

Abstract
Background and Purpose
The C-X-C chemokine receptors 3 (CXCR3) and C-X-C chemokine receptors 4 (CXCR4) are involved in various autoimmune diseases and cancers. Small antagonists have previously been shown to cross-inhibit chemokine binding to CXCR4, CC chemokine receptors 2 (CCR2) and 5 (CCR5) heteromers. We investigated whether CXCR3 and CXCR4 can form heteromeric complexes and the binding characteristics of chemokines and small ligand compounds to these chemokine receptor heteromers.
Experimental Approach
CXCR3–CXCR4 heteromers were identified in HEK293T cells using co-immunoprecipitation, time-resolved fluorescence resonance energy transfer, saturation BRET and the GPCR-heteromer identification technology (HIT) approach. Equilibrium competition binding and dissociation experiments were performed to detect negative binding cooperativity.
Key Results
We provide evidence that chemokine receptors CXCR3 and CXCR4 form heteromeric complexes in HEK293T cells. Chemokine binding was mutually exclusive on membranes co-expressing CXCR3 and CXCR4 as revealed by equilibrium competition binding and dissociation experiments. The small CXCR3 agonist VUF10661 impaired binding of CXCL12 to CXCR4, whereas small antagonists were unable to cross-inhibit chemokine binding to the other chemokine receptor. In contrast, negative binding cooperativity between CXCR3 and CXCR4 chemokines was not observed in intact cells. However, using the GPCR-HIT approach, we have evidence for specific β-arrestin2 recruitment to CXCR3-CXCR4 heteromers in response to agonist stimulation.
Conclusions and Implications
This study indicates that heteromeric CXCR3–CXCR4 complexes may act as functional units in living cells, which potentially open up novel therapeutic opportunities.
Keywords: chemokine receptor, GPCR, heteromerization, β-arrestin, radioligand binding, CXCR3, CXCR4
Introduction
Chemokines are secreted chemoattractant proteins of 8–14 kDa that direct immune cell migration through interaction with GPCRs. Over 40 chemokines and 20 chemokine receptors have been identified in humans, forming a regulatory system in which many of these receptors promiscuously bind multiple chemokines and vice versa (Scholten et al.,2012a). In addition, chemokine receptors can also form both homo- and heteromers (Scholten et al.,2012a). The relative occurrence of such GPCR complexes and transiency, however, is still a matter of debate (Dorsch et al.,2009; Hern et al.,2010). Importantly, heteromers between the CC chemokine receptors 2 (CCR2), 5 (CCR5) and C-X-C chemokine receptor 4 (CXCR4) can only bind a single chemokine with high affinity (El-Asmar et al.,2005; Springael et al.,2006; Sohy et al.,2007; 2009). This negative binding cooperativity between the heteromerized chemokine receptors is not limited to the cognate chemokines of both receptors, but extends to synthetic allosteric antagonists of these receptors (Sohy et al.,2007; 2009). Binding of these antagonists to one receptor allosterically inhibits chemokine binding to the other receptor within the heteromer, resulting in a cross-inhibition of intracellular signalling, and in vitro and in vivo chemotaxis (Sohy et al.,2007; 2009). Besides inhibiting one chemokine receptor by targeting its heteromeric partner, chemokine receptor heteromers may be therapeutically targeted by heteromer-selective ligands and/or bivalent ligands that simultaneously bind both receptors in a dimer, as previously described for, for opioid receptor heteromers (Waldhoer et al.,2005; Mathews et al.,2008; Jahnichen et al.,2010; Tanaka et al.,2010). Because GPCR heteromers are likely expressed on a more limited subset of cell types as compared with the individual GPCRs, heteromer-selective therapeutic agents have the potential to display improved efficacy and toxicity profiles.
C-X-C chemokine receptor 3 (CXCR3) and CXCR4 are expressed on activated T-cells, natural killer cells, dendritic cells and cancer cells (Vandercappellen et al.,2008; Viola and Luster,2008; Fulton,2009). CXCR4 plays an essential role in haematopoiesis, leukocyte homing/retention in secondary lymphoid tissues and recruitment to sites of inflammation, in response to local concentrations of C-X-C chemokine ligand 12 (CXCL12; Moser and Loetscher,2001). Additionally, CXCR4 and CXCL12 are upregulated in tumours by, for example hypoxia, and mediate angiogenesis, proliferation, invasion and metastasis (Vandercappellen et al.,2008; Viola and Luster,2008; Fulton,2009). Expression of CXCR3 and its ligands C-X-C chemokine ligand 9 (CXCL9), C-X-C chemokine ligand 10 (CXCL10) and C-X-C chemokine ligand 11 (CXCL11) is induced under inflammatory conditions, and has been implicated in autoimmune disease, graft-versus-host disease and transplant rejection (Lacotte et al.,2009). Moreover, CXCR3 is also involved in cancer proliferation and metastasis (Vandercappellen et al.,2008; Viola and Luster,2008; Fulton,2009).
In the present study, we show that CXCR3 and CXCR4 form heteromers at the cell surface. Negative binding cooperativity between CXCR3 and CXCR4 agonists was detected in membrane preparations of cells that co-express both receptors, but not on intact cells. However, β-arrestin2 recruitment specifically to CXCR3–CXCR4 heteromers was shown in living cells using the GPCR heteromer identification technology (GPCR-HIT) approach (Mustafa and Pfleger,2011; See et al.,2011; Mustafa et al.,2012).
Methods
Materials
The small CXCR3 ligands VUF10085 [(Johnson et al.,2007) and VUF10661 (N-(6-amino-1-((2,2-diphenyl-ethyl)amino)-1-oxohexan-2-yl)-2-(4-oxo-4-phenylbutanoyl)-1,2,3,4-tetrahydro-isoquinoline-3-carboxamide] have been previously described as AMG 487 (Johnson et al.,2007) and compound 2 (Stroke et al.,2006), respectively, and were synthesized at VU University Amsterdam (Scholten et al.,2012b). TAK-779 (N,N-dimethyl-N-[4-[[[2-(4-methylphenyl)-6,7-dihydro-5H-benzocyclohepten-8-yl]carbonyl]-amino]benzyl]-tetrahydro-2H-pyran-4-aminium chloride) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Baba et al.,1999). AMD3100 (1,1'-[1,4-phenylenebis-(methylene)]-bis-1,4,8,11-tetraazacyclotetra-decane) was obtained from Sigma-Aldrich (St-Louis, MO, USA). CXCL10 [immunoprecipitation (IP-10)], CXCL11 (I-TAC), and CXCL12 (SDF-1α) were from Peprotech (Rocky Hill, NJ, USA).
DNA constructs
Human CXCR3 (Verzijl et al.,2008) and CXCR4 were tagged at the N-terminus with FLAG (DYKDDDDK) or haemagglutinin epitope tag (HA; YPYDVPDYA) using PCR-based methods and subcloned in the pcDEF3 expression plasmid (a gift from Dr. Langer, Robert Wood Johnson Medical School, Piscataway, NJ, USA). The receptor fusion proteins CXCR3-Renilla luciferase (Rluc), CXCR3-enhanced yellow fluorescent protein (EYFP), CXCR4-Rluc, and CXCR4-EYFP have been previously described (Vischer et al.,2008). CXCR3-Rluc8 was constructed by substitution of Rluc with the optimized Rluc8 variant as previously described (Nijmeijer et al.,2010). CXCR4-Rluc8 and β-arrestin2-Venus constructs have been described previously (See et al.,2011). For sensitized emission FRET studies, CXCR3, CXCR4 and GABAB2 constructs were subcloned into the pECFP-N1 vector (Clontech Laboratories Inc., Mountain View, CA, USA) and the pVenus-N1 vector. The latter plasmid was constructed from pECFP-N1 by substitution of the sequence encoding for cyan fluorescent protein (CFP) with Venus DNA, which was obtained from pcDNA3.1-eCFP-exchange protein activated by cAMP (EPAC)-Venus (a gift from Dr. Jalink, The Netherlands Cancer Institute, Amsterdam, The Netherlands). All generated constructs were verified by DNA sequencing.
