycans, their potential role at the cell surface is thought to contribute to the orientation and organization of the molecule itself rather than to directly influence MHC interactions. For example, a global deficiency in the β1,6 N-acetylglucosaminyltransferase V (Mgat5) enzyme (Figure 1) alters T cell N-glycosylation patterns resulting in a lower activation threshold in vitro and increased incidence of autoimmune disease in vivo 46. In this case, the absence of the Mgat5 enzyme, which is critical for normal T cell N-glycan branching (Figure 1), results in a lack of galectin-mediated αβTCR lattice formation (Box 2) 46. This is now known to be a critical feature of T cell receptor signaling, in part due to the role of CD45 within the αβTCR/galectin lattice (Box 2) 47. The cytoplasmic tail of CD45 contains phosphatase activity that maintains Lck, a critical signaling molecule that helps to initiate the cascade leading to T cell activation, in a de-phosphorylated and inactive state 48. The galectin-mediated lattice is responsible for holding CD45 and the TCR signaling complex in close proximity via their O- and N-linked glycans (respectively) to prevent low-avidity T cell activation 13,46–48 (Figure 2).
Box 2. The Galectins and Cell Surface Lattices.
Galectins are β-D-galactoside binding proteins (15 identified to date [58]) that are grouped into three classes based on global structure: prototypical, tandem-repeat, and chimeric (figure).58 Cytoplasmic forms of galectins are typically found as monomers, whose function appears to rely upon protein-protein interactions more than protein-carbohydrate interactions that dominate at the cell surface. However, multimerized galectins seem to have a more potent signaling potential over their monomeric counterparts 102. Several members of the galectin family seem to be ubiquitously expressed in cells of the immune system (e.g., galectin-1), whereas others are cell-type specific (e.g., galectin-10 is specifically expressed in eosinophils, basophils, and regulatory T cells) (see [58]).
The key mechanistic feature underlying galectin activity appears to be the ability to form multivalent protein-carbohydrate associations at the cell surface 103. As most membrane proteins have more than one O- or N-linked glycan per molecule, the multivalent nature of secreted galectins allows for a network of crosslinking events known as a lattice mediated by the surface glycoprotein glycans. A galectin lattice is analogous to lipid rafts and other membrane structures that facilitate the formation of membrane domains that may be enriched for specific molecules while excluding others, only for galectins, this occurs by virtue of the fact that as secreted molecules, they contain at least two CRDs (figure). Within the immune system, clustering of surface receptors is a key to signal transduction leading to cellular activation, cellular inactivation, apoptosis, and other outcomes. As a result, galectins can both promote signaling through holding key molecules in close proximity or inhibit signaling by either excluding required molecules or through clustering inhibitory proteins with signaling molecules, as noted for αβTCR and CD45 13,47.
Galectin structural motifs.
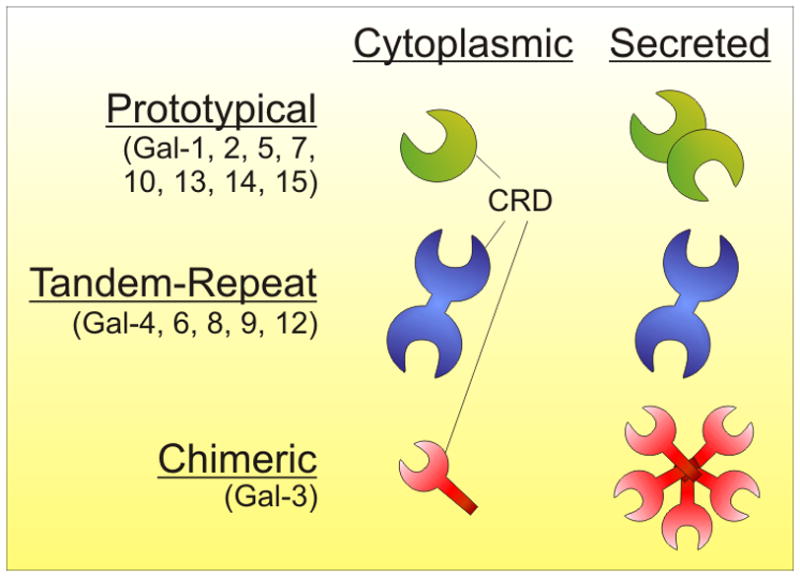
The galectin family is categorized by the structural arrangement of the carbohydrate recognition domains (CRDs). Prototypical galectins exist as monomers in the cytoplasm and are secreted as homodimers containing two CRDs (one per subunit). Tandem-repeat galectins are monomeric, yet contain two CRDs connected by a linker domain. Chimeric galectins exist as homopolymers with a variable number of subunits, each containing a single CRD.
