

Abstract
Recent studies in diabetic humans and rodent models of diabetes have identified osteopathy as a serious complication of type 1 (T1D) and type 2 (T2D) diabetes. Accumulating evidence suggests that disruption of insulin and insulin-like growth factor 1 (IGF-1) homeostasis in the diabetic condition may be responsible for the observed skeletal deficits. Indeed, replacement of insulin or IGF-1 in rodent models of T1D results in significant improvement in bone healing despite ongoing moderate to severe hyperglycemia. Insulin and IGF-1 act through distinct receptors. Mice in which the receptor for insulin or IGF-1 is selectively deleted from osteoblast lineages show skeletal deficits. Despite acting through distinct receptors, insulin and IGF-1 exert their cellular activities via conserved intracellular signaling proteins. Genetic manipulation of these signaling proteins, such as insulin receptor substrate (IRS)-1 and -2, Protein Kinase B (Akt), and MAPK/ERK kinase (MEK), has uncovered a significant role for these signal transduction pathways in skeletal homeostasis. In addition to effects on skeletal physiology via canonical signaling pathways, insulin and IGF-1 may crosstalk with wingless-int. (Wnt) and bone morphogenic protein 2 (BMP-2) signaling pathways in cells of the osteoblast lineage and thereby promote skeletal development. In this review, a discussion is presented regarding the role of insulin and IGF-1 in skeletal physiology and disruptions of this axis that occur in the diabetic condition which could underlie many of the skeletal pathologies observed.
Introduction
It is now well established that humans with type 1 (T1D) or type 2 (T2D) diabetes have an increased risk of fracture [1-6]. While decreased bone density and a state of low bone turnover have been described in those with T1D, T2D is not associated with osteopenia or osteoporosis. However, recent studies have reported that subperiosteal porosity is increased in T2D patients who fracture [7,8].
The underlying mechanisms involved in skeletal deficits observed in both T1D and T2D are poorly understood. While it is likely a multifactorial process that contributes to skeletal compromise in diabetes, as is the case with most diabetes-related complications, insulin and its homolog, insulin-like growth factor 1 (IGF-1), have been implicated in the pathogenesis of skeletal deficits attributable to diabetes (i.e., diabetic osteopathy) [8]. For instance, in humans with diabetes, there is a positive correlation between bone mineral density (BMD) by dualenergy x-ray absorptiometry (DXA) and insulin dose [9,10] or urinary C-peptide excretion, a measure of endogenous insulin production [9], suggesting that endogenous and exogenous insulin may affect skeletal homeostasis in diabetes. IGF-1 concentrations are lower in diabetic patients with osteopenia, compared to those without osteopenia, and decreased serum markers of bone formation in diabetes are associated with lower IGF-1 concentrations [11-15]. Patients with T2D who have higher IGF-1 concentrations also have higher BMD and decreased vertebral fractures [12,16]. Recently, we have shown in individuals with T1D that measures of endogenous insulin, exogenous insulin dose, and serum IGF-1 concentrations all positively correlate with osteocalcin, a marker of bone formation [17].
In this review, we will explore the current knowledge, obtained in rodent models, of how insulin and IGF-1, through their cognate receptors, regulate normal skeletal physiology, and how the actions of these peptide hormones may be critical to understanding the pathogenesis and potential treatment of diabetic osteopathy.
Insulin and IGF-1 physiology
Insulin and IGF-1 are small peptide hormones (~7.5 kD) which share a high degree of homology with proinsulin. Each possesses the ability to increase glucose disposal, insulin being significantly more potent than IGF-1 [18]. Unlike insulin, which is produced by pancreatic β-cells, IGF-1 is produced predominantly by the liver, with other tissues producing smaller amounts, and circulates at high concentrations in serum [18,19]. Insulin and IGF-1, beyond their metabolic effects, can be growth-promoting peptides which influence cellular proliferation and differentiation [18]. Unlike insulin, the interaction of IGF-1 with cell-surface receptors is tightly regulated by at least six distinct high affinity carrier proteins, the IGF-binding proteins (IGFBPs), and possibly by several low-affinity IGFBP-like molecules [18,20]. The interaction of IGF-1 with IGFBPs can prevent untoward IGF-1 effects, such as uncontrolled cellular proliferation or hypoglycemia. Conversely, disruption of the IGF:IGFBP complex is a prerequisite for IGFs to exert their mitogenic and metabolic effects through the IGF-1 receptor (IGF1R) [21].
