

Abstract
Objective:
We aimed to investigate whether HIV latency in the CNS might have adverse molecular, pathologic, and clinical consequences.
Methods:
This was a case-control comparison of HIV-1 seropositive (HIV+) patients with clinical and neuropathologic examination. Based on the levels of HIV-1 DNA, RNA, and p24 in the brain, cases were classified as controls, latent HIV CNS infection, and HIV encephalitis (HIVE). Analysis of epigenetic markers including BCL11B, neurodegeneration, and neuroinflammation was performed utilizing immunoblot, confocal microscopy, immunochemistry/image analysis, and qPCR. Detailed antemortem neurocognitive data were available for 23 out of the 32 cases.
Results:
HIV+ controls (n = 12) had no detectable HIV-1 DNA, RNA, or p24 in the CNS; latent HIV+ cases (n = 10) showed high levels of HIV-1 DNA but no HIV RNA or p24; and HIVE cases (n = 10) had high levels of HIV-1 DNA, RNA, and p24. Compared to HIV+ controls, the HIV+ latent cases displayed moderate cognitive impairment with neurodegenerative and neuroinflammatory alterations, although to a lesser extent than HIVE cases. Remarkably, HIV+ latent cases showed higher levels of BCL11B and other chromatin modifiers involved in silencing. Increased BCL11B was associated with deregulation of proinflammatory genes like interleukin-6, tumor necrosis factor–α, and CD74.
Conclusion:
Persistence of latent HIV-1 infection in the CNS was associated with increased levels of chromatin modifiers, including BCL11B. Alteration of these epigenetic factors might result in abnormal transcriptomes, leading to inflammation, neurodegeneration, and neurocognitive impairment. BCL11B and other epigenetic factors involved in silencing might represent potential targets for HIV-1 involvement of the CNS.
Infection of the CNS by HIV is associated with neurologic conditions, from HIV-associated dementia (HAD)1 to milder HIV-associated neurocognitive disorders (HAND).2 Highly active antiretroviral therapy (HAART) helps prolong the life of infected patients, reducing the incidence of HAD.3 Although cases with severe HIV-related cognitive impairment have decreased, the frequency of HIV encephalitis (HIVE), characterized by presence of infected macrophages in the CNS, microgliosis, astrogliosis, and myelin loss, has remained the same or slightly increased.4–6
Complete eradication of HIV-1 is not feasible due to persistence of latent viral forms integrated into the host genome.7 The brain represents a reservoir from which productive infections and chronic CNS injury might emerge, since resistance to HAART might allow residual viral replication,8–10 highlighting the importance of host mechanisms mediating latency. In human microglial cells, the transcription factor BCL11B (also known as CTIP2) inhibits HIV transcription by recruiting a chromatin-modifying complex to the LTR region that results in HIV silencing.11 Still, little is known about the mechanisms of HIV-1 latency in the human brain and their potential long-term effects. The persistence of latent virus in the brain might lead to cognitive impairment and neurodegeneration by gene expression alterations and proinflammatory responses.
We hypothesized that patients with latent HIV infection would display neurocognitive, neurodegenerative, and neuroinflammatory alterations similar to but milder than patients with HIVE. Increased levels of BCL11B and other components of the HIV silencing complex might deregulate neuroinflammatory cascades leading to the observed pathology in patients with latent HIV infection.
METHODS
Study population.