Cell culture and transfection
HEK293T cells were cultured in DMEM containing 10% fetal calf serum (FCS), 100 units·mL−1 penicillin and 100 μg·mL−1 streptomycin (PAA Laboratories GmbH, Cölbe, Germany) were maintained at 37°C in a humidified atmosphere containing 5% CO2. For saturation BRET, time-resolved FRET (trFRET), IP and radioligand-binding studies, HEK293T cells were transfected with indicated amounts of plasmid DNA using 25-kDa linear polyethylenimine (Polysciences, Eppelheim, Germany) as described previously (Verzijl et al.,2008). Transfected DNA was held constant by adjusting the total amount of DNA with the empty vector pcDEF3. For GPCR-HIT assays, HEK293FT cells were maintained at 37°C in 5% CO2 and complete media [DMEM containing 0.3 mg·mL−1 glutamine, 100 IU·mL−1 penicillin and 100 μg·mL−1 streptomycin (Gibco, Grand Island, NY, USA)] supplemented with 10% FCS and 400 μg·mL−1 Geneticin (Gibco). Transient transfections were carried out 24 h after seeding about 550 000 cells per well of a 6-well plate. Genejuice (Novagen, San Diego, CA, USA) transfection reagent was used according to the manufacturer's instructions. Cells were harvested with 0.05% Trypsin-EDTA (Gibco).
Saturation BRET
HEK293T cells were cultured and transfected in 96-well plates. GPCR-EYFP and GPCR-Rluc expression and saturation BRET between these receptors were measured as described previously (Vischer et al.,2008).
Time-resolved FRET (TrFRET)
HEK293T cells were transfected with plasmid DNA encoding N-terminally tagged CXCR3 and/or CXCR4. TrFRET was measured on a Novostar plate reader (BMG LabTechnologies, Offenburg, Germany) 48 h post-transfection, as described previously (van Rijn et al.,2006) with minor modifications. Briefly, following incubation with both 0.8 nM Eu3+-labelled anti-HA and 13 nM XL665-labelled anti-Flag antibodies (CisBio Bioassays, 30204 Bagnols/Ceze Cedex, France), the cells were washed and resuspended in PBS (107 cells·mL−1). Next, 50 μL of each sample was dispensed in triplicate in a white-walled 384-well microtiter plate.
Sensitized emission FRET
Imaging of HEK293T cells expressing receptor CFP or Venus fusion proteins was performed on a Leica AOBS_TCS SP2 confocal laser scanning microscope using a 63× NA 1.4 oil-immersion objective (Leica Microsystems, Rijswijk, The Netherlands) and the 458 nm line of an Ar/Kr laser. FRET was imaged by detecting sensitized emission (van Rheenen et al.,2004). FRET efficiency was determined by background subtraction, bleed-through correction, and correction of intensity (Jalink and Van Rheenen,2009). Values were measured by scaling all samples to the same level of CXCR3-CFP/CXCR3-Venus followed by detecting the intensity at different regions of interest on the cell membrane.
Co-immunoprecipitation and immunoblotting
HEK293T cells were transfected with plasmid DNA encoding N-terminally tagged receptors. 24 h after transfection, cell lysates were prepared and IP with anti-HA-agarose antibody (clone HA-7, Sigma-Aldrich) was performed according to manufacturer's instructions. Immunoprecipitated protein was eluted from anti-HA-agarose antibody by incubation with 6× sample buffer (0.35 M Tris.HCl, pH 6.8, 10.3% SDS, 30% glycerol, 0.6 M dithiothreitol, 180 μM bromophenol blue; all chemicals were obtained from Sigma-Aldrich) at room temperature (RT) for 5 min. Protein samples were resolved by 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes (AppliChem, Darmstadt, Germany). Blots were probed with the primary antibodies rat anti-HA (Roche, Indianapolis, IN, USA) or mouse anti-FLAG (Sigma-Aldrich) followed by HRP-conjugated secondary antibodies (Thermo Scientific, Rockford, IL, USA and Bio-Rad, Richmond, CA, USA) and visualized with an enhanced chemiluminescent reagent (Thermo Scientific, Rockford, IL, USA).
Radioligand binding
Preparation of HEK293T cell membrane fractions and [125I]-chemokine competition binding were performed as described previously (Verzijl et al.,2008). Cell membrane fractions were prepared 48 h post-transfection. Dissociation of [125I]-chemokine was determined using infinite dilution (Christopoulos et al.,1997). To this end, membranes were incubated in binding buffer with approximately 100 pM of radioligand at RT for 60 min, followed by centrifugation. The pellet was then resuspended in 3% of the supernatant volume and 3 μL was dispensed per well of a 96-well plate. At the indicated time points, 300 μL of binding buffer or 100 nM unlabelled chemokine or VUF10661 solution was added to the membrane suspension. Whole-cell binding experiments were performed essentially as previously described (Scholten et al.,2012b). Briefly, 24 h after transfections, cells were collected and seeded into poly-L-lysine coated 48-well plates. The next day, culture medium was replaced with ice-cold binding buffer containing 50 pM [125I]-CXCL10 or [125I]-CXCL12 in the absence or presence of 100 nM unlabelled chemokines. After 4 h the cells were washed, solubilized, and collected to measure bound radioligand in a Wallac Compugamma counter (Perkin Elmer Life Sciences, Waltham, MA, USA).
GPCR-HIT
As described previously (See et al.,2011), HEK293FT cells were transiently transfected with cDNA encoding a GPCR fused to Rluc8 (GPCR-Rluc8) and β-arrestin2 fused to Venus (β-arrestin2-Venus), along with a second GPCR that was untagged with respect to BRET signalling, or empty vector. DNA amounts of 0.1, 0.3 and 0.1 μg per well of a 6-well plate were used for GPCR-Rluc8, β-arrestin2-Venus, and untagged GPCR or empty vector, respectively. Forty-eight hours post-transfection, cells were incubated at 37°C, 5% CO2 for 2 h with 30 μM EnduRen (Promega, Madison, WI, USA) in Freestyle293 medium with 25 mM HEPES (Gibco) to ensure substrate equilibrium was reached. BRET measurements were taken at 37°C using the VICTOR Light plate reader with Wallac 1420 software (Perkin Elmer Life Sciences). Filtered light emissions were sequentially measured at 400–475 and 520–540 nm. The BRET signal was calculated by subtracting the ratio of 520–540 nm emission over 400–475 nm emission for a vehicle-treated cell sample from the same ratio for a second aliquot of the same cells treated with agonist, as described previously (Pfleger et al.,2006a,b). In this calculation, the vehicle-treated cell sample represents the background, eliminating the requirement for measuring a donor-only control sample (Pfleger et al.,2006a,b). For these BRET kinetic assays, the final pretreatment reading is presented at the zero time point (time of ligand/vehicle addition). The situation where addition of ligand specific for the untagged GPCR results in a ligand-induced BRET signal indicates β-arrestin binding specifically to a heteromer complex (See et al.,2011).
Data analysis
Statistical analysis as well as nonlinear regression analysis of saturation BRET and radioligand binding data was performed using Prism 4.03 (GraphPad Software Inc, San Diego, CA, USA).
