

SUMMARY
Genetic interaction (GI) maps, comprising pairwise measures of how strongly the function of one gene depends on the presence of a second, have enabled the systematic exploration of gene function in microorganisms. Here, we present a two-stage strategy to construct high-density GI maps in mammalian cells. First, we use ultra-complex pooled shRNA libraries (25 shRNAs/gene) to identify high-confidence hit genes for a given phenotype and effective shRNAs. We then construct double-shRNA libraries from these to systematically measure GIs between hits. A GI map focused on ricin susceptibility broadly recapitulates known pathways and provides many unexpected insights. These include a non-canonical role for COPI, a novel protein complex (SRIC) affecting toxin clearance, a specialized role for the ribosomal protein RPS25, and functionally distinct mammalian TRAPP complexes. The ability to rapidly generate mammalian GI maps provides a potentially transformative tool for defining gene function and designing combination therapies based on synergistic pairs.
INTRODUCTION
Analysis of mammalian genomic sequences provides a parts list of the proteins that comprise a cell. The remaining challenge is to define functions for these parts and understand how they act together. Work in model organisms, especially budding yeast, has demonstrated the broad utility of comprehensive genetic interaction (GI) maps in defining gene function in a systematic and unbiased manner (Collins et al., 2009; Dixon et al., 2009). GIs, which measure the extent to which the phenotype of a first mutation is modified by the presence of a second, reveal functional relationships between genes. Additionally, the pattern of GIs of a gene provides an information-rich description of its phenotype, which can be used to detect functional similarities between genes and reveal pathways without prior assumptions about cellular functions.
Systematic quantitative analysis of GIs in yeast has allowed rapid identification of new functional complexes, predicted roles for uncharacterized genes, revealed network rewiring in response to environmental changes, and demonstrated functional repurposing of complexes and interactions during evolution (Bandyopadhyay et al., 2010; Collins et al., 2009; Dixon et al., 2009; Frost et al., 2012). More recently, GI maps have also been used with great success in Gram-negative bacteria, fission yeast, and cultured cells from fruit flies (Butland et al., 2008; Frost et al., 2012; Horn et al., 2011; Ryan et al., 2012; Typas et al., 2008).
In mammalian cells, an approach for systematic mapping of GIs could have broad utility for unbiased functional annotation of the human genome as well as for targeted investigation of mammalian-specific pathways. More generally, a better understanding of the structure of GIs may clarify the complex heritability of common traits (Zuk et al., 2012). Furthermore, GIs are important in both the pathogenesis and treatment of a number of human diseases, such as cancer (Ashworth et al., 2011). For example, pairs of genes that exhibit synthetic lethality in cancer cells, but not healthy cells, are ideal targets for combination therapies aimed at limiting the emergence of drug resistance in rapidly evolving cells.
A number of challenges confront any effort to systematically quantify GIs. First, high-precision phenotypic measurements are needed to accurately determine GIs, which are quantified as the deviation of an observed double-mutant phenotype from that expected from two individual mutants. Second, GIs are typically rare (Collins et al., 2009; Dixon et al., 2009), and therefore a scalable high-throughput approach is required to generate large, high-density GI maps. At the same time, the large number of possible pairwise interactions in the human genome (~4×108) makes it necessary to focus on a subset of genes with common biological functions to create a sufficiently dense GI map to reveal meaningful insights.
Recent developments in screening technologies have laid the groundwork for systematic forward genetics in mammalian cells. Both short-hairpin (sh)RNA-based RNAi and haploid insertion approaches lend themselves to pooled screening, which, when combined with deep sequencing-based readouts (Bassik et al., 2009; Carette et al., 2011; Silva et al., 2008), allows massive multiplexing and provides a controlled, identical environment for all cells. Nevertheless, the extraction of robust biological information from genome-wide screening data is challenging (Kaelin, 2012); for RNAi-based screens in particular, the problems of false-positive hits caused by off-target effects and false-negative hits caused by ineffective RNAi agents can limit reliability. Despite these challenges, screens for modifiers of single genes have demonstrated the value of investigating GIs by RNAi (Barbie et al., 2009; Luo et al., 2009).
We have developed a scalable, high-precision pooled shRNA-based approach for robustly conducting RNAi-based screens and measuring GIs in high throughput in mammalian cells. We used our method to examine genetic modifiers of cellular susceptibility to ricin. Ricin is a member of a broad class of AB-type protein toxins that includes major human pathogens. Similar to many viral pathogens, these toxins enter cells by endocytosis and hijack intracellular trafficking pathways. While medically important in their own right, these agents have also been used with great success to probe various aspects of cell biology (Johannes and Popoff, 2008; Spooner and Lord, 2012). Since the general biology of ricin has been extensively studied, it is well-suited to evaluate screening approaches. Indeed, several recent screens have been conducted to identify factors whose depletion protects against AB-toxins (Carette et al., 2009; Guimaraes et al., 2011; Moreau et al., 2011; Pawar et al., 2011). Nonetheless, a comprehensive understanding of the pathways exploited by ricin is missing and little is known about factors whose loss enhances ricin toxicity.
In a primary genome-wide screen for modifiers of ricin susceptibility, we found ~200 known and novel factors which either sensitized or protected cells against ricin intoxication; with some interesting exceptions, these factors were remarkably well-focused on the retrograde transport pathway. We then defined functional relationships among these genes in a GI map. We could broadly recapitulate existing complexes and pathways, functionally dissect multi-protein complexes, identify new complexes with uncharacterized components, and provide unexpected insights into the functions of well-characterized genes. More broadly, this work establishes a strategy that integrates a robust method for RNAi screening with scalable, systematic analysis of GIs, which should be applicable to diverse biological problems.
RESULTS AND DISCUSSION
Strategy for Primary Screens Using Ultra-Complex shRNA Libraries
The first step in our strategy is to conduct a genome-wide screen to identify genes that function within a biological pathway of interest, and effective shRNAs that target them, using ultra-complex shRNA libraries. Ultra-complex libraries increase the likelihood of targeting each gene with several effective shRNAs, thus reducing the false-negative rate. Additionally, requiring several active shRNAs to identify a hit gene reduces the rate of false-positives, since it is unlikely that several shRNAs targeting a non-hit gene have off-target effects relevant to the phenotype of interest. Two key technical developments enable ultra-high-coverage screening: the ability to construct ultra-complex libraries using massively parallel oligonucleotide synthesis (Cleary et al., 2004; Silva et al., 2005), and the capacity of deep sequencing to monitor screening results (Bassik et al., 2009; Silva et al., 2008).
To determine the best design for a genome-wide ultra-complex shRNA library, we conducted a pilot screen with a limited library targeting ~1,000 genes with 50 shRNAs each. We chose ricin as a selective agent for our screen, since it efficiently kills cells and relies on numerous host cell factors for its toxicity. In our pilot library, we included shRNAs targeting a number of genes that were previously reported to affect ricin sensitivity. In addition, we included more than 1,000 negative control shRNAs that had the same overall base composition as the other shRNAs in the library, but that did not match the sequence of any human transcript.
We infected K562 human myelogenous leukemia cells with these libraries and subjected one half of the population to four pulses of ricin treatment, while the other half was grown in the absence of ricin. After 12 days, genomic DNA was isolated from cells of the treated and untreated populations, the shRNA-encoding cassettes were PCR-amplified, and their frequencies were quantified by deep sequencing (Figure 1A).
Figure 1. Pooled High-Coverage RNAi Screen for Ricin Resistance and Sensitization.
