

Background: The cellular prion protein (PrPC) could be a toxicity-transducing receptor for amyloid-β (Aβ) oligomers.
Results: N1, a naturally occurring fragment of PrPC, binds Aβ oligomers, inhibits their polymerization into fibrils, and suppresses their neurotoxic effects in vitro and in vivo.
Conclusion: N1 binds tightly to Aβ oligomers and blocks their neurotoxicity.
Significance: Administration of exogenous N1 or related peptides may represent an effective therapy for Alzheimer disease.
Keywords: Alzheimer Disease, Amyloid, Neurodegeneration, Neuroprotection, Prions
Abstract
A hallmark of Alzheimer disease (AD) is the accumulation of the amyloid-β (Aβ) peptide in the brain. Considerable evidence suggests that soluble Aβ oligomers are responsible for the synaptic dysfunction and cognitive deficit observed in AD. However, the mechanism by which these oligomers exert their neurotoxic effect remains unknown. Recently, it was reported that Aβ oligomers bind to the cellular prion protein with high affinity. Here, we show that N1, the main physiological cleavage fragment of the cellular prion protein, is necessary and sufficient for binding early oligomeric intermediates during Aβ polymerization into amyloid fibrils. The ability of N1 to bind Aβ oligomers is influenced by positively charged residues in two sites (positions 23–31 and 95–105) and is dependent on the length of the sequence between them. Importantly, we also show that N1 strongly suppresses Aβ oligomer toxicity in cultured murine hippocampal neurons, in a Caenorhabditis elegans-based assay, and in vivo in a mouse model of Aβ-induced memory dysfunction. These data suggest that N1, or small peptides derived from it, could be potent inhibitors of Aβ oligomer toxicity and represent an entirely new class of therapeutic agents for AD.
Introduction
Alzheimer disease (AD)3 currently afflicts 35 million individuals worldwide, and this number is expected to increase dramatically in the coming decades as the population ages (1). If effective therapies are not developed, the disease will continue to have a devastating medical and economic impact on society. AD is associated with progressive accumulation in the brain of the amyloid-β (Aβ) peptide, a proteolytic fragment of the amyloid precursor protein (2). Strong experimental evidence supports the notion that the disease process starts with the binding of soluble oligomeric assemblies of Aβ to proteins or lipids on the surface of nerve cells. These interactions are thought to be responsible for the synaptic dysfunction underlying the cognitive decline in AD (3). So far, the identity of the molecules to which oligomers bind, as well as the nature of the downstream neurotoxic pathways, has remained largely enigmatic.
Recently, the cellular prion protein (PrPC) was identified as a high affinity receptor for Aβ oligomers (4–6). Several reports also suggest that PrPC could mediate the synaptotoxic effects of Aβ oligomers (7–13), although this evidence is still controversial (6, 14–16). The two putative binding sites for Aβ oligomers identified in PrPC (residues 23–31 and 95–105) are both encompassed within the flexible N-terminal tail of the molecule (residues 23–111) (4, 5). This region is proteolytically released as part of the normal cellular processing of PrPC to produce a soluble fragment called N1 (17–19). Interestingly, an artificial secreted form of PrPC was previously reported to suppress cognitive impairment in a mouse model of AD (14). These observations suggest that, even if PrPC is not a mediator of Aβ neurotoxicity, soluble forms of PrP such as the N1 fragment could sequester oligomers in the extracellular space and block their synaptotoxic effects (20).
Here, we extend the characterization of the PrP-Aβ interaction and show that the N1 fragment is necessary and sufficient to bind Aβ oligomers and to inhibit formation of Aβ fibrils in polymerization assays. Moreover, we show that N1 potently blocks the toxic effects of Aβ oligomers in neuronal cultures, as well as in two different animal models. Our data support the notions that small peptides derived from N1 could serve as novel therapeutic agents for AD and that enhancing production of the naturally occurring N1 fragment might constitute a mechanistic strategy for treating AD.
EXPERIMENTAL PROCEDURES
Recombinant Proteins
Murine PrP cDNA was amplified by PCR with a 5′-His6 tag and tobacco etch virus cleavage sequence (MRGSHHHHHHGENLYFQG) and with a 3′-Myc tag and stop codon (EQKLISEEDL). The PCR product was cloned into the pET101 vector (Invitrogen). Constructs were generated for murine PrP23–230, N1 (residues 23–111), and C1 (residues 112–230). Proteins were expressed in BL21 Star cells (Invitrogen). PrP23–230 and C1 were recovered from inclusion bodies and purified as described previously (21). Proper folding was confirmed by circular dichroism. N1 remained soluble after expression in bacteria and was purified in the absence of denaturing agents.
Preparation of Aβ Oligomers
Aβ1–42 peptide was synthesized and purified by Dr. James I. Elliott (Keck Biotechnology Resource Laboratory, Yale University, New Haven, CT). The peptide was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma), dried, reconstituted in anhydrous dimethyl sulfoxide to a concentration of 100 mm, and then diluted in F-12 medium (Invitrogen) to a final concentration of 100 μm. After a 16-h incubation at 22 °C, each preparation was centrifuged at 14,200 × g for 15 min, and the supernatant was used for experiments.
Size Exclusion Chromatography
Aliquots (0.5 ml) of 100 μm Aβ oligomers were injected into a Superdex 75 10/300 column (GE Healthcare) and eluted with PBS at a flow rate of 0.7 ml/min using an ÄKTA FPLC system. The peptide elution profile was monitored at 220 nm.
Immunoprecipitation Assay
The PrP-Aβ binding assay was performed as described previously (22). Aβ oligomers and recombinant PrP molecules were incubated in binding buffer (50 mm Tris (pH 7.5), 200 mm NaCl, and 0.1% Nonidet P-40) at 4 °C for 4 h. Anti-Myc antibody 4A6 was cross-linked onto Dynabeads containing anti-mouse IgG (Invitrogen), and the beads were then incubated with each sample at 4 °C for 2 h. Beads were washed twice with binding buffer and resuspended in loading buffer for Western blot analysis.