Results
Identification of CXCR3–CXCR4 heteromers
Physical interactions between CXCR3 and CXCR4 were assessed by co-immunoprecipitation of differentially N-terminal epitope-tagged receptors from HEK293T cells co-expressing HA-CXCR3 and FLAG-CXCR4. To this end, cell lysates were subjected to IP with anti-HA beads. Both lysates and immunoprecipitates were resolved by SDS-PAGE and probed with anti-HA or anti-FLAG antibodies. Detection of a FLAG-immunoreactive band at 39 kDa demonstrated co-immunoprecipitation of co-expressed (i.e. co) FLAG-CXCR4 with HA-CXCR3 (Figure 1A lower panel), indicating that CXCR3 and CXCR4 are physically associated in a ligand-independent manner. Mixing of cells expressing either HA-CXCR3 or FLAG-CXCR4 (i.e. mix) prior to solubilization and anti-HA IP did not result in FLAG immunoreactivity following SDS-PAGE immunoblotting, ruling out the formation of non-specific HA-CXCR3/FLAG-CXCR4 aggregates during the solubilization process (Figure 1A lower panel). FLAG-CXCR4 was undetectable in direct immunoblots of lysates produced from cells expressing FLAG-CXCR4 in the absence (mix) or presence of HA-CXCR3 (co), which may be the consequence of low FLAG-CXCR4 expression levels (data not shown). In contrast, HA-CXCR3 was detected in lysate as well as anti-HA IP immunoblots prepared from both mixed and co-expressed cell samples (Figure 1A upper panel). To demonstrate the presence of HA-CXCR3/FLAG-CXCR4 heteromers on the cell surface, trFRET was measured on intact cells in parallel with the co-immunoprecipitation experiments. Intact cells co-expressing HA-CXCR3 and FLAG-CXCR4 (i.e. co), and mixed cells expressing either HA-CXCR3 or FLAG-CXCR4 (i.e. mix), were labelled with Eu3+-conjugated anti-HA and XL665-conjugated anti-FLAG antibodies. A higher trFRET signal was observed for cells co-expressing both receptors in comparison with cells expressing either HA-CXCR3 or FLAG-CXCR4, which were mixed prior to antibody incubation. This indicates that CXCR3 and CXCR4 indeed exist in close proximity (<10 nm) on the surface of living cells that co-express both receptors (Figure 1B). Because of poor expression of the FLAG-CXCR3, as determined by elisa (data not shown), the FLAG-CXCR3/HA-CXCR4 combination could not be investigated in either co-immunoprecipitation or trFRET experiments.
Figure 1.
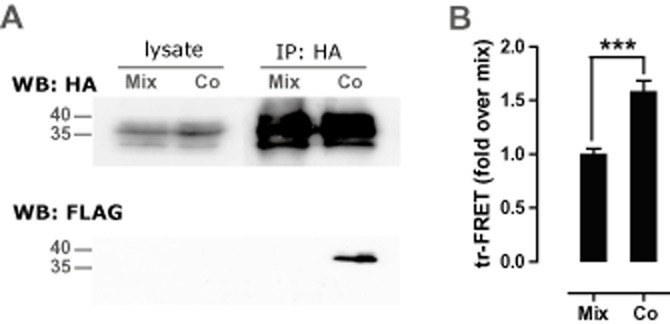
CXCR3 and CXCR4 form heteromers. HEK293T cells were transfected with HA-CXCR3 and/or FLAG-CXCR4 (500 ng/106 cells). Cells expressing HA-CXCR3 were collected and mixed (1:1) with cells expressing FLAG-CXCR4 (i.e. mix), whereas cells co-expressing HA-CXCR3 and FLAG-CXCR4 were mixed (1:1) with cells transfected with the empty vector pcDEF3 (i.e. co). (A) For co-immunoprecipitation experiments, cells were solubilized and lysates were immunoprecipitated with anti-HA beads, and both lysates and immunoprecipitates were resolved by SDS-PAGE and immunoblotted with anti-HA (top) or anti-FLAG antibodies (bottom). Immunoblots shown are from a representative experiment performed three times. (B) For trFRET analysis, cell surface expressed HA-CXCR3 and FLAG-CXCR4 were labelled with Eu3+-conjugated anti-HA and XL665-conjugated anti-Flag antibodies, respectively, and trFRET was determined by measuring emission at 665 nm 100 μs after excitation of Eu3+ at 337 nm. Specific trFRET between GPCR heteromers is given by the trFRETco/trFRETmix ratio. Pooled data from five independent experiments are shown. ***Cotransfected cells emitted a significantly higher FRET signal in comparison to the mix control (P < 0.0001). WB, Western blot.
Next, sensitized emission FRET using confocal laser scanning microscopy was used to visualize CXCR3-CXCR4 heteromers. CXCR3 and CXCR4 are colocalized at the cell surface and exist as heteromers as revealed by FRET between ECFP and Venus fluorophores that are fused to these receptors (Figure 2A). In contrast, CXCR3 and GABAB2 do not form heteromers as revealed by significantly lower FRET levels (Figure 2B), even though both receptors are colocalized at the cell surface (Figure 2A).
Figure 2.
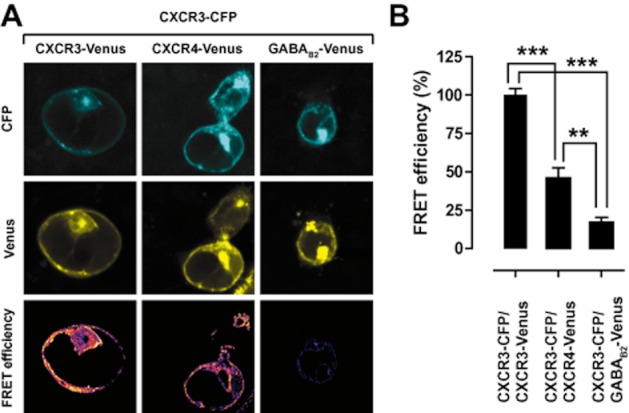
CXCR3–CXCR4 heteromers are present on the cell surface. (A) HEK293T cells were cotransfected with CXCR3–CFP and either CXCR3-Venus (left panels), CXCR4-Venus (middle panels), or GABAB2-Venus (right panels). CXCR3–CFP fluorescence images are shown in the upper panels, receptor-Venus fluorescence images are shown in the middle panels, whereas sensitized emission FRET is shown in the bottom panels. (B) Quantification of FRET efficiency from the sensitized emission FRET images.
To determine relative propensities of CXCR3 and CXCR4 to form homo- and heteromers, cells were cotransfected with a constant amount of BRET donor constructs CXCR3-Rluc or CXCR4-Rluc, and increasing amounts of BRET acceptor constructs CXCR3-EYFP or CXCR4-EYFP. Hyperbolic BRET signals were observed between CXCR4-Rluc and CXCR4-EYFP (Figure 3A), CXCR4-Rluc and CXCR3-EYFP (Figure 3B), CXCR3-Rluc and CXCR4-EYFP (Figure 3C), and CXCR3-Rluc and CXCR3-EYFP (Figure 3D). Saturation of the BRET signal with increasing EYFP/Rluc ratios indicates that close proximity (<10 nm) between Rluc and EYFP is due to specific interactions between receptor-Rluc and receptor-EYFP fusion constructs, and not the consequence of random collisions between BRET partners (Mercier et al.,2002). Saturated BRET (BRETmax) was higher between CXCR4-Rluc and CXCR4-EYFP in comparison with the other combinations. BRETmax was comparable between CXCR4-Rluc and CXCR3-EYFP, and CXCR3-Rluc and CXCR3-EYFP, whereas the CXCR3-Rluc and CXCR4-EYFP combination yielded the lowest BRETmax value. The BRET50 value of a BRET saturation curve is the EYFP/Rluc ratio resulting in half-maximal BRET, and is considered to be a measure of the relative affinity of the interacting proteins for each other. Comparable BRET50 values were obtained for all CXCR3 and CXCR4 homo- and heteromer combinations, indicating that CXCR3 and CXCR4 have comparable propensities to form homo- and heteromeric complexes (Table 1).
Figure 3.