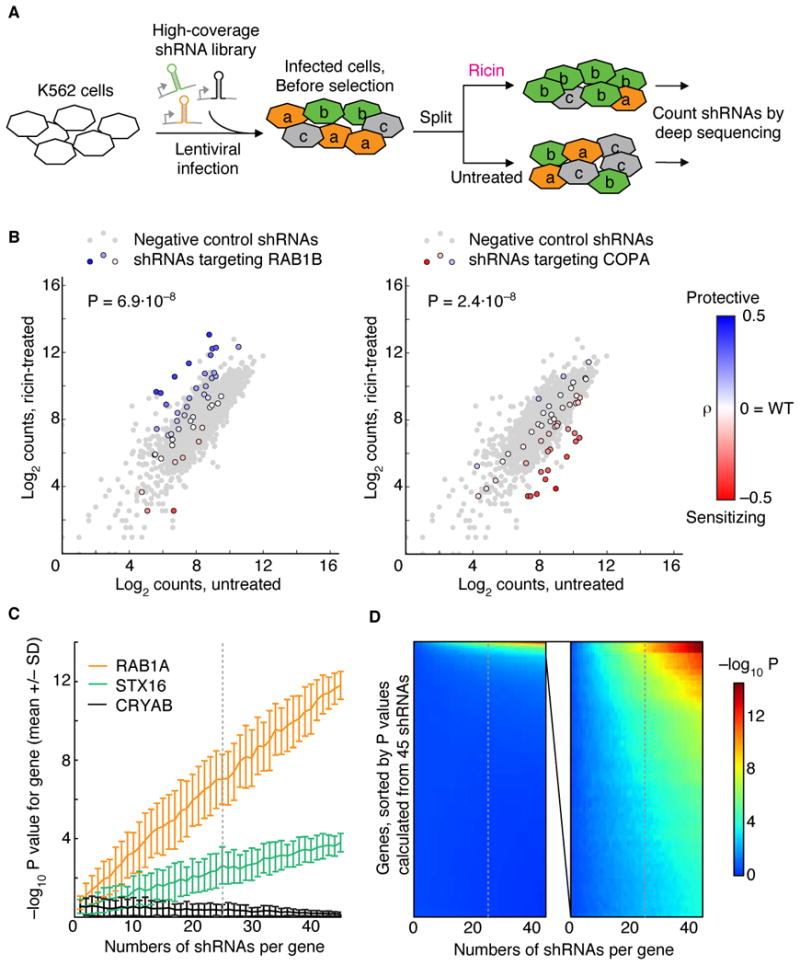
(A) Experimental strategy: A population of K562 cells was infected with a pooled high-coverage shRNA library and split into two subpopulations, one of which was treated with ricin. The frequency of shRNA-encoding constructs in each subpopulation was determined by deep sequencing.
(B) Based on the frequency in the treated and untreated subpopulations, a quantitative resistance phenotype ρ was calculated for each shRNA. Comparing the distribution of ρs for shRNAs targeting a gene of interest to the ρ distribution for negative control shRNAs using the Mann-Whitney U test yielded a P value for the gene. RAB1B knockdown protects cells from ricin (P = 6.9·10−8) whereas knockdown of COPA sensitizes cells to ricin (P = 2.4·10−8).
(C, D) Increasing the coverage of the shRNA library improves the detection of hit genes above background. P values for each gene in a test library were calculated on the basis of random subsets of the data; the number of shRNAs included per gene was varied. Random subsampling was repeated 100 times; means of −log10 P values are shown. Gray dotted lines indicated a coverage of 25 shRNAs per gene, which we chose for our genome-wide library.
(C) Means of −log10 P values +/− SD for three example genes: a strong hit (RAB1A), a moderate hit (STX16), and a non-hit (CRYAB).
(D) Means of −log10 P values for all 1,079 genes targeted by the library (left panel) and for the top 50 hits based on the P value calculated from 45 shRNAs (right panel).
Comparison of the frequency of each shRNA in the treated and untreated populations yielded an enrichment ratio. To enable direct comparisons between different experiments, we defined a metric ρ for ricin resistance, which quantifies the differential effect an shRNA has on cell growth in the presence versus absence of ricin (see Extended Experimental Procedures and Kampmann et al, MS in review). An shRNA without effect on ricin sensitivity has a ρ of 0; shRNAs conferring ricin resistance have positive ρ values; and shRNAs sensitizing cells to ricin have negative ρ values. The criterion for hit genes was based on a P value which reports on the probability that the distribution of ρs for all shRNAs targeting a given gene was significantly different from the distribution for negative control shRNAs (reflecting both random noise and off-target effects), as tested by the Mann-Whitney U test (Figure 1B). The robustness of this approach is supported by the agreement of hit genes obtained when we constructed and screened two independent shRNA libraries targeting the same genes but using different shRNA designs and target sites (Figure S1A).
To identify an appropriate complexity for a genome-wide library, we examined how the number of shRNAs targeting each gene affects the confidence of hit detection. Specifically, we calculated P values based on random subsets of shRNAs for each gene, and determined the effect of subset size on the P value for three example genes: the strong hit gene RAB1A, the weaker hit gene STX16, and the non-hit gene CRYAB (Figure 1C). In our experimental system, the ability to confidently resolve STX16 from background began at ~15 shRNAs per gene and increased steadily as more shRNAs were included. These examples are representative of the entire spectrum of genes (Figure 1D): increasing the coverage of shRNAs per gene improved the signal for hits without spuriously increasing P values for non-hits. Based on these results, we chose a coverage of 25 shRNAs per gene for a genome-wide library.
Reproducibility and Performance of Ultra-complex Libraries in a Pilot Ricin Screen
To test the ability of our screening approach to identify effective shRNAs targeting hit genes, we carried out the ricin resistance pilot screen in duplicate. The quantitative phenotypes of shRNAs targeting hit genes correlated reasonably well between replicates (Figure S1B). A main source of noise in pooled screens is thought to be Poisson sampling error originating from repeated passaging of cells through a population bottleneck (Pierce et al., 2007). Indeed, conducting a batch retest of shRNAs chosen based on primary screen results with a coverage of ~50,000 cells per shRNA species, as compared with ~1,000 cells per shRNA during the primary screen, strongly suppressed the level of observed variability (Figure S1C). In future screens, a small-scale (2 liter) bioreactor should allow one to conduct an entire primary genome-wide screen in a single batch of suspension cells with ~4,000-fold coverage of cells per shRNA.
We validated phenotypes for individual shRNAs in a pooled batch retest, or in individual competitive growth assays. These two quantitative assays gave highly correlated results (Figure S1E). Generally, the phenotypic strength of shRNAs targeting a given hit gene also correlated well with the efficiency of target mRNA knockdown (Figure S1F), suggesting that shRNAs were predominantly acting through the intended target.
A Genome-wide, High-coverage shRNA Screen for Modifiers of Ricin Toxicity Yields Diverse Hits Focused on Key Pathways
We next designed a library targeting each annotated human protein-coding gene with 25 independent shRNAs on average, as well as at least 500 negative control shRNAs per experiment. The shRNAs were grouped in 9 sublibraries of 55,000 shRNAs each, based on annotated biological functions (Extended Experimental Procedures).
For our first application of the genome-wide screening approach, we also used ricin, as it should give access to the rich biology of host pathways exploited by this toxin (Lord et al., 2005; Sandvig et al., 2010; Spooner and Lord, 2012). Specifically, ricin is internalized by endocytosis and traffics retrogradely through the secretory pathway to the ER, where its A and B subunits are dissociated. The catalytic A subunit is then retrotranslocated to the cytoplasm where it depurinates a single base in the 28S rRNA, shutting down translation and leading to apoptosis (Figure 2A).
Figure 2. Hits from a Genome-Wide Screen Recapitulate Known Ricin Biology.
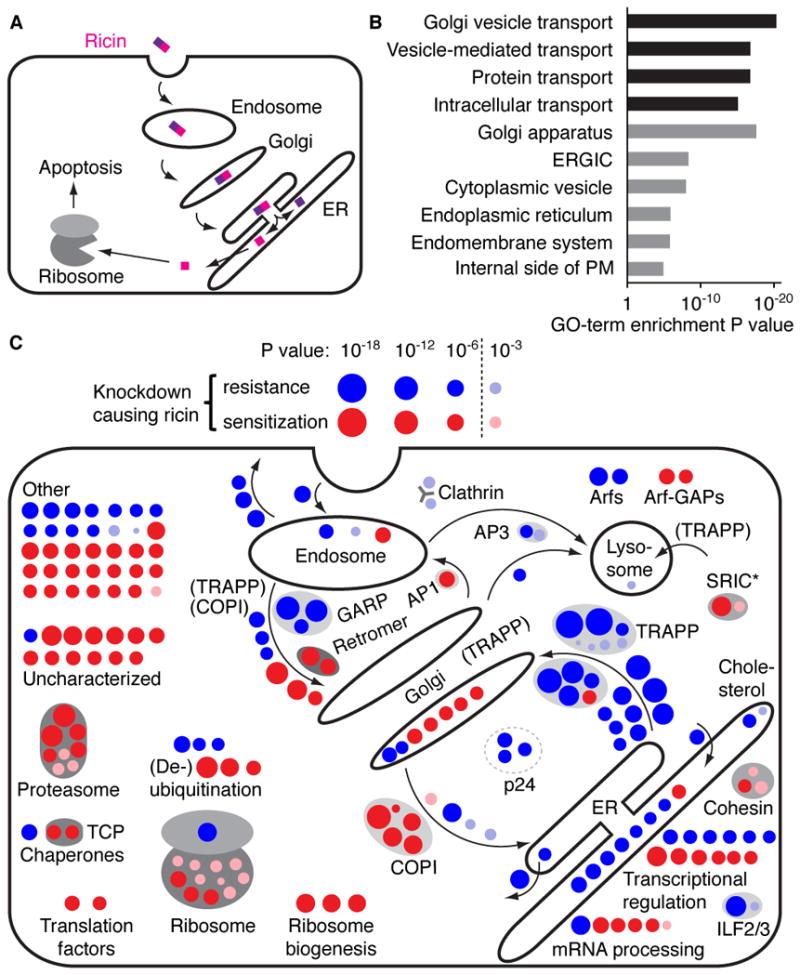
(A) Overview of ricin intoxication of mammalian cells. Ricin is taken up by endocytosis and traffics retrogradely to the ER, where ricin A and B chains dissociate. The A chain retrotranslocates to the cytoplasm and cleaves ribosomal RNA, thereby inhibiting protein synthesis and ultimately triggering apoptosis.