Downstream mediators of both insulin and IGF-1 signaling pathways are important in promoting osteogenesis
Insulin and IGF-1 signaling pathways utilize many of the same cellular proteins to achieve various cellular outcomes (Figure 1). Each ligand, through its cognate receptor, can mediate events via insulin receptor substrate (IRS)-1 and IRS-2 phosphorylation and subsequent activation of phosphatidylinositol (PI) 3-kinase [22] and by activation of the mitogen-activated protein (MAP)/ERK kinases [23]. A direct link between insulin and/or IGF-1 signaling and bone formation in vivo is supported by transgenic models which manipulate various proteins in these downstream signaling pathways (Figure 1). The growth factor receptor-bound protein 2 (Grb-2)–associated binder 1 (Gab1) is a scaffolding protein that is involved in both ERK activation as well as in regulating the PI3K-Akt signaling pathway. Osteoblast-specific elimination of Gab1 results in decreased trabecular bone, diminished bone formation, reduced strength, and reduced MAP/ERK and Akt activation in response to insulin or IGF-1 [24]. Mice null for IRS-1 and IRS-2 develop unique bone phenotypes; in vivo, IRS-2 appears to maintain dominance of bone formation over bone resorption, while IRS-1 regulates bone turnover [25,26]. Bone healing is also impaired in IRS-1 deficient mice and can be corrected with re-expression of IRS-1 within the fracture site [27]. Because IRS molecules mediate insulin and IGF receptor signaling, cross-talk downstream of IRS molecules may take place via insulin and IGF signaling in osteoblasts. Akt and forkhead transcription factors (FoxO) proteins are downstream mediators of IRS signaling within the PI3-kinase pathway. Mice null for Akt1 or both Akt1 and Akt2 have significant skeletal deficits which include delayed ossification, and even dwarfing [28,29]. In contrast, elimination of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), an antagonist of PI3-kinase activity, is associated with increased bone mineral density [30]. Activated Akt can phosphorylate several forkhead transcription factors (FoxO1, 3 and 4) and inactivate their transcriptional activity by excluding them from the nucleus to the cytoplasm. This mechanism is a major pathway in many cell types and tissues through which insulin and IGF-1 regulate gene transcription of many genes involved in cell growth, differentiation and metabolism. FoxO1, 3, and 4 are expressed in osteoblasts [31]; however, their regulation of skeletal homeostasis remains unclear. Elimination of FoxO1 in osteoblasts has the affect of decreasing bone mass and multi-tissue knockdown of FoxO1, 3 and 4 proteins decreases bone formation [32,33]. In contrast, newer data shows that deletion of FoxO1, 3, and 4 specifically in osteoblast progenitors results in an increase in vertebral and femoral BMD, as well as a striking increase in femoral cortical thickness [34,35]. In keeping with these in vivo findings, in vitro FoxO1 has been shown to directly interfere with the activities of runt-related transcription factor 2 (RUNX2), which is considered a “master regulator” of osteoblast development and whose expression is essential for normal bone formation [36,37]. In addition to the PI3K/Akt/FoxO pathway, activation of the MAP/ERK kinase pathway may also mediate insulin and IGF-1 events in bone. For instance, osteoblast specific expression of a constitutively active form of the MAP kinase, MEK1, is associated with accelerated skeletal development, enhanced skeletal size, and mineralization; whereas mice expressing a dominant negative form of MEK1 display delayed skeletal development, and these outcomes are believed to be mediated by effects on RUNX2 phosphorylation and transcriptional activity [38]. Taken together, these studies demonstrate that disruption of signal transduction pathways shared by insulin and IGF-1 receptors result in abnormalities of normal skeletogenesis.
Figure 1.
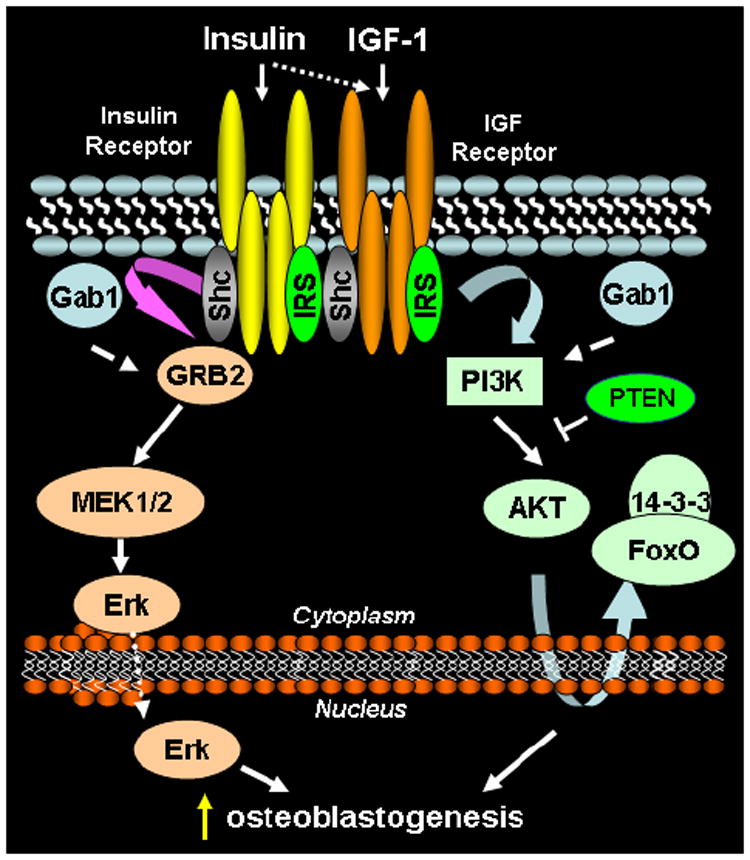
Pro-osteoblastogenic signal transduction by insulin and IGF1 receptors. Receptors at the plasma membrane bind ligands within extracellular domains, triggering receptor autophosphorylation on tyrosine residues within the cytoplasmic domain. Phosphorylated receptors recruit scaffolding proteins [src homology 2 domain-containing transforming protein C (Shc), IRS], which are subsequently phosphorylated by the receptor kinase domain. Recruitment of growth factor receptor bound protein 2 (GRB2) to the receptor-Shc complex initiates phosphorylation and activation of the MEK/ERK pathway. MEK/ERK signaling culminates with phosphorylation of osteoblastogenic transcription factors in the nucleus. IRS proteins recruit phosphatidylinositol 3-kinase to the receptor complex, resulting in kinase activation, which generates phosphatidylinositol (3,4,5) triphosphate (PIP3). PIP3 recruits AKT to the plasma membrane, resulting in AKT activation. Activated AKT phosphorylates numerous proteins, among them FoxO1, resulting in retention of FoxO1 in the cytosol, where it is unable to perform its function as a transcription factor.