Consistent with these findings, a recent study directly demonstrated that selective glycan modification of a tumor-specific αβTCR, through removal of glycan attachment sites, improved functional avidity of the T cell by improving αβTCR multimerization, decreasing TCR-MHC dissociation, and ultimately increasing the recognition of tumor cells bearing the target antigen 45. Importantly, site-directed mutagenesis of the TCR α-chain glycosylation sites alone influenced functional avidity of the T cell without conferring detectable self-antigen autoreactivity 45.
Both MHCI and MHCII molecules contain highly conserved acceptor sites of N-glycosylation across species, suggesting selective pressure that remains elusive. Historically, glycans on MHC have been thought to not play a significant role in peptide antigen binding or in interactions with the αβTCR, and the only known role for the conserved N-glycan sites in MHCI is in protein folding and trafficking to the cell surface 49,50. However, data from studies of MHCII challenge this traditional viewpoint 45,51. The N-glycosylation of MHCII molecules does not appear to play a role in early protein folding events in the ER or in trafficking of MHCII through the secretory pathway. Instead, MHCII glycosylation can directly modulate antigen binding and presentation of an atypical class of T cell-dependent carbohydrate antigens (glycoantigens) 51. Glycoantigens bind to MHCII at a relatively high affinity (Kd = 315 nM) 52 in a size- and conformation-dependent fashion 53. Unlike peptide antigens, these glycoantigens fail to associate with MHCII molecules lacking mammalian complex N-glycans 51. Expression of recombinant MHCII in cells with altered Golgi activity leads to glycan structural intermediates (e.g., high mannose and hybrid-type glycans; Figure 1) on MHCII and a subsequent failure of MHCII to associate with glycoantigens in vitro. Likewise, ablation of the Mgat2 locus (Figure 1) limits glycan branching and decreases glycoantigen presentation by MHCII leading to loss of T cell stimulatory activity 51. Incomplete MHCII glycosylation does not appear to limit the capacity of MHCII to bind and present peptide antigens 51, despite the fact that glycoantigen and peptide binding are competitive and probably share binding sites 52. Given the relatively large size of MHCII-associated N-glycans (i.e., roughly the size of an Ig domain each) 54, it is possible that the regulation of N-glycan structure, in particular the branching pattern (Box 1; Figure 1), could serve as a regulatory mechanism by which to manipulate antigen selection within the MHCII compartment. Although changes in N-glycan branching are associated with inflammatory disease (reviewed elsewhere [55–57]) and are known to alter the presentation of at least one group of T-dependent antigens 51, this intriguing possibility remains unexplored.
Galectins regulate leukocyte activity through the formation of lattices
Galectins are a highly conserved family of 15 known proteins initially identified based on their ability to bind β-D-galactosides, especially LacNAc (type I, Galβ1-3GlcNAc; type II, Galβ1-4GlcNAc) 58, found within both O- and N-glycans. These proteins function both intracellularly as modulators of the splicing machinery 59, and possibly of signaling cascades 60, as well as on the extracellular face of the cell, primarily through the formation of lattices with surface glycoproteins 61 (Box 2).
The earliest work on this family of proteins dates back to 1975 where a β-D-galactoside binding lectin (‘electrolectin’) isolated from the electric eel Electrophorus electricus was discovered 62, whereas the ability of galectins to modulate the immune response was first suggested in 1983 with the finding that electrolectin could prevent the onset of experimental autoimmune myasthenia gravis in rabbits.63 Later, human galectin-1 (then known as IML-1) was shown to inhibit experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis.64 Since the renaming of this family to ‘galectin’ 1994 12, a number of key mechanistic studies have been published. First, in 1995 65 and in the subsequent three years 66,67, studies showed that galectin-1 is an important mediator of T cell apoptosis and is involved in homeostatic maintenance of T effector cell function. It was reported that galectin-3 is important in negatively regulating T cell activation through direct interactions with glycans decorating the αβTCR 46 (Figure 2).