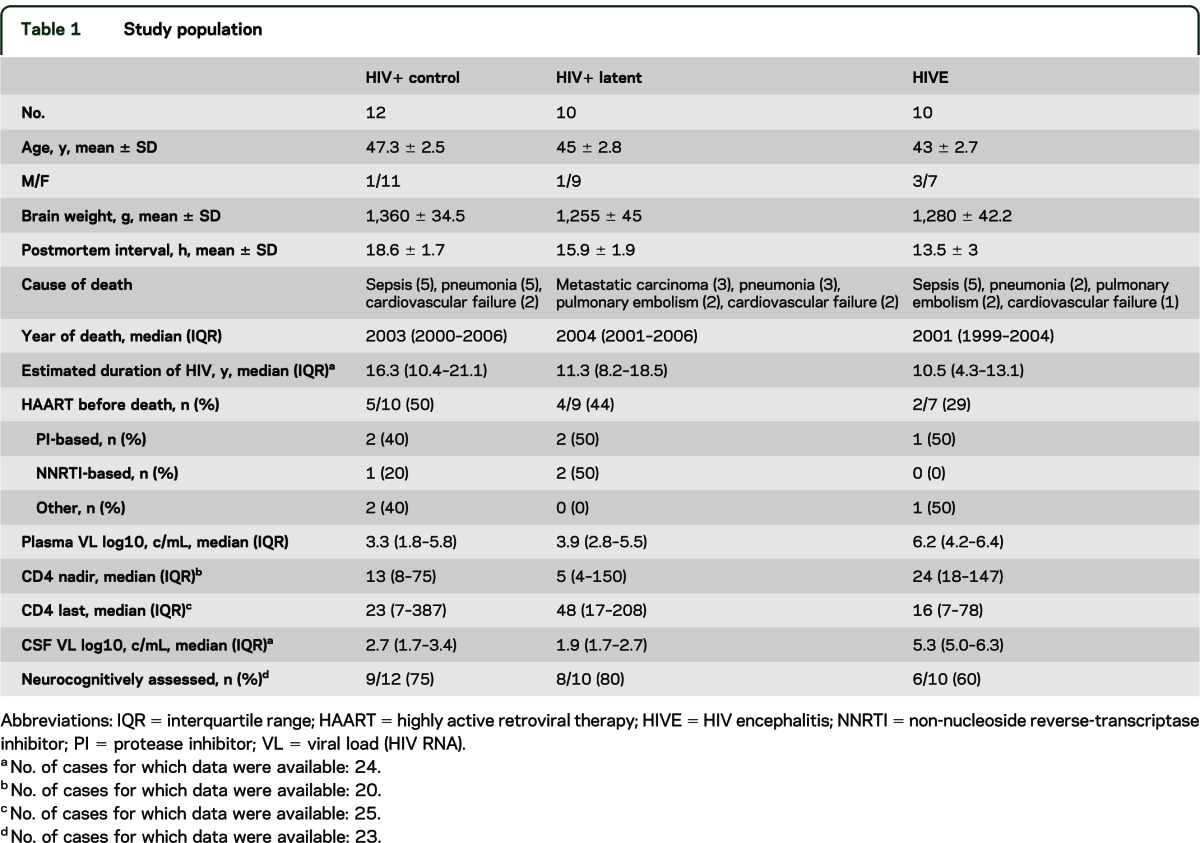
We included 32 HIV+ cases from the HIV Neurobehavioral Research Center (HNRC) and California NeuroAIDS Tissue Network at the University of California, San Diego (UCSD; table 1). Neurocognitive examination was available for 23 of the 32 cases studied. Subjects had neuromedical and neuropsychological examinations within a median of 12 months before death. Most individuals died as a result of acute bronchopneumonia or septicemia (table 1). Autopsy findings were consistent with AIDS and the associated systemic pathology found included disseminated cytomegalovirus and Mycobacterium avium infection, Pneumocystis carinii pneumonia, and hepatitis C infection. Subjects were excluded if they had a history of CNS opportunistic infections or non-HIV-related developmental, neurologic, psychiatric, or metabolic conditions that might affect CNS functioning. The methods for neuropsychological evaluation consisted of a battery of tests, including learning, memory, attention, speed of information processing, abstraction, and verbal and motor skills, as described previously.12 For this study, the neurocognitive status of cases was termed neurocognitively normal, mild to moderate neurocognitive impairment (included cases of subsyndromal/asymptomatic and minor cognitive motor disorder), and severe neurocognitive disorder (equivalent to HIV dementia).
Table 1.
Study population
Standard protocol approvals, registration, and patient consents.
We obtained postmortem brain tissue already banked at HNRC under the California NeuroAIDS Tissue Network study. Approval for the parent study was received from the local institutional review board. Written consent for research was obtained from all the patients participating in the study and donating tissue.
Analysis of HIV DNA, RNA, and p24 and category classification.
DNA and RNA were extracted from frozen human frontal cortex samples with DNeasy mini kit and RNeasy Lipid tissue mini kit (Qiagen, Hilden, Germany). Quantification of HIV-1 DNA was performed at the HIV Neurobehavioral Research Program, Neurovirology Core (UCSD), under standard protocols on 50 ng of DNA. HIV RNA in the brain was quantified by qPCR using HIV-1 pol standard (provided by the HIV Neurobehavioral Research Program, Neurovirology Core) and GAPDH standard (Invitrogen, Carlsbad, CA). Copy number of HIV-1 was normalized to 1×106 copies of GAPDH standard. Total protein homogenates were prepared from flash-frozen postmortem frontal cortex samples as described.13 HIV-1 p24 was quantified by ELISA (PerkinElmer, Waltham, MA) on homogenate aliquots containing 20 μg of total protein.