(B) GO-term enrichment analysis for top hits. Top hits were defined as the set of 73 protective genes with an FDR < 0.05 and 83 sensitizing genes with an FDR < 0.02. Non-redundant GO-terms with an FDR < 0.05 are shown; biological process (black bars), cellular component (gray bars).
(C) Visualization of top hits in cellular pathways as blue circles (protective hits) and red circles (sensitizing hits); circle area is proportional to −log10 P value. Selected hits below the top hit cutoff were included (pink and light blue circles) if they were part of a known physical complex containing a top hit, or if they were part of the GI map presented in Figure 5. Gray ovals indicate known physical complexes, the asterisk identifies the SRI complex identified in this study.
We defined a set of hit genes based on false discovery rate (FDR, Storey and Tibshirani, 2003); this set contained the 73 strongest protective hits (FDR < 0.05) and the 83 strongest sensitizing hits (FDR < 0.02) (Table S1). These hits were strongly enriched for genes related to trafficking along the secretory pathway (Figure 2B). Figure 2C displays the top hits in their canonical cellular context (see also Figure S2). A large fraction of characterized hits were either genes acting in the secretory pathway, or otherwise expected based on known ricin biology. In addition, we tagged several poorly characterized hit genes with GFP and expressed them from their native chromosomal context in BACs (Poser et al., 2008) and confirmed that they were localized to secretory pathway organelles (Figure S3). We found that many of the top hits in the screen are also known to exist in physical complexes with each other, with strong protection upon knockdown of components of COPII, TRAPP, and GARP, and strong sensitization upon knockdown of components of COPI, the ribosome, and the proteasome. Taken together, the above results illustrate the specificity and robustness of the hits identified by our approach.
Consistent with results from previous ricin screens and individual gene studies, we found that the early endocytic factors clathrin and Rab5 (Moreau et al., 2011) were required for ricin toxicity, as well as STX16, a snare protein involved in vesicle fusion at the TGN (Amessou et al., 2007). Among the most strongly enriched were components of the GARP complex, known to be required for tethering endosome-derived vesicles to the Golgi (Bonifacino and Hierro, 2011). Knockdown of several (but not all, see below) components of the vesicle tethering TRAPP complex were among the most strongly protective factors.
Surprisingly, a large number of components of the COPII machinery required for anterograde vesicle budding from the ER were strongly protective against ricin when knocked down, which has not been previously observed. It is likely that shutdown of ER-Golgi trafficking (and consequent Golgi collapse) prevents ER delivery of ricin.
Depletion of ribosomal components and ribosome biogenesis factors sensitized cells to the toxin, as expected given that ricin targets the ribosome. A notable exception was RPS25, whose knockdown was strongly protective against ricin, as discussed below.
Identification of Atorvastatin as a Small-molecule Inhibitor of Ricin Transport to the ER
One goal of RNAi-based forward genetic screens is to identify therapeutically valuable targets for small molecule inhibitors. Consistent with previous studies (Grimmer et al., 2000), our primary screen identified components of the cholesterol biosynthesis pathway, including HMG-CoA reductase (HMGCR). We observed a dose-dependent protection of ricin-treated cells by the HMGCR inhibitor atorvastatin (Figure 3A), confirming the role of HMGCR in modulating the toxicity of ricin, and demonstrating that our primary screen could identify effective pharmacological targets.
Figure 3. Characterization of Hit Genes from the Primary Screen.
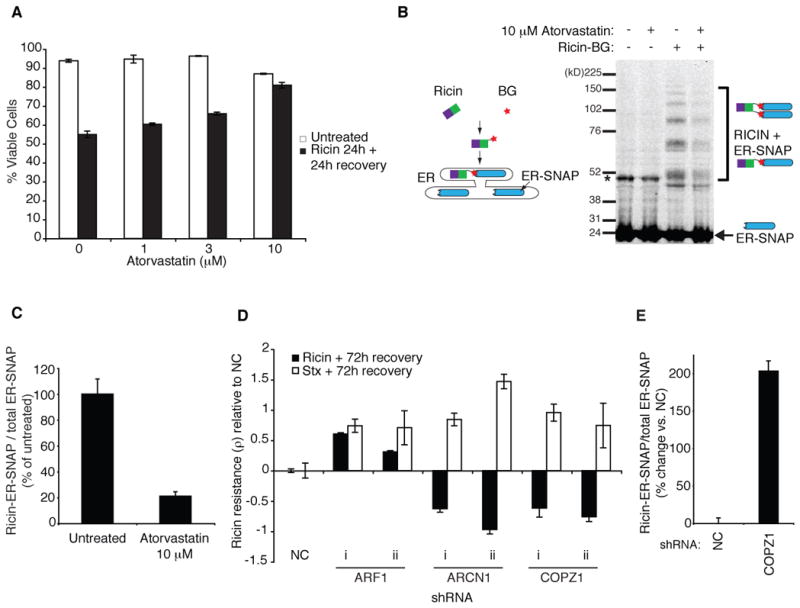
(A) K562 cells were treated with ricin in the presence or absence of atorvastatin for 24 h, and then allowed to recover in the continued presence of atorvastatin. The percentage of viable cells was quantified using flow cytometry.
(B) Cells expressing ER-localized SNAP were intoxicated with benzylguanine-labeled ricin and covalent ricin-SNAP complexes were detected by anti-SNAP Western blot.
(C) Quantification of ricin modified fraction of ER-SNAP.
(D) Raji B cells were infected with shRNAs targeting the indicated genes, and a competitive growth assay was performed in the presence of either ricin or shiga toxin.
(E) COPZ1 knockdown increases levels of ER-localized ricin as measured by the SNAP assay
To assess whether inhibition of HMGCR by atorvastatin affected delivery of ricin to the ER, we expressed an ER-targeted SNAP protein (Geiger et al., 2011) in cells and added benzylguanine (BG)-coupled ricin to measure ricin flux into the ER. Upon delivery of toxin to the ER, an irreversible bond can form between ricin-BG and ER-SNAP, which we could quantify as an increase in molecular weight by Western blot (Figure 3B). The fraction of SNAP present in ricin conjugates was reduced by ~80% upon treatment with atorvastatin (Figure 3B–C), suggesting that toxin traffic to the ER was blocked upon HMGCR inhibition.
A Paradoxical Role for COPI in Diverting Ricin from the ER
One of the more surprising results from the primary screen was a profound sensitization to ricin upon depletion of COPI components (Figure 2C, Table S1), which are normally involved in retrograde endosome-Golgi and Golgi-ER transport (Popoff et al., 2011). Several groups have observed a lack of requirement for retrograde COPI components in trafficking of ricin or Shiga toxin (Chen et al., 2002; Girod et al., 1999; Llorente et al., 2003). However, sensitization by COPI depletion or inactivation has not been described previously.
Primary hits from the ricin screen were retested in batch in a second cell type (Raji B) for their effects on sensitivity to both ricin and Shiga toxin, a similar AB toxin (data not shown). Again, we observed sensitization to ricin upon COPI knockdown, but strong protection against Shiga toxin, revealing an unexpected difference between the trafficking pathways of these two well-studied toxins. Individual shRNAs targeting COPI components ARCN1 or COPZ1 confirmed this finding (Figure 3D). This divergent set of requirements was the exception, rather than the rule: ARF1 is a representative factor that protected against both toxins (Figure 3D). COPI depletion enhanced delivery of toxin to the ER based on the SNAP assay (Figure 3E). It may be that COPI knockdown upregulates a compensatory alternative pathway, or that it normally functions in transport steps that divert ricin from ER.