Galectin binding has been linked to CD8+ T cell anergy after antigenic stimulation 68. Recent findings show that galectin-3: 1) destabilizes the immune synapse between professional antigen-presenting cells and responding T cells; 2) suppresses activation of early αβTCR signaling events; and 3) is involved in the endosomal sorting complex required for transport (ESCRT) of receptors needed for signaling and immune synapse formation.69 T cell-mediated immunity can further be regulated through the interaction of galectin-9 and Tim-3 on T helper type 1 (Th1) cells. This interaction is thought to regulate Th1 responses through inducing apoptosis of activated cells 70, and was also found to promote granulocytic myeloid-derived suppressor cell (MDSC)-mediated suppression of both CD4+ T cell proliferation and IFNγ secretion 71. Regulatory IL-10-producing dendritic cells (DCs) were also induced upon exposure of immature DCs to galectin-1. These DCs promoted T cell tolerance and induced type 1 regulatory T (Tr1) cells in an IL-27-dependent fashion 72 (Figure 2).
Using the EAE model, recent findings demonstrate that galectin-1 could be an important regulatory factor that limits autoimmune neurodegeneration. Administration of recombinant galectin-1 is known to ameliorate experimental autoimmune uveitis 73, a Th1-mediated inflammatory disease. Galectin-1 also deactivates classically activated, pro-inflammatory microglia through binding of CD45 at the cell surface 74 (Figure 2). This association appears to promote retention of CD45 at the plasma membrane, prolonging inhibitory phosphatase activity, deactivating the microglia and resulting in protection against immune-mediated neuronal damage and the onset of neurodegeneration.
The suppressive effects of galectins are not limited to tolerance or the regulation of Th1-mediated inflammation. Several other autoimmune conditions and cancers are associated with increased production of galectins and immune evasion. For example, the presence of galectins in solid tumors impairs the ability of anti-tumor T cells to effectively kill tumor cells. Although the mechanism is unclear, early studies indicate that treating mice with anti-galectin antibodies, galectin ligands, or metabolic inhibitors to eliminate host galectin binding show potential as therapeutics to overcome these suppressive effects 75,76. Beyond tumor immunology, results are less clear. Some viral infections, such as hepatitis C, induce elevated concentrations of galectins that induce Treg expansion and kill virus-infected cytotoxic T lymphocytes through crosslinking Tim-3 77. Other viral infections, such as herpes simplex virus (HSV), and some autoimmune conditions, potentially benefit from exogenous galectin therapy. In HSV-infected mice and in a mouse model of type 1-diabetes, galectin treatment promoted IL-10 secretion, triggered apoptosis of activated immune cells 78,79, impaired T cell homing, and decreased the secretion of pro-inflammatory cytokines 78. Similarly, oral GlcNAc administration enhances N-glycosylation, including galactosylation, thereby increasing the ligands available for surface galectins and inhibiting Th1/Th17 activity in EAE 80. Consistent with these data, recent reports show that a number of factors, such as host metabolism and vitamin D(3), can contribute to altered glycan synthesis and lead to autoimmunity 81,82, including EAE. In fact, mouse strains susceptible to EAE (i.e., PL/J, SJL, and NOD) show differences in N-glycan branching patterns 83 (see Figure 1), which is known to influence galectin binding affinity 84 and therefore lead to changes in galectin-mediated lattice formation and T cell-dependent neurodegeneration; however, the relevance to human multiple sclerosis remains unclear.
Although some galectins can act as suppressive molecules in inflammation and T cell responses, not all members of the galectin family follow the same pattern. For example, in contrast to the observation that galectin-3 prevents low-stringency activation of the αβTCR, galectin-8 binding to T cells appears to promote T cell proliferation, possibly through unique interactions with CD45 85 (Figure 2). The varied effects of galectins indicate differences in their binding specificities and downstream signaling pathways. It remains unclear how these glycan binding proteins have specific effects at the cell surface despite the fact that their minimal carbohydrate ligand is present on many or even most of the surface proteins on that same cell. Although binding studies are beginning to identify particular glycan ligands beyond LacNAc which are recognized by specific galectins 86, more work is needed to clarify the mechanisms by which galectins associate with their glycan targets and produce specific immune effects.
