

Summary
Over the life span of a T lymphocyte, from thymic development to death, it is subjected to a variety of stresses and stimuli. Upon receipt of each stress or stimulus, a potentially life-changing fate decision must be made, namely, whether to commit to a form of programmed cell death or to make the necessary adaptations to effectively deal with the changing environment. In our laboratory, we have identified several stresses that a T lymphocyte will encounter during a normal life span. Our studies have focused on how T cells utilize autophagy to get a grasp on the situation, or in cases in which survival is untenable, how T cells use autophagy to hasten their demise. This review focuses on the functions of T-cell autophagy in maintaining homeostasis, eliminating excess or dangerous levels of mitochondria, trimming levels of endoplasmic reticulum, and promoting a healthy metabolic level to allow cells to perform as productive components of the immune system. Additionally, the use of autophagy signaling molecules to perform autophagy-independent tasks involved in the maintenance of immune homeostasis will be discussed.
Keywords: autophagy, metabolism, homeostasis, lymphocyte signaling
Introduction
Autophagy, Greek for ‘self-eating’, is an intracellular catabolic process that involves the generation of a double-membraned structure, sequestration of various cytoplasmic components, and delivery of bound cellular matter to lysosomal compartments for degradation (1, 2). This crucial process is conserved in all eukaryotic cell types, from S. cerevisiae to H. sapiens, and has been implicated in cellular survival during periods of starvation or cytokine withdrawal, removal of extraneous or damaged organelles, turnover of long-lived proteins, and responses to an almost limitless myriad of cellular or cytotoxic stresses (3, 4). Autophagy can be divided into three main classes based on the volume of enclosed membrane, the source of the delimiting membrane, and the nature of the substrates to be catabolized. Macroautophagy, the heretofore best understood and most thoroughly characterized class of autophagy, involves the de novo synthesis of autophagosomal membranes around bulk substrates to be degraded and the stepwise fusion of these autophagosomes with degradative vesicles to form autolysosomes. Selective autophagy, a subclass of macroautophagy, involves the binding of substrates to adapter molecules for their inclusion into the nascent autophagosome (5, 6). Selective autophagy shares some characteristics with both macroautophagy and chaperone-mediated autophagy (described below). The less thoroughly studied process of microautophagy involves the small-scale blebbing and sequestration of soluble components within the immediate microenvironment of previously formed autophagosomes generated during macroautophagy (7). Chaperone-mediated autophagy (CMA) involves the import of misfolded proteins, aggregates, ubiquitinated proteins, or even organelles into pre-formed autophagosomes via adapters and receptors on the autophagosome (8).
Whereas macroautophagy is generally considered to be nonspecific, CMA is sensitive to many signaling pathways, and particular substrates can be fine-tuned by a multitude of E3 ubiquitin ligases or similar classes of adapter molecules, providing several layers of specificity. CMA is now considered to be of particular interest in the turnover of damaged organelles and the clearance of cytotoxic aggregates implicated in several human diseases, such as Parkinson's and Huntington's diseases (9, 10). In all three forms of autophagy, many families of autolysosome-localized transporters that are often specific for one class of macromolecule can convey nutrients and other recycled macromolecules back to the cytoplasm after lysosomal processing (1).
At this point in time, at least 35 autophagy gene (Atg) products that contribute to the autophagic process have been identified in yeast (1), and the number of adapter and ancillary proteins lending degrees of specificity to CMA or selective autophagy is expanding. The core autophagy machinery utilizes the coordinated activity of the class III PI3K Vps34, which produces PI(3)P, and Beclin-1, which recruits a myriad of effectors with FYVE- or GLUE-domains, to flex and elongate membranes, such as Bif, UVRAG, and Ambra (11). Although many of its direct targets remain unknown, the serine/threonine kinase Ulk1 (Atg1l), which is dependent on mammalian target of rapamycin (mTOR) inhibition by tuberous sclerosis protein 1 and 2 (TSC1/2) under periods of cytokine or nutrient starvation, is essential for autophagy induction (12, 13). The Ulk1 and PI3K class III complexes are brought together in active forms by the action of ras-related protein B (RalB) on the nascent autophagosomal membrane during nutrient starvation (14). Furthermore, elongation of the limiting membrane requires two enzymatic cascades of ubiquitin-like conjugation systems, termed such as they each involve the coordinated action of E1, E2, and E3 ligases but have no sequence homology to ubiquitin-conjugation proteins. Atg7, similar to an E1 ubiquitin priming protein, catalyzes the priming and transfer of Atg12 to Atg10 and finally to Atg5 and Atg16. This complex lines the cytoplasmic leaflet of the isolation membrane, and although the function of this process remains poorly understood, detection of Atg5-Atg12 conjugation is used to validate autophagy induction. One clue that suggests an important facet of this process is that the protein Atg16L has been implicated as a major genetic contributor to Crohn's disease in humans (15). The second cascade involves the cleavage, lipidation, and recycling of Atg8/LC3, which are accomplished through Atg4-mediated priming, Atg7-mediated E1 activity, and Atg3-mediated E2 activity. The function of this process may be related to the import of ubiquitinated aggregates or other chaperone-mediated autophagic substrates into the autophagosome (16). Detection of LC3 priming and conversion is one of the most trusted methods of validating autophagy induction (17).