Real-time PCR analysis of gene expression.
qPCR experiments were performed on iCycler iQ5 Real-Time PCR Detection System (Bio-Rad, Hercules, CA) as described.14 Total RNA was reverse transcribed using iScript cDNA Synthesis kit (Bio-Rad) with 1 μg of total RNA. Amplification was performed on a cDNA amount equivalent to 100 ng total RNA. Oligonucleotides were designed using Oligoperfect (Life Technologies, Carlsbad, CA, sequences available upon request). Calculations were done with Bio-Rad software by the comparative threshold cycle (Ct) method and expressed as 2-exp (ΔΔCt) using PPIA as internal control.
Neuropathologic analysis.
Neuropathologic assessment was performed in paraffin sections from the frontal, parietal, and temporal cortices, and the hippocampus, basal ganglia, and brainstem stained with hematoxylin & eosin and immunolabeled with antibodies against p24 (1:250, Dako, Glostrup, Denmark), Iba1 (microglial marker, 1:1,000, Wako, Richmond, VA), and glial fibrillary acidic protein (GFAP, marker of astrogliosis, 1:500, Millipore, Billerica, MA). The diagnosis of HIVE was based on the presence of HIV-p24–positive cells, microglial nodules, astrogliosis, and myelin pallor.
Immunocytochemical analysis.
Vibratome sections from midfrontal cortex (40 μm thick) from control and HIV cases were processed as described15 using antibodies against MAP2 (dendritic marker, 1:200, Millipore) and synaptophysin (presynaptic marker, 1:500, Millipore).15,16 Immunolabeled blind-coded sections were serially imaged with LSCM (MRC1024, Bio-Rad) and analyzed with Image 1.43 program (NIH), as described.17 Additional sections were immunolabeled with anti-GFAP, Iba1, BCL11B (1:250, Bethyl Labs, Montgomery, TX), interleukin (IL)-6 (1:250, Abcam, Cambridge, UK), CD74 (1:250, Abcam), and CXCR4 (1:100, Chemicon, Temecula, CA) and reacted with diaminobenzidine. Three sections (10 digital images per section at 400×) were analyzed per case to estimate the average number of immunolabeled cells per unit area (mm2) and the average intensity of the immunostaining (corrected optical density).
Immunoblot analysis.
Protein content was determined by BCA assay (Thermo Fisher Scientific, Waltham, MA). Twenty micrograms of total protein were resolved on 4%–12% Bis-Tris gels. Standard Western blotting techniques were used with antibodies: BCL11B (1:1,000, Bethyl Labs), HP1α (1:1,000, Abcam), HDAC1 (1:1,000, Active Motif), HDAC2 (1:2,000, Abcam), MECP2 (1:20,000, Active Motif), SP1 (1:1,000, Active Motif), SUV39H1 (1:1,000, Abcam), CD74 (1:50, Abcam), CXCR4 (1:1,000 Abcam), or Actin (1:1000, C4, Chemicon). Semiquantitative analysis was performed with VersaDoc imaging system and Quantity One software (Bio-Rad).
Double immunolabeling and confocal microscopy imaging.
Double-immunocytochemical analysis was performed with Tyramide Signal Amplification–Direct system (NEB Life Sciences, Ipswich, MA) to detect BCL11B. Sections were double-labeled with anti-BCL11B (1:20,000, Bethyl Labs) detected with Tyramide Red, and anti-p24, GFAP, or Iba1 detected with fluorescein isothiocyanate–conjugated secondary antibodies (1:75, Vector). Sections were processed simultaneously and experiments were performed in duplicate.
Statistical analysis.
Analyses were conducted on blind-coded samples using StatView (SAS Institute, Inc.). Comparisons among groups were performed with one-way analysis of variance with Dunnet post hoc test when comparing the HIV+ latent and HIVE groups to the HIV+ control and with post hoc Tukey-Krammer when comparing the HIV+ latent and HIVE groups. The χ2 test was utilized to compare the frequency distribution of neuromedical and neurocognitive alterations among the 3 HIV groups. All results were expressed as mean ± SEM.