C-type lectins are pro- and anti-inflammatory immunomodulators
C-type lectin receptors (CLRs) are another group of glycan-binding proteins that are important for immune recognition and regulation. CLRs were originally characterized as a family of Ca2+-dependent cell surface proteins, although this family now includes homologous but calcium-independent receptors. As a key member of the pattern recognition receptor (PRR) superfamily, CLRs bind a variety of glycan structures including mannose, fucose, galactose, and β-glucan polymers on fungi. These lectins play important roles as adhesion molecules, signaling molecules, and antigen uptake mediators (reviewed in [58,87]). Polymorphisms in several CLR genes associate with autoimmune diseases, such as type 1 diabetes and multiple sclerosis 88,89.
Some CLRs, such as the secreted RegIIIγ, have direct bactericidal properties. Systemic administration of flagellin stimulates TLR5 on CD103+CD11b+ lamina propria DCs, resulting in IL-23 production which stimulates IL-22 and RegIIIγ production by the intestinal epithelium 90 (Figure 2). By associating with glycans, RegIIIγ binds to peptidoglycan of gram-positive bacteria leading to microbial cell death 91. Although this lectin pathway increases defense against some bacterial pathogens, Helicobacter pylori was recently shown to induce RegIIIγ via production of the CagA cytotoxin, which stimulates the gastric epithelium in a STAT3-dependent fashion 92. This could be a mechanism by which gram-negative H. pylori reduces its microbial competition through the induction of gram-positive bactericidal activity 92.
In contrast to RegIIIγ, most CLRs are cell surface transmembrane proteins. Dectin-1 (also called CLEC7A) is an important CLR that recognizes β-glucans (i.e., β-linked polymers of glucose) on the surface of fungi and triggers key antimicrobial pathways, such as the respiratory burst and phagocytosis 93. Only insoluble β-glucans activate Dectin-1-dependent signaling, consistent with the recognition of fungi-attached β-glucans rather than soluble fragments 93. Dectin-1-deficient mice show greater sensitivity to dextran sulfate sodium (DSS)-induced colitis. This is linked to loss of direct contact between dectin-1 and the β-glucans found on fungi of the normal commensal flora, because DSS-induced colitis was reduced in dectin-1-deficient animals after treatment with anti-fungal antibiotics (i.e., fluconazole).94 A polymorphism in the human CLEC7A (dectin-1) gene was linked to a severe form of ulcerative colitis, suggesting that the mouse observations are directly relevant to human inflammatory bowel disease.94
CLRs represent potential targets for vaccine design. CLEC9A is a recently discovered member of the CLR family that is expressed on a small proportion of DCs, B cells, and monocytes. Human myeloid DCs expressing this receptor are able to cross-present antigens delivered via anti-CLEC9A antibodies, leading to both CD4+ and CD8+ antigen-specific T cell responses 95. Other members of this family that have been well studied are DC-SIGN and DC-ASGPR, both of which are found primarily on DCs. These molecules have become popular targets for therapeutics because of their role in the generation of antigen-specific T cell responses. Targeting antigens directly to DC-ASGPR promotes the activation and differentiation of regulatory T cells that secrete IL-10 96, while oral treatment of mice with highly mannosylated molecules target murine DCs through SIGN-R1, the mouse homolog to DC-SIGN, and leads to oral tolerance and the induction of IL-10 secretion 97. Remarkably, the signaling outcome downstream of DC-SIGN appears to be sensitive to the glycan composition of the ligand, which contradicts the common assumption that signaling through most cell surface receptors is independent of the ligand and can be studied using non-physiologic ligands (e.g., antibody-mediated crosslinking). A recent study examined the engagement of DC-SIGN by Mycobacterium tuberculosis, HIV-1, and H. pylori as well as the cytokines induced by this activation 98. Although all ligands bound to DC-SIGN, mannose-rich agonists expressed by M. tuberculosis and HIV-1 led to the expression of pro-inflammatory molecules such as IL-12 and IL-6, whereas fucose ligands from H. pylori primarily elicited only IL-10 98 (Figure 2). These data demonstrate that DC-SIGN-mediated signaling can be tuned to a pro- or anti-inflammatory status based on the glycan composition. However, DC-SIGN is not only important for the recognition of glycan ligands from microbial sources, but it also appears to play a role for the function of IgG antibodies as discussed earlier 99.
