

Abstract
Advances in genetics and genomics have fuelled a revolution in discovery-based, or hypothesis-generating, research that provides a powerful complement to the more directly hypothesis-driven molecular, cellular and systems neuroscience. Genetic and functional genomic studies have already yielded important insights into neuronal diversity and function, as well as disease. One of the most exciting and challenging frontiers in neuroscience involves harnessing the power of large-scale genetic, genomic and phenotypic data sets, and the development of tools for data integration and mining. Methods for network analysis and systems biology offer the promise of integrating these multiple levels of data, connecting molecular pathways to nervous system function.
The molecular biology revolution allowed neuroscientists to move from the study of circuits and systems to the detailed study of individual molecules of interest. However, moving from genes to an understanding of interacting signalling or metabolic pathways within cells, let alone combining these data to achieve a systems-level understanding of brain circuit function in health and disease, poses enormous challenges. The integration of data across a wide range of observations is especially important in neurobiology. In contrast to the physical sciences, the biological sciences have few guiding theories or laws (with the exception of evolution) with which to direct and prioritize investigations. At the same time, we are in the midst of a genomic and informatics revolution that permits us to view specific gene products in the context of all others. The adoption of functional genomic or molecular-systems methods that permit dynamic measurement of gene products in a highly parallel manner, coupled with an underlying systems-level knowledge of the organization of these gene products, has the potential to provide a more integrative understanding of nervous system function.
The field of neuroscience has been slow to adopt functional genomic and genetic methods and the large-scale databases and resources that ideally result from their use. For example, neuroscience has consistently lagged behind cancer biology in the adoption of new molecular and genetic methods, starting with molecular cloning and continuing to functional genomics and genetics today. There are legitimate reasons for this, including the extreme cellular heterogeneity and complexity of neural circuits relative to most non-neural tissues, and the reliance on post-mortem materials for most human studies1–4. Another obstacle is the generation of enormous amounts of data. Similarly to what occurred in the field of genetics, clusters of computationally oriented researchers have to form within or around the laboratories more concentrated on the ‘-ology’ fields (‘ologies’). This integration of computational biology or bioinformatics in modern neuroscience laboratories or groupings will become even more critical as more powerful technologies that generate many orders of magnitude more data, such as next-generation sequencing techniques, replace microarrays. An additional, sometimes unspoken, impediment to the more widespread adoption of ‘-omics’ fields (‘omics’) in neuroscience research is an underlying tension between the hypothesis-testing approach applied in the typical neurobiology laboratory and the discovery-based disciplines of genetics, genomics and proteomics (Box 1).
Box 1. In search of hypotheses.
The reluctance of neurobiologists to adopt omics approaches has many roots, including the often-stated aversion to ‘fishing expeditions’ without clearly defined hypotheses. Yet omics, or discovery-based, approaches do not eschew hypotheses; rather, they seek to elevate hypothesis testing to a new level, by allowing high-throughput hypothesis generation and prioritization. It is also notable that although functional genomics has led to a revolution in the field of cancer research, it has taken much longer to appeal to neuroscientists. Discovery-based research appears to be of more interest in disease-based fields, because the focus is on generating novel hypotheses and discovering new therapeutic and diagnostic approaches. It is only over the past two decades that disease-focused research has gradually evolved to become a considerable force in neuroscience. A corollary of this comparison is that, in general, the field of modern cancer research has been much more successful in developing new therapeutic techniques that have subsequently been translated into clinical practice71. There are many reasons for this, including access to tissue. However, advances in cancer research have also been catalysed by the core omics methods, which have not yet been as widely applied in disease-related neuroscience. The prediction is that a wider application of omics methods to neurological and psychiatric diseases14,16 in both model systems and patients will significantly accelerate advances in therapeutic development in neuroscience. One area in which this appears promising is in the detection of disease biomarkers (see page 916) based on transcriptional profiles12,13,72,73. Because brain tissue at the appropriate developmental or disease stages in humans is rarely available, molecular biomarkers do pose a particular challenge. The use of peripheral tissues, such as blood, lymphoblasts or fibroblast-derived induced pluripotent stem cells, by omics methods provides a potential solution. Published data suggest that peripheral gene expression profiles might reflect aetiological subtypes of disease on which to stratify patients for genetic studies of complex neurologic or psychiatric disorders69–71, in a loose parallel with the tissue-biopsy approach central to the cancer field.
The revolutionary nature of these genomic advances and the rapidly evolving technologies that continue to further the high-throughput omics agenda clearly distinguish this form of research from the more hypothesis-driven work performed in most neurobiology laboratories. Omics research requires not only large-scale instrumentation and multi-disciplinary teams of biologists, computer scientists, mathematicians and statisticians, but also a fundamental change in perspective5. The omics view is that an understanding of the organization and structure of the genome and the high-throughput measurements of the relationships of its elements, or gene products, provides a systematic basis on which to understand biological processes. Furthermore, significant value is placed on resource building and data sharing. Omics is not exclusive of more standard methods, and in fact is ultimately at its most powerful when combined with careful experimental validation. From this vantage point, the potential for discovery through large-scale screening and analysis provided by omics contrasts with the ability to incrementally advance science through individual hypothesis testing, one gene at a time.
The power of functional genomics in neuroscience is highlighted by several successful demonstrations of the strength of such methods to identify the molecular basis of neuronal diversity6–10, synaptogenesis11, disease biomarkers12,13, disease mechanisms and pathways13–16 and the development of user-friendly genome-scale resources17,18. These projects, along with other early proofs of principle19,20, have clearly weakened the notion of the superiority of research based on single-hypothesis testing over hypothesis generation and prioritization using omics-based or discovery-based methods as a starting point. These approaches, which provide a new framework for the rapidly growing fields of neurogenetics, neurogenomics and systems biology, and the challenges that accompany them, are the subject of this Review. We discuss the value of data sharing and provide some key examples of neuroinformatics-based or omics-based resources, highlighting areas in which genetic and functional genomic approaches have brought new biological insight to different areas of neuroscience. We conclude with a discussion of the new frontiers in biological networks and systems biology.