RESULTS
We analyzed 32 cases, whose demographic, clinical, virologic, and pathologic characteristics are presented in table 1. Based on the levels of HIV-1 DNA, RNA, and p24, 12 cases were classified as HIV+ controls, with no detectable HIV-1 DNA, RNA, or p24; 10 cases were designated as latent HIV+, with high levels of HIV-1 DNA and no detectable RNA or significant p24 (figure 1, A–C). In contrast, those cases classified as HIVE (n = 10) displayed high levels of HIV-1 DNA, RNA, and p24 (figure 1, A–C). Productive HIV infection in the latter group was accompanied by the presence of microglial nodules, astrogliosis, and myelin pallor (not shown). At the pathologic level, latent HIV cases displayed some rarefaction of the neuropil accompanied by astrogliosis, while in HIV+ controls there were no histopathologic alterations (data not shown). As shown in table 1, the proportion of subjects prescribed HAART was similar in the control and latent groups (50% vs 44%, respectively), whereas fewer subjects on the HIVE group received HAART (29%). Sustained virologic suppression on HAART was also similar in the control and latent groups (60% vs 67%), in contrast to the HIVE group, where none of the subjects showed evident viral suppression. The groups did not differ significantly with respect to age, estimated duration of HIV infection, year of death, CD4 nadir, current CD4, or plasma viral load at the visit closest to death.
Figure 1. Analysis of HIV-1 expression and neurodegenerative pathology in HIV+ controls, HIV+ latent, and HIVE brains.

HIV-1 DNA and RNA were analyzed by PCR and viral p24 protein by ELISA in frontal cortex samples from HIV+ controls (CT), HIV+ latent, and HIV encephalitis (HIVE) groups. (A) Detection of HIV-1 DNA corroborated the presence of the provirus in the brains of all infected groups. (B, C) HIV-1 active replication, as indicated by viral RNA and p24 detection, is only evident for the HIVE group. Student t test unpaired, 2-tailed, p < 0.001. (D) Immunolabeling of neuronal cells and dendritic structures with anti-MAP2 antibody. (E) Presynaptic terminals were immunostained with anti-synaptophysin antibody. (F) Astroglial cells were immunolabeled for glial fibrillary acidic protein (GFAP), and microglial cells were identified by anti-Iba1 antibody (G). Bar represents 10 μm. (H, I) Image analysis for the percentage of the area of the neuropil covered by the dendritic and synaptic markers. (J, K) Image analysis for levels of GFAP and Iba1 immunoreactivity expressed as corrected optical density per group. One-way analysis of variance with post hoc Dunnet was used to compare control vs latent and HIVE groups, *p < 0.05.
Among the 32 cases, antemortem neurocognitive diagnosis was available for 23 subjects, representing 75% of HIV+ controls (9 of 12), 80% of HIV+ latent cases (8 of 10), and 60% of HIVE cases (6 of 10) (table 1). Severe neurocognitive impairment was only reported for cases with HIVE. Mild to moderate neurocognitive impairment occurred in 6/8 (75%) of the latent cases as compared to 5/9 (56%) of the control cases. The relatively high rate of mild impairment in the control cases may reflect brain injury that occurred before HAART became available to these subjects, who were typically infected for a decade or more.
To understand the anatomical correlates to latent HIV infection, we analyzed neurodegeneration by confocal microscopy (figure 1, D–K). Latent HIV cases displayed considerable loss of MAP2 immunoreactive dendrites (figure 1, D and H) and synaptophysin immunoreactive nerve terminals in the frontal cortex (figure 1, E and I), similar to the alterations observed in HIVE cases (figure 1, D, E, H, and I). Moreover, astrogliosis (figure 1, F and J) and microgliosis were detected in the latent HIV group when compared to controls (figure 1, G and K). As expected, HIVE cases displayed the highest levels of GFAP (figure 1, F and J) and Iba1 (figure 1, G and K) accompanied by the presence of p24 immunostained microglial cells (no p24 immunoreactivity was detected in HIV controls or in latent HIV cases, not shown).
Silencing of HIV-1 transcription in microglial cell cultures is mediated by the transcriptional regulator BCL11B, which recruits a multienzymatic chromatin-modifier complex to repress gene expression.11 We therefore analyzed the levels of BCL11B and other components of the chromatin-remodeling complex in human brain samples. BCL11B protein was significantly higher in the latent group than in control and HIVE cases (figure 2, A and B). Consistently, BCL11B immunoreactivity was elevated in neuronal (figure 2, C and D) and glial cells (figure 2, C and E) in frontal cortex and white matter from latent HIV cases. Double labeling analysis confirmed that high levels of BCL11B were detected in microglial (figure 2, F and G) and astroglial cells (figure 2, H and I) only in the latent HIV cases, where p24 was not detectable, supporting the role of BCL11B in silencing HIV-1. On the contrary, in HIVE cases, p24-positive glial cells were negative for BCL11B (figure 2, G and I). No apparent changes in BCL11B mRNA levels were detected in the studied groups (figure e-1 on the Neurology® Web site at www.neurology.org), suggesting that increased BCL11B associated with latency might be regulated at the level of protein translation or stability. To evaluate whether alterations of BCL11B in the brain might translate into peripheral fluids, similar analyses were performed in CSF samples. CSF from latent HIV cases showed increased BCL11B in comparison to the HIVE group (figure 2, J and K).