Surface Plasmon Resonance
Binding studies were performed using the ProteOn XPR36 protein interaction array system (Bio-Rad) as described previously (6). Using PBS with 0.05% Tween and 1 mm EDTA as running buffer, equimolar amounts of PrP were captured on the surface of a ProteOn GLC sensor chip with anti-Myc antibody 4A6. Monomeric, oligomeric, or fibrillar Aβ1–42 preparations were then injected over the sensor chip. Nonspecific binding and buffer interactions were subtracted from each sensorgram, and the resulting curves were fitted using a Langmuir interaction model (ProteOn analysis software) to obtain binding constants.
Thioflavin T Assay
Various concentrations of recombinant N1 and monomeric Aβ1–42 were incubated in 10 μm thioflavin T (ThT) in 50 mm phosphate buffer (pH 7.2) at 37 °C for 16 h with shaking. Fluorescence was measured in a BioTek Synergy H1MF plate reader every 10 min (excitation at 440 nm and emission at 490 nm).
N1 Peptides
N1 peptides carrying a C-terminal Myc tag were synthesized by American Peptide Co. (Sunnyvale, CA). Sequences are as follows: P1, KKRPK PGGWH NQWNK PSKPK TNLKH VEQKL ISEEDL; P2, KKRPK PGGWN TGGSR YPGHN QWNKP SKPKT NLKHV EQKLI SEEDL; P3, KKRPK PGGWN TGGSR YPGQG SPGGN RYPHN QWNKP SKPKT NLKHV EQKLI SEEDL; P4, KKRPK PGGWE QKLIS EEDL; and P5, HNQWN KPSKP KTNLK HVEQK LISEE DL. Peptide purity was >90% as assessed by HPLC and mass spectrometry (data not shown).
Primary Hippocampal Culture and Analysis of Synaptic Proteins
Primary neuronal cultures were derived from the hippocampuses of 2-day-old postnatal mice and cultured as described previously (6). Briefly, neurons were plated on 35-mm dishes (600,000 cells/dish) precoated with 25 g/ml poly-d-lysine (Sigma P6407) in B27/Neurobasal-A medium supplemented with 0.5 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen). Experiments were performed 12 days after plating.
Neurons were exposed for 3 h to Aβ oligomers (1 μm) that had been preincubated for 1 h at 4 °C with N1 (1 μm) or with vehicle. Subcellular fractionation was performed as reported previously with minor modifications (23). Briefly, neurons were homogenized using a Potter-Elvehjem homogenizer in 0.32 m ice-cold sucrose buffer (pH 7.4) containing 1 mm HEPES, 1 mm MgCl2, 1 mm EDTA, 1 mm NaHCO3, and 0.1 mm PMSF in the presence of protease (Complete, Roche Applied Science) and phosphatase (Sigma) inhibitor mixtures. Samples were centrifuged at 3000 × g for 15 min to obtain a crude membrane fraction. The pellet was resuspended in buffer containing 75 mm KCl and 0.5% Triton X-100 and centrifuged at 100,000 × g for 1 h. The final pellet, referred to as the Triton-insoluble fraction, was rehomogenized in 20 mm HEPES supplemented with protease and phosphatase inhibitors and then stored at −80 °C or directly used in further experiments. The protein concentration in each sample was quantified using the Bradford assay (Bio-Rad), and Triton-insoluble fraction-extracted proteins (5 μg) were then analyzed by Western blotting (23). The primary antibodies used were anti-GluN2A and anti-GluN2B (both 1:2000; Invitrogen), anti-GluA1 and anti-GluA2 (both 1:1000; Millipore), anti-PSD95 (postsynaptic density protein 95; 1:2000; Cayman Chemical), and anti-tubulin (1:5000; Santa Cruz Biotechnology). Western blots were quantified by densitometry using Quantity One software (Bio-Rad). All experiments were repeated on six independent culture preparations (n = 6).
Caenorhabditis elegans Assay
N2 worms were obtained from the Caenorhabditis Genetic Center (University of Minnesota) and propagated on solid nematode growth medium seeded with Escherichia coli (strain OP50, Caenorhabditis Genetic Center). Nematodes (L3-L4 larval stage) were collected by washing plates with M9 buffer, transferred to tubes, centrifuged, and washed twice with 5 mm PBS (pH 7.4) to eliminate bacteria. Synthetic Aβ1–42 oligomers (0.1 μm) were incubated with 0.3 μm N1 in 5 mm PBS (pH 7.4) or with anti-Aβ antibody 4G8 (1:500 (v/v); Covance) in 5 mm PBS (pH 7.4) at 25 °C for 30 min and administered to C. elegans (100 worms/100 μl) (24, 25). After 2 h of orbital shaking, worms were transferred onto new nematode growth medium plates seeded with E. coli. The pharyngeal pumping rate was scored after 2 h of recovery by counting the number of times the terminal bulb of the pharynx contracted over a 1-min interval.
Mouse Studies
C57BL/6 mice were obtained from Charles River Laboratories (Calco, Italy). All procedures involving animals and their care were conducted according to European Union and Italian laws and policies and in accordance with the United States Department of Agriculture Animal Welfare Act and the National Institutes of Health Policy on Humane Care and Use of Laboratory Animals. Intracerebroventricular cannulation was performed as described previously (6). Briefly, mice were anesthetized with Forane (Abbott Laboratories) and mounted on a stereotaxic apparatus (model 900, David Kopf Instruments, Tujunga, CA). A 7-mm-long guide cannula was implanted into the cerebral lateral ventricle (Latera ±1.0 and Dorsal-Ventral −3.0 from the dura with an incisor bar at 0°) and secured to the skull with two stainless steel screws and dental cement. Mice were allowed 10–15 days to recover from surgery before each experiment. For treatments, Aβ oligomers (1 μm), prepared as described previously (6), and N1 (1 and 5 μm) were co-incubated on ice for 15 min before intracerebroventricular microinfusion. The novel object recognition test was then performed following a previously published protocol (6). Memory was expressed as a discrimination index (i.e. (seconds on novel object − seconds on familiar object)/(total seconds on both objects)). Animals with no memory impairment a spent longer time investigating the novel object, giving a higher discrimination index.