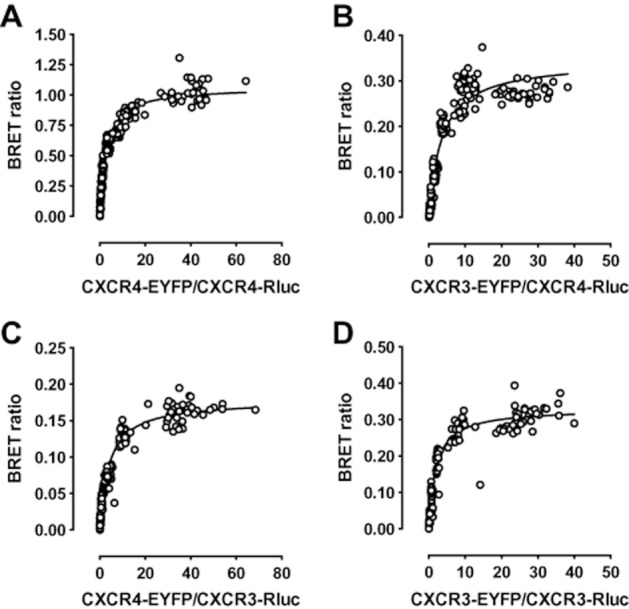
Hetero- and homomerization of CXCR3 and CXCR4. HEK293T cells were transiently cotransfected with a constant amount (150 ng/106 cells) of CXCR4-Rluc (A and B) or CXCR3-Rluc (C and D) DNA and increasing amounts of CXCR3-EYFP (B and D) or CXCR4-EYFP (A and C) (0–2200 ng/106 cells). Saturation curves were obtained by measuring BRET ratio as function of acceptor/donor ratio (i.e. EYFP/Rluc). Data were obtained from at least three independent experiments each performed in triplicate. Curves were fitted using nonlinear regression, assuming a single binding site.
Table 1.
Saturation bioluminescence resonance energy transfer between CXCR3 and CXCR4
| BRET50 | BRETmax | |
|---|---|---|
| CXCR4-Rluc/CXCR4-EYFP | 2.73 ± 0.09 | 1.06 ± 0.011 |
| CXCR4-Rluc/CXCR3-EYFP | 3.58 ± 0.17 | 0.35 ± 0.005 |
| CXCR3-Rluc/CXCR4-EYFP | 4.11 ± 0.14 | 0.18 ± 0.002 |
| CXCR3-Rluc/CXCR3-EYFP | 1.97 ± 0.09 | 0.33 ± 0.004 |
HEK293T cells were transfected with a constant amount of CXCR3-Rluc or CXCR4-Rluc DNA (150 ng/106 cells) and increasing amounts of CXCR3-EYFP or CXCR4-EYFP DNA (10–2200 ng/106 cells). BRET50 and BRETmax values ± SE were determined by fitting pooled data from three or more independent experiments to a single binding site isotherm.
The recruitment of β-arrestin2 to CXCR3 (Figure 4A) and CXCR4 (Figure 4B) was assessed using BRET with Rluc8-tagged receptors co-expressed with β-arrestin2-Venus. CXCL11 resulted in clear β-arrestin2-Venus recruitment to CXCR3-Rluc8 and no CXCL12-induced signal was observed (Figure 4A). Additionally, the CXCL11-induced β-arrestin2 recruitment to CXCR3 was not affected by co-stimulation with CXCL12 (Figure 4A). CXCL11 was used in preference to CXCL10 in these experiments as the CXCL10-induced β-arrestin2-Venus recruitment to CXCR3-Rluc8 was very weak compared with that induced by CXCL11 (data not shown), as shown previously (Scholten et al.,2012b). In contrast, CXCL12 resulted in clear β-arrestin2-Venus recruitment to CXCR4-Rluc8, with no ligand-induced signal observed with CXCL11 (Figure 4B). Combined treatment with both CXCL11 and CXCL12 in this case resulted in a similar BRET profile to that observed with CXCL12 alone (Figure 4B). In the GPCR-HIT configuration of CXCR4-Rluc8, β-arrestin2-Venus and CXCR3 (Figure 4C), the CXCL12-induced BRET signal for β-arrestin2-Venus recruitment to CXCR4-Rluc8 was smaller and more transient than that observed in the absence of co-expressed non-BRET-tagged CXCR3. Such a change in profile was observed previously when comparing β-arrestin2-Venus recruitment to CXCR4-Rluc8 with and without untagged CCR2 (See et al.,2011). A very weak signal was now observed with CXCL11, but most interesting was the BRET signal observed with both CXCL11 and CXCL12 with this configuration, which was substantially stronger than that observed with CXCL12 alone (Figure 4C). As published previously for other chemokine receptor combinations, this may be indicative of β-arrestin2 recruitment being facilitated by both types of receptor in the complex being in active conformations (See et al.,2011). Alternatively, the proximity of the donor and acceptor in these complexes may be sufficiently close to enable detection of changes in donor–acceptor distance and/or relative orientation resulting from both receptors being stabilized in active conformations instead of just one. Both scenarios are consistent with β-arrestin2 recruitment specifically to the CXCR3-CXCR4 heteromer.
Figure 4.
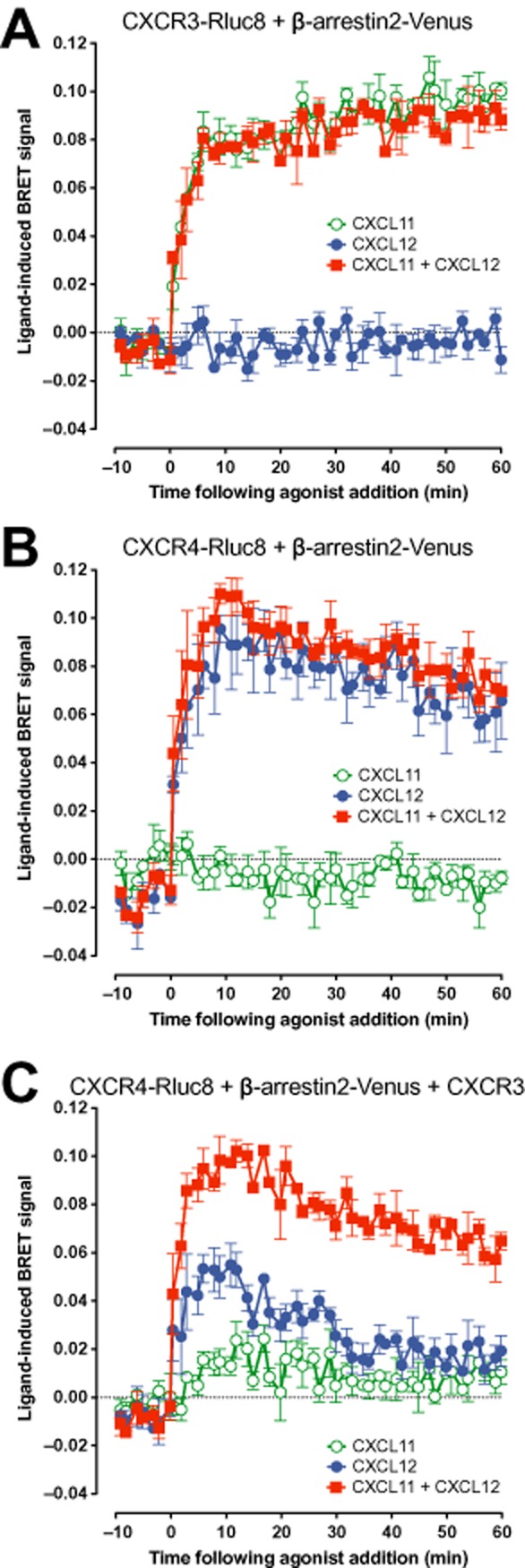
Evidence for the CXCR3-CXCR4 heteromer recruiting β-arrestin2 using the GPCR-HIT assay. eBRET kinetic profiles (Pfleger et al.,2006a) were generated with live HEK293FT cells co-expressing CXCR3-Rluc8 and β-arrestin2-Venus (A), CXCR4-Rluc8 and β-arrestin2-Venus (B) or CXCR4-Rluc8, β-arrestin2-Venus and CXCR3 (C). These cells were treated with 100 nM CXCL11, CXCL12 or both. Data are mean ± SEM of three independent experiments.
