

Significance
Many viruses encode proteases that cleave both viral and host substrates. Arteriviruses encode such a dual-specificity protease (PLP2) that removes ubiquitin from cellular proteins involved in host immunity. Based on a 3D structure of PLP2, we engineered the protease to have diminished deubiquitinating activity without affecting its activity toward its viral substrate. Viruses expressing such engineered proteases displayed a significantly weakened ability to evade host immune responses. This result demonstrates a crucial role for PLP2 in arterivirus immune evasion and opens new possibilities for developing improved attenuated virus vaccines against economically important arteriviruses and other viruses encoding similar dual-specificity proteases.
Keywords: interferon-stimulated gene 15, ISG15, +RNA
Abstract
Protein ubiquitination regulates important innate immune responses. The discovery of viruses encoding deubiquitinating enzymes (DUBs) suggests they remove ubiquitin to evade ubiquitin-dependent antiviral responses; however, this has never been conclusively demonstrated in virus-infected cells. Arteriviruses are economically important positive-stranded RNA viruses that encode an ovarian tumor (OTU) domain DUB known as papain-like protease 2 (PLP2). This enzyme is essential for arterivirus replication by cleaving a site within the viral replicase polyproteins and also removes ubiquitin from cellular proteins. To dissect this dual specificity, which relies on a single catalytic site, we determined the crystal structure of equine arteritis virus PLP2 in complex with ubiquitin (1.45 Å). PLP2 binds ubiquitin using a zinc finger that is uniquely integrated into an exceptionally compact OTU-domain fold that represents a new subclass of zinc-dependent OTU DUBs. Notably, the ubiquitin-binding surface is distant from the catalytic site, which allowed us to mutate this surface to significantly reduce DUB activity without affecting polyprotein cleavage. Viruses harboring such mutations exhibited WT replication kinetics, confirming that PLP2-mediated polyprotein cleavage was intact, but the loss of DUB activity strikingly enhanced innate immune signaling. Compared with WT virus infection, IFN-β mRNA levels in equine cells infected with PLP2 mutants were increased by nearly an order of magnitude. Our findings not only establish PLP2 DUB activity as a critical factor in arteriviral innate immune evasion, but the selective inactivation of DUB activity also opens unique possibilities for developing improved live attenuated vaccines against arteriviruses and other viruses encoding similar dual-specificity proteases.
The synthesis and posttranslational cleavage of polyproteins is a common genome expression strategy used by positive-stranded (+)RNA viruses of eukaryotes. It is used to cope with the consequences of cytoplasmic replication and the limitations of the eukaryotic translation machinery, which essentially preclude the use of (nuclear) RNA splicing and polycistronic mRNAs, respectively (1). The critical cleavage of these viral polyproteins into their functional subunits is mediated by internal virus-encoded proteases (2–5), many of which have been found to also target cellular substrates to promote virus replication or subvert host antiviral responses. Well-known examples of such dual-specificity proteases are the poliovirus 2A and hepatitis C virus NS3/4A enzymes that, in addition to the viral polyprotein, target host cell proteins involved in translation and innate immune signaling, respectively (6–10).