RESULTS
Characterization of Aβ Oligomers
To study the interaction of PrP molecules and Aβ oligomers in vitro, we used a synthetic Aβ1–42 peptide that was denatured and oligomerized following a standard protocol to generate Aβ-derived diffusible ligands, hereafter referred to as Aβ oligomers. These preparations were first characterized by size exclusion chromatography (Fig. 1A). Monomeric Aβ (blue line) migrated as a single peak, whereas Aβ oligomers (red line) eluted earlier, near the column void volume, with a second peak corresponding to the remaining monomers. These data are similar to results shown in previous studies (11, 25). To extend the characterization of Aβ oligomers, we also tested their reactivity with an oligomer-specific antibody (A11) by a nondenaturing slot blot assay (Fig. 1B). In this experiment, Aβ monomers, oligomers, and fibrils were immobilized on a nitrocellulose membrane and probed with different antibodies. As expected, we found that an oligomer-specific antibody (A11) recognized Aβ oligomers, but not monomers or fibrils. In contrast, an anti-Aβ antibody (6E10) recognized all three samples.
FIGURE 1.

Biochemical characterization of Aβ preparations. A, size exclusion chromatography of Aβ preparations eluted in PBS. In contrast to monomeric Aβ, which produced a single peak (blue line), oligomeric preparations produced two peaks (red line), one that corresponds to the residual monomer and a second, higher molecular size peak eluting near the void volume that corresponds to oligomers. An elution trace with buffer (black line) is shown for comparison. B, slot blot analysis of Aβ monomers, oligomers, and fibrils. Oligomer-directed antibody A11 recognizes Aβ oligomers, but not monomers or fibrils. In contrast, anti-Aβ antibody 6E10 recognizes all three species.
N1 Binds to Synthetic Aβ Oligomers with Nanomolar Affinity
To test whether PrPC or the N1 fragment interacts with Aβ oligomers, we incubated full-length recombinant PrPC (residues 23–230; hereafter referred to as PrP23–230) and N1 (residues 23–111), both tagged with a Myc epitope at the C terminus, with synthetic Aβ oligomers, prepared and characterized following previously defined conditions (4, 6). As a negative control, we used another Myc-tagged PrP fragment (residues 112–230; called C1), which is equivalent to the C-terminal half of PrPC that remains attached to the cell membrane after cleavage of the N1 fragment (17–19). An anti-Myc antibody (4A6) was used to immunoprecipitate the different PrP molecules. The presence of Aβ in each immunoprecipitation reaction was then tested by Western blotting. We found that Aβ oligomers coprecipitated with both PrP23–230 and N1, but not with C1 (Fig. 2). These results indicate that PrP23–230 and N1, but not C1, are able to bind Aβ oligomers.
FIGURE 2.

N1 binds to synthetic Aβ oligomers. Myc-tagged recombinant PrP23–230, N1, and C1 were incubated with Aβ oligomers and immunoprecipitated (IP) using blank beads (lanes 1, 3, and 5) or beads coated with anti-Myc antibody 4A6 (lanes 2, 4, and 6). Samples were analyzed by Western blotting (WB) using anti-Myc antibody 4A6 (upper panel) or anti-Aβ antibody 6E10 (lower panel).
To further validate these results, we employed surface plasmon resonance (SPR), a technique that allows estimation of kinetic and binding constants for protein-protein interactions (5, 6). Myc-tagged PrP23–230, N1, or C1 was captured on the surface of SPR chips that were previously coated with anti-Myc antibody 4A6. Different concentrations of synthetic Aβ oligomers (Fig. 3) or monomers or fibrils (data not shown) were then assayed for binding. We detected dose-dependent binding of Aβ oligomers, but not monomers or fibrils, to PrP23–230 and N1. These two PrP molecules showed almost identical affinities for Aβ oligomers: 30.08 × 10−9 m for PrP23–230 and 17.34 × 10−9 m for N1. In contrast, no binding was observed when C1 (Fig. 3) or anti-Myc antibody alone (data not shown) was immobilized. The binding constants (kon, koff, and KD) for the interaction of PrP23–230 or N1 with Aβ oligomers were comparable to the values we described in a previous report for brain-derived PrPC (6).
FIGURE 3.

PrP23–230 and N1 interact with Aβ oligomers with similar binding constants. Different concentrations of synthetic Aβ oligomers were injected for 3 min over sensor surfaces on which 1200 resonance units (RU) of PrP23–230, N1, or C1 had been previously captured by anti-Myc antibody 4A6, followed by a wash with buffer alone. Sensorgrams show Aβ binding in RU. Aβ oligomers bound to PrPC or N1 in a dose-dependent manner, but did not bind to C1. The data were fitted using the Langmuir equation, modeling a simple bimolecular interaction. Black lines indicate kon = 1.69 × 103 m−1 s−1, koff = 5.21 × 10−5 s−1, and KD = 30.08 × 10−9 m for PrP23–230 and kon = 4.29 × 103 m−1 s−1, koff = 7.44 × 10−5 s−1, and KD = 17.34 × 10−9 m for N1.
Of note, the concentration of Aβ oligomers in this assay is based on the concentration of monomers used as starting material. However, because each Aβ oligomer particle contains multiple subunits, the actual concentration of oligomers is lower than estimated. Therefore, the affinity of PrPC or N1 for Aβ oligomers, as calculated by SPR, is likely to be in the subnanomolar range (e.g. if each oligomeric particle includes 50 Aβ monomers, the affinity calculated by SPR is 50 times higher).
Collectively, these results demonstrate that the N-terminal region of PrPC, corresponding to the N1 fragment, is necessary and sufficient for binding to synthetic Aβ oligomers. Therefore, all subsequent experiments were conducted with N1.