IGF-1 affects on the skeleton
Research over several decades has supported a primary role for IGF-1 in anabolic bone formation [39]. However, it has only been in recent years that an essential role of IGFs in normal bone development has been confirmed through the elimination of IGF-1 and the IGF1 receptor in mice via homologous recombination [40,41]. In these animals, profound growth retardation as well as growth plate abnormalities and decreased bone calcification were observed. Later studies have further refined how elimination of IGF-1 affects bone physiology not only through the dwarfing of bones, but also by significantly decreasing bone formation rate and cortical thickness, resulting in more compact bone [42]. Studies specifically designed to examine how relative degrees of peripheral (i.e, hepatic production) IGF-1 deficiency affect bone formation have revealed only small decreases in cortical periosteal bone growth [43]. Further reductions in circulating IGF-1 concentrations (to 10-15% of controls) achieved by crossing the previously described animals with animals made null for the acid-labile subunit (ALS) of the IGFBP-3/IGF-1 complex (ALS, IGFBP-3 and IGF-1 form the major 150 kD complex responsible for carrying IGFs in the vascular compartment in mammals), results in a 10% decrease in bone mineral density and a 35% decrease in periosteal circumference and cortical thickness [43]. Thus, together these studies demonstrate that circulating (i.e., endocrine) IGF-1 can have a significant effect on several parameters of bone density and formation, as well as the overall size of the bone. Exactly how these IGF-1-mediated effects may involve anabolic effects on osteoblast activity has been recently elucidated by Zhang et al [44], who, through tissue-specific gene targeting, ablated the IGF1 receptor in mature osteoblasts using the osteocalcin promoter-Cre construct. These studies showed that mice deficient in IGF1R in mature osteoblasts were of normal size, yet demonstrated a marked decrease in cancellous bone volume, connectivity, and trabecular number, as well as a striking decrease in the rate of mineralization of osteoid matrix. Recently, the IGF1R has been eliminated in osteoblast progenitors using the osterix promoter-Cre construct. In contrast to mice lacking the IGF1R only in mature osteoblasts, those lacking the IGF1R throughout osteoblast development are growth retarded [45]. Furthermore, these mice display decreased BMD and metaphyseal deficits [45]. Thus, a significant amount of the bone-forming and mineralization actions of IGF-1 appear to be mediated via the osteoblast. Indeed, transgenic over-expression of IGF-1 in vivo under the control of the osteocalcin promoter, results in a phenotype in which bone mineral density, as measured by DXA and quantitative computed tomography, is significantly increased in transgenic mice compared with controls. Furthermore, histomorphometric measurements reveal a marked increase in femoral cancellous bone volume in mice overexpressing IGF-1 compared with controls [46]. Therefore, any alteration in the IGF system may have profound effects on anabolic bone formation.
Insulin affects on the skeleton
While many studies have clearly demonstrated that IGF-1 functions as an anabolic agent in bone, only a few studies have examined the specific role that insulin and its cognate receptor (IR) may play in regulating osteoblast physiology. Studies in vitro have shown that physiological doses of insulin promote osteoblast proliferation [47,48], collagen synthesis [49-51], alkaline phosphatase production [52,53], and glucose uptake [54,55]; nevertheless, these studies do not clarify what receptors or pathways insulin may use to promote osteogenesis. The insulin receptor is expressed in normal bone and in regenerating bone in vivo [56,57]. IR expression is detected throughout differentiation, from pre-osteoblast to mature osteoblast, in MC3T3-E1 cells (Figure 2A). IR expression is detected in early bone progenitor cells (bone marrow stromal cells and C3H10T1/2 cells) as well as in ex vivo bone cell cultures (mouse calvarial cells) (Figure 2B). Furthermore, insulin and IGF-1 can activate the PI3K and MEK/ERK pathways in osteoprogenitor cells and in osteoblastic cells (Figure 2C and 2D, respectively), suggesting that insulin signaling is operative in osteoblastic cells at various stages of differentiation. While elimination of the IR in all skeletal elements has been reported to result in no skeletal abnormalities or in diminished trabecular architecture [57,58], two recent reports queried specifically the importance of insulin meditated events in osteogenesis. Both studies knocked down IR expression specifically in osteoblasts using Cre-mediated recombination [57,59]. It was observed that knock-down of IR in osteoblasts resulted in altered bone formation [59] and in abnormal trabecular architecture [57]. While both models support a role for insulin in osteoblast development, Ferron et al reported only a partial (60%) knock-down of the IR using the Col1a1-Cre mouse, while Fuzele et al eliminated the IR only in mature osteoblasts using the osteocalcin-Cre construct [57,59]. Thus, additional informative models are needed to fully appreciate the role that insulin signaling via the IR may play throughout osteoblastogenesis and skeletal development.
Figure 2.
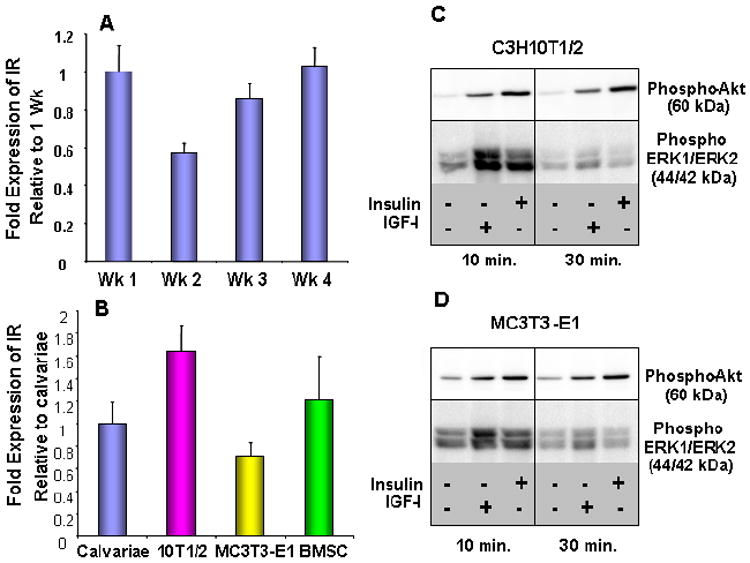
Insulin and IGF1 signal transduction is functional in osteoblast precursors in vitro. IR mRNA levels measured by quantitative RT-PCR in MC3T3-E1 cells after 1, 2, 3, or 4 weeks of differentiation with ascorbate and β-glycerol phosphate (A) and in primary calvariae, C3H10T1/2, MC3T3-E1, and bone marrow stromal cell cultures (B). Western blot detection of Akt and ERK1/2 phosphorylation in C3H10T1/2 (C) or MC3T3-E1 (D) cell lysates prepared after treating cells with 10 ng/ml of insulin or IGF-1 for the indicated times.