CXCR3 and CXCR4 agonists display negative binding cooperativity on membranes co-expressing CXCR3 and CXCR4
The CXCR3 chemokine CXCL10 and small CXCR3 agonist VUF10661 (Stroke et al.,2006; Scholten et al.,2012b) inhibited binding of [125I]-CXCL10 to CXCR3-expressing membranes in a concentration-dependent manner under equilibrium conditions (Figure 5A), which is not affected by the co-expression of CXCR4 (Figure 5B and Table 2). As expected, the CXCR4 chemokine CXCL12 was unable to displace [125I]-CXCL10 from membranes expressing only CXCR3 (Figure 5A). However, CXCL12 competed with [125I]-CXCL10 for binding to membranes co-expressing CXCR3 with CXCR4 (Figure 5B). This heterologous transinhibition of [125I]-CXCL10 binding to CXCR3/CXCR4 co-expressing membranes by CXCL12 was only partial in comparison with homologous inhibition by CXCL10 (i.e. 70 ± 8% and 100% displacement, respectively), which corresponded to the anticipated number of CXCR3 and CXCR4 being associated as homo- and heteromers. Conversely, in membranes expressing CXCR4 alone (Figure 5C), [125I]-CXCL12 was displaced by unlabelled CXCL12, but not by the CXCR3 agonists CXCL10 or VUF10661. However, on co-expression of CXCR3 with CXCR4, both CXCL10 and VUF10661 inhibited [125I]-CXCL12 binding to CXCR4 (Figure 5D). In agreement with the expected presence of both CXCR3/CXCR4 homo- and heteromers, CXCL10 and VUF10661 could only partially displace [125I]-CXCL12 (i.e. 53 ± 6% and 75 ± 3% displacement, respectively). Although the lower CXCR3 expression levels in comparison with CXCR4 might explain why CXCL10 is less effective in inhibiting CXCL12 binding to CXCR3/CXCR4 membranes than the reverse, this explanation is not supported by the more effective displacement of [125I]-CXCL12 by VUF10661.
Figure 5.
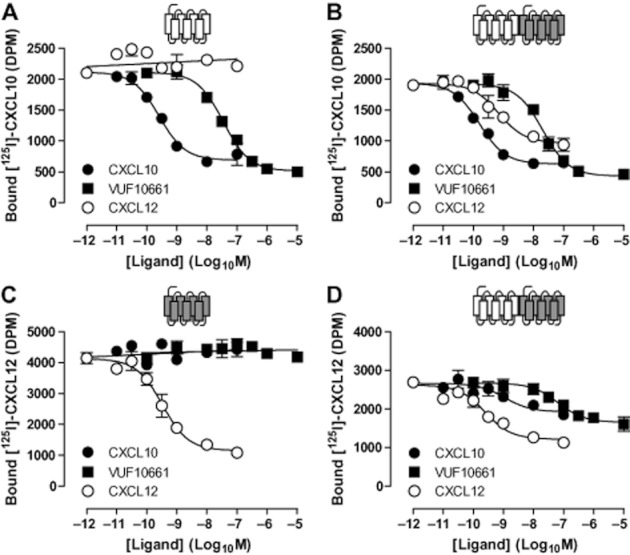
CXCR3 and CXCR4 heteromers display negative ligand binding cooperativity for endogenous and low molecular weight agonists. Membranes for [125I]-CXCL10 and [125I]-CXCL12 binding experiments were prepared from HEK293T cells transfected with 500 ng/106 cells CXCR3 DNA (A), 125 ng/106 cells CXCR4 DNA (C) or cotransfected with CXCR3 and CXCR4 DNA (B and D). Competition binding experiments were performed with approximately 50 pM of [125I]-CXCL10 (A and B) and [125I]-CXCL12 (C and D) and increasing concentrations of the CXCR3 chemokine CXCL10, small CXCR3 agonist VUF10661 and the CXCR4 chemokine CXCL12. Graphs shown are representative of three or more independent experiments performed in triplicate.
Table 2.
Binding parameters of ligands on membranes expressing CXCR3 and/or CXCR4
| Radioligand | Displacer | CXCR3 | CXCR4 | CXCR3 + CXCR4 |
|---|---|---|---|---|
| [125I]-CXCL10 | CXCL10 | 9.6 ± 0.1 | ND | 9.6 ± 0.1 |
| CXCL12 | ND | ND | 8.9 ± 0.1 | |
| VUF10661 | 7.5 ± 0.1 | ND | 7.3 ± 0.1 | |
| VUF10085 | 7.1 ± 0.1 | ND | 7.2 ± 0.1 | |
| TAK-779 | 5.7 ± 0.1 | ND | 5.7 ± 0.1 | |
| AMD3100 | ND | ND | ND | |
| Bmax (fmol·μg–1) | 0.20 ± 0.02 | ND | 0.26 ± 0.04 | |
| [125I]-CXCL12 | CXCL10 | ND | ND | 8.8 ± 0.2 |
| CXCL12 | ND | 9.5 ± 0.1 | 9.5 ± 0.2 | |
| VUF10661 | ND | ND | 6.9 ± 0.3 | |
| VUF10085 | ND | ND | ND | |
| TAK-779 | ND | ND | ND | |
| AMD3100 | ND | 6.8 ± 0.2 | 7.3 ± 0.1 | |
| Bmax (fmol·μg–1) | ND | 1.58 ± 0.15 | 1.08 ± 0.14 |
Binding parameters of the CXCR3 ligands CXCL10 (chemokine), VUF10661 (small agonist), TAK-779 and VUF10085 (antagonists) and the CXCR4 ligands CXCR4L12 (chemokine) and AMD3100 (antagonist) for their cognate receptors were determined in the absence and presence of CXCR4 and CXCR3, respectively. pIC50 values were determined using displacement of [125I]-CXCL10 and [125I]-CXCL12 from membrane preparations of HEK293T cells transfected with CXCR3, CXCR4 or both receptors. pIC50 values are given as averages ± SEM of two or more independent experiments performed in triplicate.
ND: value could not be determined.
The small-molecule CXCR3 antagonists VUF10085 (Johnson et al.,2007) and TAK-779 (Baba et al.,1999) inhibited [125I]-CXCL10 binding to membranes expressing CXCR3 alone (Figure 6A) or together with CXCR4 (Figure 6B). Calculated pIC50 values of both CXCR3 antagonists were not affected by the co-expression of CXCR4 (Table 2). The CXCR4 antagonist AMD3100 did not affect [125I]-CXCL10 binding to CXCR3-expressing membranes (Figure 6A). In contrast to the negative binding cooperativity between CXCL10 and CXCL12 at the CXCR3-CXCR4 heteromer, AMD3100 did not transinhibit [125I]-CXCL10 binding to membranes co-expressing CXCR3 and CXCR4 (Figure 6B). Similarly, [125I]-CXCL12 binding to CXCR4 in membranes expressing this receptor alone (Figure 6C) or in combination with CXCR3 (Figure 6D) was inhibited by AMD3100 with similar potencies (Table 2), whereas VUF10085 and TAK-779 could not inhibit [125I]-CXCL12 binding to either membrane preparation.
Figure 6.
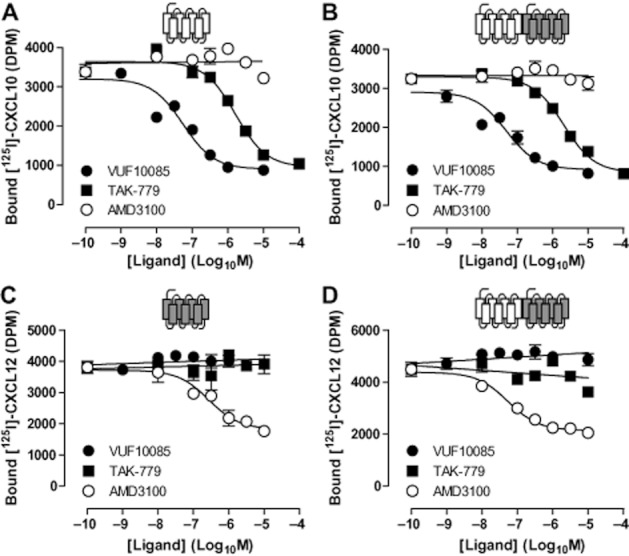
Low molecular weight antagonists of CXCR3 and CXCR4 do not have negative binding cooperativity with endogenous agonists. Membranes for [125I]-CXCL10 and [125I]-CXCL12 binding experiments were prepared from HEK293T cells transfected with 500 ng/106 cells CXCR3 DNA (A), 125 ng/106 cells CXCR4 DNA (C) or cotransfected with CXCR3 and CXCR4 DNA (B and D). Competition binding experiments were performed with approximately 50 pM of [125I]-CXCL10 (A and B) and [125I]-CXCL12 (C and D) and increasing concentrations of the CXCR3 chemokine antagonists VUF10085 and TAK-779 and the CXCR4 antagonist AMD3100. Graphs shown are representative of three or more independent experiments performed in triplicate.