N1 Binds to a Transient Population of Aβ Oligomers and Inhibits the Formation of Amyloid Fibrils
To characterize further the PrP-Aβ interaction, we tested binding of N1 (Fig. 4A) or C1 (data not shown) to different Aβ assemblies formed during polymerization into amyloid fibrils. Freshly redissolved synthetic Aβ peptide was incubated in polymerization buffer at 37 °C for 8 h. Aliquots of the polymerization reaction were collected every 2 h and immediately flowed over SPR sensor chips on which Myc-tagged PrP molecules had already been captured with an anti-Myc antibody. These analyses revealed that N1, but not C1, bound to a population of Aβ assemblies that first appeared after 2 h of incubation, with the highest levels of binding detected at 4 h (Fig. 4A). These results are consistent with the kinetics of formation of Aβ oligomers reported previously (25). No binding was detected at 8 h or at later time points (data not shown). These results indicate that the Aβ species recognized by N1 are generated transiently, early during the polymerization reaction.
FIGURE 4.
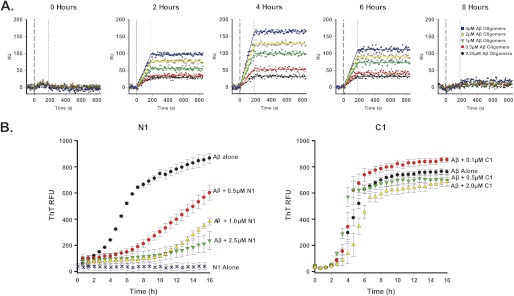
N1 binds selectively to Aβ species generated early during the polymerization process. A, solutions containing different concentrations of freshly redissolved Aβ peptide were incubated at 37 °C, and every 2 h during the polymerization process, samples were collected and injected for 3 min over sensor surfaces on which Myc-tagged recombinant N1 had previously been captured by antibody 4A6. Chips were then washed with buffer alone. Maximum amounts of PrP-binding Aβ species were present after 4 h of polymerization. B, a 20 μm aliquot of freshly redissolved Aβ peptide was incubated with 10 μm ThT at 37 °C for 20 h in presence or absence of N1 or C1. Polymerization was monitored by detecting ThT fluorescence (excitation at 440 nm and emission at 490 nm). N1 inhibited Aβ polymerization in a dose-dependent fashion, whereas C1 had no effect. RFU, relative fluorescence units.
On the basis of this evidence, we predicted that N1 could act as a soluble inhibitor of Aβ polymerization, blocking the formation of amyloid fibrils. To test this hypothesis, we utilized a ThT-based assay. ThT fluorescence shifts to red when the dye binds to β-sheet-rich protein assemblies (26). This effect is stronger for mature amyloid fibrils than for oligomeric assemblies of Aβ (27). Therefore, the formation of Aβ fibrils can conveniently be followed in real time by detecting ThT fluorescence. We incubated freshly redissolved Aβ in buffer containing 10 μm ThT in the presence of different concentrations of N1 or C1 and then monitored the fluorescence of ThT for 16 h at 37 °C. In absence of N1, Aβ polymerization proceeded with a lag phase (0–2 h) followed by rapid growth (2–6 h), reaching a plateau after 6–8 h. In presence of N1, but not C1, Aβ polymerization was delayed in a dose-dependent manner, with a significantly extended lag phase (6–16 h) (Fig. 4B). At the highest dose of N1 tested (2.5 μm, corresponding to a 1:8 molar ratio of N1 to Aβ), polymerization was almost completely inhibited. These data demonstrate that N1 binds to and stabilizes an intermediate Aβ form that appears along the polymerization pathway, blocking further maturation into amyloid fibrils.
Positively Charged Residues in Two N-terminal Regions of PrPC Dictate the Amount or Size of Aβ Oligomers Captured, but Not the Affinity of Binding
Recent reports have identified two major Aβ-binding sites in the N-terminal half of PrPC (residues 23–31 and 95–105) (4, 5). Both of these regions contain multiple basic residues, a feature that may directly determine their ability to interact with Aβ oligomers. To test the role of these positively charged residues in the PrP-Aβ interaction, we engineered N1 molecules carrying multiple substitutions that abolished the positive charges in the two Aβ-binding regions. Residues 23–27 (KKRPK) were mutated to GAAPA, and residues 95–110 (HNQWNKPSKPKTNMKH) were mutated to HNQWNAPSAPATNMAH (Fig. 5A). The interaction of this mutant form of N1 (referred to as “neutral” N1 (nN1)) with Aβ oligomers was then tested by SPR. Similar amounts of N1 and nN1 were captured on the surface of the SPR chip, as demonstrated by binding of an anti-PrP antibody (SAF32) to the chip surface (Fig. 4B). We observed significantly reduced binding of Aβ oligomers to nN1 compared with the wild-type N1 control (Fig. 5B). Surprisingly, however, N1 and nN1 showed similar dissociation constants and on/off rates for binding to Aβ oligomers (KD = 19.06 × 10−9 m for N1 and 20.75 × 10−9 m for nN1) (see Fig. 5B legend). These results suggest that the positively charged residues in the two Aβ-binding sites do not dictate the kinetics of association between N1 and Aβ oligomers or the stability of the complex, although they influence the capacity of the molecule to capture the oligomers or the size of the oligomers captured.
FIGURE 5.

Neutralization of charged residues in N1 reduces binding to Aβ oligomers without affecting binding constants. A, each of the four positively charged amino acids (indicated in red) in the two Aβ-binding regions were mutated to glycine or alanine, and the corresponding recombinant N1 mutant molecule (nN1) was expressed and purified. B, Aβ oligomers or anti-PrP antibody SAF32 was injected for 3 min over sensor surfaces on which 800 RU) of Myc-tagged recombinant N1 or nN1 had been previously captured by antibody 4A6, followed by buffer wash. The data were fitted using the Langmuir equation. Mutations in nN1 reduced the interaction with Aβ oligomers (calculated as maximum observed binding) by 56.9%. However, similar kinetic constants were measured for N1 and nN1. For N1, kon = 4.03 × 103 m−1 s−1, koff = 7.68 × 10−5 s−1, KD = 19.06 × 10−9 m, and Rmax = 490.26 RU; and for nN1, kon = 2.52 × 103 m−1 s−1, koff = 5.23 × 10−5 s−1, KD = 20.75 × 10−9 m, and Rmax = 210.92 RU.