Figure 2. BCL11B is highly expressed in the brain of latent HIV+ cases and colocalizes with glial markers.

(A) Representative immunoblot shows a duplex around 120 kDa corresponding to BCL11B. (B) Image analysis of BCL11B immunoreactive bands normalized to actin. (C) Patterns of BCL11B immunoreactivity in the frontal cortex and white matter. Arrows indicate BCL11B immunoreactivity in neurons and arrowheads in glial cells. Bar represents 10 μm. (D–E) Image analysis of BCL11B immunoreactivity levels in neuronal or glial cells expressed as corrected optical density per group. One-way analysis of variance (ANOVA) with post hoc Dunnet when compared to control, *p < 0.05; **p < 0.01; and with post hoc Tukey-Krammer when compared to latent HIV #p < 0.05. (F–H) Double labeling studies were performed in the frontal cortex with antibodies against BCL11B (red channel) and glial fibrillary acidic protein (GFAP), Iba1, or p24 (green channel) and analyzed by laser scanning confocal microscopy. (F) BCL11B colocalized with the microglial marker Iba1 (arrows) in HIV+ latent but not in HIV encephalitis (HIVE) cases. (G) Cells expressing high levels of viral p24 protein in HIVE cases exhibit the lower content of BCL11B, while latent cases show no viral expression and higher BCL11B immunoreactive signal. Arrows indicate p24-positive cells that are BCL11B-negative. (H) Colocalization of BCL11B to GFAP astroglial cells in HIV+ latent cases in comparison to HIVE cases. (I) Image analysis shows the percentage of BCL11B positive from the total cells recorded positive for Iba1, GFAP, and p24 in HIV latent or HIVE cases. Unpaired, 2-tailed, Student t test, *p < 0.05. (J) Representative blot shows BCL11B immunoreactive bands in CSF samples. (K) Densitometric analysis of BCL11B in CSF. One-way ANOVA with post hoc Dunnet to compare control (CT) vs latent and HIVE groups, *p < 0.05.
We next investigated the levels of other components of the HIV-silencing complex (figure 3). HP1α, MeCP2, and HDAC1 were significantly increased in latent cases in comparison to HIVE samples (figure 3, A, B, D, and F), while similar levels of HDAC2, SP1, and SUV39H1 were observed among all groups (figure 3, A, C, E, and G). As reported for BCL11B, upregulation of chromatin modifiers seems to operate at the protein level, without significant changes in gene expression (figure e-1).
Figure 3. The levels of chromatin-remodeling proteins are altered in HIV+ latent cases.

(A) Representative immunoblot analysis shows HP1α, MeCP2, HDAC1, HDAC2, SP1, and SUV39H1 immunoreactivity by Western blotting in frontal cortex samples. (B–G) Densitometric quantification of chromatin-remodeling protein immunoreactivity, expressed as signal to actin ratios. One-way analysis of variance with post hoc Dunnet, *p < 0.05. CT = control; HIVE = HIV encephalitis.
BCL11B is an important transcriptional regulator in the brain.14,18 Since transcription of several genes, including those involved in apoptosis and cell death, is altered in HIV-1 infected brains,19 we investigated whether this functional group of genes might represent a cluster of BCL11B targets by in silico analysis of the presence of BCL11B consensus sites.20 Interestingly, 18 out of 25 candidate genes contained at least one canonical binding site for BCL11B in their proximal promoter region (table e-1). Among them, CEBP, CD74, CXCR4, SOX9, CFLAR, and PDCD6 have the highest representation of sites, and therefore are the most likely targets for BCL11B-mediated regulation. Analysis of their relative abundance in frontal cortex samples by qPCR revealed that CEBP was reduced (figure 4A) and CD74 and CXCR4 were elevated in the HIV+ latent group (figure 4, B and C). Since these genes are involved in proinflammatory responses, we extended our analysis to include prominent inflammatory markers. We detected a decrease in IL-1 transcription (figure 4D), while significant upregulation of IL-6 was observed in latent HIV cases (figure 4E). Furthermore, CD74 and CXCR4 proteins were increased in the HIV latent group and, to a lesser extent, in HIVE cases when compared to HIV controls (figure 4, G–I). Similarly, CD74 and CXCR4 immunoreactivity were elevated in neurons and microglial cells from latent HIV and HIVE cases, in comparison to control samples (figure 4, J, L, and M). IL-6 immunoreactivity was also elevated in the astroglial cells in the latent group (figure 4, J and K).