Arteriviruses are +RNA viruses that, together with the corona- and roniviruses, belong to the order Nidovirales and include equine arteritis virus (EAV) and porcine reproductive and respiratory syndrome virus (PRRSV). EAV is the family prototype and can cause abortion in pregnant mares, pneumonia in neonatal foals, and influenza-like illness in adult horses (11). PRRSV ranks among the most important swine pathogens, and infections are characterized by reproductive failure in sows and severe respiratory disease in young pigs (12). As in all nidoviruses, the synthesis and cleavage of the replicase polyproteins (pp1a and pp1ab) are critical first steps in arterivirus infection. They are encoded by the 5′-proximal three-quarters of the 13- to 16-kb arterivirus genome and are the precursors of the nonstructural proteins (nsps) required for genome replication and transcription. In the case of EAV, at least 13 nsps are produced when the replicase polyproteins are cleaved by three virus-encoded proteases (13, 14), one of which is a papain-like protease (PLP2) located in the N-terminal region of nsp2 (15, 16). This enzyme cleaves the nsp2|nsp3 junction in pp1a and pp1ab, an event that is essential for virus replication because an EAV PLP2 active site mutant was previously found to be nonviable (17). In addition to this critical role in viral polyprotein maturation, arterivirus PLP2 was proposed to contribute to the evasion of host innate immune responses by removing ubiquitin (Ub) from cellular targets (18). Ub is an 8-kDa protein moiety that can be covalently attached to lysine residues of target proteins in a number of structurally different configurations, either by monoubiquitination or through the formation of polyubiquitin chains (19, 20). The effects of ubiquitination can range from targeting substrates for proteasomal degradation to initiating signaling cascades and—importantly—they can be reversed by deubiquitinating enzymes (DUBs), which thus allow for negative regulation of Ub-activated processes (21–24). The latter include the innate immune signaling cascades triggered by invading RNA viruses (25, 26), which ultimately lead to the transcription of genes encoding IFN-β and other proinflammatory cytokines (27, 28).
Arterivirus PLP2 and a protease domain found in the unrelated nairovirus Crimean-Congo hemorrhagic fever virus (CCHFV) were first identified as potential members of the ovarian tumor domain-containing (OTU) superfamily of DUBs on the basis of comparative sequence analysis (29). Several laboratories, including our own, subsequently confirmed that arterivirus PLP2s indeed have DUB activity that may be used to remove Ub from innate immune signaling factors to suppress the induction of an antiviral state (18, 30, 31). The potential benefits of this strategy are highlighted by the fact that proteases from virus groups as diverse as arteri-, corona-, nairo-, picorna-, hepadna-, and herpesviruses have all been implicated in DUB-based innate immune evasion (18, 32–37). Thus far, however, direct evidence linking DUB activity to the suppression of innate immune responses in virus-infected cells has not been reported for any of these proteases.
Because the DUB activity of arterivirus PLP2 depends on the same active site mediating the critical nsp2|nsp3 cleavage, it has not been possible to independently study the role of PLP2 in polyprotein processing and immune evasion in the context of virus infection. Here we present the crystal structure of EAV PLP2 in complex with Ub at 1.45-Å resolution. The complex reveals a distinctly compact conformation compared with other OTU superfamily members and the incorporation of a zinc finger within the OTU fold. Given these features, arterivirus PLP2 represents a unique subclass of zinc-dependent OTU DUBs. Importantly, the PLP2 active site is distant from its Ub-binding surface, allowing for the introduction of mutations in this region that dramatically reduced DUB activity yet did not affect nsp2|nsp3 cleavage. Compared with WT EAV, viruses carrying these mutations elicited a significantly enhanced innate immune response in primary equine cells while displaying WT replication kinetics. Taken together, our results demonstrate that PLP2 DUB activity indeed mediates innate immune suppression during arterivirus infection. The ability to selectively inactivate the PLP2 DUB function may thus contribute to the engineering of improved live attenuated vaccines against arteriviruses and other virus families encoding proteases with similar dual specificities.
Results
EAV PLP2 Adopts a Compact OTU-Domain Fold with a Unique Integral Zinc Finger.
Previously, EAV PLP2 was identified and characterized by a combination of bioinformatics analysis and site-directed mutagenesis, and two residues in particular were implicated in catalysis: Cys270 and His332 (16). Throughout this paper, amino acid numbers refer to the sequence of full-length EAV pp1a. The crystal structure of EAV PLP2 (residues 261–392; 13.6 kDa) was determined as a covalent complex with the mechanism-based inhibitor Ub(1–75)-3-bromopropylamine (Ub–3Br) (38, 39). Because the conservation of multiple cysteine residues and their demonstrated importance for protease function suggested that PLP2 could bind zinc (16), the crystal structure of the complex was determined by a multiwavelength anomalous dispersion (MAD) phasing experiment using X-ray diffraction data collected over the zinc absorption edge (Table S1). The resulting electron density map revealed residues 261–387 of PLP2 bound to a complete Ub molecule and allowed a model of the complex to be built and refined (Rwork= 0.16, Rfree= 0.18) to 1.45-Å resolution (Fig. 1 A and B).
