

Abstract
The calcium-activated chloride channel anoctamin 1 (ANO1) is located within the 11q13 amplicon, one of the most frequently amplified chromosomal regions in human cancer, but its functional role in tumorigenesis has remained unclear. The 11q13 region is amplified in ∼15% of breast cancers. Whether ANO1 is amplified in breast tumors, the extent to which gene amplification contributes to ANO1 overexpression, and whether overexpression of ANO1 is important for tumor maintenance have remained unknown. We have found that ANO1 is amplified and highly expressed in breast cancer cell lines and primary tumors. Amplification of ANO1 correlated with disease grade and poor prognosis. Knockdown of ANO1 in ANO1-amplified breast cancer cell lines and other cancers bearing 11q13 amplification inhibited proliferation, induced apoptosis, and reduced tumor growth in established cancer xenografts. Moreover, ANO1 chloride channel activity was important for cell viability. Mechanistically, ANO1 knockdown or pharmacological inhibition of its chloride-channel activity reduced EGF receptor (EGFR) and calmodulin-dependent protein kinase II (CAMKII) signaling, which subsequently attenuated AKT, v-src sarcoma viral oncogene homolog (SRC), and extracellular signal-regulated kinase (ERK) activation in vitro and in vivo. Our results highlight the involvement of the ANO1 chloride channel in tumor progression and provide insights into oncogenic signaling in human cancers with 11q13 amplification, thereby establishing ANO1 as a promising target for therapy in these highly prevalent tumor types.
Keywords: ion channel, CaCCinh-A01, TMEM16A, HNSCC, ESCC
Genomic alterations are major drivers of human cancers. Those that activate oncogenes and inactivate tumor suppressors confer a selective advantage to cancer cells. Therefore they are promising targets for anticancer therapies (1). Genome-wide analysis of human tumors has revealed a multitude of alterations within cancer cells. Thus, it is of paramount importance to discriminate experimentally between driver, bystander, and collaborative genetic and epigenetic alterations (2).
The 11q13 amplicon is one of the most frequently amplified chromosomal regions in human cancer and correlates with a poor prognosis (3–7). Although cyclin D1 (CCND1) has been considered to be the main driver of the 11q13 amplicon (8–10), it is not sufficient for malignant transformation of normal breast cells and lacks predictive value for the survival of head-and-neck squamous cell carcinoma (HNSCC) or breast cancer patients (8, 11–14). Instead, the region has been shown to harbor several independent amplification cores, indicating that other genes could be potential driving oncogenes (6, 15, 16). Fine mapping of 11q13 in HNSCC led to the identification of another gene in this amplicon, anoctamin-1 (ANO1). ANO1 has been described under multiple names [discovered on gastrointestinal stromal tumors protein 1 (DOG1), oral cancer overexpressed 2 (ORAOV2), tumor-amplified and overexpressed sequence 2 (TAOS2), and tumor-amplified and overexpressed sequence 2 (TMEM16A)] derived from its up-regulation in different cancer types, including gastrointestinal stromal tumor, squamous cell carcinoma of the upper aerodigestive tract and the esophagus (ESCC), and pancreatic cancer (3–5, 17, 18).
ANO1 has been shown to function as a calcium-activated chloride channel that is expressed in various tissues, including secretory epithelia, smooth muscles, and sensory neurons (19–21). ANO1 activity regulates airway fluid secretion, gut motility, secretory functions of exocrine glands, and vascular smooth muscle contraction and recently was reported to be involved in nociception (22, 23). Not surprisingly, knockout of ANO1 is embryonically lethal, and its dysregulation plays a critical role in several disease states, including pulmonary diseases, hypertension, and diarrhea (22). The identification of activators and inhibitors in low molecular weight compound screens suggests that chemical modification of ANO1 function is feasible, making it a promising therapeutic target (24–26). Recently, ANO1 has been found to contribute to HNSCC tumorigenesis and invasion (27–29). In contrast, the effects of ANO1 in breast cancer, its functional activity in tumorigenesis, and whether it is required for tumor maintenance have remained elusive.
In the present study we provide evidence that ANO1 is amplified, overexpressed, and contributes to breast cancer, HNSCC, and ESCC tumorigenesis by activating EGF receptor (EGFR) and calmodulin-dependent protein kinase II (CAMKII), subsequently inducing activation of AKT and mitogen-activated protein kinase 1 (MAPK) signaling. Moreover, knockdown of ANO1 in ANO1-amplified breast cancer, HNSCC, and ESCC cell lines inhibited cell proliferation, induced apoptosis, and reduced tumor growth in established xenografts. Thus, our results suggest that ANO1 is a critical oncogenic factor contributing to cell survival, proliferation, and tumor maintenance in HNSCC, ESCC, and breast cancer.
Results
ANO1 Is Amplified, Overexpressed, and Associated with a Poor Prognosis in Breast Cancer.
In a search for potential oncogenes other than CCND1 within tumors exhibiting 11q13 amplification, we analyzed comparative genomic hybridization data of primary breast tumor samples. As expected, we found a significant gain in copy number in the 11q13 region. Genomic fine mapping revealed that the most frequently and highly amplified region spans ∼5 Mb (67–72 Mb) and contains ANO1 and 15 other protein-coding genes, including fibroblast growth factor 4 (FGF4), fibroblast growth factor 9 (FGF9), cyclin D1 (CCND1), Fas (TNFRSF6)-associated via death domain (FADD), and cortactin (CTTN) (3–5, 10, 27, 30, 31) (Fig. 1A). Interestingly, ANO1 was found repeatedly within the summit of amplification (i.e., in terms of copy number and frequency), suggesting that tumors with increased ANO1 copy number have a selective advantage (Fig. 1A, arrow). Similar results were obtained in HNSCC primary tumor samples, supporting the significance of ANO1 as a potential oncogenic driver in both cancer types (Fig. S1A).
Fig. 1.

ANO1 is amplified and highly expressed in breast cancer and predicts survival. (A) Copy number (CN) variation (chromosome 11) in breast cancer tissue is shown by genomic location. Histograms depict the aggregation statistic for each chromosomal region (n = 819). Dashed lines highlight the 11q13 area shown at right as a zoom-in view. The arrow indicates the genomic location of ANO1. Data source: The Cancer Gene Atlas. (B) Box plots of ANO1 mRNA levels in non–11q13-amplified and 11q13-amplified breast tumor tissues. Normalized gene expression values (z-scores) were plotted (n = 469). Data source: The Cancer Gene Atlas. (C) Average copy number variation (the gray region depicts 95% confidence interval) in 11q13 is shown by genomic location for patients who survived the 7-y observation period (solid line) or died during the study (dashed line). The significance of the differences in copy number at the ANO1 promoter (vertical dashed line) is given above each plot. Data source: ref. 32. (D) Representative images of ANO1 expression in breast carcinoma. Note that myoepithelial cells in normal breast tissue stain positive for ANO1. (Scale bars: 5 µm.)