A Strategy for Generating High-Density GI Maps Based on Double-shRNA Screens
While our screen accurately identified genes important for ricin pathology, the large number of hits makes individual validation and characterization challenging. Indeed, the difficulty in pinpointing promising hits for in-depth follow up represents a general bottleneck for the interpretation of RNAi screens. To address this issue, we developed a strategy to systematically determine GIs between the hits based on double-knockdown phenotypes. For this purpose, we created a double-shRNA library based on effective shRNAs identified from the primary screen. shRNA-encoding cassettes were individually barcoded, pooled and ligated to obtain all pairwise combinations (Figure 4A and S4). This double-shRNA library was introduced into cells and subjected to a ricin resistance screen under the same conditions as those in the primary screen to quantify double-shRNA phenotypes.
Figure 4. Effects of Combinatorial Gene Knockdowns by Double-shRNAs.
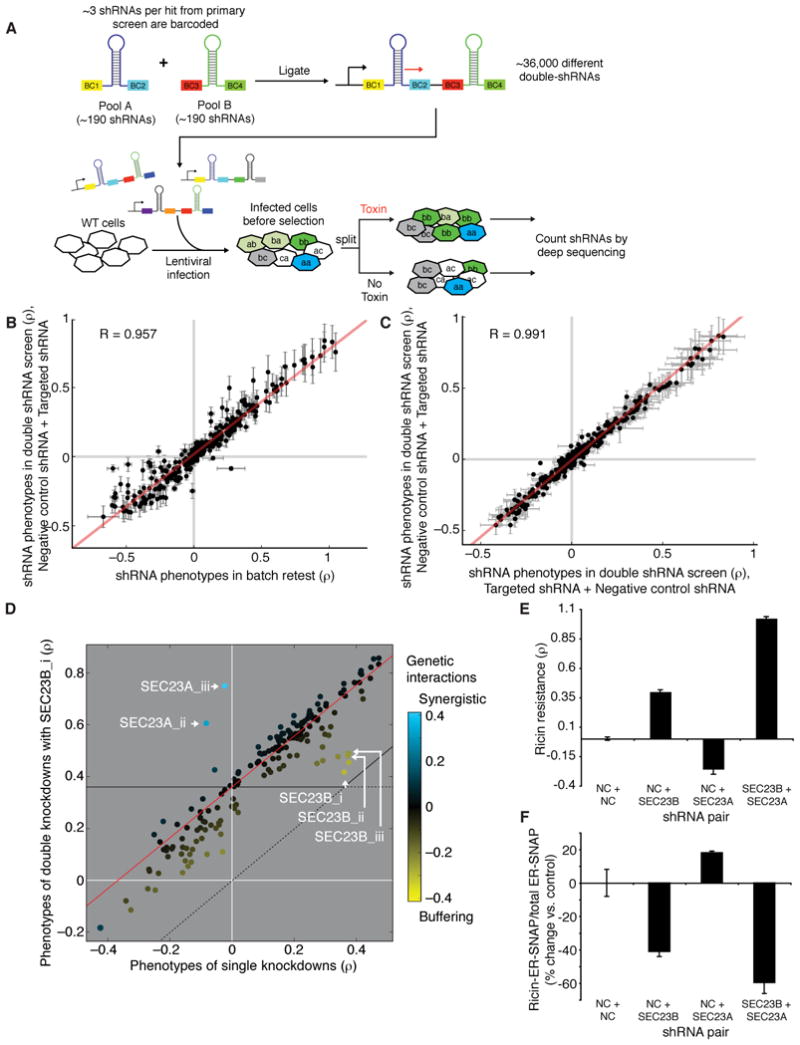
(A) Experimental strategy: Active shRNAs targeting hit genes from the primary screen were individually cloned and barcodes are added upstream and downstream of the mir30 context. Pooled ligation yielded a library of all pairwise combinations of shRNAs. Ricin resistance phenotypes of double-shRNAs were determined as for the primary screen; double-shRNA were identified by sequencing the combinatorial barcode.
(B) Reproducibility between phenotypes of individual shRNAs in a batch retest (mean of two experiments +/− spread) and the same shRNAs paired with negative control shRNAs in a double-shRNA screen (mean +/− SD for combinations with 12 different negative control shRNAs).
(C) Reproducibility between two permutations of double shRNA constructs representing (negative control + targeted) or (targeted + negative control), mean +/− SD for combinations with 12 different negative control shRNAs.
(D) Genetic interactions are calculated as deviations from the typical double-mutant phenotype. The relationship between single shRNA phenotypes and double-shRNA phenotypes in combination with an shRNA of interest (in this example SEC23B_i) is typically linear (red line). Deviations from this line are defined as genetic interactions. Buffering interactions (yellow) are closer to WT phenotype than expected, as in this case found for double-shRNAs targeting SEC23B twice. Synergistic interactions (blue) are further away from WT than expected, as in this case found for double-shRNAs targeting both isoforms of SEC23, SEC23A and SEC23B.
(E) Phenotypes for individual and combinatorial SEC23A, SEC23B knockdown measured in competitive growth assay (mean of triplicate experiments +/− SD).
(F) Quantification of ER localization of ricin measured by the SNAP assay in different knockdown strains (mean of triplicate experiments +/− SD).
In order to obtain single-shRNA phenotypes from the same screen, we included 12 negative control shRNAs in the double-shRNA library pool. Importantly, phenotypes of single shRNAs as quantified by batch retest were in excellent agreement with phenotypes of double shRNAs combining the same shRNAs with negative control shRNAs (Figure 4B). Moreover, the presence of a second shRNA and the order of shRNAs within the double-shRNA construct had minimal impact on the measured phenotypes (Figure 4C) or knockdown efficiency (Figure S5 and S6D, E).
We found that the typical phenotype of a given double shRNA could be reliably predicted by a linear relationship of the phenotypes of the two individual shRNAs (Figure 4D). GIs were thus quantified as deviations from the linear fit of this typical double-mutant phenotype. Deviations towards the phenotype of WT cells were defined as buffering GIs, and deviations away from WT were defined as synergistic GIs. As expected, two shRNAs targeting the same gene typically showed buffering GIs (e.g., SEC23B in Figure 4D), whereas synergistic GIs could be observed for some shRNAs targeting genes acting in parallel (e.g., shRNAs targeting SEC23A and SEC23B, two isoforms with partially distinct functions (Fromme et al., 2008; Schwarz et al., 2009), in Figure 4D). GIs observed in the pooled double-shRNA screen could also be reproduced in individual validation experiments. For example, SEC23A and SEC23B knockdown (whose specificity was validated by rescue experiments, Figure S6A–C) synergized to create highly ricin-resistant cells as monitored by the competitive growth assay (Figure 4E). A similar synergistic effect was seen when the amount of ricin reaching the ER was assessed by ER-SNAP assay (Figure 4F, Figure S6F).
Construction and Benchmarking of a Ricin GI Map
A major motivation for systematic GI mapping beyond the direct analysis of pairwise interactions between genes is the possibility to analyze the correlation of global GI patterns between different genes. Genes with highly correlated GI patterns tend to be functionally related (Collins et al., 2009; Dixon et al., 2009).
Correlations between shRNA GI patterns derived from our double-shRNA screens were highly reproducible between independent experimental replicates (Figure 5A). As expected, shRNAs targeting the same gene had more correlated GI patterns than other shRNAs (Figure 5B), indicating that their phenotypes were mostly due to on-target knockdown. Similarly, shRNA pairs targeting different members of the same protein complex had highly correlated GI patterns, which were clearly distinct from the bulk of shRNA pairs. This result demonstrates the ability of our approach to broadly identify genes encoding members of the same physical complex. Interestingly, shRNAs targeting a small set of genes produced GI patterns that were anti-correlated with those targeting other components of the same physical complex (Figure 5B), causing an overall bimodal distribution of intra-complex GIs. These genes also had the opposite phenotype in the primary screen: TRAPPC9 (anti-correlated with other members of the TRAPP complex), SEC23A (anti-correlated with other COPII components) and RPS25 (uncorrelated with ribosomal proteins of the large subunit). The unusual behavior of these three genes is robustly observed for all three shRNAs targeting each of them, and therefore likely to reflect the functional differences. These findings illustrate that our genetic results can functionally dissect known physical complexes, which we explore below in more detail for RPS25 and the TRAPP complex.
Figure 5. A GI Map Reveals Functionally and Physically Interacting Genes.