Concluding remarks
The advances in glycoimmunology highlighted here illustrate the inescapable conclusion that glycosylation plays a regulatory and often pivotal role in the immune system. In broad terms, pathways that influence glycan structure and activity, as well as pathways that influence glycan-binding proteins can both influence immunity. Changes in glycan structure are linked to inflammation and infection, as illustrated by regulation of IgG function by galactosylation 35,37 and sialylation 26,33,34, and by regulation of antigen presentation by MHCII glycosylation 51. However, the nature of the relationship between inflammation or infection and glycan structure, the impact on immune function, and whether glycan changes are a consequence or an underlying cause of the inflammatory cascade remain unclear 55.
While the regulation of glycan structure also directly impacts glycan-binding protein activity, the biological function of these molecules appears to be context-dependent. For example, DC-SIGN signaling is influenced by simultaneous Toll-like receptor (TLR) ligation on the same cell 100, which is consistent with the idea that a combination of PRR signaling events on the same cell converge to determine outcome. Galectin function also seems context dependent and dictated by the glycoprotein to which the galectin is bound 58, rather than by the galectin itself. A thorough understanding of how glycan-binding proteins achieve specific and differential responses under a variety of normal and disease-associated states remains a central issue.
There are at least two main challenges in the field. First, regulation of protein glycosylation is poorly understood, in particular regulation of glycosylation on a specific protein rather than the glycome of an entire cell. Golgi enzyme activity, sugar-nucleotide transporter expression, metabolism of precursor carbohydrates, and trafficking of nascent proteins through the ER and Golgi could all regulate glycosylation, but the pathways responsible for such regulation and the immunological signals they respond to are enigmatic. A second challenge is that experimentally manipulating glycan structures on a specific protein in a living cell or organism is impossible, short of simply removing the glycosylation site, which could have very different effects compared to alterations in the glycan structures themselves. A ‘systems’ approach is probably necessary to truly integrate changes in cellular glycosylation with cellular function rather than focusing on single molecules.
Approaching these questions is becoming easier due to significant technological progress dissecting glycan compositions and structures. Core facilities at the University of California San Diego (http://glycotech.ucsd.edu/), the University of Georgia Complex Carbohydrate Research Center (http://www.ccrc.uga.edu/services/index.php) and elsewhere are now available to help with glycan-related analyses on a fee-for-service basis. The Consortium for Functional Glycomics (CFG; http://www.functionalglycomics.org/) has been responsible for developing glycan binding arrays to determine glycan binding specificity, while making a large variety of purified and synthesized glycans available for investigators. The glycosylation patterns found on many common cell lines, primary cells, and tissues are freely available in the CFG-maintained databases. These technical advances coupled with increased availability of services and reagents reduce the technical threshold for entry into the intersection of glycobiology and immunity.
Acknowledgments
The Cobb laboratory would like to thank the National Institute for General Medical Sciences and the National Institutes of Health office of the Director (grants GM082916 and OD004225) as well as the American Asthma Foundation (grant 10-0187) for their continuing support of our work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Brockhausen I, et al. O-Glycans. In: Varki A, et al., editors. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; 2009. pp. 115–127. [PubMed] [Google Scholar]
- 2.Stanley P, et al. N-Glycans. In: Varki A, et al., editors. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; 2009. pp. 101–114. [PubMed] [Google Scholar]
- 3.Dodd RB, Drickamer K. Lectin-like proteins in model organisms: implications for evolution of carbohydrate-binding activity. Glycobiology. 2001;11:71R–79R. doi: 10.1093/glycob/11.5.71r. [DOI] [PubMed] [Google Scholar]
- 4.Antonopoulos A, et al. Glycosylation of mouse and human immune cells: insights emerging from N-glycomics analyses. Biochem Soc Trans. 2011;39:1334–1340. doi: 10.1042/BST0391334. [DOI] [PubMed] [Google Scholar]