Next, we assessed whether amplification of ANO1 correlates with overexpression and found that 11q13 amplification results in higher mRNA expression of ANO1 in breast and HNSCC tumors than in non–11q13-amplified tumors (Fig. 1B and Fig. S1B). As expected based on the close proximity, we found a significant correlation between the amplification of ANO1 and CCND1 in primary HNSCC and breast cancer tumor samples (Fig. S1C). However, we found no correlation between ANO1 and CCND1at the mRNA level (Fig. S1D) and only a weak correlation at the protein level (Fig. S1 E and F).
To determine whether there is an association between ANO1 amplification and clinical outcome in breast cancer patients, we analyzed a published dataset of copy number and overall survival in breast cancer patients (32) and found that amplification of ANO1 correlates with high grade disease and is a negative predictor for overall survival (Fig. 1C). In agreement with ANO1 being an important predictor for survival in breast cancer, we found a significant correlation between ANO1 expression levels and overall survival in breast cancer patients (Fig. S1H).
To examine whether the observed amplification and overexpression of ANO1 results in higher ANO1 protein levels, we stained primary breast tumors for ANO1. ANO1 staining was positive in 78% of breast tumors (Fig. 1D and Table S1). Staining of primary HNSCC and ESCC tumors for ANO1 revealed that 100% of primary HNSCC tumors and 90% of ESCC tumors are positive for ANO1 (Fig. S1G and Table S1). Thus, ANO1 is amplified and highly expressed in breast cancer and other tumors and associates with a poor prognosis.
ANO1 Is Critical for Cell Survival and Proliferation in 11q13-Amplified Breast Cancer, HNSCC, and ESCC Cells.
To find suitable models for testing the involvement of ANO1 in tumorigenesis, we analyzed a panel of established breast cancer cell lines for ANO1 amplification. A large subset of cell lines showed amplification of the same region identified in primary breast tumor samples (Fig. S1I and Table S2). Consistent with genomic amplification of ANO1, mRNA (Fig. S1J) and protein levels (Fig. S1M) were significantly higher in 11q13-amplified breast cancer cell lines than in non–11q13-amplified lines. Based on these results, we selected the ZR75-1, HCC1954, and MDA-MB-415 breast cancer cell lines for further experiments. Additionally, we profiled a set of established HNSCC and ESCC cell lines and selected FaDu (ESCC) and Te11 (HNSCC) as additional cell lines for our experiments (Fig. S1 K, L, and N).
Next, we tested whether ANO1 is important for the survival and proliferation of 11q13-amplified cancer cells by first constructing doxycycline (dox)-inducible lentiviral vectors expressing three independent shRNA constructs targeting ANO1 (shRNA_#1–#3) or a nontargeting shRNA (shRNA-NT). We then generated stable pools of ZR75-1, HCC1954, MDA-MB-415, Te11, and FaDu cells expressing these shRNAs. Induction of the expression of shRNA_#1–#3, but not shRNA-NT, led to pronounced knockdown of ANO1 in all five cell lines (Fig. S2 A and B). Knockdown of ANO1 resulted in decreased viability (Fig. 2A) and colony formation (Fig. 2B) and arrested the cells in G1 of the cell cycle (Fig. 2C). Moreover, we found increased expression of cleaved poly(ADP-ribose)polymerase and active caspase-3/9 and a reduction in B-cell lymphoma 2 (Bcl2), myeloid cell leukemia sequence 1 (Mcl-1), and survivin protein levels (Fig. 2D) upon knockdown of ANO1 in breast cancer cells, further identifying it as a protein with prosurvival activity. Similar results were obtained in Te11 and FaDu cells (Fig. S2 C–F). Thus, our results demonstrate that ANO1 expression is critical for cell survival and proliferation in 11q13-amplified breast cancer, HNSCC, and ESCC cells.
Fig. 2.
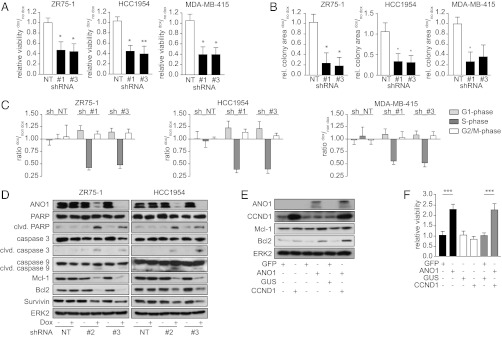
Knockdown of ANO1 decreases breast cancer cell viability and colony-formation capacity. (A and B) Bar graphs showing relative viability (A) and colony formation (B) of the indicated cancer cell lines after dox-induced knockdown of ANO1. Data were normalized to the respective non–dox-treated samples. Data are expressed as mean ± SEM, n = 5; *P < 0.05; **P < 0.01; ***P < 0.001, here and in the following panels. (C) Bar graphs showing the relative percentage of cells in the indicated phases of the cell cycle after dox-induced knockdown of ANO1. Data were normalized to the percentage of non–dox-treated cells in the respective cell-cycle phase. Data are expressed as mean ± SEM; n = 4. (D) Knockdown of ANO1 induces apoptosis. Immunoblots of lysates from the indicated cell lines after knockdown of ANO1. ERK2 was used as loading control. (E) Immunoblots of lysates from MCF10A cells stably transfected with ANO1 and/or CCND1. MCF10A cells expressing GFP and/or GUS were used as controls. (F) ANO1 increases MCF10A viability. Bar graphs showing relative viability of MCF10A cells stably transfected as in E. Data were normalized to the respective control vector cell lines. Data are expressed as mean ± SEM; n = 5.
ANO1 Is Sufficient to Promote Cell Proliferation in the Absence of 11q13 Amplification.
Having shown that ANO1 is amplified and overexpressed in breast cancer, we asked whether overexpression of ANO1 in non–11q13-amplified cells would be sufficient to promote cell viability. We generated pools of the immortalized but nontransformed breast epithelial cells MCF10A expressing ANO1, CCND1, a control vector (β-glucuronidase; GUS), or GFP (Fig. 2E). Overexpression of ANO1 significantly increased cell viability. Furthermore, we found that expression of ANO1 in MCF10A cells increased the levels of the antiapoptotic proteins Bcl2 and Mcl-1, suggesting that ANO1 has a prosurvival and antiapoptotic function. Despite its established role in inducing cell-cycle progression, cyclin D1 alone did not increase cell viability or enhance the effect of ANO1 (Fig. 2F). Thus, ANO1 is sufficient to promote cell viability in the absence of 11q13 amplification.
Chloride Channel Activity of ANO1 Is Required for Its Prosurvival Properties.