While all eukaryotic cells studied thus far seem to have some form of autophagic capacity, the stresses and signals that induce autophagy as well as the basal levels of autophagy observed in resting cells vary widely in different cell types. For example, thymic epithelial cells have a high basal level of constitutive autophagy (18). Mature naive T lymphocytes display a very low, but still detectable, level of constitutive autophagy and can upregulate autophagy in response to several stimuli, most prominently T-cell receptor (TCR) or G-protein-coupled receptor (GPCR) activation in the case of human immunodeficiency virus (HIV) binding to C-X-C chemokine receptor type 4 (CXCR4) (19-22). Generally speaking, starvation of either serum or nutrients induces a moderate level of autophagy in most cell types, whereas stimulation of several immunity-related receptors, such as the TCR or Toll-like receptors (TLRs), conveys potent autophagy induction signals. Although the precise function of autophagy varies in specialized immune cells (23), in all cell types studied, autophagy provides an alternative source of nutrients scavenged from the cytoplasm, an alternative form of cellular degradation that augments the proteasomal system, and a major pathway for intracellular remodeling to reflect changes in the developmental or metabolic requirements of the cell (2). For example, T cells, which are highly glycolytic and become even more dependent upon this inefficient process when activated, must rapidly adjust to increased energy requirements during activation and proliferation (24).
Autophagy also has grim functions, as it has been implicated as an alternative form of programmed cell death (25-27). The Lenardo group (27) has previously shown that when caspase activity is inhibited, an alternative form of programmed cell death is initiated, resembling autophagy induction, and is dependent upon Atg7 and Beclin-1. When stimulated with inducers of the intrinsic death pathway, mouse embryonic fibroblasts (MEFs) doubly deficient in the pro-apoptotic B-cell lymphoma 2 (Bcl-2) family members Bcl-2-associated X protein (Bax) and B-cell lymphoma-extra large (Bak) underwent a form of programmed cell death characterized by the presence of a large number of autophagic vacuoles. More importantly, this death was inhibited by treatment with 3-methyladenine or short interfering RNA (siRNA)-mediated knockdown of Atg5 or Beclin-1 (26). Moreover, the use of the pan-caspase inhibitor zVAD to inhibit caspase function likewise induced autophagic cell death that was inhibited by siRNA-mediated knockdown of Atg7 and Beclin-1 (27). However, both of these studies relied on the inhibition of classical apoptosis, which is normally insignificant under physiological conditions, with the notable exception being in oncogenesis. A more clinically relevant form of autophagic cell death occurs in T cells exposed to but not infected with HIV. The HIV glycoprotein gp120 binds to CXCR4 on T lymphocytes and induces autophagy on a grand scale. T cells undergo apoptosis in response to this stimulation, and silencing of Beclin-1 or Atg7 restores T-cell viability (21, 28).
In this review, we focus on the role of autophagy in the development and homeostasis of T and B lymphocytes, citing evidence for the requirements for autophagy in the degradation of specific substrates and organelles. Another focus is on the metabolic requirements of T cells and how autophagy contributes to fulfilling them. Finally, we discuss the roles of autophagic signaling molecules, including Vps34, Beclin-1, TSC-1, and mTOR, and how many of these molecules function differently in lymphocytes than in other cell types.
Autophagy induction in T and B lymphocytes
Though small in size (10-20 μM) and replete for cytoplasmic volume, lymphocytes are the essential mediators of the adaptive immune system. Thus, it was somewhat surprising to note that autophagy genes including Vps34, Atg3, Atg5, Atg7, Beclin-1, LC3, and p62 were expressed in both T and B lymphocytes (19, 20, 29). Both fluorescence microscopy of LC3 and transmission electron microscopy (TEM) of highly characteristic double-membraned structures have been utilized to visualize autophagic structures in T lymphocytes, not only after starvation, stimulation with certain cytokines (30), or TCR and BCR engagement (20, 22, 31), but also at basal levels in naive, antigen-inexperienced cells (19, 22, 32). Perhaps autophagy is crucial in these cells due to space limitations, which require elegant organelle organization and a tightly regulated metabolic environment. Additionally, maintaining open lanes of shipping for proper vesicular trafficking and organelle identity in limited cytoplasmic straits are now considered an indispensable role for autophagy (33).