- 5.North SJ, et al. Mass spectrometry in the analysis of N-linked and O-linked glycans. Curr Opin Struct Biol. 2009;19:498–506. doi: 10.1016/j.sbi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill S, et al. Proteoglycans: key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat Rec (Hoboken) 2010;293:968–981. doi: 10.1002/ar.21094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanover JA, et al. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 8.Hart GW, et al. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang D, et al. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91:221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei B, et al. Interplay between carbohydrate and lipid in recognition of glycolipid antigens by natural killer T cells. Ann N Y Acad Sci. 2012;1253:68–79. doi: 10.1111/j.1749-6632.2011.06435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sloane JA, et al. A clear and present danger: endogenous ligands of Toll-like receptors. Neuromolecular Med. 2010;12:149–163. doi: 10.1007/s12017-009-8094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barondes SH, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76:597–598. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- 13.Earl LA, et al. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J Biol Chem. 2010;285:2232–2244. doi: 10.1074/jbc.M109.066191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley P, Guidos CJ. Regulation of Notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. 2009;230:201–215. doi: 10.1111/j.1600-065X.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- 15.Stanley P, Okajima T. Roles of glycosylation in Notch signaling. Curr Top Dev Biol. 2010;92:131–164. doi: 10.1016/S0070-2153(10)92004-8. [DOI] [PubMed] [Google Scholar]
- 16.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 17.Tsuboi S, Fukuda M. Roles of O-linked oligosaccharides in immune responses. Bioessays. 2001;23:46–53. doi: 10.1002/1521-1878(200101)23:1<46::AID-BIES1006>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crocker PR, et al. CD33-related siglecs as potential modulators of inflammatory responses. Ann N Y Acad Sci. 2012;1253:102–111. doi: 10.1111/j.1749-6632.2011.06449.x. [DOI] [PubMed] [Google Scholar]
- 20.Grigorian A, et al. Interleukin-2, Interleukin-7, T cell-mediated autoimmunity, and N-glycosylation. Ann N Y Acad Sci. 2012;1253:49–57. doi: 10.1111/j.1749-6632.2011.06391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawashima H, Fukuda M. Sulfated glycans control lymphocyte homing. Ann N Y Acad Sci. 2012;1253:112–121. doi: 10.1111/j.1749-6632.2011.06356.x. [DOI] [PubMed] [Google Scholar]
- 22.Paulson JC, et al. Siglecs as sensors of self in innate and adaptive immune responses. Ann N Y Acad Sci. 2012;1253:37–48. doi: 10.1111/j.1749-6632.2011.06362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sperandio M. The expanding role of alpha2–3 sialylation for leukocyte trafficking in vivo. Ann N Y Acad Sci. 2012;1253:201–205. doi: 10.1111/j.1749-6632.2011.06271.x. [DOI] [PubMed] [Google Scholar]
- 24.Vasta GR, et al. Diversity in recognition of glycans by F-type lectins and galectins: molecular, structural, and biophysical aspects. Ann N Y Acad Sci. 2012;1253:E14–E26. doi: 10.1111/j.1749-6632.2012.06698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold JN, et al. Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics. 2008;8:3284–3294. doi: 10.1002/pmic.200800163. [DOI] [PubMed] [Google Scholar]
- 26.Kaneko Y, et al. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 27.Shields RL, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 28.Masuda K, et al. Enhanced binding affinity for FcgammaRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol Immunol. 2007;44:3122–3131. doi: 10.1016/j.molimm.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Davies J, et al. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol Bioeng. 2001;74:288–294. [PubMed] [Google Scholar]
- 30.Coxon A, et al. Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- 31.Diaz de ST, et al. Expression of FcgammaRIII is required for development of collagen-induced arthritis. Eur J Immunol. 2002;32:2915–2922. doi: 10.1002/1521-4141(2002010)32:10<2915::AID-IMMU2915>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Scallon BJ, et al. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. 2007;44:1524–1534. doi: 10.1016/j.molimm.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Anthony RM, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anthony RM, et al. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karsten CM, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med. 2012 doi: 10.1038/nm.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 37.Holland M, et al. Hypogalactosylation of serum IgG in patients with ANCA-associated systemic vasculitis. Clin Exp Immunol. 2002;129:183–190. doi: 10.1046/j.1365-2249.2002.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alavi A, et al. Immunoglobulin G glycosylation and clinical outcome in rheumatoid arthritis during pregnancy. J Rheumatol. 2000;27:1379–1385. [PubMed] [Google Scholar]
- 39.Parekh RB, et al. Galactosylation of IgG associated oligosaccharides: reduction in patients with adult and juvenile onset rheumatoid arthritis and relation to disease activity. Lancet. 1988;1:966–969. doi: 10.1016/s0140-6736(88)91781-3. [DOI] [PubMed] [Google Scholar]