To determine the effect of ANO1 knockdown on chloride flux, we used a manual patch-clamp assay or the planar patch (QPatch) platform and found that ANO1 knockdown led to a significant reduction in calcium-dependent chloride currents (Fig. S3 A–D). Therefore, as expected, genetic ablation of ANO1 results in a significant loss of chloride channel activity.
We next used calcium-activated chloride channel inhibitor (CaCCinh)-A01, a low molecular weight inhibitor of ANO1 activity (25), to test whether the chloride channel activity of ANO1 is required for proliferation. Treatment of ZR75-1, HCC1954, and MDA-MB-415 cells with CaCCinh-A01 reduced cell viability (Fig. 3A) and inhibited colony formation (Fig. 3B). Similar results were obtained for FaDu and Te11 cells (Fig. S3 E and F). The sensitivity to CaCCinh-A01 correlated with both ANO1 amplification and expression levels (Table 1 and Fig. S1), suggesting that ANO1 biochemical activity is required for the promotion of cell viability in cell lines overexpressing ANO1.
Fig. 3.

Inhibition of ANO1 function decreases breast cancer cell viability and colony formation. (A and B) Bar graphs showing relative viability (A) or colony formation (B) of breast cancer cell lines after inhibition of ANO1 with CaCCinh-A01. Data were normalized to the respective DMSO-treated samples. Data are expressed as mean ± SEM; n = 5. (C) Bar graphs showing relative viability of MCF10A cells stably transfected with wild-type ANO1, the pore-mutants ANO1-R621E, or ANO1-K668E, respectively. Data were normalized to the GFP control vector cell line. Data are expressed as mean ± SEM; n = 5. (D) Bar graphs showing relative viability of MCF10A modified as in C after treatment with the indicated concentrations of CaCCinh-A01. Data were normalized within the same cell line to the DMSO-treated cells. Data are expressed as means ± SEM; n = 5. *P < 0.05; **P < 0.01; ***P < 0.001.
Table 1.
IC50 values for the inhibition of cell viability in HSNCC, ESCC, and breast cancer cell lines by CACCinh-A01 in correlation with ANO1 amplification status
| Cell line | Copy no. ANO1 | IC50 CaCCinh-A01 (µM) |
| KYSE70 | n.a. | >20 |
| KYSE150 | n.a. | >20 |
| KYSE450 | n.a. | 16 |
| Te1 | n.a. | 16 |
| Te9 | n.a. | 17 |
| FaDu | 14 | 8.5 |
| Te11 | 9 | 2.9 |
| MCF7 | n.a. | 20 |
| Hs578t | n.a. | 20 |
| BT549 | n.a. | 20 |
| ZR75-1 | 3 | 8.0 |
| HCC1954 | 3 | 8.0 |
| MDA-MB-415 | 7 | 8.0 |
Data represent the mean of three independent experiments. n.a., not amplified.
The residues R621 and K668 of ANO1 map to a highly conserved region between transmembrane domains TM5 and TM6. Mutation of these residues to glutamate reduced permeability for anions while promoting cation permeability (19). In contrast to the effect of wild-type ANO1, overexpression of similar levels of the R621 or K668 mutants in MCF10A cells had no effect on cell viability (Fig. 3C and Fig. S3G). These results confirm that biochemical activity of ANO1 is required for its effects on cell viability. Treatment with CaCCinh-A01 decreased cell viability in MCF10A-ANO1 wild-type cells in a concentration-dependent manner with an IC50 similar to its effect on ANO1-amplified breast cancer cell lines (IC50 ∼8 µM) described above. At the same time, CaCCinh-A01 had no effect on the viability of MCF10A cells overexpressing ANO1 mutants, confirming the selectivity of the inhibitor (Fig. 3D). These data suggest that ANO1 overexpression in non–11q13-amplified cells establishes an addiction to ANO1 biochemical activity, which sensitizes the cells to the inhibition of ANO1.
ANO1 Is Required for HNSCC, ESCC, and Breast Cancer Tumor Growth and Maintenance in Vivo.
The effects of ANO1 on the growth and maintenance of 11q13-amplified cancers in vivo are unknown. To test the effects of ANO1 knockdown after overt tumor development, pools of two breast cancer cell lines (ZR75-1 and HCC1954) expressing shRNA-NT or shRNA targeting ANO1 were injected into immunodeficient mice. The growth of tumors arising from shRNA-NT cells with or without dox and from cells with ANO1 shRNA but without dox was similar. In contrast, dox treatment resulting in ANO1 knockdown significantly impaired the growth of xenografts expressing ANO1 shRNA (Fig. 4 A and B). Again, similar results were obtained in HNSCC (Te11) and ESCC (FaDu) cell lines (Fig. 4 C and D). These results show that ANO1 is critical for tumor growth and maintenance in 11q13-amplified breast cancer, HNSCC, and ESCC cells.
Fig. 4.

Inhibition of ANO1 reduces tumor growth and maintenance. (A and B) Growth curves of breast cancer tumors with or without knockdown of ANO1. Cells expressing shRNA-NT or shRNA_3 were orthotopically injected into SCID/beige mice, and dox treatment was started when tumors were palpable. Data are expressed as mean ± SEM; n = 5–8; P < 0.001. (C and D) Growth curves of HNSCC and ESCC tumors with or without knockdown of ANO1. nu/nu mice were s.c. injected with cells expressing shRNA-NT or shRNA_2 and were fed with dox-containing food when average tumor volume reached 100 mm3. Data are expressed as mean ± SEM; n = 8; P < 0.001.
ANO1 Regulates EGFR- and Calcium-Dependent Signaling Pathways That Promote Cancer Cell Viability.
To define the biochemical mechanisms underlying the effects of ANO1 in breast cancer, we performed an antibody array on ZR75-1 and HCC1954 breast tumor lysates after ANO1 knockdown. We found that EGFR phosphorylation was strongly inhibited after ANO1 knockdown in ZR75-1 and HCC1954 breast tumor lysates (Fig. S4A). Subsequent analysis by immunoblotting validated these findings in ZR75-1 and HCC1954 breast tumor lysates and cell lysates (Fig. 5 A and B). In addition, the phosphorylation of ERK1/2, AKT, and v-src sarcoma viral oncogene homolog (SRC) was dramatically decreased (Fig. 5 A and B and Fig. S4B). Consistently, knockdown of ANO1 in HNSCC (Te11) and ESCC (FaDu) cells resulted in a similar inhibition of EGFR, ERK1/2, AKT, and SRC phosphorylation, supporting the hypothesis that the inhibition of EGFR signaling is a general rather than breast cancer-specific consequence of ANO1 knockdown (Fig. 5C and Fig. S4C). Furthermore, treatment of ZR75-1 and HCC1954 cells with CaCCinh-A01 decreased EGFR phosphorylation, suggesting that the chloride channel activity of ANO1 is important for its role in regulating EGFR activation (Fig. 5D). Together, these results suggest that ANO1 inhibition reduces EGFR signaling, contributing to a reduction in AKT, ERK1/2, and SRC phosphorylation and a decrease in cell viability.