CXCR3-CXCR4 heteromerization alters ligand-binding kinetics
Transinhibition of CXCL12 equilibrium binding to CXCR4 by both CXCL10 and VUF10661 suggests an allosteric mode of action between agonist-occupied CXCR3 and CXCR4, rather than steric hindrance between these agonists to bind receptor heteromers. To confirm allosteric interactions between the CXCR3 and CXCR4 ligand binding sites, [125I]-CXCL12 dissociation rates from CXCR4 and CXCR3-CXCR4 heteromers were determined in the absence or presence of CXCR3 agonists, using an infinite radioligand dilution approach (Christopoulos et al.,1997). To this end, membranes were pre-equilibrated with [125I]-CXCL12, and free [125I]-CXCL12 was removed by centrifugation. Dissociation kinetics of bound [125I]-CXCL12 was measured upon 100-fold dilution in binding buffer in the absence or presence of unlabelled chemokines. Dissociation of [125I]-CXCL12 from membranes expressing CXCR4 alone was significantly accelerated in the presence of 100 nM unlabelled CXCL12 as compared with the basal dissociation rate in binding buffer (Figure 7A and Table 3). This could be due to negative binding cooperativity within CXCR4 homomers, but could also be due to direct competition with [125I]-CXCL12 that prevents re-binding of dissociated [125I]-CXCL12. In agreement with equilibrium binding (Figure 5C), both CXCR3 agonists did not affect CXCL12 dissociation in the absence of CXCR3 (Figure 7A). Interestingly, CXCR3 co-expression accelerated basal CXCL12 dissociation from CXCR4, which was further accelerated by 100 nM CXCL10 or 10 μM VUF10661 (Figure 7B and Table 3). The very rapid dissociation of [125I]-CXCL10 from CXCR3 in the absence or presence of CXCR4 did not allow the measurement of accelerated effects of unlabelled ligands within this short time frame (data not shown).
Figure 7.
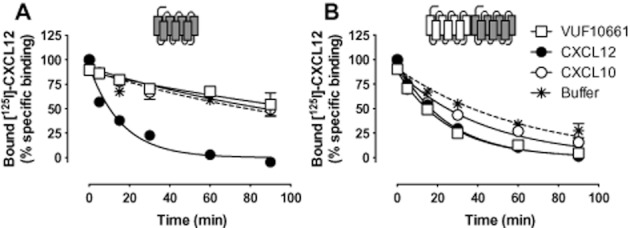
Heteromerization of CXCR3 and CXCR4 increases the dissociation rate of CXCL12. [125I]-CXCL12 dissociation half-life was determined in HEK293T membranes expressing CXCR4 (A) alone or (B) in combination with CXCR3, in the absence (asterisk with dotted line) and presence of the CXCR3 endogenous agonist CXCL10 (open circles), the small CXCR3 agonist VUF10661 (open squares), and the CXCR4 chemokine CXCL12 (closed circles). Representative graphs of three or more independent experiments performed in triplicate are shown.
Table 3.
[125I]-CXCL12 dissociation half-life from CXCR4 is decreased in the presence of CXCR3
| t1/2 ± SEM (min) | ||
|---|---|---|
| CXCR4 | CXCR3 + CXCR4 | |
| Buffer | 93 ± 15 | 56 ± 8 |
| 100 nM CXCL10 | 81 ± 9 | 32 ± 8# |
| 100 nM CXCL12 | 12 ± 1* | 24 ± 6* |
| 100 nM VUF10661 | 83 ± 25 | 16 ± 3* |
Results shown are average ± SEM of three or more independent experiments performed in triplicate.
t1/2 differs significantly from t1/2 in presence of vehicle (P < 0.05).
t1/2 differs significantly from t1/2 in membranes expressing CXCR4 alone (P < 0.05).
Negative binding cooperativity between CXCR3 and CXCR4 chemokines is not apparent in intact cells
High-affinity agonist binding to chemokine receptors requires the coupling of G proteins (De Lean et al.,1980; Springael et al.,2006), as revealed by decreased membrane binding of CXCL12 to CXCR4 and CXCL10 to CXCR3 upon G protein uncoupling using GTPγS and pertussis toxin, respectively (Cox et al.,2001; Nijmeijer et al.,2010). Hence, negative cooperativity observed in equilibrium binding experiments on isolated membrane fractions between two agonists that interact with different receptors, may be the consequence of G protein scavenging, if these receptors couple to the same and limited pool of G proteins (Chabre et al.,2009; Birdsall,2010). In contrast, to our observations on isolated membranes, CXCL10 and CXCL12 do not affect each other's equilibrium binding on intact cells co-expressing CXCR3 and CXCR4 at comparable levels (Figure 8).
Figure 8.
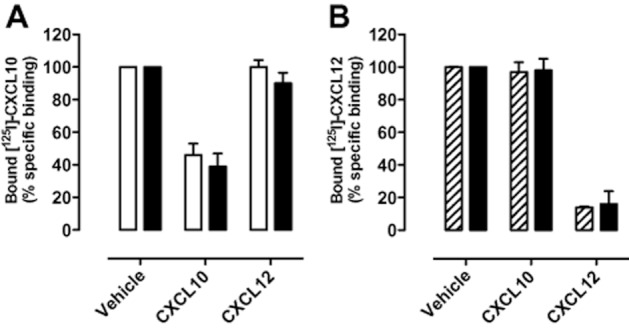
Chemokines display no negative binding cooperativity on intact cells. Binding of approximately 50 pM [125I]-CXCL10 (A) or [125I]-CXCL12 (B), in the absence or presence of 100 nM unlabelled CXCL10 or CXCL12, was measured on intact HEK293T cells transiently transfected with 500 ng CXCR3 DNA/106 cells (open bars), 125 ng CXCR4 DNA/106 cells (hatched bars), or cotransfected with CXCR3 and CXCR4 DNA (closed bars). Graphs show mean ± SEM of two or more independent experiments performed in triplicate.