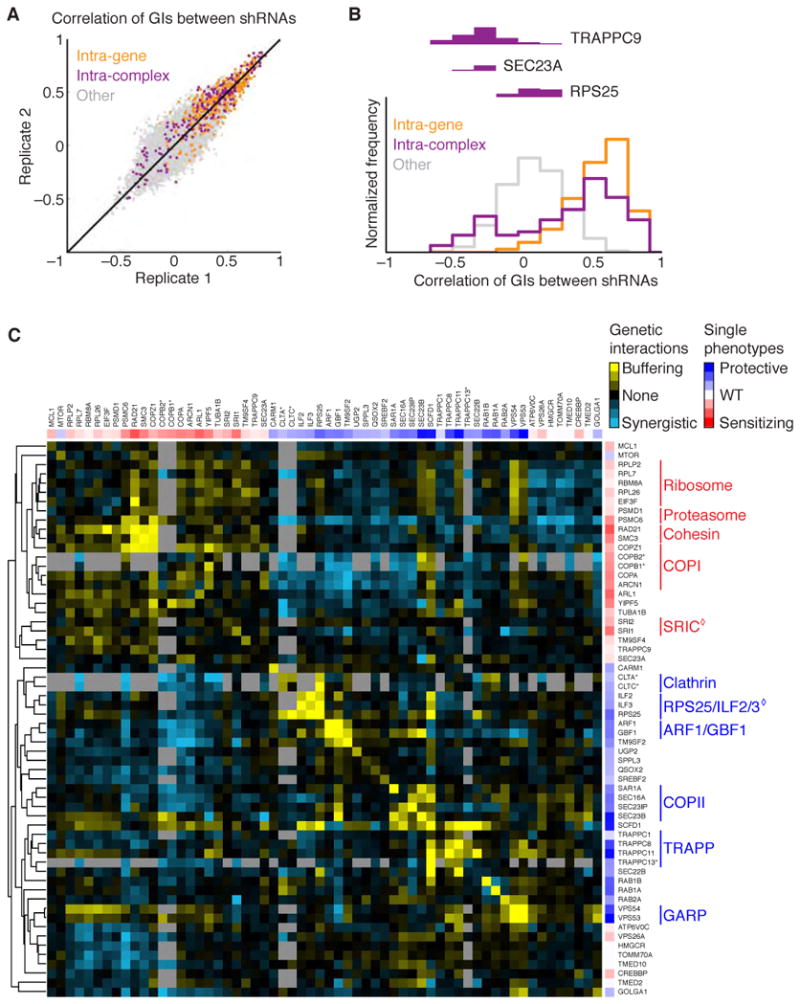
(A, B) Correlations of GI patterns between shRNA pairs: shRNAs targeting the same gene in orange, shRNAs targeting different genes in previously known physical complexes in purple, other pairs of shRNAs in gray.
(A) Reproducibility of GI correlations between shRNA pairs in two experimental replicates.
(B) High inter-gene and inter-complex correlation of GIs. Distribution of correlation coefficients between shRNA pairs are shown for the three classes of shRNA pairs. The anti-correlated part of the bimodal distribution of intra-complex shRNA pairs is fully accounted for by pairs including shRNAs targeting TRAPPC9, SEC23A, and RPS25.
(C) GIs for all gene pairs were calculated (shown as a yellow-cyan heatmap) and genes were clustered hierarchically based on the correlation of their GIs. Individual phenotypes are indicated by sidebars using a red-blue heatmap. Genes marked with asterisks were imported from a separate double-shRNA screen we conducted with a partially overlapping gene set. Known physically or functionally interacting groups of genes are labeled by vertical lines; diamonds mark interactions defined in this study.
A possible source of noise in an RNAi-based GI map is the fact that an effective on-target shRNA can have partial off-target effects, which can confound its GI pattern. To minimize this effect, we required each gene in the GI map to be targeted by at least two (and typically three) shRNAs whose GI patterns were sufficiently correlated (Kampmann et al, MS in review), and averaged the GIs of these highly correlated shRNAs for each gene. Using these stringent criteria, the resulting GI map (Figure 5C) encompassed pairwise interactions between 60 genes, each represented by 3 shRNAs on average, and was based on the pooled measurement of > 36,000 double-shRNA phenotypes. The main limitation for increasing the scale of GI maps is the availability of highly validated shRNAs, as a single bioreactor run can measure >500,000 shRNA pairs.
Functional Predictions from the Ricin GI Map
Hierarchical clustering of genes based on the correlation of their GIs was remarkably successful at recapitulating a number of well-characterized complexes, including the COPI and COPII vesicle coats, clathrin, GARP and the ribosome, as well as complexes with unknown roles in ricin biology, such as the cohesins (Figure 5C). In addition, the map demonstrated clustering of functionally interacting proteins, such as the small GTPase ARF1 and its guanine nucleotide exchange factor GBF1.
The GI map also lead to numerous novel functional predictions, three of which are highlighted below.
An unexpected role for ribosomal protein RPS25
Remarkably, we found that RPS25 knockdown conferred ricin resistance. By contrast, all other ribosomal hits sensitized cells to ricin, as expected, since ribosome inactivation is the basis for ricin cytotoxicity. The GI map provided a clue to the divergent role of RPS25: RPS25 formed a cluster with ILF2 and ILF3 (Figure 5C). ILF2/3 encode heterodimeric nucleic acid binding proteins with roles in transcription, mRNA stability, and translational control (Barber, 2009). ILF2/3 knockdown protected against ricin, and we confirmed that the shRNAs against RPS25 and ILF2/3 acted through their intended target genes (Figure S7). As expected for proteins in a physical complex, shRNAs targeting ILF2 showed buffering GIs with shRNAs targeting ILF3 (Figure 6A, B). Surprisingly, we also observed very strong buffering interactions between ILF2/3 and RPS25, which was consistent for all combinations of the 9 shRNAs targeting ILF2, ILF3, and RPS25 (Figure 6B).
Figure 6. Novel Interactions Predicted From the GI Map: RPS25/ILF2/3 and SRIC.
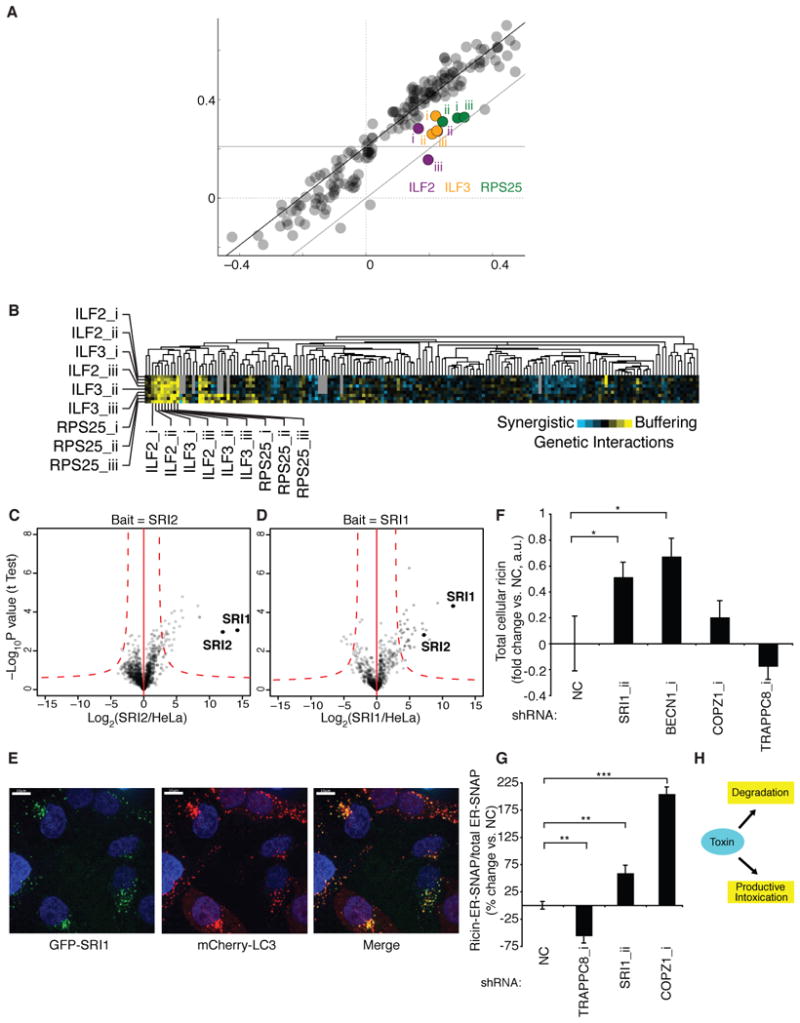
(A) Buffering genetic interactions between shRNAs targeting ILF3, the ribosomal subunit RPS25, and ILF2/ILF3.