Effects of insulin and IGF-1 on diabetic osteopathy
In rodent models of T1D, skeletal architecture, bone quality and bone integrity are compromised [see reference [60] for review of studies]. These alterations are associated with poor bone formation and regeneration [56]. Furthermore, at a molecular level, T1D impacts bone formation by down-regulating RUNX2 [61,62], and genes known to be targets of RUNX2 activity (e.g., osteocalcin, matrix metalloproteinases 9 and 13, integrin-binding sialoprotein, collagen, phosphate regulating endopeptidase homolog, X-linked (PHEX), dentin matrix acidic phosphoprotein 1 (DMP-1), alkaline phosphatase, osteopontin, vitamin D receptor, and ameloblastin) [62]. These data suggest that osteoblastogenesis is impaired from early stages of osteoblast commitment and may therefore explain the profound lack of new bone formation, compromised skeletal microarchitecture, and diminished bone strength observed in diabetic models.
In rodent models of diabetes, insulin therapy is consistently capable of improving many of the histomorphometric and biomechanical properties of bone, as well as the biochemical abnormalities observed in diabetic rodents [56,63-65]. Moreover, RUNX2 and RUNX2-regulated osteogenic genes are in large part normalized in insulin-treated diabetic animals, suggesting that insulin may directly or indirectly regulate bone formation through a pro-osteogenic pathway involving RUNX2 expression and RUNX2 downstream targets [62]. Critical to the argument that insulin may have direct effects on osteoblastogenesis during diabetes, local insulin delivery can normalize mineralization, callus bone content, and biomechanical properties in the healing fracture callus of the diabetic rat, despite persistent systemic hyperglycemia and systemic hypoinsulinemia [66].
While much research supports a primary role for IGF-1 in anabolic bone formation in rodents as just described [39,67,68], its role in the pathogenesis or reversal of diabetic osteopathy has been examined in only a limited way. In the Biobreeding (BB) rat model of diabetes, infusion with IGF-I dramatically increased tibial epiphyseal width and overall bone growth despite persistent hyperglycemia, suggesting that stimulatory effects on bone may be independent of metabolic affects of IGF-I [69]. Recently, we have explored the impact of IGF-I treatment in a mouse model of T1D on bone regeneration and bone strength. Regenerate bone was assessed by distraction osteogenesis [56]. IGF-I treatment significantly improved regenerate bone formation in this model. Furthermore, significant reductions in trabecular thickness, yield strength and peak force, were also improved with IGF-I treatment in diabetes [70]. These findings demonstrate that despite persistent hyperglycemia and insulinopenia, IGF-I therapy can promote new bone formation and improve biomechanical properties of bone in T1D.
Potential signaling pathways critical to insulin and IGF-1 induced bone formation in diabetic osteopathy
In the diabetic bone, other pro-osteogenic pathways may be disrupted, such as the Wnt-signaling pathway [71]. The current understanding of these anabolic pathways in skeletal development and homeostasis suggests that insulin and IGF-1 signaling could crosstalk with two major pro-osteogenic pathways that ultimately regulate RUNX2 activity: the canonical Wnt signaling pathway and the BMP-2 signaling pathway. The importance of the canonical Wnt signaling pathway in determination of bone mass has been extensively documented and confirmed in both animal models and in human genetic conditions in which loss-of-function or gain-of-function mutations in specific components of this pathway either disrupt or accentuate bone formation and/or osteoblastogenesis [72]. Key elements of this pathway are diagrammed in (Figure 3). Briefly, Wnts (secreted lipid modified proteins) are known to bind to a receptor complex consisting of lipoprotein receptor-related proteins 5 or 6 (LRP5/LRP6) with frizzled [72]. In the absence of Wnt ligand, a “destruction complex” consisting of glycogen synthase kinase 3 (GSK-3), Axin, and tumor suppressor adenomatous polyposis coli protein (APC) mediates the phosphorylation of β-catenin, resulting in its proteolysis. In the presence of Wnt ligand, phosphorylation and inactivation of GSK-3 occurs, inhibiting the constitutive phosphorylation and subsequent enzymatic degradation of β-catenin. β-catenin then accumulates in the cytoplasm, translocates into the nucleus, where it associates with the human T-cell factor 1 (TCF-1), mouse lymphoid enhancer factor (LEF-1) family of transcription factors and initiates the expression of Wnt target genes, including RUNX2. GSK-3 is a multifunctional kinase involved in numerous cellular functions, including regulation of insulin-dependent glycogen synthesis. Specifically, insulin, via the PI3K/AKT pathway, inhibits GSK-3 activity in skeletal muscle [73], promoting glycogen synthesis. While there remains debate over cross-signaling via the insulin/IGF-1 pathway and the Wnt signaling pathway through GSK-3 [74], studies have now shown that IGF-1 signaling can enhance Wnt protein production and activate β-catenin [75]. Furthermore, inhibitors of Wnts can partially inhibit IGF-1 actions [75]. Bone morphogenetic protein (BMP)-2 induced osteogenesis is another major pathway contributing to bone formation (Figure 3). The interaction of BMPs with BMP receptors leads to the phosphorylation of Smads and ultimately to distal-less homeobox 5 (Dlx5) transcription. Dlx5 then independently regulates the osteogenic transcription factors, RUNX2. In mesenchymal stem cells, MAPK serves as a point of convergence for mediating up-regulation of osterix, a RUNX2 target gene and major promoter of osteogenesis, via BMP-2 and IGF-1 signaling [76]. Thus, these examples serve to demonstrate the great potential for insulin and IGF-1 signaling to synergize with other signaling pathways involved in promoting osteogenesis.