As previously mentioned, autophagosomes can be detected within freshly harvested primary murine T and B lymphocytes. While both the number and size of autophagosomes increase upon withdrawal of either serum or amino acids, this increase is almost wholly dependent upon the phosphatidylinositol 3′-kinase (PI3K) pathway, as general PI3K inhibitors, such as 3-MA and wortmannin, abate this increase, suggesting that the starvation-mediated induction of autophagy is regulated by mTOR and PI3K activity in lymphocytes (19, 32). In vitro stimulation of the TCR for 24-48 h further enhances autophagy, as measured both by detection of the processing of LC3 from an unconjugated form (LC3 I) to a lipidated form (LC3 II) and visualization of LC3 II punctae formation by fluorescence microscopy (19, 20). Many of the findings related to autophagy induction in murine T cells hold true for human T cells as well. For example, TEM examination of purified human CD8+ peripheral blood mononuclear cells (PBMCs) showed that chronic restimulation with a mitogen results in a large (3-4-fold) induction of autophagy after long-term culture (34). This TCR activation-induced autophagy is also responsive to general PI3K inhibitors, such as 3-MA, as well as c-Jun N-terminal kinase (JNK) inhibitors, and can be rapidly enhanced by administration of rapamycin (affecting mTOR) and zVAD (affecting caspase activity), suggesting a role for both of these pathways, as well as PI3K activity and mitogen activated protein kinase (MAPK) pathways (34). The involvement of this diverse array of pro-survival pathways in T-cell autophagy should come as no surprise, as autophagy plays an overwhelmingly cytoprotective role in T cells. Additionally, T-cell activation requires extensive cytoplasmic reorganization and mobilization of energy resources to effect a functional immune response (35).
One major contradiction to the pro-survival paradigm, however, is that autophagy is highly induced by the binding of the HIV envelope glycoprotein gp120 to CXCR4 on the surface of uninfected bystander CD4+ T cells (21). This autophagy is induced on such a massive scale that it has been implicated in the type II, caspase-independent programmed cell death (PCD type II) that occurs when apoptosis is inhibited in these bystander cells. Of note, silencing of the autophagic genes Atg7 or Beclin-1 or inhibition of autophagy induction or autophagosome-lysosome fusion by 3-MA or bafilomycin, respectively, completely inhibited CXCR4 engagement-mediated apoptosis and autophagy (21). The ability of CXCR4 engagement to induce autophagy strongly suggests a role for GPCRs in the induction of autophagy; more specifically, chemokine receptors may serve to modulate the function of lymphocytes entering specific microenvironments by fine-tuning their autophagy levels. The precise role of endogenous chemokine receptor ligands in inducing T-lymphocyte autophagy remains unknown, but we speculate that chemokine receptor ligation acts as both a pro-survival signal and a mechanism of fine-tuning metabolism as T cells enter differing tissue environments.
Autophagy induction in B lymphocytes shares many characteristics with T lymphocyte autophagy induction. Autophagy can be induced in B cells through prolonged BCR stimulation through surface-bound IgM, leading to apoptosis (31). This autophagy was inhibited by CD40 engagement, as was apoptosis. The CD40-mediated inhibition of both autophagy and apoptosis in this context may be mediated by the induction of Bcl-2, as Bcl-2 has been shown to bind to both Bax and Beclin-1 (36). JNK-mediated signals prevent Bcl-2 from binding Beclin-1, allowing Beclin-1 to associate with Vps34 and induce autophagy. However, if apoptotic signals are not inhibited by autophagic induction, further JNK signaling can dissociate Bcl-2 from Bax, leading to apoptosis (37-40). BCR-mediated autophagy is also inhibited by wortmannin, once again suggesting roles for PI3K in autophagy induction in B lymphocytes.
Autophagy in lymphocyte development
Autophagy is also important for the development of B cells. Studies utilizing a GFP-Beclin-1 fusion protein demonstrated that Beclin-1 was expressed at very high levels in pro-B cells, but was downregulated through the pre-B transition (41). Interestingly, these findings are analogous to the observation of high levels of Atg5, Beclin-1, and LC3 in double negative (DN) thymocytes but not double positive (DP) thymocytes. These changes in the expression of autophagic mediators may make DP thymocytes and pre-B cells even more dependent upon lymphocyte receptor-mediated pro-survival signaling by removing the layer of cytotoxic protection provided by autophagy (20). These findings correlate well with the observation that Atg5 is dispensable for later B-cell development, as the only phenotype observed in mice in which Atg5 was deleted under the control of a CD19-cre transgene was a marked decrease in self-renewing B1 cells (42). However, in an Atg5-null fetal liver chimera system, the numbers of pre-B cells and immature B cells were dramatically reduced, providing evidence that autophagy-sufficient lymphoid precursors are necessary to fill niches and develop into the corresponding B-lineage cells (20). Together, these findings suggest a stage-specific requirement for autophagy in B cells that affects the development and survival of self-renewing populations but not mature, circulating B2 cells.
Even more recent evidence has confirmed that hematopoietic lymphocyte precursors are more dependent upon autophagy, not only for intracellular homeostasis and proper organelle quality control but also to maintain a steady population not susceptible to leukemic transformations. When Atg7 was deleted in hematopoietic stem cells (HSCs), a rapid, myeloproliferative disease developed, and yet paradoxically, Atg7-deficient HSCs were insufficient to reconstitute an irradiated mouse or to support either lymphoid or myeloid development (43). Therefore, autophagy is essential to maintain a healthy self-renewing population of cells capable of populating the T and B-lymphocyte compartments.