(B) Correlation and buffering genetic interactions between shRNAs targeting ILF2, ILF3 and RPS25 in an shRNA-based genetic interaction map.
(C, D) The poorly characterized, genetically correlated proteins SRI1 and SRI2 interact physically, as shown by reciprocal co-immunoprecipitation and MS.
(E) GFP-SRI1 partially colocalizes with the autophagosome/lysosome marker mCherry-LC3 in HeLa cells.
(F) Total cellular ricin levels after intoxication, as quantified by western blotting, are increased upon knockdown of degradation-related genes and SRI1 (which sensitizes to ricin). The asterisk indicates statistically significant differences (P < 0.05, Student’s t test).
(G) SRI1 and COPZ1 knockdown increase levels of ER-localized ricin as measured by the SNAP assay, whereas TRAPPC8 knockdown decreases levels of ER-localized ricin. The asterisks indicate statistically significant differences (**, P < 0.01; ***, P < 0.001; Student’s t test).
(H) Model: Ricin partitions between degradation and productive intoxication pathways; inhibition of degradation increases productive intoxication.
Previous literature has implicated both RPS25 and ILF2/3 in translational control: RPS25 has been shown to be required for translation of IRES-containing mRNAs in cricket paralysis virus (Landry et al., 2009), while ILF2/3 can bind viral IRES and control translation (Merrill and Gromeier, 2006). Therefore it is tempting to speculate that ILF2/3 and RPS25 may work together to control translation of certain transcripts that affect ricin sensitivity, possibly under particular stress conditions.
Identification of the SRI (sensitization to ricin) complex
One unexpected prediction was the interaction between WDR11 and C17orf75, two poorly characterized genes. Both sensitized cells to ricin when depleted, exhibited highly correlated profiles in the GI map, and showed buffering interactions with each other, which is often a signature for genes encoding proteins in the same pathway or physical complex. We found indeed that the encoded proteins interacted in reciprocal immunoprecipitation experiments (Figure 6C–D). Since the proteins appear to form a complex and sensitize cells to ricin when depleted, we propose that they be named SRI1 and SRI2 (Sensitization to RIcin), for WDR11 and C17orf75, respectively.
Previously SRI1 (WDR11) was suggested to interact with a transcription factor (Kim et al., 2010), as well as to impact flux through the autophagy pathway (Behrends et al., 2010). Consistent with the latter observation, we found that GFP-tagged SRI1 partially colocalized with the autophagosome marker LC3 (Figure 6E). This suggests a potential role for SRI in toxin degradation. Indeed, depletion of SRI1 or BECN1, a regulator of autophagy, caused an increase in total cellular ricin (Figure 6F). By contrast, other genes that sensitized (COPZ1) or protected (TRAPPC8) cells against ricin had an insignificant effect on total toxin levels (although they do affect toxin delivery to the ER, Figure 6G). When degradation pathways are inhibited, more ricin can enter the productive intoxication pathway (Figure 6H), which provides a potential explanation for the observed increase in delivery of toxin to the ER upon depletion of SRI1 (Figure 6G). Nonetheless, further study will be required to define the precise role of this complex.
Functional dissection of the mammalian TRAPP complex
Two of the most strongly protective hits from our primary screen, C4orf41 and KIAA1012, were poorly characterized at the onset of our studies. In our GI map, these genes formed a highly correlated cluster connected by buffering GIs with another poorly characterized gene, C5orf44, and with TRAPPC1, a member of the TRAPP complex, a highly conserved multi-subunit complex involved in ER-Golgi, endosome-Golgi, and autophagosome transport (Barrowman et al., 2010). Based on this pattern, we predicted that C4orf41, KIAA1012 and C5orf44 function as TRAPP complex components. To test this, we GFP-tagged and immunoprecipitated these components (Figure 7A–B). We could identify most TRAPP components described to date in both immunoprecipitations, as well as C5orf44. C4orf41 and KIAA1012 were previously identified as TRAPP3 interactors in a high-throughput immunoprecipitation study (Gavin et al., 2002), and concurrent with our studies, were independently identified as TRAPP components and designated TRAPPC8 and TRAPPC11, respectively (Scrivens et al., 2011). Additionally, C5orf44 was recently shown to exhibit homology to yeast Trs65 and physically interact with other TRAPP components (Choi et al., 2011). Based on these observations, we designate C5orf44 as TRAPPC13.
Figure 7. Functional Dissection of the TRAPP Complex.
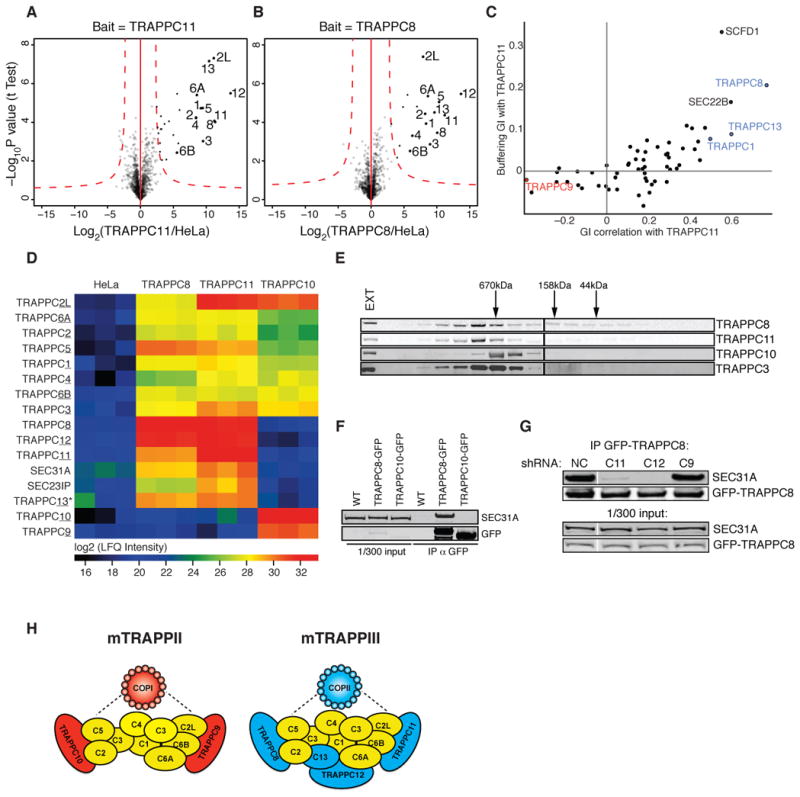
(A–B) All TRAPP complex members (other than TRAPPC9/10) specifically co-immunoprecipitate with TRAPPC11 (A) and TRAPPC8 (B), as quantified by mass spectrometry.
(C) Correlation of genetic interactions with TRAPPC11 and buffering genetic interaction with TRAPPC11 is shown for each gene included in the genetic interaction map. TRAPP complex members are shown in green, the functionally related SEC22B in green. TRAPPC9 shows a strongly anti-correlated genetic interaction pattern when compared to other TRAPP complex members.
(D) Abundance (quantified as LFQ) of each TRAPP subunit in the immunoprecipitation is indicated by color scale.
(E) Extracts from K562 cells were fractionated by size exclusion chromatography on a superose 6 column. Western blot could detect co-migration of TRAPPC8 and TRAPPC11, which were larger in size than TRAPPC10. The core component TRAPPC3 migrated with both components. EXT=unfractionated extract.
(F) Immunoprecipitation of TRAPPC8 or TRAPPC10 tagged with GFP showed specific association of TRAPPC8 with SEC31A.
(G) Association of GFP-TRAPPC8 with SEC31A was assessed by immunoprecipitation in extracts from cells stably expressing shRNAs targeting the indicated TRAPP components.
(H) Hypothetical model for mammalian TRAPP complexes. We propose that at least two complexes exist, which contain a core set of proteins (yellow) and unique subunits, either TRAPPC9/10 (mTRAPPII) or TRAPPC8/11/12/13 (mTRAPPIII), which associate with COPI or COPII vesicles, respectively.
In yeast, several TRAPP complexes have been identified (Barrowman et al., 2010) with distinct roles in ER-Golgi traffic (TRAPPI), intra-Golgi and endosome-Golgi traffic (TRAPPII), and autophagy (TRAPPIII). In mammalian cells, TRAPP has been suggested to form a single large complex (Scrivens et al., 2011), and it has been unclear whether this complex is responsible for all observed TRAPP activities.