- 40.Ercan A, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010;62:2239–2248. doi: 10.1002/art.27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, et al. Fc-glycosylation of IgG1 is modulated by B-cell stimuli. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones MB, et al. Anti-inflammatory IgG Production Requires Functional P1 Promoter in beta-Galactoside alpha2,6-Sialyltransferase 1 (ST6Gal-1) Gene. J Biol Chem. 2012;287:15365–15370. doi: 10.1074/jbc.M112.345710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oefner CM, et al. Tolerance induction with T cell-dependent protein antigens induces regulatory sialylated IgGs. J Allergy Clin Immunol. 2012 doi: 10.1016/j.jaci.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 44.Reinherz EL, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 45.Kuball J, et al. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. The Journal of Experimental Medicine. 2009;206:463–475. doi: 10.1084/jem.20082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demetriou M, et al. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- 47.Chen IJ, et al. Lateral Compartmentalization of T Cell Receptor Versus CD45 by Galectin-N-Glycan Binding and Microfilaments Coordinate Basal and Activation Signaling. J. 2007;282:35361–35372. doi: 10.1074/jbc.M706923200. [DOI] [PubMed] [Google Scholar]
- 48.Grigorian A, et al. T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol Rev. 2009;230:232–246. doi: 10.1111/j.1600-065X.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang W, et al. A role for UDP-glucose glycoprotein glucosyltransferase in expression and quality control of MHC class I molecules. Proceedings of the National Academy of Sciences. 2011;108:4956–4961. doi: 10.1073/pnas.1102527108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wearsch PA, et al. Essential glycan-dependent interactions optimize MHC class I peptide loading. Proceedings of the National Academy of Sciences. 2011;108:4950–4955. doi: 10.1073/pnas.1102524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryan SO, et al. MHCII glycosylation modulates Bacteroides fragilis carbohydrate antigen presentation. J Exp Med. 2011;208:1041–1053. doi: 10.1084/jem.20100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cobb BA, Kasper DL. Characteristics of carbohydrate antigen binding to the presentation protein HLA-DR. Glycobiology. 2008;18:707–718. doi: 10.1093/glycob/cwn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kreisman LS, et al. Structure and function relations with a T-cell-activating polysaccharide antigen using circular dichroism. Glycobiology. 2007;17:46–55. doi: 10.1093/glycob/cwl056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudd PM, et al. Roles for glycosylation of cell surface receptors involved in cellular immune recognition. J Mol Biol. 1999;293:351–366. doi: 10.1006/jmbi.1999.3104. [DOI] [PubMed] [Google Scholar]
- 55.Kreisman LS, Cobb BA. Infection, Inflammation, and Host Carbohydrates: A Glyco-Evasion Hypothesis. Glycobiology. 2012;22:1019–1030. doi: 10.1093/glycob/cws070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryan SO, Cobb BA. Roles for major histocompatibility complex glycosylation in immune function. Semin Immunopathol. 2012;34:425–441. doi: 10.1007/s00281-012-0309-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryan SO, Cobb BA. Host glycans and antigen presentation. Microbes Infect. 2012;14:894–903. doi: 10.1016/j.micinf.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity. 2012;36:322–335. doi: 10.1016/j.immuni.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 59.Dagher SF, et al. Identification of galectin-3 as a factor in pre-mRNA splicing. Proc Natl Acad Sci U S A. 1995;92:1213–1217. doi: 10.1073/pnas.92.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karlsson A, et al. Galectin-3 activates the NADPH-oxidase in exudated but not peripheral blood neutrophils. Blood. 1998;91:3430–3438. [PubMed] [Google Scholar]
- 61.Pace KE, et al. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163:3801–3811. [PubMed] [Google Scholar]
- 62.Teichberg VI, et al. A beta-D-galactoside binding protein from electric organ tissue of Electrophorus electricus. Proc Natl Acad Sci U S A. 1975;72:1383–1387. doi: 10.1073/pnas.72.4.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levi G, et al. Prevention and therapy with electrolectin of experimental autoimmune myasthenia gravis in rabbits. Eur J Immunol. 1983;13:500–507. doi: 10.1002/eji.1830130613. [DOI] [PubMed] [Google Scholar]
- 64.Offner H, et al. Recombinant human beta-galactoside binding lectin suppresses clinical and histological signs of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1990;28:177–184. doi: 10.1016/0165-5728(90)90032-i. [DOI] [PubMed] [Google Scholar]
- 65.Perillo NL, et al. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 66.Rabinovich GA, et al. Specific inhibition of lymphocyte proliferation and induction of apoptosis by CLL-I, a beta-galactoside-binding lectin. J Biochem. 1997;122:365–373. doi: 10.1093/oxfordjournals.jbchem.a021762. [DOI] [PubMed] [Google Scholar]
- 67.Rabinovich GA, et al. Activated rat macrophages produce a galectin-1-like protein that induces apoptosis of T cells: biochemical and functional characterization. J Immunol. 1998;160:4831–4840. [PubMed] [Google Scholar]