Discussion and conclusions
The chemokine receptors CXCR3 and CXCR4 are implicated in the pathogenesis of autoimmune disease and various cancers, and are gaining recognition as attractive targets for therapeutic intervention (Vandercappellen et al.,2008; Viola and Luster,2008; Wijtmans et al.,2008). Interestingly, chemokines and small antagonists that specifically interact with either CCR2 or CCR5 inhibited CXCL12-induced CXCR4 activity, and vice versa, provided that these chemokine receptors are co-expressed (Sohy et al.,2007; 2009). This cross-inhibition is observed in both recombinant and native cells, and is the consequence of negative binding cooperativity between chemokine receptors that are situated in heteromeric complexes. Because CXCR3 and CXCR4 are co-expressed on various immune and cancer cell types, we investigated whether these chemokine receptors heteromerize and display negative ligand-binding cooperativity. In the present study, we demonstrated that CXCR3 and CXCR4 form heteromers using the following five different technologies: co-immunoprecipitation, trFRET, FRET, saturation BRET and/or GPCR-HIT. The detection of CXCR4 homomers is in agreement with previous studies (Issafras et al.,2002; Babcock et al.,2003; Percherancier et al.,2005; Sohy et al.,2007; 2009; Hamatake et al.,2009; Luker et al.,2009). On the other hand, the detection of CXCR3 homomers and CXCR3-CXCR4 heteromers is in conflict with the conclusions of a recent study in which homo- and heteromerization of CXCR3 and CXCR4 was evaluated using BRET analyses (Hamatake et al.,2009). In that study, various combinations of chemokine receptors fused to either Rluc or GFP were co-expressed in cells at equal luminescence and fluorescence levels, respectively. High BRET was observed between CXCR4-Rluc and CXCR4-GFP, whereas the significantly lower BRET levels between CXCR3/CXCR3, CXCR3/CXCR4, CXCR2/CXCR2, CXCR2/CXCR4 and CCR5/CXCR4 were considered to be marginal. On the basis of these results, Hamatake et al. (Hamatake et al.,2009) concluded that only CXCR4 is situated in higher-order complexes. Indeed, we and others observed higher BRET levels for CXCR4 homomers as compared with CXCR4 heteromers and homomers between CCR2 (Percherancier et al.,2005), CCR5 (Sohy et al.,2009) and CXCR3 in saturation BRET experiments. However, BRET levels per se cannot be used as a quantitative measure of the relative number of complexes being formed between different receptor combinations, as BRET is not only determined by numbers of complexes, but also heavily depends on the close proximity and relative orientation between the BRET donor (i.e. Rluc) and acceptor (i.e. GFP or EYFP) proteins within each of the different receptor complexes. On the other hand, the relative propensity of receptors to form complexes can be extracted from saturation BRET analysis as the BRET50 value, which corresponds to the BRET acceptor/donor ratio of receptor fusion proteins resulting in 50% of the saturated BRET signal (Mercier et al.,2002). The BRET50 values were comparable for CXCR3–CXCR3, CXCR3–CXCR4 and CXCR4–CXCR4 complexes in our experiments, suggesting that CXCR3 and CXCR4 have comparable probability to form homo- and heteromers. Likewise, comparable BRET50 values have been observed for homo- and heteromers between chemokine receptors CCR2 and CCR5 (El-Asmar et al.,2005; Springael et al.,2006), CCR2 and CXCR4 (Percherancier et al.,2005; Sohy et al.,2007), CCR5 and CXCR4 (Contento et al.,2008; Sohy et al.,2009), CXCR1 and CXCR2 (Wilson et al.,2005), and CXCR4 and CXCR7 (Levoye et al.,2009). Importantly, chemokine receptor homo- and heteromerization were confirmed in these studies by other biophysical, biochemical and/or pharmacological evidence.
Negative binding cooperativity has been described within heteromers of CCR2, CCR5 or CXCR4 (El-Asmar et al.,2005; Springael et al.,2006; Sohy et al.,2007; 2009). In this study, we showed that negative binding cooperativity also occurs within CXCR3–CXCR4 heteromers, as binding of the chemokines CXCL10 and CXCL12 is mutually exclusive on membranes co-expressing these receptors when using trace concentrations of radiolabelled chemokine (100 pM). Moreover, inhibition of [125I]-CXCL12 binding to CXCR4 by the small CXCR3 agonist VUF10661 suggests that the observed negative binding cooperativity is not due to steric hindrance, but rather results from agonist-induced conformational changes transmitted from one receptor to the other within CXCR3–CXCR4 heteromers. Such transmission of conformational changes across receptor pairs has been directly shown for the norepinephrine-occupied α2A-adrenergic receptor upon morphine binding to the associated μ-opioid receptor within the heteromer using intramolecular FRET analysis (Vilardaga et al.,2008). Hence, agonist binding to CXCR3 constrains CXCR4 to a conformation with lower affinity for CXCL12 and vice versa. Negative binding cooperativity within CXCR3–CXCR4 heteromers results in partial reduction in radiolabelled chemokine binding in equilibrium binding experiments, which reflects the proportion of CXCR3-CXCR4 heteromers relative to homomers of both receptor types. This is in agreement with the comparable propensities of CXCR3 and CXCR4 to form homo- and heteromers as observed in our saturation BRET analyses. Interestingly, negative binding cooperativity within CXCR3–CXCR4 heteromers is limited to agonists, which contrasts with previous observations on CCR2–CXCR4 and CCR5–CXCR4 heteromers, in which small antagonists cross-inhibited both chemokine binding and chemokine-induced in vitro and in vivo activity (Sohy et al.,2007; 2009). This discrepancy may be related to distinct interfaces between various chemokine receptor pairs, resulting in different efficiencies with which a conformational change in one protomer is conveyed to the other. However, antagonist binding to the μ-opioid receptor did not induce conformational changes across heteromeric receptor pairs as revealed by an unaltered intramolecular FRET signal in the α2A-adrenergic receptor (Vilardaga et al.,2008), which is in line with our observations.
The validity of negative binding cooperativity detected in membrane preparations expressing two GPCRs has recently been questioned (Chabre et al.,2009). Based on a catalytic model, Chabre et al. (Chabre et al.,2009) argued that G protein coupling to an agonist-occupied receptor is almost irreversible in equilibrium binding assays on membrane preparations, as free GTP is not available to occupy the empty nucleotide binding pocket in the G protein upon GDP release in this experimental setup. Because the pool of shared G proteins might be smaller than the total number of cognate receptors, this agonist-induced or constitutive G protein scavenging by one of the receptor subtypes results in a permanent depletion of G-proteins from other receptors (Nijmeijer et al.,2010). As a consequence, the latter GPCRs often display low agonist binding affinity, which is observed as apparent negative binding cooperativity between two agonists of co-expressed GPCRs, but in fact does not necessarily require receptor heteromerization. In this respect, the absence of negative binding cooperativity between antagonists and chemokines on CXCR3/CXCR4 membrane preparations might also be explained as further support for the ‘G protein scavenging’ model. Sohy et al. (Sohy et al.,2009) confirmed negative binding cooperativity between CCR2, CCR5 and CXCR4 chemokine receptor heteromers in both recombinant and native intact cells in which GTP is readily available in the cytoplasm. In contrast, CXCL10 and CXCL12 did not affect each other's binding to intact HEK293T cells that co-express CXCR3 and CXCR4. Indeed, negative binding cooperativity on membrane preparations may involve heteromeric CXCR3-CXCR4 complexes present in intracellular compartments that are not accessible in binding assays on intact cells. However, the presence of CXCR3-CXCR4 heteromers on the cell surface as detected by (tr)FRET and GPCR-HIT assays is not in support of this hypothesis. Furthermore it could be argued that the results of the GPCR-HIT studies, again in live cells, are also more consistent with a lack of negative cooperativity as we observe greater BRET signals with dual agonist treatment, although we have previously suggested that these findings could be reconciled because the significant conformational changes believed to result in negative binding cooperativity may also influence the ability of the heteromer to recruit β-arrestin2 (See et al.,2011). The contradiction between intact cells and isolated membranes indeed suggests G protein scavenging in the latter format as proposed by Chabre et al. (Chabre et al.,2009). Yet, this ‘G protein scavenging’ model cannot explain the increased dissociation rate of CXCL12 from membranes co-expressing CXCR3 and CXCR4 in the presence of CXCR3 agonists. This decreased affinity of CXCR4 for bound CXCL12 can only be explained by allosteric interactions between CXCR4 and CXCR3 upon binding of agonists to the latter, as initial binding of radiolabelled CXCL12 takes place in the absence of these CXCR3 agonists, and is consequently not hampered by G-protein availability. To distinguish direct allosteric interactions between these receptors from downstream crosstalk events such as G protein scavenging (Vischer et al.,2011), intramolecular FRET approaches might be applied on CXCR3 and CXCR4 in the future (Vilardaga et al.,2008). At present, however, we cannot explain the observed discrepancy in CXCR3/CXCR4 binding cooperativity.
In summary, we show that CXCR3 and CXCR4 form heteromers at the cell surface using multiple experimental approaches. CXCR3 co-expression increased the dissociation of CXCL12 from CXCR4 membranes, which is further accelerated by CXCR3 agonists. Although specific CXCR3 and CXCR4 agonists inhibit each other's equilibrium binding on isolated membranes as well, this apparent negative binding cooperativity was not observed on intact cells.
Acknowledgments
This work was supported by the Top Institute Pharma (project number D1-105: the GPCR forum). KDGP is an Australian Research Council (ARC) Future Fellow (FT100100271) and WCJ is supported by ARC Project Grant DP120101297. We thank Dr. S. Jähnichen and Razia Alimahomed (Leiden/Amsterdam Center for Drug Research, Division of Medicinal Chemistry, Faculty of Science, VU University Amsterdam) for technical assistance.