Fig. 1.

Structure of the EAV PLP2-Ub complex and superposition with yeast OTU1 and CCHFV OTU. (A) Structure of EAV PLP2 (blue) bound to Ub (orange) showing the two-domain fold. (B) 90° rotation of complex shown in A. (C) Electron density for the C4 zinc finger motif. Blue density is a maximum-likelihood weighted 2Fo-Fc map contoured at 1.0σ. Green density about the zinc atom (gray) is a Fo-Fc omit map contoured at 5.0σ. (D) The catalytic triad of the EAV PLP2 active site. Blue density is a maximum-likelihood weighted 2Fo-Fc map contoured at 1.0σ. The cysteine nucleophile (Cys270) is covalently linked to Ub via the 3CN linker, which replaces Gly76 of Ub. Asn263, which orients the imidazole ring of His332, occurs in two alternate conformations. (E) Superposition of EAV PLP2 (blue), CCHFV OTU (cyan), and yeast OTU1 (red). PLP2 shares a conserved core of two central helices and a four-stranded β-sheet with CCHFV OTU and yeast OTU1. Topology diagrams for EAV PLP2, CCHFV OTU, and yeast OTU1 are shown in F–H, respectively. The region that is conserved among the enzymes is outlined (dashed box). Structural images were prepared using PyMOL (84).
The protease adopts a compact, two-domain fold with a shallow Ub-binding surface that directs the C terminus of the bound Ub molecule (the distal Ub in a Ub dimer) toward a solvent exposed active site that includes Cys270 and His332 (Fig. 1 A and B). Domain I of PLP2 (residues 267–307 and 365–387) consists of a three-helix bundle (α1, α2, α4) packed against a two-stranded antiparallel sheet (β2↑ β6↓). Domain II centers on a four-stranded β-sheet (β1↑ β5↓ β4↑ β3↑) and an α-helix (α3) that together pack against helices α1 and α2 of domain I. Domain II comprises the majority of the Ub-binding surface, which is stabilized by four cysteine residues (Cys319, 349, 354, and 356) that coordinate a zinc ion with tetrahedral geometry (Fig. 1C). Their arrangement forms a C4 zinc finger that resembles a C-terminal type zinc necklace motif (40). A large insertion between positions C1 (Cys319) and C2 (Cys349), which includes His332, appears to extend the stabilizing effect of the zinc finger throughout much of domain II. A fifth cysteine (Cys344) is located near the zinc ion but does not coordinate with it, consistent with other zinc necklace motifs that have been described (40) and with previous findings showing that a Cys344 to alanine mutation had no effect on catalytic activity of PLP2 (16). Three of the cysteines (Cys319, 349, and 354) are fully conserved in arteriviruses, and mutational analysis of these residues and Cys356 demonstrated zinc binding to be essential for catalytic activity (16). Given its distance from the active site, however (∼25 Å; Fig. 1B), the zinc atom appears to play a structural role as opposed to participating in catalysis. Expression of PLP2 in Escherichia coli grown in the absence of zinc (M9 medium) yielded insoluble protein, supporting the idea that the role of the zinc finger is structural and is likely required for correct folding of the protease.
Consistent with OTU DUBs and papain-like cysteine proteases in general, the PLP2 active site contains a catalytic cysteine nucleophile (Cys270) and histidine (His332) residue, along with an asparagine (Asn263) that hydrogen bonds with the imidazole ring of His332 (Fig. 1D). As expected, the side chain of Cys270 is covalently coupled to the C terminus of Ub via the 3-propylamine (3CN) modification, mimicking the acyl-enzyme intermediate step of the catalytic reaction and confirming the identity of Cys270 as the catalytic nucleophile.