Figure 4. Altered expression of BCL11B-regulated proinflammatory targets in HIV+ latent cases.

(A–C) Quantification of BCL11B-downstream targets CEBP, CXCR4, and CD74 mRNAs by qPCR in frontal cortex samples. (D–F) Quantification of the relative abundance of inflammatory markers mRNA by qPCR revealed alterations in the HIV+ latent and the HIV encephalitis (HIVE) groups, suggestive of proinflammatory processes. One-way analysis of variance (ANOVA) with post hoc Dunnet to compare control (CT) vs latent and HIVE groups, *p < 0.05. (G) Representative immunoblot analysis of CXCR4 and CD74 proteins in frontal cortex samples. (H, I) Densitometric analysis of CXCR4 and CD74 immunoreactivity expressed as a ratio to actin. One-way ANOVA with post hoc Dunnet to compare CT vs latent and HIVE groups, *p < 0.05. (J) Immunodetection of interleukin (IL)–6, CD74, and CXCR4 in neuronal and glial cells in frontal cortex sections. Bar represents 10 μm. (K–M) Image analysis of neuronal or glial cells shows corrected optical density of each studied protein per group.
DISCUSSION
We investigated the neuropathologic and molecular alterations associated with CNS latent HIV-1 infections, defined as cases displaying proviral DNA in the brain with no productive infection. Our results suggest that patients with latent HIV display cognitive deficits, neurodegenerative alterations, and neuroinflammatory changes, supporting the notion that viral latency is not innocuous for the brain and might represent a distinct condition with particular pathologic features. Although HIV+ latent and HIVE cases displayed similar clinical and neurodegenerative traits, the extent of the cognitive and pathologic alterations was greater in the HIVE group. At the molecular level, latent cases showed increased BCL11B protein levels, a transcriptional regulator that mediates silencing of the integrated provirus, while this epigenetic modulator of HIV infection was reduced in HIVE cases.
Previous studies suggest that CNS is an important reservoir for HIV where proviral DNA and productive infections can be detected.8,21 Although HAART greatly benefited infected patients, it is unable to cure the infection due to the persistence of long-lived transcriptionally silent proviruses that can integrate into the host genome in subpopulations of susceptible cells, generating viral reservoirs in peripheral blood and organs. The brain, in particular, represents a sanctuary for HIV-1 latency, where the provirus can persist due to the variable and poor penetration of antiretroviral drugs.22,23
While viral replication was considered necessary for inflammation and neurodegeneration, recent studies showed a high prevalence of HAND even in long-standing aviremic patients.24 We provide here a novel explanation for brain damage in the absence of viral replication, mediated by deregulation of BCLL1B that contributes to sustain HIV-1 latency and might also alter the expression of inflammation-related genes.
Neuroinflammatory responses play important roles in neurodegeneration in HIV+ latent and HIVE groups, but based on BCL11B analysis, the pathways leading to inflammation might differ among groups. In HIVE, it has been proposed that HIV-infected monocytes cross the blood–brain barrier early on during infection and subsequently initiate a cascade of inflammatory mechanisms through the release of progeny virus/viral protein and cytokines/chemokines.25,26 Migrating infected monocytes express IL-1, IL-6, tumor necrosis factor (TNF)–α, tumor growth factor–β, GM-CSF, and prostaglandin E2,27 which bind glia receptors and activate additional inflammatory genes through a positive feedback mechanism. In addition to neuroinflammation mediated by physiologic cellular molecules, monocyte-expressed HIV proteins, vpr, tat, nef, and gp120 activate interferon, apoptosis, and MAPK pathways in microglia and astrocytes contributing to chronic inflammation and neurotoxicty.28–31 In contrast, we propose that in patients with latent HIV infections, increased levels of BCL11B might directly trigger neuroinflammatory cascades by transcriptional deregulation of CD74, IL-6, and TNF-α. Although we cannot exclude that the cases we classified here as HIV+ latent could have previously been HIVE manifestations in which effective HAART suppressed viral replication, this possibility seems unlikely, since 50% of the subjects of the latent group never received HAART and the remaining only followed the treatment intermittently, favoring the idea that absence of viral RNA resulted from bona fide silencing mechanisms.