Autophagy in T-cell homeostasis and death
Although the stage-specific requirements for autophagy in the earliest stages of hematopoietic cell development seem to be acute, autophagy also plays a more generalized role in T-cell homeostasis and long-term survival. Autophagy is a crucial cell survival strategy in mature peripheral naive T cells. It is essential that T cells are able to adjust to long and indefinite periods of starvation, as they must adjust to the differing microenvironments of secondary lymphoid organs, blood, and lymphatic ducts. T lymphocytes are acutely sensitive to apoptosis, and the levels of anti-apoptotic Bcl-2 family members, such as Bcl-2 and induced myeloid leukemia cell differentiation protein (Mcl-1) are important for the survival of DN and SP thymocytes as well as mature naive T cells (44). Additionally, Bcl-X is important for the survival of DP thymocytes and activated mature T cells. The expression of these proteins is highly regulated through cytokine signaling during development (45-47). Therefore, it is not surprising that cytokine signals, which are critical for the maintenance of lymphocyte homeostasis, play substantial roles in homeostatic autophagy.
In the absence of either of the core autophagy proteins Atg5 (20, 48) or Atg7 (29), thymocyte development was essentially normal, with slight but reproducible reductions in thymocyte numbers and no evident increase in apoptosis ex vivo. In contrast, mice with autophagy-deficient T cells displayed dramatic reductions in peripheral T cells. Decreases of greater than 50% were often observed in mature CD4+ and CD8+ T-lymphocyte numbers. Moreover, the remaining mature T cells rapidly succumbed to apoptosis ex vivo, even in the presence of high levels of pro-survival homeostatic cytokines, and failed to efficiently proliferate in response to TCR stimulation (29). Therefore, autophagy provides one or more non-redundant contributions to the survival of naive T lymphocytes beyond the homeostatic cytokine signaling and constant TCR-to-major histocompatibility complex (MHC) contact that are required for T-cell survival. Notably, the addition of the metabolic substrate methyl pyruvate, which can feed directly into the glycolytic pathway, had no significant effect on the survival of mature autophagy-deficient T lymphocytes (Heather Pua and You-Wen He, unpublished observations). In contrast, it was shown that methyl pyruvate can restore cytokine production by autophagy-deficient T cells (22), suggesting a function of autophagy in energy production and other essential processes in T lymphocytes.
Kovacs et al.(49) demonstrated recently the importance of the class III PI3K complex protein Beclin-1 in the survival and polarization of T cells during the induction of experimental autoimmune encephalitis (EAE). Using CD4-cre to mediate the deletion of Beclin-1, the authors showed that while peripheral T-cell numbers were reduced to numbers similar to those observed in an Atg7-deficient mouse, the turnover of mitochondria by autophagy (mitophagy) was unaffected in T cells (49). In this model, Beclin-1-deficient T cells rapidly die by apoptosis after TCR stimulation, and this has been attributed to the inability of the cells to eliminate the pro-apoptotic molecules pro-caspase-3, pro-caspase-8, and Bcl-2-like protein 11 (Bim) via Beclin-1. However, autophagy was not directly measured in this study, so the results are still open to some interpretation. Considering the dual roles of Beclin-1 in autophagy and apoptosis, these findings suggest a critical role for Beclin-1 in T-cell apoptosis.
Autophagy has been implicated as a crucial mechanism for the removal of damaged or extraneous organelles in several systems. In neurons, parkin serves as an E3 ubiquitin ligase that targets mitochondria with damaged or sheared membranes for mitophagy (50, 51). This process also extends to hematopoietic cells, where the BH3-only protein Nix has been demonstrated to be necessary for mitophagy induction in developing erythrocytes, leaving them completely devoid of mitochondria (52) through a process dependent on the core autophagy machinery (53). Autophagy in T lymphocytes has also been shown to prevent death by removing excess organelles in vivo. Work from our laboratory has demonstrated a requirement for maturing T lymphocytes to decrease their mitochondrial mass. As mature CD4+ or CD8+ SP thymocytes egress from the thymus, their tolerance for mitochondria decreases as they take on a mature, naive phenotype that is considered relatively quiescent. Autophagy-deficient T cells are not able to undergo mitophagy and can be found in the periphery containing increased numbers of mitochondria and displaying elevated levels of reactive oxidative species (ROS) (29), which are toxic and can induce apoptosis (54). The increase in mitochondrial mass in autophagy-deficient T cells might explain why these cells have increased metabolic activity (Fig. 1). Further support for this idea is provided by the observation that the expression of mitochondrial proteins is increased in autophagy-deficient T lymphocytes, and genes related to mitochondrial maintenance are transcriptionally upregulated (29, 48). The signal(s) that induce this potent mitophagy in lymphocytes remain to be identified; however, engagement of the sphingosine-1-phosphate receptor 1 (S1PR1) is an interesting candidate, as it mediates both autophagy (55), and the process of thymic egress, which is temporally correlated with the autophagic removal of mitochondria in developing T cells.
Fig. 1. Atg7 T cells have increased levels of activated S6K and increased levels of inhibitory 4EBP-1.