Figure 3.
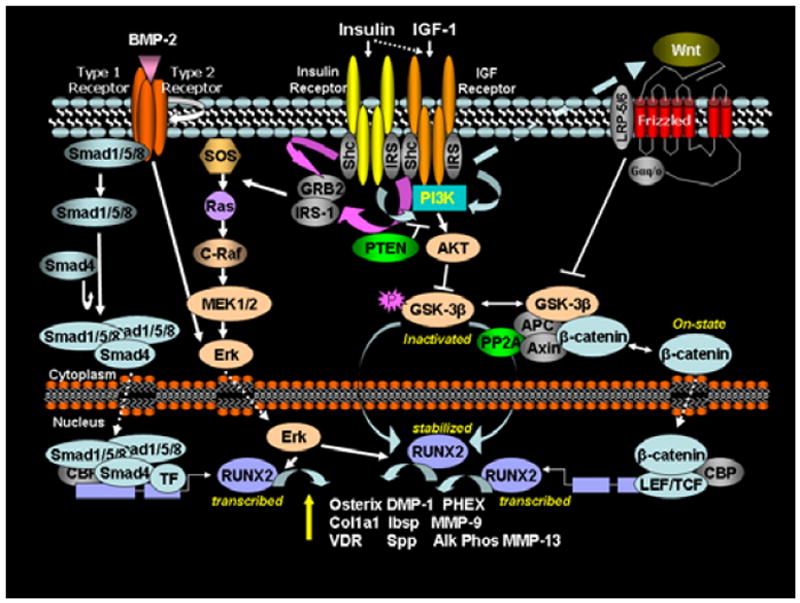
Potential cross-talk between insulin/IGF-1, BMP2, and Wnt signaling pathways. BMP2 receptor signaling and the MEK/ERK branch of the insulin/IGF-1 signaling pathway converge on ERK1/2 kinase, which can modulate the activity of the master osteoblastogenic transcription factor, Runx2. Wnt and the PI3-K/Akt branch of the insulin/IGF-1 signaling pathway converge on GSK-3b, inhibiting GSK-3b kinase activity and reducing its constitutive phosphorylation of β-catenin. Unphosphorylated β-catenin is not readily degraded and can accumulate in the nucleus, where it acts as a pro-osteoblastogenic transcription factor by regulating transcription of Runx2 and other genes.
Summary and Significance
Insulin and IGF-1 may exert independent effects on skeletal homeostasis, yet they are highly homologous peptides and can cross-signal through insulin receptors and IGF-1 receptors, and they share downstream mediators. (Table 1) summarizes various mouse models in which components of insulin and/or IGF-I signaling have been independently assessed in skeletogenesis. Furthermore, both insulin and IGF-I may crosstalk with other pro-osteogenic pathways (e.g., Wnt and BMP-2). The relative contributions of insulin dysregulation (i.e., hypoinsulinemia in T1D vs. hyperinsulinemia in T2D) and/or IGF-1 deficiency (T1D) to skeletal integrity in diabetes remains largely unexplored. Thus, investigations into the specific effects of insulin and IGF-1 on osteoblastogenesis and bone formation as well as the relative contribution of insulin and IGF-1 to diabetic osteopathy are critical to understanding how manipulation of these hormones may improve skeletal health in persons with diabetes.
Table 1.
Various mouse models in which components of insulin and/or IGF-I signaling have been independently assessed in skeletogenesis.
| Perturbations in Insulin/IGF-1 signaling resulting in diminished skeletogenesis | |||||||
|---|---|---|---|---|---|---|---|
| Gene(s) modifed1 | Genetic approach2 | Phenotype | Ref. | ||||
| Dwarf | Trabecular Bone | Cortical Bone | BMD | Bone Strength | |||
| Gab1 | C (OC-Cre) | NR3 | ↓ | ↓ | NR | ↓ | 24 |
| IRS1 | G | Yes | ↓ | ↓ | ↓ | NR | 26 |
| IRS2 | G | No | ↓ | ↓ | ↓ | NR | 25 |
| Akt1 | G | Yes | ↓ | ↓ | ↓ | NR | 29 |
| Akt1 & Akt2 | G | Yes | NR | NR | NR | NR | 28 |
| FoxO1 | C (Col1a1-Cre) | No | ↓ | ↓ | ↓ | NR | 32 |
| FoxO1, 3 & 4 | MT (Mx-Cre) | No | ↓ | ↓ | ↓ | NR | 33 |
| IGF1R | C (OC-Cre) | No | ↓ | ↔ | ↓ | NR | 44 |
| IGF1R | C (Osx-Cre) | Yes | ↓ | NR | NR | NR | 45 |
| IR | C (Col1a1-Cre) | NR | NR | NR | ↓ | NR | 59 |
| IR | C (OC-Cre) | NR | ↓ | NR | NR | NR | 57 |
| MEK1 | TG (dominant/negative) | Yes | NR | NR | ↓ | NR | 38 |
| Perturbations in Insulin/IGF-1 signaling resulting in enhanced skeletogenesis | |||||||
| MEK1 | TG (active) | No (large) | NR | NR | ↑ | NR | 38 |
| PTEN | C (OC-Cre) | NR | ↑ | ↑ | ↑ | NR | 30 |
| FoxO1, 3 & 4 | C (Osx-Cre) | NR | NR | ↑ | ↑ | NR | 34 |
Pathways presented in Figure 1
Genetic Approach: C= conditional knockout; G=global knockout; MT = multiple tissue knockout; TG = transgenic over-expression
NR = Not reported
Acknowledgments
The authors wish to thank Hannah Coleman for technical assistance. This work was supported by grants from the Children’s University Medical Group fund of the Arkansas Children’s Hospital Research Institute (to K.M.T.), the Martha Ann Pugh Diabetes Research Fund (to K.M.T.), the Arkansas Biosciences Institute (to J.L.F.), and in part by National Institutes of Health Grants R01DK055653 (to J.L.F.), and C06RR16517 (to Arkansas Children’s Hospital Research Institute).