Public data sharing and resource generation
Omics data provide a platform for small-scale and large-scale in silico discovery and hypothesis generation that often goes beyond the scope of the original experiment. This notion supports the proposition that data from microarray studies have most value in the public domain, where they can be mined or combined with other studies21. However, proteomic and transcriptomic data are inherently less generic than sequence data, so obstacles such as variable sample preparation, experimental annotation and platform differences prevent a single fully unified data resource. The development of a framework, MIAME (minimal information about a microarray experiment), allowed the creation of centralized repositories for these data created on multiple platforms in laboratories around the world (Box 2). A growing number of studies that have capitalized on such databases, including those cited above and others22–27, have demonstrated that the widespread availability of these proteomic and transcriptomic data are of great benefit. One caveat with any resource based on compilation from multiple sources is that the data are only as good as the experimental and sample phenotype annotation. As microarray-based hybridization platforms for expression analysis give way to sequencing-based approaches, these data will become more generic, diminishing, but not eliminating, cross-platform compatibility issues.
Box 2. Major public databases.
The Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo), supported by the US National Institutes of Health, is the most widely used open-access repository for storing and uploading unfiltered, unmanipulated microarray and sequencing data. ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae), housed at the European Bioinformatics Institute, is the other major public repository of microarray data. A large non-public repository of microarray data is the Stanford Microarray Database (http://smd.stanford.edu). Investigators can easily access archived data and implement their analyses on these data sets, allowing continued analysis of any deposited data set using future methodologies. The ability to query several parameters from a vast number of deposited samples across many species and tissues should allow for the development of interconnected data sets at multiple expression levels to build a wider view of expression in a given species and between species. The Celsius database (http://celsius.genomics.ctrl.ucla.edu) is one such attempt, and combines nearly 100,000 publicly available Affymetrix microarray data sets for global analysis of particular gene co-expression patterns74. ArrayExpress provides a portal through which to query highly annotated meta-summary data from approximately 1,000 microarray experiments and nearly 30,000 microarrays. Similarly valuable proteomic and protein-interaction databases exist75–78, including lab-based systems such as the Organelle Map Database79 (http://141.61.102.16/ormd/), which organizes 1,405 proteins into ten specific organelles on the basis of correlation profiling. Considerable work is going into centralizing and curating proteomics data; databases include Peptide Atlas (http://www.peptideatlas.org) and The Human Proteinpedia80 (http://www.humanproteinpedia.org), which involves the efforts of over 70 laboratories and provides the most integrated, searchable portal to many data types from diverse platforms, ranging from cytochemistry to mass spectrometry.
In parallel with the growth of generic data repositories, standardized resources built by coupling high-throughput analyses with bioinformatics tools provide a clear demonstration of the value of large-scale omics data. The development of such resources requires significant foresight, planning and investment, but their worth is unquestionable if they are properly managed. Additionally, just as for DNA sequences and the genome databases, errors exist. Quality control must be balanced with throughput. Ideally, such resources would provide access for users to post corrective data or suggest annotations that would enhance the accuracy of the resource in real time.
Use of these resources to reanalyse public data is often a necessary feature of the well-rounded systems biology approach, raising the issue of the primacy of data analysis versus data generation. There is a clear tension between biological advances obtained in this way and the generation of new data, the latter usually carrying more weight in terms of novelty and degree of difficulty. Often, data analysis is not considered to be as much of an advance as the generation of new data, no matter how narrow the new data may be. However, a new perspective must be adopted to take into account studies that analyse data generated by others or in the public domain, to lead to new levels of understanding, or provide database tools around these data, even if no ‘new biology’ is done. The notion of what constitutes an experiment must expand to include analysis only, in addition to standard bench work. Similarly, we need to ensure that the next generation of neuroscientists has the quantitative skills to allow them to take full advantage of these opportunities. Here we highlight a few examples of advances based either on the generation of new omics data resources or on the analysis of data from such resources to provide fresh biological insights.
Surveying the nervous system transcriptome
Knowledge of the spatial and temporal expression of every gene in the nervous system is a natural, neuroscience-specific extension of the human genome project. Thus, a number of different approaches have been undertaken to identify neuroanatomical patterns of expression on a global scale. The most complete gene expression resource for neuroscientists is the Allen Brain Atlas (ABA; http://www.brain-map.org), which is a neuroanatomical repository of gene expression information in both mouse and human brain. Using high-throughput, standardized in situ hybridization to display messenger RNA expression in a given section of brain tissue, the Allen Institute for Brain Science has generated an interactive guide into the entire known brain transcriptome. Users can query the maps by gene or brain structure and generate three-dimensional overlays of any permutation of gene and structure. Such a tool can be useful in determining the developmental and regional specificity of a particular gene, as well as the combinatorial expression of groups of genes. GenePaint (http://www.genepaint.org) and the Brain Gene Expression Map (BGEM; http://www.stjudebgem.org) are two other databases of mouse gene expression based on in situ hybridization at various developmental stages. Both contain more embryonic data than the ABA, but they are more limited in scope and do not reference a three-dimensional atlas.
The first project involving the Allen Institute focused on adult mouse brain in one strain (C57BL/6J)18, but since then its research has expanded to encompass a variety of adult and developing mouse data sets and will soon also contain a first-generation data set from adult human brain. Additional data-mining tools that take advantage of the digital nature of the data include the Anatomic Gene Expression Atlas, which integrates all of the data from the ABA and the Allen Reference Atlas to build three-dimensional maps of correlated gene expression patterns. For example, this level of analysis permits the querying of different cortical layers and laminar gene expression to find clusters of co-expressed genes.