Lymph nodes of WT, Atgf/fLck-cre, and Vps34f/fLck-cre mice, aged 5-7 weeks, were harvested. Freshly isolated cell suspensions were permeabilized with 0.1% saponin and stained with anti-phosphoS6 Kinase (Thr421/Ser424) or anti-phospho4EBP-1 (Thr36/Thr45). Shown here are representative histograms from CD4+CD44lo or CD8+CD44lo naive cell populations.
In addition to mitochondria, autophagy can also degrade other membranes and membrane-bound structures. Pexophagy, or autophagy-mediated peroxisomal turnover, has been described in cellular systems to reduce redox activity and eliminate cytotoxic ROS (56). Moreover, in T lymphocytes, the endoplasmic reticulum (ER) is carefully regulated in a manner similar to that of mitochondria. In Atg7f/fLck-cre mice, T-lymphocyte ER membrane is expanded, and calcium storage is highly dysregulated (57). Indeed, aberrant membrane structures resembling ER membrane are highly enriched as are resident ER proteins, such as calnexin, and ER stress markers. Consistent with expanded ER and defective calcium flux, these Atg7-deficient cells display a defective proliferative response to mitogen (57). The steady-state calcium storage is surprisingly higher in the ER of these cells, leading to an imbalance between intracellular and extracellular Ca2+, which causes calcium channels to be open for a shorter period of time during TCR stimulation (57). Interestingly, the SERCA pump inhibitor thapsigargin, which prevents the uptake of cytoplasmic Ca2+ to the ER, rescues this defect, demonstrating that reduced calcium storage restores the calcium influx potential of autophagy-deficient T cells (57). It remains to be determined whether ER turnover is regulated by autophagy during periods of cellular stress or whether the ER membrane is naturally used in T lymphocytes as a constant membrane donor for autophagosomes, thus leading to a steady state of calcium storage over the life of a T cell. However, using an inducible deletion system for the core autophagy protein Atg3 (Atg3f/fER-cre), we have recently shown that ER expansion takes place over a period of weeks after deletion, suggesting that a constant program of membrane trimming is required to counteract steady-state ER membrane synthesis (58) (Fig. 2). Notably, death in inducible Atg3-deficient T lymphocytes is the result of an accumulation of defects, as a significant increase in apoptosis was not observed until more than three weeks after the deletion of Atg3 (58).
Fig. 2. Autophagy limits mitochondrial and endoplasmic reticulum expansion.
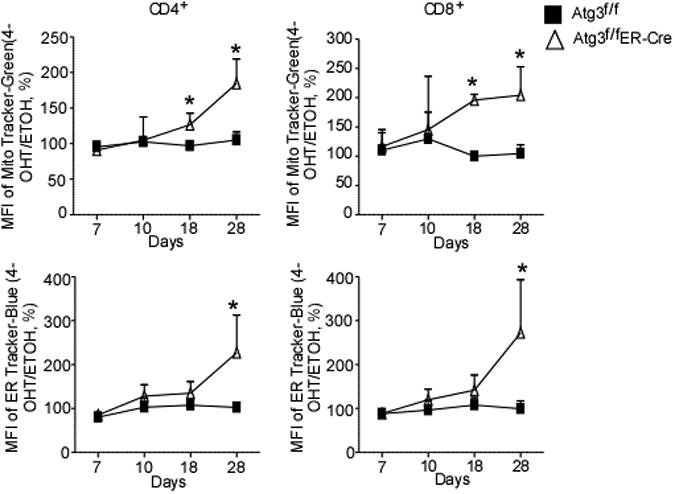
Splenocytes from Atg3f/fLck-Cre or 4-OH Tamoxifen–treated Atg3f/fER-Cre splenocytes and Atg3f/f control splenocytes were incubated with 100 nM Mito-Tracker Green (Invitrogen, Molecular Probes) or 1 mM ER-Tracker Blue-White DPX (Invitrogen, Molecular Probes) in RPMI 1640 medium at 37°C for 30 min. The geometric mean intensity of fluorescence was used to represent the volume of mitochondria or ER. One tailed, paired, Student's t tests were performed. *=p<.05
These findings demonstrate that autophagy is essential for turning over excess organelles and their associated membranes. It remains to be determined whether cytoplasmic targets or aggregates other than the previously mentioned pro-death effector molecules are targeted to autophagosomes in lymphocytes, either in the context of TCR signaling or during naive homeostasis.
How is autophagy unique in lymphocytes?
Many fine points of autophagy induction in T lymphocytes set them apart from other cell types. Recently, O'Brien and colleagues demonstrated that T lymphocytes from mice deficient in TSC1, a critical regulatory protein of mTOR in the TORC1 complex, had only slightly elevated levels of constitutive autophagy, and no increase was observed in autophagic levels upon nutrient withdrawal or TCR activation (59). These findings suggest that mTOR activity is not nearly as important for autophagy induction in T lymphocytes as it is in other cell types. However, TSC1-deficient T cells displayed abnormal mitochondrial homeostasis, reduced mitochondrial volume, and decreased ROS production compared to wild type murine T lymphocytes (59), suggesting that mitochondrial autophagy (mitophagy) was enhanced in these cells. It is interesting to observe that although overall autophagy levels did not seem to be affected by the overactivation of the critical metabolic sensor mTOR, a more specific mitochondrial turnover pathway was severely impacted.