Footnotes
Publisher's Disclaimer: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Meyer HE, Tverdal A, Falch JA. Risk factors for hip fracture in middle-aged Norwegian women and men. Am J Epidemiol. 1993;137:1203–1211. doi: 10.1093/oxfordjournals.aje.a116622. [DOI] [PubMed] [Google Scholar]
- 2.Nicodemus KK, Folsom AR. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes care. 2001;24:1192–1197. doi: 10.2337/diacare.24.7.1192. [DOI] [PubMed] [Google Scholar]
- 3.Janghorbani M, Feskanich D, Willett WC, Hu F. Prospective study of diabetes and risk of hip fracture: the Nurses’ Health Study. Diabetes care. 2006;29:1573–1578. doi: 10.2337/dc06-0440. [DOI] [PubMed] [Google Scholar]
- 4.Espallargues M, Sampietro-Colom L, Estrada MD, Sola M, del Rio L, et al. Identifying bone-mass-related risk factors for fracture to guide bone densitometry measurements: a systematic review of the literature. Osteoporos Int. 2001;12:811–822. doi: 10.1007/s001980170031. [DOI] [PubMed] [Google Scholar]
- 5.Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166:495–505. doi: 10.1093/aje/kwm106. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. Jama. 2011;305:2184–2192. doi: 10.1001/jama.2011.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strotmeyer ES, Cauley JA, Schwartz AV, Nevitt MC, Resnick HE, et al. Diabetes is associated independently of body composition with BMD and bone volume in older white and black men and women: The Health, Aging, and Body Composition Study. J Bone Miner Res. 2004;19:1084–1091. doi: 10.1359/JBMR.040311. [DOI] [PubMed] [Google Scholar]
- 8.Thrailkill KM, Lumpkin CK, Jr, Bunn RC, Kemp SF, Fowlkes JL. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am J Physiol Endocrinol Metab. 2005;289:E735–745. doi: 10.1152/ajpendo.00159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukunaga Y, Minamikawa J, Inoue D, Koshiyama H. Does insulin use increase bone mineral density in patients with non-insulin-dependent diabetes mellitus? Arch Intern Med. 1997;157:2668–2669. doi: 10.1001/archinte.157.22.2668a. [DOI] [PubMed] [Google Scholar]
- 10.Weinstock RS, Goland RS, Shane E, Clemens TL, Lindsay R, et al. Bone mineral density in women with type II diabetes mellitus. J Bone Miner Res. 1989;4:97–101. doi: 10.1002/jbmr.5650040114. [DOI] [PubMed] [Google Scholar]
- 11.Kemink SA, Hermus AR, Swinkels LM, Lutterman JA, Smals AG. Osteopenia in insulin-dependent diabetes mellitus; prevalence and aspects of pathophysiology. J Endocrinol Invest. 2000;23:295–303. doi: 10.1007/BF03343726. [DOI] [PubMed] [Google Scholar]
- 12.Jehle PM, Jehle DR, Mohan S, Bohm BO. Serum levels of insulin-like growth factor system components and relationship to bone metabolism in Type 1 and Type 2 diabetes mellitus patients. J Endocrinol. 1998;159:297–306. doi: 10.1677/joe.0.1590297. [DOI] [PubMed] [Google Scholar]
- 13.Moyer-Mileur LJ, Slater H, Jordan KC, Murray MA. IGF-1 and IGF-binding proteins and bone mass, geometry, and strength: relation to metabolic control in adolescent girls with type 1 diabetes. J Bone Miner Res. 2008;23:1884–1891. doi: 10.1359/jbmr.080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leger J, Marinovic D, Alberti C, Dorgeret S, Chevenne D, et al. Lower bone mineral content in children with type 1 diabetes mellitus is linked to female sex, low insulin-like growth factor type I levels, and high insulin requirement. J Clin Endocrinol Metab. 2006;91:3947–3953. doi: 10.1210/jc.2006-0711. [DOI] [PubMed] [Google Scholar]
- 15.Mastrandrea LD, Wactawski-Wende J, Donahue RP, Hovey KM, Clark A, et al. Young women with type 1 diabetes have lower bone mineral density that persists over time. Diabetes care. 2008;31:1729–1735. doi: 10.2337/dc07-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Yano S, et al. Serum insulin-like growth factor-I level is associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes mellitus. Osteoporos Int. 2007;18:1675–1681. doi: 10.1007/s00198-007-0430-0. [DOI] [PubMed] [Google Scholar]
- 17.Thrailkill KM, Jo CH, Cockrell GE, Moreau CS, Lumpkin CK, Jr, et al. Determinants of undercarboxylated and carboxylated osteocalcin concentrations in type 1 diabetes. Osteoporos Int. 2011 doi: 10.1007/s00198-011-1807-7. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 19.Ohlsson C, Mohan S, Sjogren K, Tivesten A, Isgaard J, et al. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009;30:494–535. doi: 10.1210/er.2009-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rechler MM. Insulin-like growth factor binding proteins. Vitam Horm. 1993;47:1–114. doi: 10.1016/s0083-6729(08)60444-6. [DOI] [PubMed] [Google Scholar]
- 21.Bunn RC, Fowlkes JL. Insulin-like growth factor binding protein proteolysis. Trends Endocrinol Metab. 2003;14:176–181. doi: 10.1016/s1043-2760(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 22.Shepherd PR, Kahn BB. Glucose transporters and insulin action--implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- 23.Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, et al. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994;269:13689–13694. [PubMed] [Google Scholar]
- 24.Weng T, Mao F, Wang Y, Sun Q, Li R, et al. Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. J Cell Sci. 2010;123:682–689. doi: 10.1242/jcs.058396. [DOI] [PubMed] [Google Scholar]