The Ability of N1 to Bind Aβ Oligomers Is Influenced by Residues 32–94
To further characterize the interaction of N1 with Aβ oligomers, we synthesized a series of N1-derived peptides carrying different deletions between the two predicted binding sites (residues 23–31 and 95–105), referred to as P1 (Δ51–94), P2 (Δ41–94), and P3 (Δ32–94). Two additional peptides corresponding to the individual binding sites, called P4 and P5 (residues 23–31 and 95–111, respectively), were used as controls (Fig. 6A).
FIGURE 6.

Binding of N1 to Aβ oligomers is strongly influenced by residues 32–94. A, schematic of N1 peptides. Peptides were synthesized with a series of deletions between the two predicted binding sites (residues 23–31 and 95–105): P1 (Δ51–94), P2 (Δ41–94), and P3 (Δ32–94). Two additional peptides corresponding to each individual binding site, called P4 and P5 (residues 23–31 and 95–111, respectively), were used as controls. B, Aβ oligomers were injected for 4 min over sensor surfaces on which 320 RU of Myc-tagged N1, 150 RU of P1, 115 RU of P2, 85 RU of P3, 30 RU of P4, and 60 RU of P5 had been previously captured by anti-Myc antibody 4A6, followed by buffer wash. Deletions between the two binding sties (residues 23–31 and 95–105) progressively reduced binding to Aβ oligomers: 39% for P1, 72% for P2, and for 92% for P3. P4 and P5 did not bind a significant amount of Aβ oligomers. Curves for binding to N1, P1, and P2 were fitted using the Langmuir equation, modeling a simple bimolecular interaction. White lines indicate kon = 1.60 × 103 m−1 s−1, koff = 6.54 × 10−5 s−1, and KD = 40.9 × 10−9 m for N1; kon = 8.71 × 103 m−1 s−1, koff = 1.11 × 10−4 s−1, and KD = 12.8 × 10−9 m for P1; and kon = 5.21 × 103 m−1 s−1, koff = 1.37 × 10−4 s−1, and KD = 26.3 × 10−9 m for P2. Signals for P3, P4, and P5 were too low to be fit.
To analyze the relative ability of the different N1 peptides to bind Aβ oligomers, we immunocaptured Myc-tagged recombinant N1 and synthetic peptides in parallel lanes of a SPR chip. Aβ oligomers were then flowed across the different lanes, and the interactions were measured simultaneously (Fig. 6B). We found that deleting the octarepeat region (P1, Δ51–94) decreased binding to Aβ oligomers by ∼39%. Binding was further diminished by deleting 10 additional residues (P2, Δ41–94; 72% reduction) and almost completely abolished by extending the deletion to residue 32 (P3, Δ32–94, 92% decrease). Similarly, binding was barely detectable for peptides corresponding to each individual binding site (P4 and P5, 96 and 99%, respectively). These data demonstrate that binding of N1 to Aβ oligomers depends critically on residues between the two binding sites (residues 23–31 and 95–105).
N1 Blocks Aβ Oligomer-mediated Loss of Postsynaptic Markers in Hippocampal Neurons
To test the relevance of the N1-Aβ interaction at a biological level, we sought to determine whether N1 could rescue the toxic effects of Aβ oligomers in primary neuronal cultures, as measured by detecting the levels of five postsynaptic marker proteins. Postnatal mouse hippocampal neurons were kept in culture for 12 days and then incubated for 3 h with Aβ oligomers. The amounts of several glutamate receptor subunits and other postsynaptic markers were analyzed by Western blotting of the Triton-insoluble fractions. A representative Western blot is shown in Fig. 7A. A 3-h treatment with Aβ oligomers (1 μm) induced an ∼60% loss of GluN2A and GluN2B, two subunits of the NMDA receptor (Fig. 7B, panels i and ii). We also observed a 73% reduction of GluA1 and a 70% reduction of GluA2, subunits of the AMPA receptor (Fig. 7B, panels iii and iv), as well as a 70% reduction of PSD95 (panel v). The levels of these proteins were not significantly affected by treatment with 1 μm N1 alone. However, preincubation of Aβ oligomers with N1 (in a 1:1 molar ratio) for 1 h significantly rescued the levels of all of these synaptic markers: GluN2A and GluN2B levels were restored by 65 and 73%, respectively; GluA1 and GluA2 by 66 and 104%, respectively; and PSD95 by 55%. The level of a control protein (tubulin) was not affected by either Aβ oligomers or N1 (Fig. 7B, panel vi). These results indicate that the interaction with N1 antagonizes the Aβ oligomer-mediated loss of synaptic markers.
FIGURE 7.

N1 antagonizes the loss of postsynaptic makers of Aβ oligomers in cultured neurons. Cultures of hippocampal neurons were treated for 3 h with vehicle (control (CTR)), N1 alone (1 μm), Aβ oligomers alone (1 μm), or Aβ oligomers preincubated with N1. Postsynaptic marker proteins were then measured by Western blotting. A, representative Western blots. B, quantification of several independent experiments. Aβ oligomers induced a loss of postsynaptic markers, which was significantly attenuated by preincubation with N1. **, p < 0.01 for control versus Aβ oligomers (two-way analysis of variance and Bonferroni post hoc test); ##, p < 0.01 for Aβ oligomers versus Aβ oligomers + N1; $, p < 0.05 for N1 versus Aβ oligomers + N1. Values are means ± S.E. (n = 6). Tubulin levels were not affected by Aβ oligomer treatment (p > 0.05 (two-way analysis of variance); n = 6).