HIV-1 latency requires a successful integration event, where the host chromatin environment plays a key role.8 In microglial cells, BCL11B assembles a multienzymatic complex by recruiting SP1, HP1α, HDAC1, HDAC2, and SUV39H to the viral LTR region, and establishing a heterochromatic environment at the viral insertion site silencing HIV-1 transcription.11 In the present work, we investigated for the first time the status of BCL11B and associated epigenetic factors in the human brain in the context of HIV-1 infection. We report increased BCL11B, HP1α, MECP2, and HDAC1 in latent HIV infection, most probably due to posttranscriptional mechanisms, including protein translation or turnover. BCL11B is a common regulator of gene transcription in the brain14 implicated in the negative regulation of BDNF signaling, which is altered in several neurodegenerative disorders.18 Our results suggest that recruitment of BCL11B to the viral insertion sites during latency might alter the transcription of its target proinflammatory genes, triggering chronic inflammatory responses. We suggest that the presence of silent provirus in the brain might trigger chronic pathology and might be associated with neurocognitive impairment in latently infected patients.
Diagnosis of HIV-1 infection in the CNS can be achieved by detection of HIV in the CSF; still, the presence of latent viral forms can only be corroborated postmortem, hindering efforts to detect resident virus earlier to determine possible therapeutic interventions. An important outcome of the present work is the observation of abnormal BCL11B levels in the CSF of latently infected patients, in correlation with alterations in the brain. Further investigation will determine the potential use of BCL11B as biomarker of silent HIV-1 infections in the CNS.
The recent discovery of involvement of histone methylases and deacetylases in HIV latency fueled research aimed to reactivate latent reservoirs and to facilitate eradication by targeting actively replicating virus.32 Valproic acid and trichostatin A were unable to reactivate HIV in latently infected primary CD4 T cells,33 raising concerns about their potential use in neuroAIDS. Modulation of BCL11B could represent an alternative therapeutic strategy for eradication of reservoirs.
Taken together, our results suggest that latent HIV-1 infection in the CNS might be associated with increased levels of chromatin modifiers, including BCL11B. Alteration of these epigenetic factors might activate proinflammatory cascades associated with neurodegeneration and neurocognitive impairment. BCL11B and other silencing factors might represent novel targets to ameliorate the effects of HIV-1 infections in the CNS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Jerel Fields for contributions to the Discussion.
GLOSSARY
- GFAP
glial fibrillary acidic protein
- HAART
highly active antiretroviral therapy
- HAD
HIV-associated dementia
- HAND
HIV-associated neurocognitive disorders
- HIVE
HIV encephalitis
- HNRC
HIV Neurobehavioral Research Center
- IL
interleukin
- TNF
tumor necrosis factor
- UCSD
University of California, San Diego
Footnotes
Supplemental data at www.neurology.org
Editorial, page 1363
AUTHOR CONTRIBUTIONS
Dr. Desplats and Dr. Masliah designed the study. Dr. Smith performed the DNA and RNA studies. Dr. Everall, Dr. Letendre, Dr. Ellis, Dr. Cherner, and Dr. Grant clinically assessed the patients. Dr. Desplats, Dr. Masliah, Dr. Dumaop, Dr. Smith, and Dr. Adame performed the experimental work. Dr. Desplats and Dr. Masliah analyzed the data and wrote the manuscript. Dr. Desplats, Dr. Masliah, Dr. Grant, Dr. Ellis, and Dr. Letendre contributed to the discussion.