Work from our laboratory has demonstrated that the class III PI3K Vps34 is also dispensable for autophagy induction in T lymphocytes. Using a conditional deletion system in which mice with a floxed Vps34 allele were crossed to mice with an Lck-cre transgene (Vps34f/fLck-cre or Vps34-deficient), CD4+ CD8+ double positive (DP) thymocytes lacking Vps34 were produced. Although the numbers of both thymocytes and peripheral T lymphocytes were reduced by as much as 80% in Vps34f/fLck-cre mice and the survival of these T lymphocytes was severely impaired, they showed little similarity to autophagy-deficient (Atg3-, Atg5-, or Atg7-conditionally deleted) T cells (20, 32, 58). Mitochondrial mass was not increased above wildtype (WT) levels, nor were reactive oxygen species increased, indicating that metabolic output and mitochondrial function were intact. Similarly, Vps34-deficient T lymphocytes efficiently proliferated when subjected to TCR stimulation and produced normal levels of IL-2 (32). As a further indication that the metabolic function of Vps34-deficient T cells was not perturbed, these cells did not upregulate activated S6K or display increased inhibitory phosphorylation of 4EBP, whereas these proteins were highly phosphorylated in Atg7f/fLck-cre T cells, indicating an abnormally active metabolism (Figure 1, lower panel). Finally, the anti-apoptotic protein Bcl-2, which is highly upregulated in Atg7-deficient T cells, was significantly downregulated in Vps34-deficient T cells. Conversely, Mcl-1, which is expressed at normal levels in Atg7f/fLck-cre T cells, was significantly upregulated in Vps34f/fLck-cre T cells. Thus, Vps34f/fLck-cre T cells showed no phenotypic similarity to autophagy-deficient T cells (32).
No differences were detected in basal, starvation-induced, and TCR-induced macroautophagy in Vps34f/fLck-cre T cells. Assessment of basal autophagy by transmission electron microscopy and LC3 fluorescence microscopy indicated equal numbers of characteristic double-membraned structures corresponding to autophagosomes and electron-dense double-membraned structures corresponding to autolysosomes and equal numbers and sizes of LC3-positive structures, respectively, in wildtype and Vps34-deficient T cells. Furthermore, microscopic analysis of endogenous LC3 punctae formation (Fig. 3A) and Western blot analysis of LC3 processing revealed no differences in autophagy levels between wildtype and Vps34-deficient T cells cultured under conditions of amino acid or serum withdrawal. However, autophagy induction was completely abrogated by the administration of general PI3K inhibitors, such as wortmannin or 3-methyladenine (3MA), suggesting a role for other classes of PI3K in the process (32). This is not completely surprising, given studies demonstrating a similar role for the class I PI3K p110β in autophagy induction in murine embryonic fibroblasts (60). Additionally, Vps34 deletion in certain classes of neurons leaves the autophagic pathway intact (61, 62). Similarly, when Vps34f/fLck-cre and wildtype T cells were stimulated with anti-CD3 and anti-CD28 for 24 or 72 h, no differences in macroautophagy levels were detected between the two cell types by endogenous LC3 fluorescence microscopy. Finally, to test whether CMA induction remained intact, Vps34f/fLck-cre T cells were subjected to starvation or TCR stimulation, and p62 levels were measured. No significant difference was detected in the degradation of p62, indicating that one major form of CMA or selective autophagy remained functional in cells lacking Vps34 (32). Thus, Vps34, a protein critical for autophagy induction in yeast and certain types of cells from higher eukaryotes, is dispensable for autophagy induction in T lymphocytes.
Fig. 3. Vps34f/fLck-cre T cells have normal autophagy but abnormal vesicular trafficking.

(A). Splenocytes from Vps34f/f(WT) or Vps34f/fLck-cre(Vps34) were isolated and CD4+ T cells sorted. Cells were washed in HBSS to remove bound nutrients and remove residual medium. 24 h post-starvation, cells were permeabilized with 0.1% saponin and stained with anti-LC3 (MBL, Woburn, MA). Autophagosomes were visualized by fluorescence microscopy and analyzed using Metamorph (Molecular Devices, Sunnyvale, CA) to quantify pixel sizes and Autoquant X2 (MediaCybernetics, Bethesda, MD) to analyze LC3 punctate structures. (B). SSC ratios (Vps34/WT) of indicated T-cell lineages, taken from 10 independent measurements, analyzed on a FACSCanto (BD Biosciences, San Jose, CA).