N1 Blocks the Disruptive Effects of Aβ Oligomers in Animal Models
To validate these results in vivo, we turned to two different animal models of Aβ toxicity. The first utilizes the nematode C. elegans. The pharyngeal pumping rate of C. elegans can be rapidly reduced by sublethal doses of chemical stressors. A previous report showed that both rhythmic contraction and relaxation of the pharyngeal muscle in C. elegans are significantly impaired (∼50%) by feeding the nematodes with synthetic Aβ oligomers (25). Although it remains uncertain whether the biochemical mechanisms underlying Aβ toxicity in worms and humans are identical, this model has been previously utilized as a surrogate assay for Aβ toxicity. Here, we employed this assay to test the anti-Aβ effect of N1. Consistent with previous observations, the pumping rate of the worms was significantly impaired when they were fed 0.1 μm Aβ oligomers (Fig. 8A). N1 alone at >0.5 μm showed a slight but significant inhibitory effect on the pumping rate (5 and 10% inhibition at 0.5 and 1 μm, respectively) (data not shown), whereas no effect was observed when N1 was administered at <0.5 μm. However, preincubation of Aβ oligomers with 0.3 μm N1 robustly antagonized the oligomer-dependent effect on the pumping rate (Fig. 8A). The inhibitory effect of N1 was comparable to that of anti-Aβ antibody 4G8 (25) (data not shown). These results demonstrate that N1 counteracts the toxic effects of Aβ oligomers in vivo using C. elegans as a model system.
FIGURE 8.

N1 antagonizes the toxic effects of Aβ oligomers in animal models. A, freshly dissolved Aβ1–42 (100 μm) was incubated at 25 °C for 5 h, diluted to 10 μm in 5 mm PBS (pH 7.4), and incubated or not for an additional 30 min with 0.3 μm N1 in PBS. Samples were then administered to worms. Control worms were fed vehicle (control (CTR)) or 0.3 μm N1 alone. The pharyngeal pumping rate was scored after 2 h of recovery by counting the number of times the terminal bulb of the pharynx contracted over a 1-min interval (pumps/min). Values are means ± S.E. (n = 30). Two-way analysis of variance showed that N1 significantly reduced the inhibitory effect of Aβ on the pumping rate (p < 0.01). **, p < 0.001 for Aβ versus vehicle; °°, p < 0.001 for Aβ versus Aβ + N1 (Bonferroni test). B, the effect of Aβ oligomers (1 μm) on memory was investigated in male C57BL/6 mice using a novel object recognition task after intracerebroventricular injections (7.5 μl) in the presence or absence of N1. Bars show the discrimination index (mean ± S.E.) for exploration of familiar versus novel objects. Mice treated with Aβ oligomers (n = 19) spent an equal time investigating both objects, whereas vehicle-injected control mice (n = 21) spent significantly more time investigating the novel object. *, p < 0.05 (Student's t test). The memory impairment effect was abrogated when Aβ oligomers were incubated with N1 before injection (for 1 μm N1, n = 8; and for 5 μm N1, n = 17). One-way analysis of variance (F2,41 = 4.7; p = 0.015) and Tukey's post hoc test (#, p < 0.05) revealed a significant recovery of memory in mice receiving N1 at 5 μm compared with Aβ oligomer-treated mice.
To further substantiate these observations, we evaluated the protective effect of N1 on the memory dysfunction caused by intracerebroventricular injection of synthetic Aβ oligomers in mice using a novel object recognition task. This assay was recently applied to demonstrate that synthetic Aβ oligomers induce memory impairment in mice in a PrPC-independent fashion (6). Here, we hypothesized that, regardless of whether PrPC mediates the neurotoxicity of Aβ oligomers, the N1 fragment could sequester oligomers in the extracellular space and prevent their toxic effects. To test this hypothesis, synthetic Aβ oligomers (1 μm) were incubated with various amounts of N1 (0, 1, or 5 μm) for 15 min before microinfusion into the lateral ventricles of C57BL/6 mice. Animals were initially trained in an arena containing two objects that they could explore freely (familiarization phase) and, 24 h later, were exposed to one familiar and one new object (test phase). As expected, mice treated with Aβ oligomers were unable to distinguish the new object (e.g. no significant difference in the percentage of time spent investigating new and old objects) (Fig. 8B) compared with vehicle-injected controls. However, preincubation of Aβ oligomers with N1 rescued the behavioral impairment in a dose-dependent fashion (Fig. 8B). These results demonstrate that N1 blocks the Aβ oligomer-induced memory impairment in vivo.
DISCUSSION
Despite uncertainty regarding the role of cell-surface PrPC in mediating the toxic effects of Aβ oligomers, the ability of these two molecules to interact with high affinity is widely agreed upon (28). In this study, we have presented a detailed characterization of the binding interaction between Aβ oligomers and the major, soluble, physiologically generated, N-terminal fragment of PrPC (N1). We showed that N1 fully retains the ability to bind Aβ oligomers, whereas the entire C-terminal half of PrPC is dispensable. N1 binds selectively to transient Aβ species forming during the polymerization of this peptide into amyloid fibrils, and it thereby inhibits the polymerization process. We have also reported that the binding capacity, but not the affinity, of N1 for Aβ oligomers is influenced by positively charged residues in the two Aβ-binding sites (residues 23–31 and 95–105) and the distance between the two sites. Finally, we demonstrated that N1 is a potent inhibitor of Aβ toxicity based on its ability to prevent the detrimental effects of Aβ oligomers in cultured hippocampal neurons, C. elegans, and mice. Collectively, these data argue that, whether or not the neurotoxicity of Aβ oligomers is elicited in a PrPC-dependent fashion, N1-based compounds may represent novel tools for preventing their neurotoxic effects.
How Does PrP Interact with Aβ Oligomers?
Previous studies have identified two distinct regions in PrPC involved in the binding of Aβ oligomers. The first site (residues 95–105) was mapped by employing a combination of deletion mutants and antibody treatments (4), whereas the second (residues 23–27) was identified using SPR and EPR spectroscopy (5). Synthetic peptides corresponding to each individual binding site failed to inhibit aggregation Aβ oligomers (20). Our SPR data confirm that peptides containing the two individual binding sites alone or combined do not interact with Aβ oligomers, suggesting that additional regions in the N terminus of PrPC may contribute to the binding. Furthermore, our data suggest that efficient binding requires separation of the two sites by additional amino acids. Future studies will address whether this phenomenon depends only on the distance separating the two sites, whether there is a sequence-specific contribution of particular residues in the linker region, or both.