Fig. 5.

ANO1 induces proliferation by activating both EGFR- and calcium-dependent signaling mechanisms. (A) Immunoblots of lysates from breast cancer tumors at the study end point as described in Fig. 4. (B) Immunoblots of lysates from breast cancer cell lines as indicated. (C) Immunoblots of lysates from Te11 (HNSCC) and FaDu (ESCC) cells as indicated. (D) Immunoblots of lysates from breast cancer cells treated for 6 h with 10 µM CaCCinh-A01 and/or 20 ng/mL EGF as indicated. (E) Bar graphs showing relative viability of breast cancer cell lines after treatment with 10 µM CaCCinh-A01 and/or 20 ng/mL EGF or 10 µM carbachol as indicated. Data were normalized to the respective vehicle-treated samples. Data are expressed as mean ± SEM; n = 5; ***P < 0.001; ns, not significant. (F) Immunoblots of lysates from breast cancer cells treated for 6 h with 10 µM CaCCinh-A01 and/or 10 µM carbachol as indicated. (G) Immunoblots of lysates from MCF10A cells stably expressing WT-ANO1 or the GFP vector control and treated for 6 h with DMSO or 100 nM AEE788 as indicated. (H) Immunoblots of lysates from MCF10A cells stably expressing wt-ANO1 or the GFP-empty vector control and treated for 6 h with DMSO or 5 µM KN93 as indicated. (I) Bar graphs showing relative viability of MCF10A cells expressing wild-type ANO1 or GFP-empty vector control after treatment with DMSO or 100 nM AEE788 and/or 5 µM KN93 as indicated. Data were normalized to the values of vector control cells and are expressed as mean ± SEM; n = 5; **P < 0.01; ***P < 0.001; ns, not significant.
To test whether the decrease in EGFR activation after knockdown of ANO1 is caused by a reduction in autocrine EGFR-ligand secretion, we measured the levels of EGF and TGF-α in the supernatant of HCC1954 and ZR75-1 cells after knockdown of ANO1. The levels of secreted EGF and TGF-α were reduced upon ANO1 knockdown (Fig. S4 E and F). These results suggest that knockdown of ANO1 reduces EGFR signaling by decreasing autocrine EGFR-ligand secretion in breast cancer cells. Next, we asked whether the reconstitution of EGFR signaling is sufficient to rescue the effect of ANO1 inhibition on cell viability. Treatment of ZR75-1 and HCC1954 cells with 20 ng/mL EGF was sufficient to reverse the inhibitory effect of CaCCinh-A01 on EGFR phosphorylation (Fig. 5D). Notably, although 20 ng/mL of EGF restored EGFR phosphorylation, the inhibitory effect of CaCCinh-A01 on cell viability was reversed only partially, suggesting that additional mechanisms are involved in the effect of ANO1 on cell viability (Fig. 5E). Given that the chloride channel activity of ANO1 is important for its oncogenic function, we hypothesized that chloride-driven activation of calcium-dependent pathways also contributes to the effects of ANO1 on cell viability. Intracellular calcium can activate the calcium/calmodulin-dependent protein kinase kinase (CAMKK) and lead to the phosphorylation and activation of CaMKII (33). To test this hypothesis, we first assessed the phosphorylation of CaMKII and found it to be dramatically reduced by ANO1 inhibition (Fig. 5 D and F). Next, we treated the cells with carbachol, a cholinergic agonist that leads to intracellular calcium release via activation of the acetylcholine receptor. Similar to EGF, carbachol restored CaMKII phosphorylation but only partially rescued the inhibitory effect of CaCCinh-A01 on cell viability (Fig. 5 E and F). Notably, combined EGF and carbachol treatments completely rescued the inhibitory effect of CaCCinh-A01 application on cell viability. Taken together, these results indicate that ANO1 regulates cell viability by modulation of both EGFR and CaMKII signaling.
Finally, we examined whether overexpression of ANO1 enhances the phosphorylation of EGFR and CAMKII. Immunoblotting revealed an increase in EGFR, SRC, and CAMKII phosphorylation in lysates of MCF10A cells overexpressing ANO1 compared with control cells (Fig. 5G and Fig. S4D). Notably, MCF10A cells overexpressing ANO1 and ANO1-amplified HNSCC, ESCC, and breast cancer cells were more sensitive to SRC inhibition (Table S3). Consistent with the observed reduction of EGFR-ligand secretion after knockdown of ANO1, overexpression of ANO1 in MCF10A cells led to increased secretion of EGF and TGF-α (Fig. S4G). Treatment of MCF10A-ANO1 cells with either the EGFR inhibitor AEE788 or with the CAMKII inhibitor KN93 reduced EGFR and CAMKII phosphorylation, respectively, to the levels of the parental cells (Fig. 5 G and H) but only partially reversed ANO1 promotion of cell viability (Fig. 5I). Consistently, ANO1-amplified HNSCC and ESCC cell lines were more sensitive than nonamplified lines to several EGFR inhibitors (Table S4). Notably, the combination of the EGFR and CAMKII inhibitors completely abrogated the effect of ANO1 overexpression on cell viability (Fig. 5I). Last, we analyzed the expression of ANO1 and the phosphorylation of EGFR and CAMKII in lysates of primary human breast tumors. Consistent with our results in human breast cancer cell lines, expression of ANO1 correlated with the phosphorylation of EGFR and CAMKII in primary human breast tumor samples (Fig. S4H). In summary, these findings suggest that ANO1 promotes oncogenesis in ANO1-amplified and -overexpressed cancers by activating EGFR- and calcium-dependent signaling pathways.
Discussion
11q13 is a frequently amplified chromosomal region in several human cancers with poor prognosis (3–7). Although CCND1 is considered to be the main tumor-promoting gene in this amplicon, it does not have predictive value for the survival of HNSCC and breast cancer patients (8, 11–14). The 11q13 amplicon harbors several independent amplification cores, indicating the presence of additional driving oncogenes in this region (6, 15, 16, 34). The calcium-activated chloride channel ANO1, located within the 11q13 amplicon, is known to be overexpressed in several cancers and recently has been reported to promote oncogenesis in HNSCC by activating MAPK (28). In this study, we provide evidence that ANO1 contributes to breast cancer tumorigenesis. We show that ANO1 is amplified and overexpressed in a significant number of primary breast tumors and cell lines at the genomic, RNA, and protein levels. Knockdown or pharmacological inhibition of ANO1 decreased cell proliferation and increased apoptosis in several human breast cancer cell lines. Furthermore, knockdown of ANO1 after overt tumor development in four cell lines grown as xenografts reduced tumor growth, suggesting that ANO1 is an essential oncogenic factor in breast cancer. Notably, overexpression of ANO1 in nontransformed human mammary cells was sufficient to increase their viability, indicating that overexpression of ANO1 in the absence of the 11q13 amplicon is sufficient to increase viability. ANO1 previously was linked to HNSCC, ESCC, and prostate carcinoma. Our findings that ANO1 is amplified and highly expressed in breast cancer as well as in HNSCC and ESCC and that it is essential for tumor maintenance validate ANO1 as a potential driver in these cancer types.