These tools have been used28 to study hippocampal molecular anatomy, further subdividing the CA3 region of the hippocampus into nine distinct regions, each with its own genomic signature presumably underlying different functionalities. Using both computational and manual methods of analysis, the hippocampus was recurrently subdivided into smaller subsections based on overlapping patterns of gene expression. This subdivision allowed the generation of novel three-dimensional modelling of the hippocampus, as well as functional analysis of gene expression within a given subsection. The validity of the approach was confirmed using a combination of retrograde labelling and in situ hybridization. Thus, using the publicly available ABA, this study verified that hippocampal circuitry is driven, at least in part, by regional gene expression. These findings are critical for future experiments examining hippocampal function in targeted transgenic model systems.
The Gene Expression Nervous System Atlas (GENSAT; http://www.gensat.org) is a multifaceted resource that includes a public database of gene expression in the central nervous system of both the developing mouse and the adult mouse, based on bacterial artificial chromosome (BAC)-transgenic reporter mice29. These mice, which are publicly available, provide unique, high-spatial-resolution information on the morphological pattern of expression of specific genes in vivo. The genetic labelling of individual and specific neuronal populations has implications for a wide range of investigations in fields from physiology30 to functional genomics9. For example, such techniques can be used to purify neurons, in combination with other approaches such as retrograde injections, fluorescence-activated cell sorting, manual dissections or immunopanning, to uncover cell-class-specific neuronal gene expression profiles6,7,9,10.
On a related theme, the recently developed ‘bacTRAP’ technology uses BAC-transgenic mice with green fluorescent protein fused to a ribosomal protein under the direction of a specific cell-type promoter to permit translating-ribosome affinity purification (TRAP)8. All mRNAs in the process of translation, which are thus attached to ribosomes, can be isolated from a specific cell type by using an antibody against green fluorescent protein. This approach avoids the potential for stress-induced or injury-induced changes in gene expression and is amenable to high-throughput analysis, once the mice are grown. An impressive 24 lines of bacTRAP mice generated deep expression profiles31, providing thousands of new markers and cell-specific targets for further investigation. Future studies will be needed to validate this approach and determine its improvement over the other well-studied methods mentioned above. All of these investigations of purified neuronal or glial populations demonstrate how the generation of large-scale microarray data from a single study facilitates further studies into the identification of neuronal classes.
Resources such as the ABA and GENSAT, which are based on anatomical expression patterns, provide high spatial resolution but are inherently qualitative, so atlases of gene expression based on quantitative expression profiling are important adjuncts. However, most current quantitative atlases, such as BioGPS (http://biogps.gnf.org), and those curated at centralized browsers such as the GEO or GeneCards (http://www.genecards.org), have very low spatial resolution, limiting their use to the most basic analyses. A notable exception is the Cerebellar Development Transcriptome Database (http://www.cdtdb.brain.riken.jp), which approaches a systems level of analysis by integrating not only high-resolution spatial imaging but also temporal and tissue-specific patterns of expression, connecting genes with overlapping gene ontologies, and linking to other databases such as the ABA, GenePaint and the BGEM. Finally, quantitative areal maps of the adult mouse brain using microarray profiling of dissected brain regions have yielded important data and emphasized the enormous differences in gene expression and regulation between different inbred mouse strains20,32,33, which is an important consideration for those using mice as a model system.
Recent work34 moves towards the idea of combining genome-wide expression data with higher-resolution neuroanatomical correlates, by creating a first-generation map of gene expression in the fetal human brain. Thirteen brain regions from both hemispheres in four fetal brains were assessed on Affymetrix exon arrays. Extensive experimental validation, bioinformatic analysis and data mining were performed, confirming regional patterns of expression, identifying previously unknown splice isoforms and gene groupings related to regional specificity, and showing an association of regionally enriched patterns with highly evolving regulatory elements. Network analysis of gene co-expression relationships was also used to organize these data. Notably, several of the genes that co-expression network analysis related to specific cortical areas were not detected by standard analysis of differential expression. These data and the accompanying database provide an early quantitative foundation for the field of developmental neurobiology that will serve as a reference for comparisons between model systems and the human brain.
Exploring the synaptic proteome
Synaptic transmission is a fundamental component of nervous system function, and its dysfunction is implicated in virtually every neurological or psychiatric disease. Thus, identification and functional annotation of its molecular components provide a foundational resource for neuroscience that is as important as more ubiquitous cellular organelle-related proteomes or transcriptomes are for biology in general35,36. Proteomics is probably the method of choice for identification of specific synaptic components, because unless more complex experimental designs that include network analysis25,37 are used, transcriptional profiling cannot usually provide organelle-specific data. This work requires a long-term approach38, as it typically involves laborious purification of different synaptic components by means of immunoprecipitation, sub-cellular fractionation or other methods, followed by mass spectrometry analysis37,39–41. So far, more than 1,000 synapse-related proteins have been identified, a level of complexity that was initially unexpected42.
One exemplary study11 of the synaptic proteome represents the vanguard of genetics, genomics and systems biology applied to neuroscience. In it, genome databases were mined to compare the postsynaptic proteome across 19 species, and the expansion of the synaptic proteome was found to correspond to known evolutionary hierarchies; the largest expansion in the number of synaptic proteins was observed during the transition between the invertebrate and vertebrate lineages. It was concluded that the regionalization of the cortex at both the anatomical and the functional levels is associated with evolution in synaptic diversity at the proteomic level. By combining gene expression and immunochemical data from their own laboratory and from multiple public-domain data sources including the BGEM43 and an adult-mouse microarray expression atlas44, the authors determined that genes of more recent origin show the greatest regional variation in expression in mammalian brain. These results are of particular interest, as the newer genes are involved in complex processes such as extracellular signalling and scaffolding at the synapse. Whether the anatomical differences are due to changes in DNA cis-regulatory sequence or instead are a consequence of antecedent differences in anatomy and physiology remains to be determined. Nevertheless, this tour-de-force study, essentially driven by the mining of previously existing data sets from multiple publicly available data sources, complemented by wet-bench experimentation, provides a new list of molecular tools for exploring synaptic structure–function relationships, their correspondence with specific brain circuits and the emergence of higher cognition.