Our SPR data show, surprisingly, that basic residues within the two Aβ-binding sites do not influence the affinity of the interaction between N1 and Aβ oligomers. However, in absence of the positively charged residues, a lower mass of Aβ oligomers interacts with N1. This effect could be explained if smaller Aβ oligomers were captured by the neutralized PrP sites. Therefore, the presence of positively charged residues in the two Aβ-binding sites could determine the size of the oligomers that interact with PrPC. Our data also suggest that binding between N1 and Aβ oligomers does not depend on a simple electrostatic interaction between negatively charged residues on Aβ and positively charged residues on PrPC. Rather, a conformational rearrangement of the flexible N-terminal domain of PrPC could govern the interaction with Aβ oligomers. This observation is further supported by our observation that the binding ability of the N terminus is influenced by the length of the region separating the two binding regions. Further studies will clearly be needed to elucidate this hypothesized mechanism.
Our observation that N1 binds to Aβ species generated early during the polymerization process is consistent with evidence that neither monomeric nor fibrillar forms of Aβ have significant affinity for PrPC and that binding is specific for oligomers. Importantly, N1 is capable of markedly suppressing the Aβ polymerization process itself, as measured by ThT binding, suggesting that interaction with N1 blocks or slows further growth of oligomers into longer fibrils. Similar effects on Aβ polymerization have also been reported recently by Nieznanski et al. (20) using longer portions of the PrP molecule.
N1 Neutralizes Toxic Assemblies of Aβ
Previous studies reported that N1 exerts a neuroprotective effect in neuronal cells by reducing p53-mediated cell death (29). Recently, N1 was shown to protect also against the cytotoxic effects of Aβ monomers and oligomers (30). This effect was possibly explained by the ability of N1 to activate intracellular cell survival mechanisms involving the Akt/p53 pathway. Another recent study provided direct experimental support for the notion that N1 interacts directly with Aβ oligomers, blocking their neurotoxicity in transformed cell lines (based on cell viability assays) (20). Here, we have shown for the first time that N1 counteracts Aβ effects in primary neurons and suppresses Aβ toxicity in two different animal models (C. elegans and mice). These effects were seen when N1 was mixed with preformed Aβ oligomers, even at low stoichiometric ratios, suggesting that N1 blocks the interaction of a specific oligomeric form of Aβ with cellular receptors responsible for toxicity. Whether such receptors include PrPC itself remains to be determined.
In principle, attempts to develop Aβ-directed agents would be facilitated by knowing the structure of the toxic Aβ species at high resolution. Unfortunately, research has been hampered by the fact that Aβ assemblies are often structurally heterogeneous and metastable. In this study, we observed that N1 stabilizes a particular subset of Aβ assemblies and inhibits the neurotoxic effects of Aβ preparations in vitro and in vivo. Such a rescue effect provides evidence that Aβ species recognized by N1 are neurotoxic. Therefore, as also suggested by a recent study (8), N1-based compounds may allow the isolation of toxic Aβ species from biological samples (e.g. cerebrospinal fluid) or quantitation of the amount of toxic species in clinical samples. Moreover, the biophysical characterization of the complex formed by synthetic Aβ oligomers and N1 could provide important insights regarding the oligomerization pathway and the structure of toxic Aβ oligomers, laying the groundwork for the rational design of anti-AD therapeutics.
N1-based Therapies for Treatment of Neurodegenerative Diseases
Soluble Aβ oligomers represent a primary pharmacological target for reducing synaptic dysfunction and cognitive decline in AD (31). For example, anti-Aβ antibodies have been extensively tested in animal models and human patients, and although they have not proven effective in clinical trials, this may be because they have not been administered early enough in the disease process (32). Our data indicate that N1 interacts with Aβ oligomers with an affinity comparable to anti-Aβ antibodies and blocks their toxicity in a variety of experimental settings, including cells, worms, and mice. Therefore, development of N1-based compounds (e.g. short N1-derived peptides) may represent a novel pharmacological approach for treatment of AD.
N1 is a naturally occurring soluble fragment that is generated by endogenous proteolytic processing of membrane-bound PrPC at the α-site (residues 111 and 112) (18). Up to 50% of the PrPC in cells and brain is cleaved at this site. There is uncertainty about the identity and cellular location of the relevant proteases, although cell-surface ADAM proteases represent one set of candidates (17). Importantly, α-cleavage of PrPC can be stimulated pharmacologically, e.g. by activators of protein kinase C (33). It is possible that such agents could have therapeutic effects in AD by increasing production of the neuroprotective N1 fragment.
A recent study provided evidence that PrPC could bind not only Aβ oligomers but also other β-sheet-rich protein assemblies (34). This unexpected property of PrPC seems to rely on the same N-terminal domains that are involved in binding to Aβ oligomers (residues 23–31 and 95–105). Therefore, it is possible that N1-derived compounds will possess the ability to block the neurotoxic effects of several other aggregated proteins linked to different neurodegenerative diseases.
Acknowledgments
We thank Frederick P. Bowman and Dr. Elena S. Klimtchuk for help with protein purification and circular dichroism, respectively. We also thank Flamma S.p.A. (Bergamo, Italy) for the kind gift of N-(9-fluorenyl)methoxycarbonyl (Fmoc) amino acids.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS065244 and R01 NS040975 (to D. A. H.). This work was also supported by the Eben Ellison Foundation (to D. A. H.), Cassa di Risparmio delle Provincie Lombarde (CARIPLO, Italy) Grants 2009-2425 (to T. B.) and 2009-2543 (to L. D.), the Alzheimer's Drug Discovery Foundation (ADDF; to T. B.), Telethon Foundation Grant GGP10120 (to L. D.), and Alzheimer's Association Grant NIRG-10-171655 (to L. D.).
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- PrPC
- cellular prion protein
- ThT
- thioflavin T
- SPR
- surface plasmon resonance
- nN1
- neutral N1
- RU
- resonance units.