Fold analysis of the PLP2 structure using the DALI server (41) revealed that its closest structural homologs indeed belong to the OTU superfamily of DUBs (29, 42). The most significant matches were to yeast Otu1 (38) (Z-score: 5.1) and the viral OTU protease from CCHFV (43–45) (Z-score: 4.6; Fig. 1E), followed by Otubain1 from human (46) and Caenorhabditis elegans (47) (Z-scores: 4.1 and 4.0, respectively) and the OTU domain of human DUBA (48) (Z-score: 3.9). The sequence identity of the PLP2 regions that aligned with these OTU proteases was low (ranging from 9% for yeast Otu1 to 21% for human Otubain1); however, they accounted for ∼60% of the total PLP2 structure and superposed well with the equivalent regions in the above proteins, with an average rmsd of ∼2.8 Å. The greatest structural similarity between PLP2 and members of the OTU superfamily occurs at the active site and adjoining channel that binds the C-terminal RLRGG-tail of Ub.
PLP2 Zinc Finger Motif Plays a Central Role in Ub Binding.
Arterivirus PLP2 and nairovirus OTU enzymes differ from eukaryotic OTU DUBs in that they also remove the Ub-like antiviral protein IFN stimulated gene 15 (ISG15) from target proteins, a process also known as deISGylation (18, 49). ISG15 conjugation has been postulated to interfere with proper viral protein function, possibly through steric hindrance, although the exact mechanism underlying its antiviral activity is unknown (50). For CCHFV OTU, cross-reactivity with ISG15 arises primarily from a unique β-hairpin on the Ub-binding surface. The hairpin modifies the surface so that the viral enzyme binds the β-grasp folds of Ub and the C-terminal Ub-like domain of ISG15 in an orientation that is rotated nearly 75° with respect to that observed for Ub bound to a representative eukaryotic OTU DUB from yeast (Otu1) (43, 44). Surprisingly, the β-hairpin is absent in EAV PLP2 and replaced by helix α3 of the zinc finger motif (Fig. 2A). However, in keeping with the role of the β-hairpin in CCHFV OTU, residues of helix α3 bind to the hydrophobic Ile44 patch of Ub, a site commonly targeted by Ub-binding proteins (51), and they also assist in positioning Ub in a rotated manner equivalent to that observed for CCHFV OTU (Fig. 2 B and C).
Fig. 2.
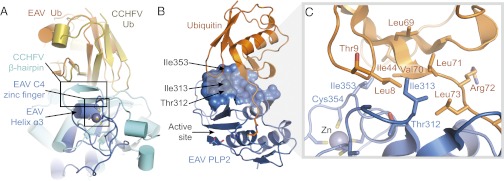
Ub-binding surface of EAV PLP2. (A) Superposition of EAV PLP2 (blue) and CCHFV OTU (cyan) in complex with Ub. Both enzymes grasp Ub in a similar orientation, with the C4 zinc finger motif of EAV PLP2 replacing the β-hairpin of CCHFV OTU. (B) EAV PLP2 (blue) bound to Ub (orange), showing the 612-Å2 Ub-binding surface. Residues targeted for mutational analysis to disrupt DUB activity are indicated by arrows. (C) Close-up of the EAV PLP2 Ub-binding surface. EAV PLP2 residue Ile353 forms van der Waals interactions with Ile44, Leu8, and Val70 of Ub, whereas residues Thr312 and Ile313 interact with an additional hydrophobic patch on Ub (formed by residues Leu8, Val70, Leu71, and Ile73) closer to the active site of PLP2.
Structure-Guided Decoupling of PLP2 Deubiquitinase and Polyprotein Cleavage Activities.