From a systems perspective, a daunting combination of additional levels of electrophysiological and anatomical phenotypes will be required to relate molecular pathways operating at the synapse to cellular function and, subsequently, to complex circuits. As a step in this direction, multi-electrode recording has been combined with microarrays to correlate genome-wide mRNA expression with synaptogenesis and synaptic activity in dissociated hippocampal neurons cultured on multi-electrode grids45. By conducting a time-course analysis of these parameters together with morphological measurements, the authors found that gene expression changes occurred first, followed by concurrent changes in electrical activity and synaptic maturation. These data suggest that the program of gene expression initiating synaptogenesis is independent of neuronal network activity, a hypothesis that they test in vitro by blocking neuronal activity and assessing key gene expression changes. This work stops short of a functional assessment of any of the novel genes identified. However, by coupling multiplexed physiological measurements and global expression profiling by microarrays it prefigures future studies, in which data from both neural and gene expression networks must be integrated to bridge systems and molecular neuroscience.
Integrating genetic and phenotypic data
Another approach to adding systems-level structure to transcriptome data is the analysis of these data in concert with genetic and phenotypic data to integrate across all three levels of observation. Thus, the advent of expression quantitative trait locus (eQTL) analysis is a major advance in integrating large-scale genomic or genetic data sets to understand a model system or cohort of patients at a systems level. In eQTL mapping, gene expression data are used as a phenotype on which to base quantitative genetic association mapping (Fig. 1), the rationale being that gene expression is a more proximal, intermediate quantitative phenotype to the underlying genetic risk than heterogeneous behavioural or anatomical phenotypes. Because large samples are necessary to provide statistical power for whole genome-wide association, peripheral tissues such as blood cells were used in the initial pioneering studies in humans46. Recent studies in animal models demonstrate its utility in brain samples47,48. A very promising avenue is the combination of gene network analyses of expression for eQTL analysis as first demonstrated in ref. 49 and, more recently, ref. 50, both of which studied phenotypes related to metabolism. Another intriguing recent paper merges a different form of gene network analysis with structural chromosomal variation identified in patients with autism, suggesting a role for genes related to glycobiology in this disorder51.
Figure 1. Correlating genetic polymorphism and gene expression data.

Investigation of whole-genome single-nucleotide polymorphism (SNP) data from different phenotypic subgroups is typically used to perform genetic association based on diagnostic categories such as dementia. By treating gene expression data as a quantitative phenotype for genetic association, these two data sets can be combined to identify genetic loci that control quantitative variation in gene expression (which are known as eQTLs). Here, an analysis of mRNA and DNA from three types of brain sample (healthy individuals, patients with Alzheimer’s disease and those with early-onset Alzheimer’s disease) is depicted. The heat map (centre top) depicts the expression levels of all genes as determined by microarray analysis. The yellow box highlights genes with expression variations across the patient groups. The plot beneath shows DNA data from the same patients assessed for genetic polymorphisms (negative logarithm of the P value versus position on a chromosome); two SNPs (1 and 2) are found to correlate with a subset of patients (genotype A). Combining these data (right), the different genotypes are found to correlate with differences in gene expression.
One comprehensive, user-friendly interface for eQTL analysis in mouse is the database and toolkit provided by WebQTL17 (http://www.genenetwork.org). WebQTL allows integration of genetic polymorphism data from several strains of mice and rats, gene expression data from microarrays, and neurobiological traits based on neuroanatomy, pharmacology and behaviour. These tools have been implemented to provide proof-of-principle evidence of the utility of this approach; behavioural, microarray and genotyping data from a recombinant inbred mouse strain have been recombined47 to uncover significant QTLs correlating with gene expression and neuronal phenotypes, including, most interestingly, synapse function. Numerous QTLs were mapped to the majority of specific transcripts, which included a number of loci that appear to be ‘master’ transcriptome regulators, because they account for the expression of hundreds of genes. This study also uncovered QTLs that have polymorphisms associated with phenotypes of a specific behaviour. These links indicate that QTL-related transcript regulation can have wide-ranging effects on numerous functions, from the synapse to behaviour. Most of the transcriptional data in WebQTL is from whole brain, and the resource would benefit greatly from transcriptome analysis at higher spatial resolution, because significant variation occurs regionally48. However, this work connects genetic polymorphisms to RNA levels and to variation in both anatomy and behaviour, demonstrating the power of systems-level approaches to uncover complex neurobiological interactions.
QTL analysis has been extensively employed in rodents, but so far only two studies have combined gene expression data from human brain with genetic polymorphism data52,53. In the first study, nearly 200 pathologically proven normal cerebral cortical samples were merged with genome-wide SNP data53. Although this is a small number from the standpoint of genetic association in complex disease, the difficulty and cost associated with profiling human tissue makes this an important demonstration of the approach. Furthermore, this study identified a large number of potential cis-QTLs that, once replicated, can serve as a basis for understanding the direct functional consequences of human genetic variation on brain gene expression in disease. Supporting this notion, these data were used in a recent comparison with eQTLs identified in a parallel study of the brains of sufferers of Alzheimer’s disease, identifying several novel and known candidates for this complex brain disease52. The next step for these, and similar, studies is replication of the disease-association or eQTL-association findings in an independent cohort.
Accelerating discovery through next-generation sequencing
The advent of next-generation sequencing has raised multiple possibilities for the study of all layers of regulation leading to gene expression, changing the way we think about designing functional genomic experiments. Early studies clearly indicated that sequence data surpasses microarrays for studying gene expression, in terms of both depth of coverage and analysis of splicing, among other factors54,55. This technology can be used to quantitatively query not only mRNA expression but also RNA splicing, microRNA expression, epigenetic modifications, DNA binding, copy number variations and genetic deletions, insertions and mutations. Bridging these different outputs to generate a complete picture, from DNA to modified and bound DNA to precursor mRNA to mature mRNA, challenges even the most seasoned systems biologist. In addition, combining these data with proteomic data sets will completely revolutionize our understanding of the central dogma of molecular biology in action in any given cell. Another feature of next-generation sequencing that surpasses microarrays is the ability to assay species for which arrays do not exist, and eliminate the bias inherent in cross-species comparisons on microarrays56. The major challenges in the use of next-generation sequencing revolve around data storage and handling, but they are not unlike those encountered in the early years of microarray technology (Box 3), despite the amount of data involved being several orders of magnitude greater than that gained from microarrays. The relative platform independence of next-generation sequencing, leading to its more generic nature, will lower barriers for data sharing, meta-analysis and integration.