Glossary
- CFP
cyan fluorescent protein
- CXCL10
C-X-C chemokine ligand 10
- CXCL11
C-X-C chemokine ligand 11
- CXCL12
C-X-C chemokine ligand 12
- CXCL9
C-X-C chemokine ligand 9
- CXCR3
C-X-C chemokine receptor 3
- CXCR4
C-X-C chemokine chemokine receptor 4
- eBRET
extended BRET
- EYFP
enhanced yellow fluorescent protein
- FCS
fetal calf serum
- GABAB2
GABA-B receptor 2
- HA
haemagglutinin epitope tag
- HIT
heteromer identification technology
- IP
immunoprecipitation
- Rluc
Renilla luciferase
- RT
room temperature
- trFRET
time-resolved FRET
Conflicts of interest
In addition to being Head of the Laboratory for Molecular Endocrinology–GPCRs, Western Australian Institute for Medical Research and Centre for Medical Research, University of Western Australia, K.D.G.P. is Chief Scientific Officer of Dimerix Bioscience Pty Ltd, a spin-out company of the University of Western Australia that has been assigned the rights to the ‘GPCR-HIT’ technology. K.D.G.P. has a minor shareholding in Dimerix.
References
- Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 1999;96:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. 2003;278:3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- Birdsall NJ. Class A GPCR heterodimers: evidence from binding studies. Trends Pharmacol Sci. 2010;31:499–508. doi: 10.1016/j.tips.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Chabre M, Deterre P, Antonny B. The apparent cooperativity of some GPCRs does not necessarily imply dimerization. Trends Pharmacol Sci. 2009;30:182–187. doi: 10.1016/j.tips.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Lanzafame A, Ziegler A, Mitchelson F. Kinetic studies of co-operativity at atrial muscarinic M2 receptors with an ‘infinite dilution’ procedure. Biochem Pharmacol. 1997;53:795–800. doi: 10.1016/s0006-2952(96)00814-3. [DOI] [PubMed] [Google Scholar]
- Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, et al. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci U S A. 2008;105:10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MA, Jenh CH, Gonsiorek W, Fine J, Narula SK, Zavodny PJ, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylyl cyclase-coupled b-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bunemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat Methods. 2009;6:225–230. doi: 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
- El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67:460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- Fulton AM. The chemokine receptors CXCR4 and CXCR3 in cancer. Curr Oncol Rep. 2009;11:125–131. doi: 10.1007/s11912-009-0019-1. [DOI] [PubMed] [Google Scholar]
- Hamatake M, Aoki T, Futahashi Y, Urano E, Yamamoto N, Komano J. Ligand-independent higher-order multimerization of CXCR4, a G-protein-coupled chemokine receptor involved in targeted metastasis. Cancer Sci. 2009;100:95–102. doi: 10.1111/j.1349-7006.2008.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci U S A. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issafras H, Angers S, Bulenger S, Blanpain C, Parmentier M, Labbe-Jullie C, et al. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J Biol Chem. 2002;277:34666–34673. doi: 10.1074/jbc.M202386200. [DOI] [PubMed] [Google Scholar]
- Jahnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci U S A. 2010;107:20565–20570. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalink K, Van Rheenen J. FilterFRET: quantitative imaging of sensitized emission. In: Gadella TW Jr, editor. Fret and Flim Techniques. Vol. 33. Burlington, MA: Academic Press; 2009. pp. 289–349. [Google Scholar]
- Johnson M, Li AR, Liu J, Fu Z, Zhu L, Miao S, et al. Discovery and optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett. 2007;17:3339–3343. doi: 10.1016/j.bmcl.2007.03.106. [DOI] [PubMed] [Google Scholar]
- Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- Luker KE, Gupta M, Luker GD. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23:823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews JL, Fulton BS, Negus SS, Neumeyer JL, Bidlack JM. In vivo characterization of (–)(–)MCL-144 and (+)(–)MCL-193: isomeric, bivalent ligands with mu/kappa agonist properties. Neurochem Res. 2008;33:2142–2150. doi: 10.1007/s11064-008-9752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- Moser B, Loetscher P. Lymphocyte traffick control by chemokines. Nat Immunol. 2001;2:123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- Mustafa S, Pfleger KD. G protein-coupled receptor heteromer identification technology: identification and profiling of GPCR heteromers. J Lab Autom. 2011;16:285–291. doi: 10.1016/j.jala.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Mustafa S, See HB, Seeber RM, Armstrong SP, White CW, Ventura S, et al. Identification and profiling of novel alpha1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J Biol Chem. 2012;287:12952–12965. doi: 10.1074/jbc.M111.322834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijmeijer S, Leurs R, Smit MJ, Vischer HF. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J Biol Chem. 2010;285:29632–29641. doi: 10.1074/jbc.M110.115618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Dromey JR, Dalrymple MB, Lim EM, Thomas WG, Eidne KA. Extended bioluminescence resonance energy transfer (eBRET) for monitoring prolonged protein-protein interactions in live cells. Cell Signal. 2006a;18:1664–1670. doi: 10.1016/j.cellsig.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein–protein interactions. Nat Protoc. 2006b;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Langeslag M, Jalink K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86:2517–2529. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Chazot PL, Shenton FC, Sansuk K, Bakker RA, Leurs R. Oligomerization of recombinant and endogenously expressed human histamine H(4) receptors. Mol Pharmacol. 2006;70:604–615. doi: 10.1124/mol.105.020818. [DOI] [PubMed] [Google Scholar]
- Scholten D, Canals M, Maussang D, Roumen L, Smit M, Wijtmans M, et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol. 2012a;165:1617–1643. doi: 10.1111/j.1476-5381.2011.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten D, Canals M, Wijtmans M, de Munnik S, Nguyen P, Verzijl D, et al. Pharmacological characterisation of a small-molecule agonist for the chemokine receptor CXCR3. Br J Pharmacol. 2012b;166:898–911. doi: 10.1111/j.1476-5381.2011.01648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See HB, Seeber RM, Kocan M, Eidne KA, Pfleger KD. Application of G protein-coupled receptor-heteromer identification technology to monitor beta-arrestin recruitment to G protein-coupled receptor heteromers. Assay Drug Dev Technol. 2011;9:21–30. doi: 10.1089/adt.2010.0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohy D, Parmentier M, Springael JY. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. 2007;282:30062–30069. doi: 10.1074/jbc.M705302200. [DOI] [PubMed] [Google Scholar]
- Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of ‘selective’ antagonists. J Biol Chem. 2009;284:31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Stroke IL, Cole AG, Simhadri S, Brescia MR, Desai M, Zhang JJ, et al. Identification of CXCR3 receptor agonists in combinatorial small-molecule libraries. Biochem Biophys Res Commun. 2006;349:221–228. doi: 10.1016/j.bbrc.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J Am Chem Soc. 2010;132:15899–15901. doi: 10.1021/ja107447w. [DOI] [PubMed] [Google Scholar]
- Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- Verzijl D, Storelli S, Scholten DJ, Bosch L, Reinhart TA, Streblow DN, et al. Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J Pharmacol Exp Ther. 2008;325:544–555. doi: 10.1124/jpet.107.134783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol. 2008;4:126–131. doi: 10.1038/nchembio.64. [DOI] [PubMed] [Google Scholar]
- Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- Vischer HF, Nijmeijer S, Smit MJ, Leurs R. Viral hijacking of human receptors through heterodimerization. Biochem Biophys Res Commun. 2008;377:93–97. doi: 10.1016/j.bbrc.2008.09.082. [DOI] [PubMed] [Google Scholar]
- Vischer HF, Watts AO, Nijmeijer S, Leurs R. G protein-coupled receptors: walking hand-in-hand, talking hand-in-hand? Br J Pharmacol. 2011;163:246–260. doi: 10.1111/j.1476-5381.2011.01229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, et al. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci U S A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijtmans M, Verzijl D, Leurs R, de Esch IJ, Smit MJ. Towards small-molecule CXCR3 ligands with clinical potential. ChemMedChem. 2008;3:861–872. doi: 10.1002/cmdc.200700365. [DOI] [PubMed] [Google Scholar]
- Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem. 2005;280:28663–28674. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]