REFERENCES
- 1. Alzheimer's Association (2012) 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 8, 131–168 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe D. J. (2011) Alzheimer's disease. Cold Spring Harb. Perspect. Biol. 3, a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selkoe D. J. (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S., Yadav S. P., Surewicz W. K. (2010) Interaction between human prion protein and amyloid-β (Aβ) oligomers: role of N-terminal residues. J. Biol. Chem. 285, 26377–26383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., Chiesa R., Gobbi M., Salmona M., Forloni G. (2010) Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Um J. W., Nygaard H. B., Heiss J. K., Kostylev M. A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E. C., Strittmatter S. M. (2012) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. You H., Tsutsui S., Hameed S., Kannanayakal T. J., Chen L., Xia P., Engbers J. D., Lipton S. A., Stys P. K., Zamponi G. W. (2012) Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kudo W., Lee H. P., Zou W. Q., Wang X., Perry G., Zhu X., Smith M. A., Petersen R. B., Lee H. G. (2012) Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freir D. B., Nicoll A. J., Klyubin I., Panico S., Mc Donald J. M., Risse E., Asante E. A., Farrow M. A., Sessions R. B., Saibil H. R., Clarke A. R., Rowan M. J., Walsh D. M., Collinge J. (2011) Interaction between prion protein and toxic amyloid-β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barry A. E., Klyubin I., Mc Donald J. M., Mably A. J., Farrell M. A., Scott M., Walsh D. M., Rowan M. J. (2011) Alzheimer's disease brain-derived Amyloid-β-mediated inhibition of LTP in Vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung E., Ji Y., Sun Y., Kascsak R. J., Kascsak R. B., Mehta P. D., Strittmatter S. M., Wisniewski T. (2010) Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calella A. M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I. M., Aguzzi A. (2010) Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kessels H. W., Nguyen L. N., Nabavi S., Malinow R. (2010) The prion protein as a receptor for amyloid-β. Nature 466, E3–E4; discussion E4–E5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cissé M., Sanchez P. E., Kim D. H., Ho K., Yu G. Q., Mucke L. (2011) Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 31, 10427–10431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vincent B., Paitel E., Saftig P., Frobert Y., Hartmann D., De Strooper B., Grassi J., Lopez-Perez E., Checler F. (2001) The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J. Biol. Chem. 276, 37743–37746 [DOI] [PubMed] [Google Scholar]
- 18. Chen S. G., Teplow D. B., Parchi P., Teller J. K., Gambetti P., Autilio-Gambetti L. (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 270, 19173–19180 [DOI] [PubMed] [Google Scholar]
- 19. Harris D. A., Huber M. T., van Dijken P., Shyng S. L., Chait B. T., Wang R. (1993) Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 32, 1009–1016 [DOI] [PubMed] [Google Scholar]
- 20. Nieznanski K., Choi J. K., Chen S., Surewicz K., Surewicz W. K. (2012) Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J. Biol. Chem. 287, 33104–33108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zahn R., von Schroetter C., Wüthrich K. (1997) Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417, 400–404 [DOI] [PubMed] [Google Scholar]
- 22. Cissé M., Halabisky B., Harris J., Devidze N., Dubal D. B., Sun B., Orr A., Lotz G., Kim D. H., Hamto P., Ho K., Yu G. Q., Mucke L. (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gardoni F., Schrama L. H., Kamal A., Gispen W. H., Cattabeni F., Di Luca M. (2001) Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J. Neurosci. 21, 1501–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stravalaci M., Beeg M., Salmona M., Gobbi M. (2011) Use of surface plasmon resonance to study the elongation kinetics and the binding properties of the highly amyloidogenic Aβ(1–42) peptide, synthesized by depsi-peptide technique. Biosens. Bioelectron. 26, 2772–2775 [DOI] [PubMed] [Google Scholar]
- 25. Stravalaci M., Bastone A., Beeg M., Cagnotto A., Colombo L., Di Fede G., Tagliavini F., Cantù L., Del Favero E., Mazzanti M., Chiesa R., Salmona M., Diomede L., Gobbi M. (2012) Specific recognition of biologically active amyloid-β oligomers by a new surface plasmon resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J. Biol. Chem. 287, 27796–27805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saeed S. M., Fine G. (1967) Thioflavin T for amyloid detection. Am. J. Clin. Pathol. 47, 588–593 [DOI] [PubMed] [Google Scholar]
- 27. Reinke A. A., Gestwicki J. E. (2011) Insight into amyloid structure using chemical probes. Chem. Biol. Drug Des. 77, 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biasini E., Turnbaugh J. A., Unterberger U., Harris D. A. (2012) Prion protein at the crossroads of physiology and disease. Trends Neurosci. 35, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guillot-Sestier M. V., Sunyach C., Druon C., Scarzello S., Checler F. (2009) The α-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 284, 35973–35986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guillot-Sestier M. V., Sunyach C., Ferreira S. T., Marzolo M. P., Bauer C., Thevenet A., Checler F. (2012) α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid-β (Aβ)-associated cell death. J. Biol. Chem. 287, 5021–5032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 32. Panza F., Frisardi V., Solfrizzi V., Imbimbo B. P., Logroscino G., Santamato A., Greco A., Seripa D., Pilotto A. (2012) Immunotherapy for Alzheimer's disease: from anti-β-amyloid to tau-based immunization strategies. Immunotherapy 4, 213–238 [DOI] [PubMed] [Google Scholar]
- 33. Alfa Cissé M., Louis K., Braun U., Mari B., Leitges M., Slack B. E., Fisher A., Auberger P., Checler F., Vincent B. (2008) Isoform-specific contribution of protein kinase C to prion processing. Mol. Cell. Neurosci. 39, 400–410 [DOI] [PubMed] [Google Scholar]
- 34. Resenberger U. K., Harmeier A., Woerner A. C., Goodman J. L., Müller V., Krishnan R., Vabulas R. M., Kretzschmar H. A., Lindquist S., Hartl F. U., Multhaup G., Winklhofer K. F., Tatzelt J. (2011) The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]