STUDY FUNDING
Supported by NIH grants NS057096, AG03197, AG5131, MH62962 (to E.M.), and MH076681, MH79881, MH45294, MH5974, MH58164, and DA12065, California NeuroAIDS Tissue Network U01 MH83506 (to I.G.). This work was also funded by the HIV Neurobehavioral Research Center (HNRC) CSPAR Developmental Core Grant HNRC-859 (P.D.). The HNRC is supported by Center award MH62512 from NIMH.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fauci AS. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 1988;239:617–622 [DOI] [PubMed] [Google Scholar]
- 2.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 2007;8:33–44 [DOI] [PubMed] [Google Scholar]
- 3.Price RW, Spudich S. Antiretroviral therapy and central nervous system HIV type 1 infection. J Infect Dis 2008;197(suppl 3):S294–S306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everall I, Vaida F, Khanlou N, et al. Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol 2009;15:360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langford TD, Letendre SL, Larrea GJ, Masliah E. Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol 2003;13:195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS 2000;14:69–74 [DOI] [PubMed] [Google Scholar]
- 7.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 2003;22:1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res 2008;6:388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Letendre S, Marquie-Beck J, Capparelli E, et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol 2008;65:65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redel L, Le Douce V, Cherrier T, et al. HIV-1 regulation of latency in the monocyte-macrophage lineage and in CD4+ T lymphocytes. J Leukoc Biol 2010;87:575–588 [DOI] [PubMed] [Google Scholar]
- 11.Marban C, Suzanne S, Dequiedt F, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J 2007;26:412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore DJ, Masliah E, Rippeth JD, et al. Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS 2006;20:879–887 [DOI] [PubMed] [Google Scholar]
- 13.Patrick C, Crews L, Desplats P, et al. Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am J Pathol 2011;178:1646–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desplats PA, Lambert JR, Thomas EA. Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington's disease. Neurobiol Dis 2008;31:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masliah E, Ge N, Morey M, DeTeresa R, Terry RD, Wiley CA. Cortical dendritic pathology in human immunodeficiency virus encephalitis. Lab Invest 1992;66:285–291 [PubMed] [Google Scholar]
- 16.Achim CL, Heyes MP, Wiley CA. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brains. J Clin Invest 1993;91:2769–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mucke L, Abraham CR, Ruppe MD, et al. Protection against HIV-1 gp120-induced brain damage by neuronal expression of human amyloid precursor protein. J Exp Med 1995;181:1551–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang B, Di Lena P, Schaffer L, Head SR, Baldi P, Thomas EA. Genome-wide identification of bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS One 2011;6:e23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everall I, Salaria S, Roberts E, et al. Methamphetamine stimulates interferon inducible genes in HIV infected brain. J Neuroimmunol 2005;170:158–171 [DOI] [PubMed] [Google Scholar]
- 20.Avram D, Fields A, Senawong T, Topark-Ngarm A, Leid M. COUP-TF (chicken ovalbumin upstream promoter transcription factor)-interacting protein 1 (CTIP1) is a sequence-specific DNA binding protein. Biochem J 2002;368:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiley CA, Achim CL, Christopherson C, et al. HIV mediates a productive infection of the brain. AIDS 1999;13:2055–2059 [DOI] [PubMed] [Google Scholar]
- 22.Banerjee A, Nonnemacher MR, Wigdahl B. HIV latency and reactivation: role in neuropathogenesis. In: Meucci O, ed. Chemokine Receptors and NeuroAIDS: Beyond Co-Receptor Function and Links to Other Neuropathologies. New York: Springer; 2010:87–118 [Google Scholar]
- 23.McPhail ME, Robertson KR. Neurocognitive impact of antiretroviral treatment: thinking long-term. Curr HIV/AIDS Rep 2011;8:249–256 [DOI] [PubMed] [Google Scholar]
- 24.Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010;24:1243–1250 [DOI] [PubMed] [Google Scholar]
- 25.Irish BP, Khan ZK, Jain P, et al. Molecular mechanisms of neurodegenerative diseases induced by human retroviruses: a review. Am J Infect Dis 2009;5:231–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koenig S, Gendelman HE, Orenstein JM, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986;233:1089–1093 [DOI] [PubMed] [Google Scholar]
- 27.Roulston A, Lin R, Beauparlant P, Wainberg MA, Hiscott J. Regulation of human immunodeficiency virus type 1 and cytokine gene expression in myeloid cells by NF-kappa B/Rel transcription factors. Microbiol Rev 1995;59:481–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasyanov A, Tamamura H, Fujii N, Xiong H. HIV-1 gp120 enhances giant depolarizing potentials via chemokine receptor CXCR4 in neonatal rat hippocampus. Eur J Neurosci 2006;23:1120–1128 [DOI] [PubMed] [Google Scholar]
- 29.Lu SM, Tremblay ME, King IL, et al. HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 2011;6:e23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Power C, Hui E, Vivithanaporn P, Acharjee S, Polyak M. Delineating HIV-associated neurocognitive disorders using transgenic models: the neuropathogenic actions of Vpr. J Neuroimmune Pharmacol 2012;7:319–331 [DOI] [PubMed] [Google Scholar]
- 31.Yang B, Akhter S, Chaudhuri A, Kanmogne GD. HIV-1 gp 120 induces cytokine expression, leukocyte adhesion, and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. Microvasc Res 2009;77:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison BE, Majdzadeh N, D'Mello SR. Histone deacetylases: focus on the nervous system. Cell Mol Life Sci 2007;64:2258–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahu GK, Cloyd MW. Latent HIV in primary T lymphocytes is unresponsive to histone deacetylase inhibitors. Virol J 2011;8:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.