We did, however, find a role for Vps34 in vesicular trafficking and T-cell survival. Flow cytometric analysis of side scatter showed significantly increased granularity in all T-lineage cells from Vps34f/fLck-cre mice (Fig. 3B). This observation was indicative of abnormal vesicular trafficking, which was observed as a dramatic increase in membrane structures visualized by TEM. Vesicles containing the GLUE-domain-bearing protein Vps36 were highly aggregated in Vps34f/fLck-cre T cells, indicating a lack of inward vesicular maturation. Moreover, levels of the interleukin 7 receptor α (IL-7Rα) subunit were significantly lower on freshly isolated Vps34f/fLck-cre T cells than on wildtype T cells, and Vps34f/fLck-cre T cells could not maintain or upregulate IL-7Rα on withdrawal of this crucial pro-survival cytokine. Importantly, Vps34f/fLck-cre T cells could not maintain enough IL-7Rα on the cell surface to generate a full pro-survival signaling cascade upon binding of IL-7. When cell surface receptors were proteolytically cleaved by pronase, WT T cells efficiently upregulated IL-7Rα within 2 h, indicating the presence of an intracellular IL-7Rα pool that was not sensitive to the translation inhibitor cyclohexamide but was highly sensitive to the microfilament inhibitor cytochalasin B. In contrast, Vps34f/fLck-cre T cells could not upregulate surface IL-7Rα at earlier time points and had to synthesize the receptor de novo to generate a significant level of the receptor (32). Thus, Vps34 is indispensable for the intracellular recycling of IL-7Rα to the surface of naive T lymphocytes through a completely autophagy-independent mechanism.
Concluding remarks
Autophagy remains a vibrant and dynamic field in the study of the immune system. Using several murine knockout models of Atg genes, our laboratory has focused on the effects of autophagy on the clearance of excess organelles and membranes that when accumulated at high levels in T lymphocytes are cytotoxic both in vitro and in vivo (Fig. 4). In the case of mitochondria, there is a stage-specific requirement for organelle clearance as mature thymocytes leave for the secondary lymphoid organs. In the case of the ER and its associated membrane, regulation by autophagy seems to occur at a basal rate over the life span of the lymphocyte. Much work remains to identify the soluble, cytoplasmic components that require autophagy for turnover and to determine whether their autophagy-mediated turnover requires specific adapter proteins. It might be more than idle speculation to assume that much of the autophagy of specific cytoplasmic components now classified as macroautophagy may someday be found to be a form of chaperone-mediated autophagy or specific autophagy, with new adapters and chaperones being discovered all the time. This is an attractive thought that would give elegant specificity to the autophagic process, allowing nearly limitless control over exactly which proteins, not to mention which fractions of a given protein, are shuttled into the autophagosome.
Fig. 4. Autophagy's diverse role in lymphocyte homeostasis.

Autophagy-inducing signals from TCR-, BCR-, CXCR4-, and S1P1R-mediated signals induce autophagy in lymphocytes. The two ubiquitin-like conjugation systems are crucial for autophagy progression and membrane elongation, to maintain mitochondrial and ER levels. Vps34 is dispensable for autophagy but crucial for IL-7Rα turnover. Beclin-1 contributes to pro-death effector molecule turnover. The TSC complex regulates both mTOR activity and T lymphocyte mitophagy, but not basal or activation-induced autophagy. In the absence of an intact apoptotic response, autophagy mediates a pro-death response.
We have also focused on the regulation of autophagy by common signals encountered by T lymphocytes. TCR signaling, which mediates a potent induction of autophagy, requires the coordinated activity of PI3K signaling and JNK signaling. However, either Vps34 activity is not involved in autophagy induction in T cells or compensatory mechanisms exist (32). Whether other PI3K proteins, such as other isoforms of class I PI3K, which are potently induced by TCR signaling, or class II PI3K, which is poorly characterized, are involved in T-cell autophagy induction is unknown. Additionally, TSC1, an important regulator of mTOR activity, does not seem to play a role in starvation-induced autophagy in T cells, despite the fact that rapamycin treatment can induce T-cell autophagy (32). The fact that Vps34 is also not required for T-cell starvation-induced autophagy calls into question the central role of mTOR in autophagy in T cells.
Additionally, what downstream targets of the T-cell receptor are responsible for activation-mediated autophagy? Perhaps the coordinated activity of NFAT, NFκB, or MAPK signals are required. Furthermore, how autophagy contributes to a robust T-cell proliferative response is unclear. The degradation of certain inhibitors of proliferation is one attractive possibility. Clearly, more work is necessary to determine exactly which targets are processed by autophagy and which are proteosomally degraded. Following this line of evidence, targeting autophagy might provide interesting therapies for T cells to overcome anergy induced by chronic infections.
Acknowledgments
We thank Dr. Claire Gordy for her thoughtful reading of this manuscript. The work in the authors' laboratory is supported by NIH grants AI73947, AI074754 and AI074944. The authors have no conflicts of interest to declare.