Our data revealed a clear functional distinction between different TRAPP components. We found only a subset of TRAPP components as strongly protective hits, while other components had either no phenotype or, in the case of TRAPPC9, were mildly sensitizing (Table S1, Figure S7E). Moreover, the genetic interaction pattern of TRAPPC9 showed a striking anti-correlation with TRAPPC11 (Figure 7C), suggesting that complexes containing these proteins are distinct, and have opposing roles in ricin trafficking. Indeed, we found that immunoprecipitation of either TRAPPC8 or TRAPPC11 pulled down the COPII components SEC31A and SEC23IP as well as the other known TRAPP components, with the prominent exception of TRAPPC9 and TRAPPC10 (Figure 7D). Similarly, previous immunoprecipitation experiments found that TRAPPC9 did not recover TRAPPC8 (Zong et al., 2011). Conversely, we found that immunoprecipitation of TRAPPC10 pulled down core TRAPP components, but not TRAPPC8/11/12/13, SEC31 or SEC23IP (Figure 7D). Based on this, we examined the migration properties of the various TRAPP components by size exclusion chromatography. These studies directly established the existence of two physically distinct complexes: a larger complex containing TRAPPC8 and TRAPPC11 and a smaller one containing TRAPPC10 (Figure 7E).
To further define mammalian TRAPP complexes, we examined their interactions with COPII components. The yeast TRAPPI complex is a COPII vesicle tethering factor (Sacher et al., 2001), and COPII and TRAPPC3 interact in yeast and mammalian cells (Cai et al., 2007). Consistent with this, GFP-labeled TRAPPC8 and TRAPPC11 colocalized with SEC31A (Figure S7F). Our finding that TRAPPC8 and TRAPPC11, but not TRAPPC10, co-immunoprecipitated the COPII component SEC31A (Figure 7F), suggests that differential interaction with COPII may functionally distinguish the two mammalian TRAPP complexes. Indeed, knockdown of TRAPC11 or TRAPC12 but not TRAPPC9 disrupted the interaction of TRAPPC8 with SEC31A (Figure 7F). Thus the two distinct mammalian TRAPP complexes, defined by the presence of TRAPPC9/10 or TRAPPC8/11/12/13, differentially interact with COPII (Figure 7H); we refer to these as mTRAPPII and mTRAPPIII, respectively.
The two TRAPP complexes seem to have opposing roles in ricin transport. Since we observe protection against ricin with COPII or mTRAPPIII knockdowns, and these components interact physically, it is tempting to speculate that this complex functions similarly to yeast TRAPPI in COPII vesicle tethering. Additionally, TRAPPC8 knockdown has been reported to impact flux through the autophagy pathway (Behrends et al., 2010), and we observed a mild enhancement of toxin degradation upon TRAPPC8 knockdown (Figure 6H), raising the possibility that mTRAPPIII functions in both COPII-mediated trafficking and autophagy. By contrast, TRAPPC9/10 was previously reported to interact with COPI components (Yamasaki et al., 2009). Moreover, we find that both COPI and TRAPPC9 knockdown sensitize cells to ricin suggesting that mTRAPPII may function in tethering of COPI vesicles. More generally, our findings establish that there are functionally distinct mammalian TRAPP complexes and lay the groundwork for a mechanistic understanding of their specialized functions.
Perspective
Building on previous pooled shRNA strategies (e.g. Moffat et al., 2006; Silva et al., 2005), we have developed an integrated platform to functionally dissect complex biological processes in mammalian cells using high-density genetic interaction maps. Our strategy opens mammalian cell biology to the types of systematic genetic analyses that have been highly successful in microorganisms (Collins et al., 2009; Dixon et al., 2009).
Our first application of the platform elucidated key cellular pathways and revealed how they modulate ricin susceptibility. Our studies of the TRAPP complex, in particular, illustrate how genetic and physical interactions provide complementary approaches to understand the functions of multiprotein complexes as our studies revealed two functionally distinct mammalian TRAPP complexes.
A key aspect of our primary screening platform is the ability to identify hit genes based on the likelihood that shRNAs act through the intended target gene rather than solely the strength and reproducibility of observed shRNA phenotypes. This is facilitated by the use of ultra-complex shRNA libraries that include a large number of negative controls. Our approach also provides a principled way to benchmark shRNA library design and screening systems based not only on the strength of on-target mRNA knockdown, but also by the ability to distinguish true hits from background (e.g., off-target effects or statistical noise). Using this criterion, we are currently exploring modifications to the experimental strategy and shRNA design of our ultra-complex libraries. Another important feature of ultra-complex libraries is that they target each gene with a wide spectrum of shRNAs with different knockdown strengths, effectively creating an allelic series. This will facilitate the study of essential genes, as well as gene dosage effects. While our genetic interaction maps are currently based on shRNAs identified in a primary screen, our growing library of validated shRNAs will soon enable mapping of interactions between genes that do not have an individual phenotype, and the detection of synergistic genetic interactions between them. Ongoing efforts by several groups to identify effective shRNAs (Cheung et al., 2011; Fellmann et al., 2011; Marcotte et al., 2012) will greatly facilitate the construction of larger GI maps.
Our approach should be broadly applicable to the study of complex biological systems. While we present a pooled screening strategy based on cell growth and viability, other phenotypic readouts that physically separate cell populations can be used, such as fluorescence-activated cell sorting or migration assays. In addition, the ability to rapidly generate and screen a double-shRNA library will allow one to explore conservation and rewiring of genetic interactions in diverse cell types and under different conditions (Bandyopadhyay et al., 2010 and Kampmann et al, MS in review).
The systematic exploration of genetic interactions in human cells also has broad medical relevance, especially for cancer biology and therapy. Functional surveys of genes in cancer cells can distinguish oncogenic drivers from mere passengers. Genetic interactions are thought to be crucial determinants of properties of individual cancer cells (Ashworth et al., 2011), such as their resistance to therapeutic agents. A better understanding of resistance pathways in specific genetic backgrounds could pave the way for personalized combination therapies that preemptively block the cancer’s escape routes. More generally, as demonstrated for HIV, combination therapy is a promising strategy to counter the problem of rapidly evolving drug resistance in tumors. The ability to identify rare synthetic lethal interactions between huge numbers of gene pairs maximizes the opportunity to identify pairs of drugs that synergistically target a disease state.
EXPERIMENTAL PROCEDURES
shRNA libraries
To express shRNAs from a PolII promoter in a miR30-derived context, we adapted strategies developed by the Hannon and Elledge groups (Paddison et al., 2004; Silva et al., 2005). Construction of pooled libraries was conducted essentially as previously described (Bassik et al., 2009). The genome-wide library was divided into 9 sublibraries with 55,000 shRNA each and targeted each human protein-coding gene with ~25 independent shRNAs. Each sublibrary also contained 500 or more negative control shRNAs, which were designed to match the base composition of targeted shRNAs within the same sublibrary, without targeting any transcript in the human genome.
Ricin resistance screening
For pooled screens, cells were seeded at 0.5 × 106/ml at a representation of 1000 cells/library element, and treated with 0.5 ng/ml ricin (Vector labs), which reduced cell number by ~50% compared with untreated cells, due to a combination of cell death and reduced growth rate. This selective pressure represents a compromise between stronger selection, which can increase the dynamic range of observed phenotypes, and weaker selection, which reduces population bottlenecks and thus reduces Poisson sampling noise. After 24 h, ricin was washed out. Each day during the screen, cells were diluted to to 0.5 × 106/ml. After 2–3 days of recovery when treated cells were again doubling at WT rate, a new cycle of ricin treatment was initiated (total of 4 pulses). For competitive growth assays, cells were infected with lentivirus encoding individual shRNAs. After 3 days, cells were seeded in 24-well plates at 0.5 × 106/ml and treated with 0.5 ng/ml ricin. After 24 h, ricin was washed out, and cells were adjusted to 0.5 × 106/ml. Percentages of mCherry-positive cells were assessed by FACS 24–48h later.
Supplementary Material
Research Highlights.