Given the distance of helix α3 from the PLP2 active site (Fig. 1B), we hypothesized that mutations could be introduced into this region of the Ub-binding surface that would selectively disrupt PLP2 DUB activity without affecting EAV polyprotein cleavage at the nsp2|nsp3 junction (presumably RLIGG↓WIY). Although this sequence closely resembles the C-terminal tail of Ub (RLRGG↓), we postulated that the nsp2 sequence immediately upstream of the nsp2|nsp3 junction does not adopt a Ub-like fold and that the majority of the PLP2 Ub-binding surface is therefore not required for its cleavage. To test our hypothesis, we used the crystal structure to select three positions within the PLP2 Ub-binding surface (Thr312, Ile313, and Ile353) and engineered a panel of (combined) mutations (Fig. 2 B and C). Ile353 is located at the C-terminal end of helix α3 next to C3 (Cys354) of the zinc finger motif. It projects directly into the Ile44 patch of Ub where it makes extensive van der Waals interactions with Ile44, Val70, and Leu8. Given that Ile353 is located on the Ub-binding surface, we aimed to disrupt Ub binding by introducing various other residues at this position, including large bulky residues such as arginine and tryptophan. Thr312 and Ile313 are located closer to the active site, where they make additional hydrophobic interactions with Leu8, Leu71, and Leu73 of Ub. In an attempt to further disrupt the interaction between PLP2 and Ub, the mutations Thr312Ala and Ile313Val were also combined with changes at position Ile353. Given the close proximity of Ile313 to Leu73 of the RLRGG motif of Ub, mutation to valine was chosen to minimize adverse effects on nsp2|nsp3 cleavage.
Before proceeding to infection experiments with mutant viruses, we used ectopic expression of PLP2 and an in vitro enzymatic assay to characterize the effect of various mutations at the positions described above on polyprotein processing and DUB activity. To this end, mutations were introduced into a mammalian expression vector encoding a self-cleaving nsp2-3 polyprotein carrying an N-terminal HA-tag. On expression of nsp2-3 in HEK293T cells, WT PLP2 mediated efficient cleavage of the nsp2|nsp3 junction (Fig. 3A). As expected, a PLP2 active site mutant (C270A/H332A) did not display any processing of the nsp2|nsp3 site, and only the nsp2-3 precursor was detected. Seven single-site mutants, in which the Ub-binding surface was targeted by replacement of Thr312 or Ile353, displayed WT levels of nsp2|nsp3 cleavage, suggesting that their polyprotein processing activity was not notably affected. In addition, combinations of mutations at positions Thr312, Ile313, and Ile353 were tested, with similar results (Fig. 3A). Longer exposure times did reveal some nsp2-3 precursor, even in the case of WT PLP2, but this amount only marginally increased when two or three mutations were combined (Fig. 4B).
Fig. 3.

Decoupling of the polyprotein processing and DUB activities of EAV PLP2. (A) HEK293T cells were transfected with plasmids encoding nsp2-3 containing WT or mutant PLP2. After 16 h at 37 °C, cells were lysed, and results were analyzed by Western blot. Proteolytic processing of the nsp2|nsp3 junction by WT PLP2 resulted in the release of HA-tagged nsp2 from the nsp2-3 precursor. (B) HEK293T cells were transfected with a combination of plasmids encoding nsp2-3 containing WT or mutant PLP2 and FLAG-Ub. Expression of FLAG-Ub leads to FLAG-tagged ubiquitination of a wide range of cellular proteins, which can be visualized on Western blot using an anti-FLAG antibody. Ub, ubiquitin.
Fig. 4.