- 68.Demotte N, et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414–424. doi: 10.1016/j.immuni.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 69.Chen HY, et al. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc Natl Acad Sci U S A. 2009;106:14496–14501. doi: 10.1073/pnas.0903497106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 71.Dardalhon V, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol. 2010;185:1383–1392. doi: 10.4049/jimmunol.0903275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ilarregui JM, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol. 2009;10:981–991. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 73.Toscano MA, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. 2006;176:6323–6332. doi: 10.4049/jimmunol.176.10.6323. [DOI] [PubMed] [Google Scholar]
- 74.Starossom SC, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37:249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cedeno-Laurent F, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates antitumor immunity. J Invest Dermatol. 2012;132:410–420. doi: 10.1038/jid.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Antonopoulos A, et al. Loss of effector function of human cytolytic T lymphocytes is accompanied by major alterations in N- and O-glycosylation. J Biol Chem. 2012;287:11240–11251. doi: 10.1074/jbc.M111.320820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mengshol JA, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS One. 2010;5:e9504. doi: 10.1371/journal.pone.0009504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rajasagi NK, et al. Galectin-1 reduces the severity of herpes simplex virus-induced ocular immunopathological lesions. J Immunol. 2012;188:4631–4643. doi: 10.4049/jimmunol.1103063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perone MJ, et al. Suppression of autoimmune diabetes by soluble galectin-1. J Immunol. 2009;182:2641–2653. doi: 10.4049/jimmunol.0800839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grigorian A, et al. N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J Biol Chem. 2011;286:40133–40141. doi: 10.1074/jbc.M111.277814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mkhikian H, et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun. 2011;2:334. doi: 10.1038/ncomms1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Surolia I, et al. Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature. 2010;466:243–247. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee SU, et al. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J Biol Chem. 2007;282:33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- 84.Dennis JW, et al. Adaptive regulation at the cell surface by N-glycosylation. Traffic. 2009;10:1569–1578. doi: 10.1111/j.1600-0854.2009.00981.x. [DOI] [PubMed] [Google Scholar]
- 85.Tribulatti MV, et al. Galectin-8 provides costimulatory and proliferative signals to T lymphocytes. J Leukoc Biol. 2009;86:371–380. doi: 10.1189/jlb.0908529. [DOI] [PubMed] [Google Scholar]
- 86.Song X, et al. Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem Biol. 2009;16:36–47. doi: 10.1016/j.chembiol.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garcia-Vallejo JJ, van KY. Endogenous ligands for C-type lectin receptors: the true regulators of immune homeostasis. Immunol Rev. 2009;230:22–37. doi: 10.1111/j.1600-065X.2009.00786.x. [DOI] [PubMed] [Google Scholar]
- 88.Hakonarson H, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 89.Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kinnebrew MA, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cash HL, et al. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee KS, et al. Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3gamma via gastric STAT3 activation. PLoS One. 2012;7:e30786. doi: 10.1371/journal.pone.0030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goodridge HS, et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schreibelt G, et al. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood. 2012;119:2284–2292. doi: 10.1182/blood-2011-08-373944. [DOI] [PubMed] [Google Scholar]
- 96.Li D, et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med. 2012;209:109–121. doi: 10.1084/jem.20110399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Y, et al. Oral tolerance to food-induced systemic anaphylaxis mediated by the C-type lectin SIGNR1. Nat Med. 2010;16:1128–1133. doi: 10.1038/nm.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gringhuis SI, et al. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol. 2009;10:1081–1088. doi: 10.1038/ni.1778. [DOI] [PubMed] [Google Scholar]
- 99.Anthony RM, et al. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105:19571–19578. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gringhuis SI, et al. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. 2007;26:605–616. doi: 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 101.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 102.Earl LA, et al. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology. 2011;21:6–12. doi: 10.1093/glycob/cwq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rabinovich GA, et al. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol. 2007;17:513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]