- 25.Akune T, Ogata N, Hoshi K, Kubota N, Terauchi Y, et al. Insulin receptor substrate-2 maintains predominance of anabolic function over catabolic function of osteoblasts. J Cell Bio. 2002;159:147–156. doi: 10.1083/jcb.200204046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata N, Chikazu D, Kubota N, Terauchi Y, Tobe K, et al. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J Clin Invest. 2000;105:935–943. doi: 10.1172/JCI9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimoaka T, Kamekura S, Chikuda H, Hoshi K, Chung UI, et al. Impairment of bone healing by insulin receptor substrate-1 deficiency. J Biol Chem. 2004;279:15314–15322. doi: 10.1074/jbc.M312525200. [DOI] [PubMed] [Google Scholar]
- 28.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawamura N, Kugimiya F, Oshima Y, Ohba S, Ikeda T, et al. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PloS One. 2007;2:e1058. doi: 10.1371/journal.pone.0001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Bruxvoort KJ, Zylstra CR, Liu J, Cichowski R, et al. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc Natl Acad Sci U S A. 2007;104:2259–2264. doi: 10.1073/pnas.0604153104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almeida MJ. Unraveling the role of FoxOs in bone - Insights from mouse models. Bone. 2011;49:319–327. doi: 10.1016/j.bone.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, et al. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010;11:147–160. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ambrogini E, Almeida M, Martin-Millan M, Paik JH, Depinho RA, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136–146. doi: 10.1016/j.cmet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambrogini E, Han L, Bartell S, Shelton R, Deloose A, et al. Deletion of the FoxO1, 3, and 4 genes from the committed osteoblast progenitors expressing Osterix increases Wnt signaling and bone mass. ASBMR Annual Meeting; Toronto. 2010. abstract 1073. [Google Scholar]
- 35.Iyer S, Ambrogini E, Han L, Bartell S, Warren A, et al. Combined deletion of the transcription factors FoxO1, 3, and 4 from osteoblast progenitors expressing osterix causes bone anabolism and postpones the adverse effects of aging on bone. ASBMR Annual Meeting; San Diego, CA. 2011. Abstract 1168. [Google Scholar]
- 36.Stein GS, Lian JB, van Wijnen AJ, Stein JL, Montecino M, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23:4315–4329. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 37.Fulzele K, DiGirolamo DJ, Liu Z, Xu J, Messina JL, et al. Direct actions of insulin on osteoblasts revealed by Cre-mediated disruption of the IGF-1 receptor. 28th Annual Meeting of the Society for Bone and Mineral Research; Philadelphia, PA, USA. 2006. Abstract # 1048. [Google Scholar]
- 38.Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176:709–718. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC. Growth hormone and bone. Endocr Rev. 1998;19:55–79. doi: 10.1210/edrv.19.1.0324. [DOI] [PubMed] [Google Scholar]
- 40.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 41.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 42.Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, et al. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res. 2001;16:2320–2329. doi: 10.1359/jbmr.2001.16.12.2320. [DOI] [PubMed] [Google Scholar]
- 43.Sjogren K, Sheng M, Moverare S, Liu JL, Wallenius K, et al. Effects of liver-derived insulin-like growth factor I on bone metabolism in mice. J Bone Miner Res. 2002;17:1977–1987. doi: 10.1359/jbmr.2002.17.11.1977. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 45.Pang L, Wu X, Lei W, Qiu T, Li F, et al. IGF-I released during osteoclastic bone resorption induces osteoblast differentiation of BMSCs in the coupling process. ASBMR Annual Meeting; Toronto. 2010. abstract 1115. [Google Scholar]
- 46.Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–2682. doi: 10.1210/endo.141.7.7585. [DOI] [PubMed] [Google Scholar]
- 47.Hashizume M, Yamaguchi M. Stimulatory effect of beta-alanyl-L-histidinato zinc on cell proliferation is dependent on protein synthesis in osteoblastic MC3T3-E1 cells. Mol Cell Biochem. 1993;122:59–64. doi: 10.1007/BF00925737. [DOI] [PubMed] [Google Scholar]
- 48.Wergedal JE, Baylink DJ. Characterization of cells isolated and cultured from human bone. Proc Soc Exp Biol Med. 1984;176:60–69. doi: 10.3181/00379727-176-41843. [DOI] [PubMed] [Google Scholar]
- 49.Canalis EM, Dietrich JW, Maina DM, Raisz LG. Hormonal control of bone collagen synthesis in vitro. Effects of insulin and glucagon Endocrinology. 1977;100:668–674. doi: 10.1210/endo-100-3-668. [DOI] [PubMed] [Google Scholar]
- 50.Pun KK, Lau P, Ho PW. The characterization, regulation, and function of insulin receptors on osteoblast-like clonal osteosarcoma cell line. J Bone Miner Res. 1989;4:853–862. doi: 10.1002/jbmr.5650040610. [DOI] [PubMed] [Google Scholar]
- 51.Rosen DM, Luben RA. Multiple hormonal mechanisms for the control of collagen synthesis in an osteoblast-like cell line, MMB-1. Endocrinology. 1983;112:992–999. doi: 10.1210/endo-112-3-992. [DOI] [PubMed] [Google Scholar]