Ultracomplex shRNA library reduces false positives/negatives in genome-wide screens
Pooled double-shRNA strategy systematically maps genetic interactions between hits
Application of 2-step strategy identifies pathways controlling ricin susceptibility
The resulting map uncovers functionally distinct mammalian TRAPP complexes
Acknowledgments
We thank M. Augsburg, O. Chen, C. Chu, S. Churchman, S. Collins, R. Geiger, A. Heidersbach, N. Ingolia, M. Leuscher, J. Lund, L. Mitic, W. Patena, M. Pham, and D. Root, M. Schuldiner, A. Ssykor, N. Stern-Ginossar, K. Thorn, and X. Wang, for technical advice and assistance, S. Collins, A. Frost, M. Jonikas, C. Jan, G. Ku, N. Shariat, P. Temkin, M. von Zastrow, D. Acosta-Alvear and members of the Weissman lab for helpful discussions and critical reading of the manuscript. The work is supported by the Jane Coffin Childs Memorial Fund and the UCSF Program for Breakthrough Biomedical Research (MK) the Leukemia and Lymphoma Society (MB), The German medical genome research grant FKZ01GS0861 (MM, AAH), the European Community’s FP7/2007-2013 under grant agreement 241548 (MitoSys Project, AAH), and the HHMI (JSW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amessou M, Fradagrada A, Falguieres T, Lord JM, Smith DC, Roberts LM, Lamaze C, Johannes L. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. J Cell Sci. 2007;120:1457–1468. doi: 10.1242/jcs.03436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–38. doi: 10.1016/j.cell.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Mehta M, Kuo D, Sung MK, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–1389. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. The NFAR’s (nuclear factors associated with dsRNA): evolutionarily conserved members of the dsRNA binding protein family. RNA Biol. 2009;6:35–39. doi: 10.4161/rna.6.1.7565. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman J, Bhandari D, Reinisch K, Ferro-Novick S. TRAPP complexes in membrane traffic: convergence through a common Rab. Nat Rev Mol Cell Biol. 2010;11:759–763. doi: 10.1038/nrm2999. [DOI] [PubMed] [Google Scholar]
- Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM, Schuldiner M, Weissman JS, McManus MT. Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat Methods. 2009;6:443–445. doi: 10.1038/nmeth.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Hierro A. Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 2011;21:159–167. doi: 10.1016/j.tcb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Babu M, Diaz-Mejia JJ, Bohdana F, Phanse S, Gold B, Yang W, Li J, Gagarinova AG, Pogoutse O, et al. eSGA: E. coli synthetic genetic array analysis. Nat Methods. 2008;5:789–795. doi: 10.1038/nmeth.1239. [DOI] [PubMed] [Google Scholar]
- Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature. 2007;445:941–944. doi: 10.1038/nature05527. [DOI] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol. 2011;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Hu T, Mikoryak C, Draper RK. Retrograde transport of protein toxins under conditions of COPI dysfunction. Biochim Biophys Acta. 2002;1589:124–139. doi: 10.1016/s0167-4889(02)00163-5. [DOI] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Davey M, Schluter C, Pandher P, Fang Y, Foster LJ, Conibear E. Organization and assembly of the TRAPPII complex. Traffic. 2011;12:715–725. doi: 10.1111/j.1600-0854.2011.01181.x. [DOI] [PubMed] [Google Scholar]
- Cleary MA, Kilian K, Wang Y, Bradshaw J, Cavet G, Ge W, Kulkarni A, Paddison PJ, Chang K, Sheth N, et al. Production of complex nucleic acid libraries using highly parallel in situ oligonucleotide synthesis. Nat Methods. 2004;1:241–248. doi: 10.1038/nmeth724. [DOI] [PubMed] [Google Scholar]
- Collins SR, Weissman JS, Krogan NJ. From information to knowledge: new technologies for defining gene function. Nat Methods. 2009;6:721–723. doi: 10.1038/nmeth1009-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu Rev Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, Xu Q, Hengartner MO, Elledge SJ, Hannon GJ, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41:733–746. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromme JC, Orci L, Schekman R. Coordination of COPII vesicle trafficking by Sec23. Trends in cell biology. 2008;18:330–336. doi: 10.1016/j.tcb.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Frost A, Elgort MG, Brandman O, Ives C, Collins SR, Miller-Vedam L, Weibezahn J, Hein MY, Poser I, Mann M, et al. Functional Repurposing Revealed by Comparing S. pombe and S. cerevisiae Genetic Interactions. Cell. 2012;149:1339–1352. doi: 10.1016/j.cell.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol. 2011;13:1305–1314. doi: 10.1038/ncb2339. [DOI] [PubMed] [Google Scholar]
- Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM, Lord JM, Nilsson T, Pepperkok R. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol. 1999;1:423–430. doi: 10.1038/15658. [DOI] [PubMed] [Google Scholar]
- Grimmer S, Iversen TG, van Deurs B, Sandvig K. Endosome to Golgi transport of ricin is regulated by cholesterol. Mol Biol Cell. 2000;11:4205–4216. doi: 10.1091/mbc.11.12.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes CP, Carette JE, Varadarajan M, Antos J, Popp MW, Spooner E, Brummelkamp TR, Ploegh HL. Identification of host cell factors required for intoxication through use of modified cholera toxin. J Cell Biol. 2011;195:751–764. doi: 10.1083/jcb.201108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T, Sandmann T, Fischer B, Axelsson E, Huber W, Boutros M. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell. 2008;135:1175–1187. doi: 10.1016/j.cell.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science. 2012;337:421–422. doi: 10.1126/science.1225787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010;87:465–479. doi: 10.1016/j.ajhg.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009;23:2753–2764. doi: 10.1101/gad.1832209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente A, Lauvrak SU, van Deurs B, Sandvig K. Induction of direct endosome to endoplasmic reticulum transport in Chinese hamster ovary (CHO) cells (LdlF) with a temperature-sensitive defect in epsilon-coatomer protein (epsilon-COP) J Biol Chem. 2003;278:35850–35855. doi: 10.1074/jbc.M303425200. [DOI] [PubMed] [Google Scholar]
- Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr Top Microbiol Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MK, Gromeier M. The double-stranded RNA binding protein 76:NF45 heterodimer inhibits translation initiation at the rhinovirus type 2 internal ribosome entry site. J Virol. 2006;80:6936–6942. doi: 10.1128/JVI.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Moreau D, Kumar P, Wang SC, Chaumet A, Chew SY, Chevalley H, Bard F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev Cell. 2011;21:231–244. doi: 10.1016/j.devcel.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Cleary M, Silva JM, Chang K, Sheth N, Sachidanandam R, Hannon GJ. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat Methods. 2004;1:163–167. doi: 10.1038/nmeth1104-163. [DOI] [PubMed] [Google Scholar]
- Pawar V, De A, Briggs L, Omar MM, Sweeney ST, Lord JM, Roberts LM, Spooner RA, Moffat KG. RNAi screening of Drosophila (Sophophora) melanogaster S2 cells for ricin sensitivity and resistance. J Biomol Screen. 2011;16:436–442. doi: 10.1177/1087057110397890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SE, Davis RW, Nislow C, Giaever G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc. 2007;2:2958–2974. doi: 10.1038/nprot.2007.427. [DOI] [PubMed] [Google Scholar]
- Popoff V, Adolf F, Brugger B, Wieland F. COPI budding within the Golgi stack. Cold Spring Harb Perspect Biol. 2011;3:a005231. doi: 10.1101/cshperspect.a005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Roguev A, Patrick K, Xu J, Jahari H, Tong Z, Beltrao P, Shales M, Qu H, Collins SR, et al. Hierarchical modularity and the evolution of genetic interactomes across species. Mol Cell. 2012;46:691–704. doi: 10.1016/j.molcel.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro-Novick S. TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Molecular cell. 2001;7:433–442. doi: 10.1016/s1097-2765(01)00190-3. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Torgersen ML, Engedal N, Skotland T, Iversen TG. Protein toxins from plants and bacteria: probes for intracellular transport and tools in medicine. FEBS Lett. 2010;584:2626–2634. doi: 10.1016/j.febslet.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, Paw BH, Hopfner KP, Holzmann K, Russo R, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nature genetics. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- Scrivens PJ, Noueihed B, Shahrzad N, Hul S, Brunet S, Sacher M. C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking. Mol Biol Cell. 2011;22:2083–2093. doi: 10.1091/mbc.E10-11-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, Elledge SJ, Hannon GJ, Chang K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner RA, Lord JM. How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr Top Microbiol Immunol. 2012;357:19–40. doi: 10.1007/82_2011_154. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A, Menon S, Yu S, Barrowman J, Meerloo T, Oorschot V, Klumperman J, Satoh A, Ferro-Novick S. mTrs130 is a component of a mammalian TRAPPII complex, a Rab1 GEF that binds to COPI-coated vesicles. Molecular biology of the cell. 2009;20:4205–4215. doi: 10.1091/mbc.E09-05-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong M, Wu XG, Chan CW, Choi MY, Chan HC, Tanner JA, Yu S. The adaptor function of TRAPPC2 in mammalian TRAPPs explains TRAPPC2-associated SEDT and TRAPPC9-associated congenital intellectual disability. PLoS One. 2011;6:e23350. doi: 10.1371/journal.pone.0023350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.