Box 3. The challenges of next-generation sequencing.
Next-generation sequencing will be revolutionary in the amount and content of data generated, but there are many obstacles to surmount. Extensive comparisons of sequencing data have not been published demonstrating whether there are batch effects in data due to sample preparation, library generation, flow cell preparation or machine run. Few studies have compared the commercial platforms for either gene expression or gene regulation81,82. Data storage and analysis are currently a much larger challenge than data generation. Many researchers have devised their own algorithms for analysing either the raw or the filtered data. But this will change as an official consensus is reached on what constitutes acceptable data in terms of basic features such as quality scores and alignment algorithms. Also, better genome annotations and increased read lengths will aid in improving data interpretation. Ultimately, the advances of sequencing will permit the testing of what is actually expressed without preconception, taking the microarray-based approach many steps farther.
Moving from lists to networks
Even the most elegant of the multilevel functional genomic approaches essentially involve the analysis of overlapping lists of microarray or other data. In most transcriptomic or proteomic studies, data are organized in order from most differentially expressed to least. This is useful because these very large data sets need to be put into a form that allows them to be analysed and understood. However, such simple levels of data organization cannot and do not represent even a small fraction of the potential information inherent in the data. Furthermore, the use of standard pathway tools limits the analysis to known relationships. Fortunately, we are just beginning to appreciate that the data itself has an underlying structure, and acknowledgement of this network structure as a general biological principle is opening new avenues of complementary investigation57,58.
The demonstration that transcriptome data can be organized into networks based on expression correlation, the application of graph theory, and robust statistical methods to develop weighted gene co-expression network analysis (WGCNA; http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork) raised expression profiling to the level of systems biology by elucidating the relationships among all of the elements being studied27,59 (Fig. 2). Several studies have now demonstrated that such networks derived from human or animal brain represent a reproducible and robust structure (for example refs 22–24), and that network position has significant functional implications. Similarly to earlier work in simpler organisms, this work has shown that proteomic and transcriptomic data derived from complex tissue such as brain show a high level of correspondence24,25. Such structure can be used to inform a new level of neuroscientific investigation that is not possible using standard analysis of differential expression22–25.
Figure 2. WGCNA schematic.
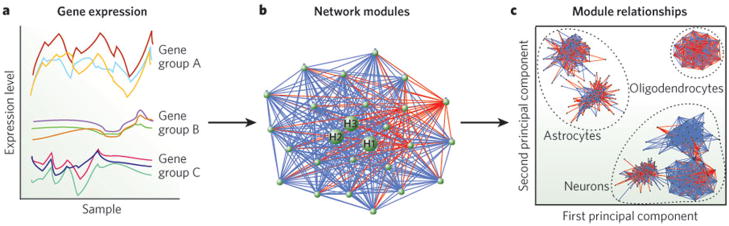
The underlying structure of a molecular network can be identified from high-dimensionality data sets such as those obtained from proteomic techniques or microarrays. This network structure can be used to guide research. a, Co-expression of groups of molecules across samples is measured to build networks, which comprise highly related clusters, or modules (for instance gene groups A, B and C here). b, A network module displaying the interconnection of genes. A gene’s position within the network has significant functional implications. Hub genes are the most connected, or central, genes within each module (depicted here as H1, H2 and H3). Each gene is depicted as a green node; blue lines indicate positive correlations; and red lines indicate negative correlations. c, The multidimensional scaling plot of the first and second principal components of all of the modules in a network demonstrates the meta-module structure, which clusters into functional groups such as, in this example, different central-nervous-system cell subtypes.
For example, one of the first such studies23 showed that gene networks could be used to provide a unifying method of identifying transcriptional targets of human brain evolution in the context of the neutral model of evolution for the transcriptome60. More recently, it has been demonstrated that the human cerebral cortex transcriptome is organized into a robust network and shown how its modular nature could be used to drive functional understanding and discovery in many directions, including the identification of markers for human adult neural stem cells24. One of the more remarkable observations here, especially with relevance to the discussion of neuronal heterogeneity and the need for individual cell study, is that from whole tissue WGCNA can recover modules that represent the transcriptional programs of the major cell classes in human brain, an example of in silico tissue dissection24. Importantly, these analyses relied on several public data sources, including the ABA and a resource provided by the transcriptional analysis of purified neurons, astrocytes and oligodendrocytes7.
These same network methods have recently been applied to provide a platform-independent comparison of pathways altered in normal ageing and dementia22. Data from two different microarray studies were combined and reanalysed to identify overlapping network modules corresponding to the synapse and mitochondria, illustrating common mechanisms shared by normal ageing and Alzheimer’s disease. Similarly, reanalysis25 of single-cell expression data10 has confirmed the initial findings and led to new biological insights, including a major transcriptional distinction between two classes of mitochondria: those located in the synapse and neuronal processes, and those located in the cell body. Both of these studies show how network methods can elucidate organelle-specific or cellular-component-specific expression profiles without the need for their purification (another form of in silico dissection), and further demonstrate the value of data placed in the public domain for subsequent reanalysis or use by others.
Proteomic networks can be constructed either through the investigation of actual protein–protein interactions or by the correlation of protein levels across samples or observations. The coupling15 of a high-throughput proteomic screen for protein interactions with bioinformatic analyses has been used to uncover an interaction network specific to ataxia. A screen for protein partners of ataxia-associated proteins was performed using the yeast two-hybrid system and a human-brain complementary DNA library, ultimately validating interactions in silico and in vivo. These interactions were expanded by culling known protein–protein interactions from public databases and the literature to create a final interaction network containing nearly 7,000 protein–protein pairs among almost 4,000 proteins. This network is an important resource for researchers studying neurodegeneration; it has already either provided confirmation of specific interactions or the impetus for their study61,62.