- 52.Canalis E. Effect of hormones and growth factors on alkaline phosphatase activity and collagen synthesis in cultured rat calvariae. Metabolism. 1983;32:14–20. doi: 10.1016/0026-0495(83)90149-x. [DOI] [PubMed] [Google Scholar]
- 53.Kream BE, Smith MD, Canalis E, Raisz LG. Characterization of the effect of insulin on collagen synthesis in fetal rat bone. Endocrinology. 1985;116:296–302. doi: 10.1210/endo-116-1-296. [DOI] [PubMed] [Google Scholar]
- 54.Hahn TJ, Westbrook SL, Sullivan TL, Goodman WG, Halstead LR. Glucose transport in osteoblast-enriched bone explants: characterization and insulin regulation. J Bone Miner Res. 1988;3:359–365. doi: 10.1002/jbmr.5650030317. [DOI] [PubMed] [Google Scholar]
- 55.Ituarte EA, Halstead LR, Iida-Klein A, Ituarte HG, Hahn TJ. Glucose transport system in UMR-106-01 osteoblastic osteosarcoma cells: regulation by insulin. Calcif Tissue Int. 1989;45:27–33. doi: 10.1007/BF02556657. [DOI] [PubMed] [Google Scholar]
- 56.Thrailkill KM, Liu L, Wahl EC, Bunn RC, Perrien DS, et al. Bone formation is impaired in a model of type 1 diabetes. Diabetes. 2005;54:2875–2881. doi: 10.2337/diabetes.54.10.2875. [DOI] [PubMed] [Google Scholar]
- 57.Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Irwin R, Lin HV, Motyl KJ, McCabe LR. Normal bone density obtained in the absence of insulin receptor expression in bone. Endocrinology. 2006;147:5760–5767. doi: 10.1210/en.2006-0700. [DOI] [PubMed] [Google Scholar]
- 59.Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nyman JS, Even JL, Jo CH, Herbert EG, Murry MR, et al. Increasing duration of type 1 diabetes perturbs the strength-structure relationship and increases brittleness of bone. Bone. 2011;48:733–740. doi: 10.1016/j.bone.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu H, Kraut D, Gerstenfeld LC, Graves DT. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology. 2003;144:346–352. doi: 10.1210/en.2002-220072. [DOI] [PubMed] [Google Scholar]
- 62.Fowlkes JL, Bunn RC, Liu L, Wahl EC, Coleman HN, et al. Runt-related transcription factor 2 (RUNX2) and RUNX2-related osteogenic genes are down-regulated throughout osteogenesis in type 1 diabetes mellitus. Endocrinology. 2008;149:1697–1704. doi: 10.1210/en.2007-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Follak N, Kloting I, Merk H. Influence of diabetic metabolic state on fracture healing in spontaneously diabetic rats. Diabetes Metab Res Rev. 2005;21:288–296. doi: 10.1002/dmrr.537. [DOI] [PubMed] [Google Scholar]
- 64.Follak N, Kloting L, Wolf E, Merk H. Delayed remodeling in the early period of fracture healing in spontaneously diabetic BB/OK rats depending on the diabetic metabolic state. Histol Histopathol. 2004;19:473–486. doi: 10.14670/HH-19.473. [DOI] [PubMed] [Google Scholar]
- 65.Hou JC, Zernicke RF, Barnard RJ. Effects of severe diabetes and insulin on the femoral neck of the immature rat. J Orthop Res. 1993;11:263–271. doi: 10.1002/jor.1100110214. [DOI] [PubMed] [Google Scholar]
- 66.Gandhi A, Beam HA, O’Connor JP, Parsons JR, Lin SS. The effects of local insulin delivery on diabetic fracture healing. Bone. 2005;37:482–490. doi: 10.1016/j.bone.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 67.Fowlkes JL, Thrailkill KM, Liu L, Wahl EC, Bunn RC, et al. Effects of systemic and local administration of recombinant human IGF-I (rhIGF-I) on de novo bone formation in an aged mouse model. J Bone Miner Res. 2006;21:1359–1366. doi: 10.1359/JBMR.060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niu T, Rosen CJ. The insulin-like growth factor-I gene and osteoporosis: a critical appraisal. Gene. 2005;361:38–56. doi: 10.1016/j.gene.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 69.Zapf J. Growth promotion by insulin-like growth factor I in hypophysectomized and diabetic rats. Mol Cell Endocrinol. 1998;140:143–149. doi: 10.1016/s0303-7207(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 70.Fowlkes JL, Nyman JS, Jo CH, Wahl EC, Liu L, et al. Osteopromoting effects of rhIGF-I in a mouse model of type1 diabetes. Endocr Rev. 2011;32:P1–215. [Google Scholar]
- 71.Hie M, Iitsuka N, Otsuka T, Tsukamoto I. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activity. Int J Mol Med. 2011;28:455–462. doi: 10.3892/ijmm.2011.697. [DOI] [PubMed] [Google Scholar]
- 72.Manolagas SC, Almeida M. Gone with the Wnts: {beta}-catenin, TCF, FOXO, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21:2605–2614. doi: 10.1210/me.2007-0259. [DOI] [PubMed] [Google Scholar]
- 73.Patel S, Doble B, Woodgett JR. Glycogen synthase kinase-3 in insulin and Wnt signalling: a double-edged sword? Biochemical Society transactions. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Reports. 2010;2:82. doi: 10.3410/B2-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang L, Shao YY, Ballock RT. Thyroid hormone-mediated growth and differentiation of growth plate chondrocytes involves IGF-1 modulation of beta-catenin signaling. J Bone Miner Res. 2010;25:1138–1146. doi: 10.1002/jbmr.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Celil AB, Hollinger JO, Campbell PG. Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling. J Cellular Biochem. 2005;95:518–528. doi: 10.1002/jcb.20429. [DOI] [PubMed] [Google Scholar]