Amplification of ANO1 recently has been described to be a negative predictor for survival in HNSCC (28). We provide evidence that amplification of ANO1 in primary breast tumor correlates with poor survival and grade of disease. These findings are in agreement with previous studies identifying the 11q13 amplicon as a high-risk marker for poor survival in breast cancer (34).
Mechanistically, we found that the chloride channel activity of ANO1 is essential for its effect on cell viability. Evidence is provided by the fact that mutants of ANO1 with impaired chloride channel activity failed to increase cell viability and that an ANO1 inhibitor that abrogates its chloride channel activity blocked the promoting effects of ANO1 on cell viability. The roles of ion channels in tumorigenesis are not well understood. Although the expression and activity of some membrane channels [e.g., K+ channel: Kv11.1 (hERG1); Ca+ channel: Cav, SOC] have been linked to cancer progression, their impact on oncogenic signaling pathways has remained largely elusive (35).
In the present study, we show that ANO1 promotes cell viability in breast cancer as well as in HNSCC and ESCC models by activating EGFR- and calcium-dependent pathways. Inhibition of the chloride channel activity or knockdown of ANO1 decreased EGFR phosphorylation and subsequently inhibited AKT, SRC, and ERK activation. Furthermore, depletion of ANO1 or inhibition of its biochemical activity blocked CAMKII activation, perhaps also contributing to decreased AKT and ERK phosphorylation as reported previously (33, 36–38). Consistent with these findings, overexpression of ANO1 in an ANO1-negative cell line promoted cell growth and led to the phosphorylation of both EGFR and CAMKII, indicating the activation of both pathways by ANO1 overexpression. Notably, the inhibition of both EGFR and CAMKII abrogated the ANO1 effects on cell viability, whereas activation of both pathways was sufficient to rescue the inhibitory effect of ANO1 knockdown.
Several studies have demonstrated the involvement of ion channels in the activation of EGFR signaling, and changes in intracellular calcium levels have been shown to stimulate EGFR phosphorylation and AKT/SRC/MAPK signaling (39–44). It also has been shown that membrane depolarization activates PI3K signaling and AKT phosphorylation in epithelial cells (45). Our findings that knockdown of ANO1 or its inhibition by CaCCinh-A01 decreases chloride channel activity suggest that ANO1 activates EGFR phosphorylation via imbalanced intracellular ion homeostasis, membrane depolarization, and/or activation of further ion channels. Changes in intracellular ion homeostasis regulate both expression and shedding of EGFR ligands from membrane-bound precursors (43), providing a possible explanation for the observed decrease in EGFR-ligand secretion after knockdown of ANO1 in 11q13-amplified breast cancer cell lines. However, we could not detect any secretion of EGF or TGF-α in HNSCC cells, indicating that the mechanisms leading to ANO1-dependent activation of EGFR might be cell-type specific. Activation of EGFR itself can trigger a variety of ionic changes in the cell, including a transient membrane hyperpolarization and an increase in cytoplasmic calcium concentration (46). Furthermore, EGF stimulation has been shown to mediate calcium-activated chloride channel activation via activation of SRC (47) and to increase ANO1 expression in epithelial cells, suggesting a positive feedback mechanism between EGFR/SRC signaling and ANO1 (47, 48). SRC recently has been shown to interact with ANO1 and can itself modulate the activation of EGFR, providing an additional explanation for the ANO1-dependent activation of EGFR signaling (49, 50).
EGFR is a known contributor to HNSCC, ESCC, and breast cancer tumorigenesis and is one the most important therapeutic targets in HNSCC. High levels of EGFR and the activation of PI3K signaling have been correlated with poor prognosis in HNSCC and breast cancer, and the prognostic significance of EGFR phosphorylation has been described recently for both cancer types (51–54). However, EGFR amplification and activating mutations are rare and account for only a small subset of HNSCC cases (55, 56). Our finding that ANO1 enhances EGFR signaling could provide an explanation for the elevated activation of EGFR and PI3K signaling observed in these tumors. Combined with the observation that HNSCC and ESCC cell lines with amplified ANO1 are more sensitive to EGFR inhibition than nonamplified lines, these results suggest ANO1 overexpression as a predictive marker for the response to EGFR-targeting agents in HNSCC and possibly in breast cancer therapy.
In summary, our study establishes ANO1 as a key tumor-promoting factor in 11q13-amplified breast cancer and other malignancies, highlights the importance of chloride channels in cancer, and provides mechanistic insight in their role in tumorigenesis. Most importantly, our findings open up potential opportunities for therapeutic intervention in several prevalent cancers.
Methods
Cell Culture.
Te11, Te1, Te9, and KYSE70 cells were maintained in Roswell Park Memorial Institute medium (RPMI); KYSE150 and KYSE450 cells were maintained in 45% (vol/vol) F-12/45% (vol/vol) RPMI; and FaDu cells were maintained in Eagle's Minimum Essential Medium supplemented with 10% (vol/vol) FBS at 37 °C, 5% CO2. ZR75-1, HCC1954, and MDA-MB-415 (MDA-415) cells were propagated in RPMI with 10% (vol/vol) FBS and 1% (vol/vol) penicillin/streptomycin (PenStrep) at 37 °C, 5% CO2. For experiments, the serum concentration was reduced to 0.5% (vol/vol) to avoid masking the effects of growth factors present under full-serum conditions. MCF10A cells were obtained from J. Brugge (Harvard Medical School, Boston, MA) and propagated in DMEM/F-12 medium (Invitrogen) supplemented with 5% (vol/vol) horse serum (HyClone), 20 ng/mL human recombinant EGF (Peprotech), 0.5 µg/mL hydrocortisone (Sigma), 100 ng/mL cholera toxin (Sigma), 10 µg/mL insulin (Sigma), 100 IU/mL penicillin, and 100 µg/mL streptomycin. Te1, Te9, and Te11 cells were purchased form the RIKEN cell bank; KYSE70, KYSE150, and KYSE450 cells were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (the German cell bank), and all other cell lines were obtained from the American Type Culture Collection.
Compounds.