A more recent example of the combination of network methods and proteomic analysis identified a large number of postsynaptic protein complexes that show significant overlap with putative schizophrenia-susceptibility genes37. Five interconnected protein clusters were found by combining in vivo tandem affinity purification of PSD-95 (also known as DLG4) and mass spectrometry with network analysis. Almost half of the proteins in the clusters were related to at least one neurological disease, and, remarkably, 70% of the constituents of one of the clusters were specifically linked to schizophrenia. These and other early protein network analyses in brain should fuel future investigations and database development to catalogue and organize the entire neuronal proteome.
Conclusions and future directions
Many of the systems-level approaches that have been described here involve omics-level analysis of large data sets that span one or two dimensions of biological experimentation (Fig. 3). The most ambitious systems approach would see the integration of enormous data sources across multiple levels of genotypic, genomic, proteomic, epigenetic and phenotypic data (Fig. 3). Great efforts and powerful tools will be needed to achieve this in neuroscience, including the standardization of complex measurements of animal and human nervous system phenotypes and the development of accompanying ontologies63. Most forms of molecular data can be made into relatively generic forms, but translating complex phenotypes, from neuronal morphologies to the cognitive and behavioural profiles of neuropsychiatric disease entities, will require far more groundwork. Furthermore, although there is a compelling rationale to study nervous system phenotypes as they evolve over time, in many cases current funding and review processes are barriers to the collection of longitudinal data.
Figure 3. The systems biology approach to high-dimensional data sets allows integration of multiple layers of data.

a, The traditional experimental approach to the complexity of neuronal systems and diseases usually stretches across one or two layers of information. Typically, efforts are directed towards genetic data (such as sequence variants and epigenetic modifications), genomic data (such as gene expression) or phenotypic data (such as electrophysiological and clinical data). The systems biology approach seeks to consider all of these aspects at the same time, through the creation of comprehensive relational databases. The identification of a higher structure in high-dimensional data sets (for example by using network methods) facilitates the connection between different types of information (for example between genetic data and genomic data). b, Illustration of a potential systems-level integration of regional brain gene expression, coupled with network-based analysis methods and imaging data, to provide insights into brain connectivity. This is a stylized visualization of the combination of diffusion tensor imaging data for language areas70 with gene expression and WGCNA analysis to reveal integration of gene co-expression across brain areas (BA, Brodmann area), as well as novel brain-region wiring. The green lines and dashed red lines indicate information flow in both directions and can be extrapolated to suggest excitatory and inhibitory interconnections. The integration of network analysis, gene expression data and imaging analysis will elucidate the relationships among key genetic drivers in distinct regions and their relationship to brain regional connectivity in normal conditions and in disease. Each gene is depicted as a node (green or pink), with hub genes represented by pink nodes. Blue lines indicate positive correlations, and red lines indicate negative correlations. Lines between Brodmann areas indicate real and potential interactions through white-matter tracts.
However, even one data dimension, such as knowledge of transcriptome organization by means of network analysis, can promote large conceptual leaps by providing a new view of gene function, independent of the proteome or genome. For example, we have observed groups of genes annotated by gene ontology as mitochondrial, ribosomal and proteasomal within single co-expression modules in several data sets. From a proteomics standpoint these organelles are distinct, but the high co-expression of genes within them suggests that they are part of a highly coordinated and interconnected system that spans cellular compartments as they are typically defined. Here transcriptional network analysis provides a new, systems-level view of gene function within cellular pathways that was not readily apparent from proteomic or genomic data alone.
Another frontier in systems-level analysis in neuroscience is highlighted by the necessary scale and complexity of endeavours analysing human phenotype data from a genetic perspective. Recent work combines text mining64,65 with genetic analysis and modelling to integrate across disease phenotypes66,67. In a remarkable study67, the co-morbidity among 161 different medical conditions abstracted from 1.5 million medical records was analysed in the context of a basic probabilistic model of inheritance. This work not only detected known genetic relationships between diseases, but also showcased the power of such methods to detect unexpected genetic relationships between human disorders considered to be distinct. For example, it predicted significant causal overlap between disorders such as bipolar disorder, autism and schizophrenia, as well as a shared genetic connection among autism and autoimmune and infectious disorders. In the future, such work should be greatly facilitated by detailed and standardized clinical phenotype ontologies, as well as integration with other levels of quantifiable phenotypic data, from molecular biomarkers such as gene expression and epigenetic profiles to neuroimaging data.
The application of systems-level analyses to neuroimaging data alone, based on a theoretical framework similar to WGCNA, has also begun to reveal remarkable insights into human brain networks68. This work shows a robust relationship between specific aspects of brain functional networks and structural connectivity69. Analogously to gene-based networks, particular functional modules and key hub regions can be identified on the basis of maps of functional or structural connectivity. At this point, the possibility of connecting such networks to the panoply of potential genetic, genomic or environmental factors that regulate them may seem distant. However, there is likely to be a reasonable systems biology solution. Standardized network analyses based on graph theory, such as WGCNA, could be used to integrate anatomically based brain networks with gene expression or proteomic networks from profiling of the same brain regions from post-mortem samples (Fig. 3b). This type of analysis would reach a level of integration of ‘neuro-omic’ and neuroimaging data that goes far beyond the current state of genotype–phenotype correlations. Such studies would be greatly accelerated by the requirement that data from all published neuroimaging studies be made publicly available in a usable, normalizable form, similar to omics data.