References
- 1.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dice JF, et al. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin Cell Biol. 1990;1:449–455. [PubMed] [Google Scholar]
- 6.Tuttle DL, Lewin AS, Dunn WA., Jr Selective autophagy of peroxisomes in methylotrophic yeasts. Eur J Cell Biol. 1993;60:283–290. [PubMed] [Google Scholar]
- 7.Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci USA. 1983;80:2179–2183. doi: 10.1073/pnas.80.8.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuervo AM, Gomes AV, Barnes JA, Dice JF. Selective degradation of annexins by chaperone-mediated autophagy. J Biol Chem. 2000;275:33329–33335. doi: 10.1074/jbc.M005655200. [DOI] [PubMed] [Google Scholar]
- 9.Xilouri M, Stefanis L. Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med. 2011;13:e8. doi: 10.1017/S1462399411001803. [DOI] [PubMed] [Google Scholar]
- 10.Nijholt DA, De Kimpe L, Elfrink HL, Hoozemans JJ, Scheper W. Removing protein aggregates: the role of proteolysis in neurodegeneration. Curr Med Chem. 2011;18:2459–2476. doi: 10.2174/092986711795843236. [DOI] [PubMed] [Google Scholar]
- 11.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 12.Hara T, Mizushima N. Role of ULK-FIP200 complex in mammalian autophagy: FIP200, a counterpart of yeast Atg17? Autophagy. 2009;5:85–87. doi: 10.4161/auto.5.1.7180. [DOI] [PubMed] [Google Scholar]
- 13.Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bodemann BO, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–267. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massey DC, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn's disease. Autophagy. 2007;3:649–651. doi: 10.4161/auto.5075. [DOI] [PubMed] [Google Scholar]
- 16.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–236. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 19.Li C, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 20.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espert L, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357. doi: 10.4049/jimmunol.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle. 2004;3:1124–1126. [PubMed] [Google Scholar]
- 27.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 28.Espert L, Codogno P, Biard-Piechaczyk M. What is the role of autophagy in HIV-1 infection? Autophagy. 2008;4:273–275. doi: 10.4161/auto.5211. [DOI] [PubMed] [Google Scholar]
- 29.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe K, Ichinose S, Hayashizaki K, Tsubata T. Induction of autophagy by B cell antigen receptor stimulation and its inhibition by costimulation. Biochem Biophys Res Commun. 2008;374:274–281. doi: 10.1016/j.bbrc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 32.McLeod IX, Zhou X, Li QJ, Wang F, He YW. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J Immunol. 2011;187:5051–5061. doi: 10.4049/jimmunol.1100710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Botelho RJ. Changing phosphoinositides “on the fly”: how trafficking vesicles avoid an identity crisis. Bioessays. 2009;31:1127–1136. doi: 10.1002/bies.200900060. [DOI] [PubMed] [Google Scholar]
- 34.Gerland LM, et al. Autolysosomes accumulate during in vitro CD8+ T-lymphocyte aging and may participate in induced death sensitization of senescent cells. Exp Gerontol. 2004;39:789–800. doi: 10.1016/j.exger.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012 doi: 10.1016/j.it.2012.01.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe K, Tsubata T. Autophagy connects antigen receptor signaling to costimulatory signaling in B lymphocytes. Autophagy. 2009;5:108–110. doi: 10.4161/auto.5.1.7278. [DOI] [PubMed] [Google Scholar]
- 38.Maiuri MC, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 39.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3:17–27. doi: 10.1007/s13238-011-1127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arsov I, et al. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008;15:1385–1395. doi: 10.1038/cdd.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller BC, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–414. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 43.Mortensen M, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dzhagalov I, Dunkle A, He YW. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol. 2008;181:521–528. doi: 10.4049/jimmunol.181.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang N, Hartig H, Dzhagalov I, Draper D, He YW. The role of apoptosis in the development and function of T lymphocytes. Cell Res. 2005;15:749–769. doi: 10.1038/sj.cr.7290345. [DOI] [PubMed] [Google Scholar]
- 47.Dunkle A, Dzhagalov I, He YW. Mcl-1 promotes survival of thymocytes by inhibition of Bak in a pathway separate from Bcl-2. Cell Death Differ. 2010;17:994–1002. doi: 10.1038/cdd.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephenson LM, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–635. doi: 10.4161/auto.5.5.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovacs JR, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19:144–152. doi: 10.1038/cdd.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5:706–708. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- 51.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mortensen M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA. 2010;107:832–837. doi: 10.1073/pnas.0913170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildeman DA, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 55.Chang CL, Ho MC, Lee PH, Hsu CY, Huang WP, Lee H. S1P(5) is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am J Physiol Cell Physiol. 2009;297:C451–C458. doi: 10.1152/ajpcell.00586.2008. [DOI] [PubMed] [Google Scholar]
- 56.Grunau S, et al. The phosphoinositide 3-kinase Vps34p is required for pexophagy in Saccharomyces cerevisiae. Biochem J. 2011;434:161–170. doi: 10.1042/BJ20101115. [DOI] [PubMed] [Google Scholar]
- 57.Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186:1564–1574. doi: 10.4049/jimmunol.1001822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186:5313–5322. doi: 10.4049/jimmunol.1002404. [DOI] [PubMed] [Google Scholar]
- 59.O'Brien TF, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011;41:3361–3370. doi: 10.1002/eji.201141411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dou Z, et al. The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. J Cell Biol. 2010;191:827–843. doi: 10.1083/jcb.201006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou X, et al. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc Natl Acad Sci USA. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou X, Wang F. Effects of neuronal PIK3C3/Vps34 deletion on autophagy and beyond. Autophagy. 2010;6:798–799. doi: 10.4161/auto.6.6.12511. [DOI] [PMC free article] [PubMed] [Google Scholar]