CaCCinh-A01 (Specs), KN93 (Tocris, Sigma Aldrich) and AEE788, gefitinib, erlotinib, BIBX1382, lapatinib, saracatinib, dasatinib, and bosutinib (Tocris) were dissolved in DMSO to a final concentration of 10 mM. Carbamoylcholine chloride (carbachol; Sigma Aldrich) was dissolved in ethanol to a final concentration of 10 mM. Cells were treated with the indicated concentrations of inhibitors or matching volumes of DMSO and/or ethanol.
Lentivirus Preparation and Generation of Stable Cell Lines.
For lentiviral production, HEK 293T cells were cotransfected using Fugene (Promega) with pLKO1-tet on ANO1 shRNA or pLKO1-tet on nonsilencing shRNA and packaging plasmid mix (Sigma).The culture medium was replaced with fresh medium after 16 h, and supernatants were collected 48 h and 72 h after transfection. For generation of stable lines, 0.8 × 106 cells per well were infected with lentiviruses in the presence of 8 μg/mL Polybrene (Applied Bioanalytical) at a multiplicity of infection of 20. Cells stably expressing the shRNA were selected with 1.5–2 µg/mL puromycin. MCF10A cells were transduced with the lentiviral internal ribosome entry site (IRES)-GFP vector (pLKO-IRES-GFP) expressing human ANO1 (ac-splice-form) cDNA and/or the lentiviral pSD69-human phosphoglycerate kinase 1 (hPGK)-Puro-GUS vector human CCND1 cDNA or the empty vector. Cells stably expressing pSD69-hPGK-Puro-GUS were selected with 1.5 µg/mL puromycin, and GFP-expressing cells were selected using one to three rounds of cell sorting by FACS.
Copy Number Analysis.
Genomic DNA was isolated using Qiagen’s DNeasy Kit according to the manufacturer’s instructions. RT-PCR analysis was performed as described using a probe for ANO1: Hs04399219_cn, CCND1: Hs69455941_cn, and RNAseP (4401631; Invitrogen). Copy number was calculated using Copy Caller v1.0 freeware (Applied Biosystems), normalized to RNAseP and expressed relative to normal human tissue (human placenta, female D3035; Sigma).
Bioinformatics Analysis.
Clinical annotation, copy number (SNP 6.0; Affymetrix), and expression (Illumina HiSeq) calls for breast cancer and HNSCC tissue samples were obtained from ref. 32 or The Cancer Genome Atlas (https://tcga-data.nci.nih.gov/tcga/) using the Data Matrix functionality and cgdsr package, available from cBIO Cancer Genomics Portal (ww.cbioportal.org/). Breast cancer and HNSCC cell line data came from the Cancer Cell Line Encyclopedia effort, a collaboration among Novartis Institutes for Biomedical Research, the Broad Institute, and the Genomics Institute of the Novartis Foundation (57). All data were formatted, filtered, and analyzed in R. Plots were created using the ggplot2 package and ref. 58.
Cell-Viability and Colony-Formation Assays.
For measurement of cell viability, 3 × 103 cells per well were seeded into a 96-well plate, adhered overnight, and treated with the indicated concentrations of inhibitor or solvent for 72 h. Cell viability was assessed using the Cell Proliferation Reagent WST-1 (Roche) or Cell Titer Glo (Promega). Colony-formation assays were performed by seeding 1,000 cells per well in six-well plates (for breast cancer cell lines) or 1,000 Te11 or FaDu cells per well in 24-well plates. Cells were allowed to adhere overnight before treatment with CaCCinh-A01 or DMSO as indicated. Colonies were stained after 10–18 d with 0.2% (wt/vol) crystal violet in PBS/4% (vol/vol) formalin. Colony area was quantified using the Odyssey scanner and software (LI-COR Biosciences).
Cell-Cycle Analysis.
Cells were detached using trypsin-EDTA, resuspended in growth medium, and counted. Then 1 × 106 cells and supernatant were washed in PBS, fixed in 70% (vol/vol) ethanol for 60 min at 4 °C, washed twice, and resuspended in PI buffer (PBS supplemented with 50 μg/mL propidium iodide, 10 μg/mL RNase A, 0.1% (wt/vol) sodium citrate, and 0.1% (vol/vol) Triton X-100). At least 2 × 104 cells per sample were analyzed with a FACScan flow cytometer (Becton Dickinson).
Conventional Patch-Clamp and QPatch Recordings.
Whole-cell currents were measured using conventional and planar (QPatch; Sophion Bioscience) patch-clamp electrophysiology (59). For conventional patch-clamp recordings, cells were plated onto glass coverslips. All measurements were performed at room temperature (21–23 °C) 24 h after plating. Currents were recorded using an Axopatch 200B patch-clamp amplifier, low-pass filtered at 2 kHz, and subsequently digitized at 10 kHz with a Digidata 1322A and pClamp 9.0 data acquisition software (Molecular Devices). For QPatch recordings, cells were harvested in serum-free medium containing trypsin inhibitor. QPatch was operated in accordance with the manufacturer’s specification. Gigaseal, whole-cell configuration, and series resistance compensation were established using QPatch software (Sophion Biosciences). Cells were measured 72 h after dox addition. The voltage protocol used for QPatch recordings combined a voltage step with a voltage ramp protocol from a holding potential of −70 mV. The protocol was repeated every 30 s. The same buffers were used for both conventional and QPatch recordings. The extracellular recording solution contained (in mM): NaCl (156), Hepes (10), glucose (10), CaCl2 (5), at pH 7.4 adjusted with NaOH. The intracellular solution contained (in mM): N-methyl-D-glucamine (130), EGTA (20), Hepes (10), MgCl2 (1), 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) (10), Mg ATP (2), CaCl2, at pH 7.2 adjusted with HCl; osmolarity adjusted to 320 with sucrose. The pipette solution contained either 338 nM or 1 µM free Ca2+.
Immunoblotting.
Cells were lysed with RIPA buffer [50 mM Tris⋅HCl (pH 8), 150 mM NaCl, 1% (vol/vol) Nonidet P-40, 0.5% (wt/vol) sodium deoxycholate, 0.1% (vol/vol) SDS] supplemented with 1× protease inhibitor mixture (Complete Mini; Roche), 0.2 mM sodium orthovanadate, 20 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride. Lysates from xenografts and human primary breast tumors were prepared by lysing kryo-homogenized tumor powder in RIPA buffer. Whole-cell lysates were subjected to SDS/PAGE and immunoblotting. Membranes were incubated with antibodies as indicated and exposed to secondary HRP-/IRDye-coupled antibodies (LI-COR Biosciences). The following primary antibodies were used: ANO1 (SP31; Abcam), Tubulin (Sigma), ERK2 (Santa Cruz), phoshpo-CAMKII (pCAMKII) (T286; Abcam) and phospho-EGFR (pEGFR) (Y1086; Epitomics); all other primary antibodies were from Cell Signaling Technology.