In many ways, the omics revolution provides an example of the enormous value of public data sharing in making efficient use of our relatively limited resources to fuel scientific and biomedical advances. The value of these data highlights the urgent need for common language and measurements in addition to incentives and portals for data sharing from even the smallest-scale studies. In parallel, it also speaks to the need for large-scale collaborative endeavours that collect data in a way that facilitates such sharing and permits integration across multiple disciplines. This is not to suggest that omics should or could necessarily replace ology in any way. However, omics approaches offer the possibility of a new foundation for ologies to build on, by allowing for the testing of many hypotheses in parallel and providing a systems-level context for data interpretation that is necessary to further our understanding of brain function.
Acknowledgments
This Review would not have been possible without help from members of the Geschwind laboratory, especially G. Coppola, who helped with several figures and provided critical comments on the manuscript; M. Oldham, for his pioneering use of WGCNA in the brain; and D. Crandall of the Mental Retardation Research Centre media core at the University of California, Los Angeles, for assistance with Fig. 3. We are also grateful to our collaborators S. Horvath, S. Nelson and P. Mischel, for their generosity of time and expertise. We apologize to the authors of the many outstanding studies we were not able to cite owing to space limitations. We acknowledge support from the US National Institutes of Health (grants NIMH R37 MH60233-06A1 and NINDS U24 NS52108), the US National Institute on Aging, and the Dr Miriam & Sheldon G. Adelson Medical Research Foundation programme on neural repair and rehabilitation for our work in functional genomics (D.H.G.); and the A. P. Giannini Foundation Medical Research Foundation and NARSAD (G.K.).
Footnotes
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Coppola G, Geschwind DH. Microarrays and the microscope: balancing throughput with resolution. J Physiol (Lond) 2006;575:353–359. doi: 10.1113/jphysiol.2006.112623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson SB, Hempel C, Sugino K. Probing the transcriptome of neuronal cell types. Curr Opin Neurobiol. 2006;16:571–576. doi: 10.1016/j.conb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Mirnics K, Pevsner J. Progress in the use of microarray technology to study the neurobiology of disease. Nature Neurosci. 2004;7:434–439. doi: 10.1038/nn1230. [DOI] [PubMed] [Google Scholar]
- 4.Geschwind DH. Mice, microarrays, and the genetic diversity of the brain. Proc Natl Acad Sci USA. 2000;97:10676–10678. doi: 10.1073/pnas.97.20.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hood L, Heath JR, Phelps ME, Lin B. Systems biology and new technologies enable predictive and preventative medicine. Science. 2004;306:640–643. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 6.Arlotta P, et al. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 7.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nature Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- 10.Sugino K, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nature Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 11.Emes RD, et al. Evolutionary expansion and anatomical specialization of synapse proteome complexity. Nature Neurosci. 2008;11:799–806. doi: 10.1038/nn.2135. In this paper, a combination of genomics and proteomics is used to identify synaptic proteins that have changed with evolution and study how they might relate to brain anatomy and function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagasaka Y, et al. A unique gene expression signature discriminates familial Alzheimer’s disease mutation carriers from their wild-type siblings. Proc Natl Acad Sci USA. 2005;102:14854–14859. doi: 10.1073/pnas.0504178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura Y, et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 14.Karsten SL, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Lim J, et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. This paper was the first to demonstrate the utility of microarray analysis to uncover new genes and properties associated with neuropsychiatric disease. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Williams RW, Manly KF. WebQTL: web-based complex trait analysis. Neuroinformatics. 2003;1:299–308. doi: 10.1385/NI:1:4:299. [DOI] [PubMed] [Google Scholar]
- 18.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 19.Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg R, et al. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geschwind DH. Sharing gene expression data: an array of options. Nature Rev Neurosci. 2001;2:435–438. doi: 10.1038/35077576. [DOI] [PubMed] [Google Scholar]
- 22.Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci USA. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oldham MC, et al. Functional organization of the transcriptome in human brain. Nature Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. This paper demonstrates that the brain transcriptome in its normal state has a reproducible structure that can be used to guide discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winden K, et al. The organization of the transcriptional network in specific neuronal classes. Mol Syst Biol. 2009;5:291. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 27.Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004;14:1085–1094. doi: 10.1101/gr.1910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson CL, et al. Genomic anatomy of the hippocampus. Neuron. 2008;60:1010–1021. doi: 10.1016/j.neuron.2008.12.008. This paper is an example of the power of using tools such as the ABA as a reference together with other wet-lab tools to uncover new neuroanatomical connections, in this case new hippocampal subdivisions. [DOI] [PubMed] [Google Scholar]
- 29.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 30.Okaty BW, Miller MN, Sugino K, Hempel CM, Nelson SB. Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J Neurosci. 2009;29:7040–7052. doi: 10.1523/JNEUROSCI.0105-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cowley MJ, et al. Intra- and inter-individual genetic differences in gene expression. Mamm Genome. 2009;20:281–295. doi: 10.1007/s00335-009-9181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nadler JJ, et al. Large-scale gene expression differences across brain regions and inbred strains correlate with a behavioral phenotype. Genetics. 2006;174:1229–1236. doi: 10.1534/genetics.106.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson MB, et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kislinger T, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 36.Brunner E, et al. A high-quality catalog of the Drosophila melanogaster proteome. Nature Biotechnol. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez E, et al. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol Syst Biol. 2009;5:269. doi: 10.1038/msb.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson CN, Grant SG. High throughput protein expression screening in the nervous system — needs and limitations. J Physiol (Lond) 2006;575:367–372. doi: 10.1113/jphysiol.2006.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor–adhesion protein signaling complexes. Nature Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 40.Takamori S, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 41.Trinidad JC, et al. Quantitative analysis of synaptic phosphorylation and protein expression. Mol Cell Proteomics. 2008;7:684–696. doi: 10.1074/mcp.M700170-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Croning MD, Marshall MC, McLaren P, Armstrong JD, Grant SG. G2Cdb: the Genes to Cognition database. Nucleic Acids Res. 2009;37:D846–D851. doi: 10.1093/nar/gkn700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magdaleno S, et al. BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006;4:e86. doi: 10.1371/journal.pbio.0040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zapala MA, et al. Adult mouse brain gene expression patterns bear an embryologic imprint. Proc Natl Acad Sci USA. 2005;102:10357–10362. doi: 10.1073/pnas.0503357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valor LM, Charlesworth P, Humphreys L, Anderson CN, Grant SG. Network activity-independent coordinated gene expression program for synapse assembly. Proc Natl Acad Sci USA. 2007;104:4658–4663. doi: 10.1073/pnas.0609071104. This paper exemplifies the combination of multiple layers of functional data — in this case neuronal activity recordings and morphological measurements — with gene expression data to directly uncover how changes in function and gene expression relate to each other over time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheung VG, et al. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–1369. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesler EJ, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature Genet. 2005;37:233–242. doi: 10.1038/ng1518. This paper provides an early example of combining data across multiple levels of function, factoring genotypes, phenotypes and gene expression in mouse to identify systems-level interactions. [DOI] [PubMed] [Google Scholar]
- 48.Hovatta I, et al. DNA variation and brain region-specific expression profiles exhibit different relationships between inbred mouse strains: implications for eQTL mapping studies. Genome Biol. 2007;8:R25. doi: 10.1186/gb-2007-8-2-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghazalpour A, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Zwaag B, et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE. 2009;4:e5324. doi: 10.1371/journal.pone.0005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Webster JA, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myers AJ, et al. A survey of genetic human cortical gene expression. Nature Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 54.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu F, et al. Comparison of hybridization-based and sequencing-based gene expression technologies on biological replicates. BMC Genomics. 2007;8:153. doi: 10.1186/1471-2164-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Preuss TM, Caceres M, Oldham MC, Geschwind DH. Human brain evolution: insights from microarrays. Nature Rev Genet. 2004;5:850–860. doi: 10.1038/nrg1469. [DOI] [PubMed] [Google Scholar]
- 57.Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nature Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 58.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási A-L. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. This paper is a seminal demonstration of the higher-order organization of metabolism across phylogeny. [DOI] [PubMed] [Google Scholar]
- 59.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 60.Khaitovich P, et al. A neutral model of transcriptome evolution. PLoS Biol. 2004;2:e132. doi: 10.1371/journal.pbio.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lam YC, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 62.Canterini S, Bosco A, De Matteis V, Mangia F, Fiorenza MT. THG-1pit moves to nucleus at the onset of cerebellar granule neurons apoptosis. Mol Cell Neurosci. 2009;40:249–257. doi: 10.1016/j.mcn.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 63.Bilder RM, et al. Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009 Jan 20; doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rzhetsky A, et al. GeneWays: a system for extracting, analyzing, visualizing, and integrating molecular pathway data. J Biomed Inform. 2004;37:43–53. doi: 10.1016/j.jbi.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 65.Rodriguez-Esteban R, Iossifov I, Rzhetsky A. Imitating manual curation of text-mined facts in biomedicine. PLoS Comput Biol. 2006;2:e118. doi: 10.1371/journal.pcbi.0020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iossifov I, Zheng T, Baron M, Gilliam TC, Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res. 2008;18:1150–1162. doi: 10.1101/gr.075622.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. This paper demonstrates that with enough phenotypic information it is possible to build modelling networks that predict the underlying genetic overlap among neuropsychiatric diseases with previously distinct aetiologies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bullmore E, Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nature Rev Neurosci. 2009;10:186–198. doi: 10.1038/nrn2575. [DOI] [PubMed] [Google Scholar]
- 69.Honey CJ, Kotter R, Breakspear M, Sporns O. Network structure of cerebral cortex shapes functional connectivity on multiple time scales. Proc Natl Acad Sci USA. 2007;104:10240–10245. doi: 10.1073/pnas.0701519104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rilling JK, et al. The evolution of the arcuate fasciculus revealed with comparative DTI. Nature Neurosci. 2008;11:426–428. doi: 10.1038/nn2072. [DOI] [PubMed] [Google Scholar]
- 71.Mischel PS, Cloughesy TF, Nelson SF. DNA-microarray analysis of brain cancer: molecular classification for therapy. Nature Rev Neurosci. 2004;5:782–792. doi: 10.1038/nrn1518. [DOI] [PubMed] [Google Scholar]
- 72.Tang Y, Lu A, Aronow BJ, Sharp FR. Blood genomic responses differ after stroke, seizures, hypoglycemia, and hypoxia: blood genomic fingerprints of disease. Ann Neurol. 2001;50:699–707. doi: 10.1002/ana.10042. [DOI] [PubMed] [Google Scholar]
- 73.Thomas EA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Day A, Carlson MR, Dong J, O’Connor BD, Nelson SF. Celsius: a community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. doi: 10.1186/gb-2007-8-6-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDowall MD, Scott MS, Barton GJ. PIPs: human protein–protein interaction prediction database. Nucleic Acids Res. 2009;37:D651–D656. doi: 10.1093/nar/gkn870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB — a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–D628. doi: 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatr-Aryamontri A, Zanzoni A, Ceol A, Cesareni G. Searching the protein interaction space through the MINT database. Methods Mol Biol. 2008;484:305–317. doi: 10.1007/978-1-59745-398-1_20. [DOI] [PubMed] [Google Scholar]
- 78.Mathivanan S, et al. An evaluation of human protein-protein interaction data in the public domain. BMC Bioinformatics. 2006;7 (suppl 5):S19. doi: 10.1186/1471-2105-7-S5-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foster LJ, et al. A mammalian organelle map by protein correlation profiling. Cell. 2006;125:187–199. doi: 10.1016/j.cell.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 80.Mathivanan S, et al. Human Proteinpedia enables sharing of human protein data. Nature Biotechnol. 2008;26:164–167. doi: 10.1038/nbt0208-164. [DOI] [PubMed] [Google Scholar]
- 81.Linsen SE, et al. Limitations and possibilities of small RNA digital gene expression profiling. Nature Methods. 2009;6:474–476. doi: 10.1038/nmeth0709-474. [DOI] [PubMed] [Google Scholar]
- 82.Passalacqua KD, et al. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]