PathScan and Receptor Tyrosine Kinase Arrays.
Cell lysates were analyzed using the PathScan receptor tyrosine kinase (RTK) Signaling Antibody Array Kit (Cell Signaling Technology) and the Human Phospho-RTK Array Kit (R&D Systems) according to the manufacturers’ instructions. Signals were normalized to the respective positive controls on the array.
EGF and TGF-α ELISA.
Human EGF and TGF-α in the supernatant were measured by ELISA (Invitrogen and R&D Systems, respectively) according to the manufacturers’ instructions. Cells were cultured for 24 h in starving conditions, and values were normalized to total cell number at the end of the experiment.
Animal Experiments.
All work involving laboratory animals was carried out in strict accordance with the Swiss and US federal, state, local and institutional guidelines governing the use of laboratory animals in research and were approved by the Swiss Cantonal Veterinary Office of Basel or the American Association for Laboratory Animal Science and the Novartis Institutes for BioMedical Research Institutional Animal Care and Use Committee (IACUC). For HNSCC xenograft assays, 7-wk-old female outbred athymic nu/nu mice (Taconic) were inoculated s.c. in the dorsal axillary region with 2 × 106 FaDu_shRNA or 5 × 106 Te-11_shRNA cells plus 25% (vol/vol) Matrigel. Breast cancer xenograft assays were carried out with 7- to 9-wk-old SCID/beige and SCID/NOD mice (Jackson Labs). For orthotopic engraftment of the ZR75-1 and HCC1954 cell lines, 2 × 106 cells were suspended in a 100-μL mixture of Basement Membrane Matrix Phenol Red-free (BD Biosciences) and PBS 1:1 and were injected into mouse mammary gland 4 or between mammary glands 2 and 3. For growth of the ZR75-1 cell line, mice were switched to estrogen-containing drinking water 2 d before injection. When the average tumor volume reached 100 mm3, some animals were switched to food containing dox at 400 ppm. Tumor volume and animal weight were determined every 3 or 4 d.
Immunohistochemistry.
Surgically resected tissue samples were procured under Institutional Review Board approval by Maine Medical Center Tissue Bank, Maine Medical Center Pathology Department, Portland, ME. Primary breast tumors were obtained with the appropriate informed consent. Human tissue microarray for esophageal squamous cell carcinoma (ESC961; Pantomics), for head-and-neck tumors (HN802; BioMax), normal tissues (FDA955; BioMax), and sections of archival formalin-fixed, paraffin-embedded human tumor specimen were deparaffinized and immunostained with a 1:50 dilution of anti-ANO1 antibody (SP31; Abcam) using the Ventana Discovery XT system. UltraMap HRP-conjugated secondary antibody was used, followed by 3,3'-diaminobenzidine and hematoxylin counterstaining. Staining was assessed by a pathologist, and the intensity of staining was scored as negative (0), mild (1+), moderate (2+), and strong (3+).
Statistical Analysis.
All data are expressed as means ± SEM. Statistical analyses were performed in GraphPad Prism using the Student t test or ANOVA with Tukey’s post test as appropriate. The Mann–Whitney test was used to calculate the significance of the difference in copy number in Fig. 1C.
Supplementary Material
Acknowledgments
We thank members of the M.B.-A. laboratory and Nancy Hynes (Friedrich Miescher Institute for Biomedical Research) for advice and discussions. We are grateful to Y. Kuhn and C. Peters (Saint-Louis Clinic) for providing primary tumors. Research in the M.B-A. laboratory is supported by the Novartis Research Foundation, the European Research Council (ERC Starting Grant 243211-PTPsBDC), the Swiss Cancer League, and the Krebsliga beider Basel. A. Bill is a Presidential Postdoctoral Fellow at the Novartis Institutes for BioMedical Research.
Footnotes
Conflict of interest statement: A. Bill, C.R., I.C., M.R., P.R., C.T.G., K.W., E.G., M.O.P., S.L., H.C., M.G., L.W., S.F., J. Borawski, J. Baffoe, M.L. and L.A.G. are employees of Novartis Institutes for Biomedical Research.
*This Direct Submission article had a prearranged editor.
See Author Summary on page 4176 (volume 110, number 11).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217072110/-/DCSupplemental.
References
- 1.Jonkers J, Berns A. Oncogene addiction: Sometimes a temporary slavery. Cancer Cell. 2004;6(6):535–538. doi: 10.1016/j.ccr.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West RB, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107–113. doi: 10.1016/S0002-9440(10)63279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kashyap MK, et al. Genomewide mRNA profiling of esophageal squamous cell carcinoma for identification of cancer biomarkers. Cancer Biol Ther. 2009;8(1):36–46. doi: 10.4161/cbt.8.1.7090. [DOI] [PubMed] [Google Scholar]
- 5.Carles A, et al. Head and neck squamous cell carcinoma transcriptome analysis by comprehensive validated differential display. Oncogene. 2006;25(12):1821–1831. doi: 10.1038/sj.onc.1209203. [DOI] [PubMed] [Google Scholar]
- 6.Ormandy CJ, Musgrove EA, Hui R, Daly RJ, Sutherland RL. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003;78(3):323–335. doi: 10.1023/a:1023033708204. [DOI] [PubMed] [Google Scholar]
- 7.Jönsson G, et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res. 2010;12(3):R42. doi: 10.1186/bcr2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigo JP, et al. Distinctive clinicopathological associations of amplification of the cortactin gene at 11q13 in head and neck squamous cell carcinomas. J Pathol. 2009;217(4):516–523. doi: 10.1002/path.2462. [DOI] [PubMed] [Google Scholar]
- 9.Sawey ET, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell. 2011;19(3):347–358. doi: 10.1016/j.ccr.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuuring E. The involvement of the chromosome 11q13 region in human malignancies: Cyclin D1 and EMS1 are two new candidate oncogenes—a review. Gene. 1995;159(1):83–96. doi: 10.1016/0378-1119(94)00562-7. [DOI] [PubMed] [Google Scholar]
- 11.Debnath J, et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111(1):29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 12.Velasco-Velázquez MA, et al. Examining the role of cyclin D1 in breast cancer. Future Oncol. 2011;7(6):753–765. doi: 10.2217/fon.11.56. [DOI] [PubMed] [Google Scholar]
- 13.Gleich LL, Li YQ, Wang X, Stambrook PJ, Gluckman JL. Variable genetic alterations and survival in head and neck cancer. Arch Otolaryngol Head Neck Surg. 1999;125(9):949–952. doi: 10.1001/archotol.125.9.949. [DOI] [PubMed] [Google Scholar]
- 14.Vielba R, et al. p53 and cyclin D1 as prognostic factors in squamous cell carcinoma of the larynx. Laryngoscope. 2003;113(1):167–172. doi: 10.1097/00005537-200301000-00031. [DOI] [PubMed] [Google Scholar]
- 15.Holm K, et al. Characterisation of amplification patterns and target genes at chromosome 11q13 in CCND1-amplified sporadic and familial breast tumours. Breast Cancer Res Treat. 2012;133(2):583–594. doi: 10.1007/s10549-011-1817-3. [DOI] [PubMed] [Google Scholar]
- 16.Jin C, et al. Molecular cytogenetic characterization of the 11q13 amplicon in head and neck squamous cell carcinoma. Cytogenet Genome Res. 2006;115(2):99–106. doi: 10.1159/000095228. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci USA. 2002;99(17):11369–11374. doi: 10.1073/pnas.172285799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Lu M, Liu B, Huang Y, Wang K. Inhibition of Ca(2+)-activated Cl(-) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012;326(1):41–51. doi: 10.1016/j.canlet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Yang YD, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caputo A, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 22.Huang F, Wong X, Jan LY. International Union of Basic and Clinical Pharmacology. LXXXV: Calcium-activated chloride channels. Pharmacol Rev. 2012;64(1):1–15. doi: 10.1124/pr.111.005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho H, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15(7):1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- 24.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286(3):2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73(3):758–768. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, Namkung W, Verkman AS, Sharma PK. Novel 5-substituted benzyloxy-2-arylbenzofuran-3-carboxylic acids as calcium activated chloride channel inhibitors. Bioorg Med Chem. 2012;20(14):4237–4244. doi: 10.1016/j.bmc.2012.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayoub C, et al. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br J Cancer. 2010;103(5):715–726. doi: 10.1038/sj.bjc.6605823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duvvuri U, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13):3270–3281. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruiz C, et al. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS ONE. 2012;7(8):e43265. doi: 10.1371/journal.pone.0043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishizuka T, et al. Gene amplification profiling of esophageal squamous cell carcinomas by DNA array CGH. Biochem Biophys Res Commun. 2002;296(1):152–155. doi: 10.1016/s0006-291x(02)00836-7. [DOI] [PubMed] [Google Scholar]
- 31.Chung CC, et al. Fine mapping of a region of chromosome 11q13 reveals multiple independent loci associated with risk of prostate cancer. Hum Mol Genet. 2011;20(14):2869–2878. doi: 10.1093/hmg/ddr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hicks J, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16(12):1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396(6711):584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- 34.Curtis C, et al. METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arcangeli A, et al. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr Med Chem. 2009;16(1):66–93. doi: 10.2174/092986709787002835. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt JM, Smith S, Hart B, Fletcher L. CaM Kinase control of AKT and LNCaP cell survival. J Cell Biochem. 2011 doi: 10.1002/jcb.24020. [DOI] [PubMed] [Google Scholar]
- 37.Swulius MT, Waxham MN. Ca(2+)/calmodulin-dependent protein kinases. Cell Mol Life Sci. 2008;65(17):2637–2657. doi: 10.1007/s00018-008-8086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cipolletta E, et al. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology. 2010;151(6):2747–2759. doi: 10.1210/en.2009-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen LB, Greenberg ME. Stimulation of growth factor receptor signal transduction by activation of voltage-sensitive calcium channels. Proc Natl Acad Sci USA. 1996;93(3):1113–1118. doi: 10.1073/pnas.93.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tahara S, et al. Potassium channel blocker activates extracellular signal-regulated kinases through Pyk2 and epidermal growth factor receptor in rat cardiomyocytes. J Am Coll Cardiol. 2001;38(5):1554–1563. doi: 10.1016/s0735-1097(01)01558-3. [DOI] [PubMed] [Google Scholar]
- 41.Tsai W, Morielli AD, Peralta EG. The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J. 1997;16(15):4597–4605. doi: 10.1093/emboj/16.15.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanou N, et al. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J Biol Chem. 2012;287(18):14524–14534. doi: 10.1074/jbc.M112.341784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horiuchi K, et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18(1):176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bradshaw JM. The Src, Syk, and Tec family kinases: Distinct types of molecular switches. Cell Signal. 2010;22(8):1175–1184. doi: 10.1016/j.cellsig.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Chatterjee S, et al. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol. 2012;302(1):H105–H114. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peppelenbosch MP, Tertoolen LG, den Hertog J, de Laat SW. Epidermal growth factor activates calcium channels by phospholipase A2/5-lipoxygenase-mediated leukotriene C4 production. Cell. 1992;69(2):295–303. doi: 10.1016/0092-8674(92)90410-e. [DOI] [PubMed] [Google Scholar]
- 47.Jeulin C, Seltzer V, Bailbé D, Andreau K, Marano F. EGF mediates calcium-activated chloride channel activation in the human bronchial epithelial cell line 16HBE14o-: Involvement of tyrosine kinase p60c-src. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L489–L496. doi: 10.1152/ajplung.90282.2008. [DOI] [PubMed] [Google Scholar]
- 48.Mroz MS, Keely SJ. Epidermal growth factor chronically upregulates Ca(2+)-dependent Cl(-) conductance and TMEM16A expression in intestinal epithelial cells. J Physiol. 2012;590(Pt 8):1907–1920. doi: 10.1113/jphysiol.2011.226126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc Natl Acad Sci USA. 1995;92(15):6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Cornejo P, et al. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc Natl Acad Sci USA. 2012;109(26):10376–10381. doi: 10.1073/pnas.1200174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hama T, et al. Prognostic significance of epidermal growth factor receptor phosphorylation and mutation in head and neck squamous cell carcinoma. Oncologist. 2009;14(9):900–908. doi: 10.1634/theoncologist.2009-0058. [DOI] [PubMed] [Google Scholar]
- 52.Wheeler S, et al. Tumor epidermal growth factor receptor and EGFR PY1068 are independent prognostic indicators for head and neck squamous cell carcinoma. Clin Cancer Res. 2012;18(8):2278–2289. doi: 10.1158/1078-0432.CCR-11-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magkou C, et al. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast Cancer Res. 2008;10(3):R49. doi: 10.1186/bcr2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du L, Shen J, Weems A, Lu SL. Role of phosphatidylinositol-3-kinase pathway in head and neck squamous cell carcinoma. J Oncol. 2012;2012:450179. doi: 10.1155/2012/450179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 56.Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012;122(6):1951–1957. doi: 10.1172/JCI59889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wickham H. The split-apply-combine strategy for data analysis. J Stat Softw. 2011;40(1):1–29. [Google Scholar]
- 59.Mathes C, Friis S, Finley M, Liu Y. QPatch: The missing link between HTS and ion channel drug discovery. Comb Chem High Throughput Screen. 2009;12(1):78–95. doi: 10.2174/138620709787047948. [DOI] [PubMed] [Google Scholar]