PLP2 Ub-binding surface mutations attenuate inhibition of IFN-β promoter activation. (A) Luciferase-based reporter assay to assess the effect of various Ub-binding surface mutations on the inhibition of IFN-β promoter activity by PLP2. HEK293T cells were transfected with a combination of plasmids encoding firefly luciferase under control of the IFN-β promoter, renilla luciferase, RIG-I(2CARD), and nsp2-3 containing WT or mutant PLP2. Results were obtained in three independent experiments. Error bars represent SDs, and P values are relative to WT. (B) Lysates obtained in each of the three experiments used for A were mixed in a 1:1:1 ratio and analyzed by Western blot for the expression of nsp2-3.
Next, we assayed the effect of these mutations on PLP2 DUB activity by transfecting mammalian cells with plasmids encoding FLAG-tagged Ub and nsp2-3 carrying WT or mutant PLP2. FLAG-tagged ubiquitination of a wide range of cellular targets could be visualized by Western blot analysis using an anti-FLAG antibody (Fig. 3B). As expected, expression of WT PLP2 strongly decreased the accumulation of Ub-conjugates, whereas expression of the active site mutant (C270A/H332A) had a negligible effect. Fig. 3B presents the results obtained with a selection of PLP2 Ub-binding surface mutants with the most pronounced effect on DUB activity, some of which approached the level of the active site mutant. In contrast, these Ub-binding surface mutations had only minor effects on the deISGylating activity of PLP2 (Fig. S1).
To corroborate our findings from the expression system showing that PLP2 DUB activity could be selectively removed without disturbing nsp2|nsp3 cleavage, an in vitro activity assay of recombinant PLP2 produced in E. coli was performed using the fluorescently labeled substrates Ub-aminomethylcoumarin (Ub-AMC) or RLRGG-AMC, representing the C-terminal peptide motif of Ub. By comparing the activity of PLP2 mutants against Ub-AMC (which requires the Ub-binding surface) versus their activity against RLRGG-AMC (which binds to the active site region only), PLP2 mutants with a selective reduction in DUB activity could be identified. Indeed, compared with WT enzyme, mutants I353W and I353R exhibited ∼10- and 40-fold reductions in specificity (kcat/Km) toward Ub, respectively; in contrast, their activity toward RLRGG-AMC was essentially unaltered (Table 1). Expression of PLP2 containing additional mutations at Thr312 and Ile313 yielded insoluble protein in E. coli, preventing analysis of these mutants. Nevertheless, the results of the in vitro activity assay further confirmed the successful decoupling of the polyprotein processing and DUB activities of EAV PLP2 by specifically targeting key residues of the Ub-binding surface.
Table 1.
Effect of Ub-binding surface mutations on the substrate specificity of PLP2
| PLP2 enzyme | Substrate |
|
| RLRGG-AMC (kcat/Km) (M−1⋅s−1) | Ub-AMC (kcat/Km) (M−1⋅s−1) | |
| WT | 45 ± 11 | 17,000 ± 4,000 |
| I353R | 65 ± 7 | 413 ± 177 |
| I353W | 155 ± 12 | 1741 ± 850 |
Ability of PLP2 to Suppress Innate Immune Signaling Depends on Its DUB Activity.
The effect of the various PLP2 Ub-binding surface mutations on innate immune signaling was initially assessed in the context of ectopic nsp2-3 expression using a luciferase-based IFN-β promoter activity assay. For this, HEK293T cells were transfected with a combination of plasmids encoding firefly luciferase under control of the IFN-β promoter, renilla luciferase as an endogenous control, constitutively active RIG-I(2CARD) to induce innate immune signaling (52), and EAV nsp2-3 containing either WT or mutant PLP2. In this assay, reporter gene expression was reduced to ∼20% on coexpression of WT PLP2, whereas 80% of the level of the untreated control was retained on expression of the PLP2 active site mutant (C270A/H332A; Fig. 4A). Compared with WT PLP2, all mutants included in Fig. 4A were significantly impaired in their inhibitory activity (P < 0.01), with mutants I353R, I353W, T312A/I313V/I353R, and T312A/I313V/I353W displaying inhibition levels similar to that of the active site mutant (P > 0.05). Western blot analysis confirmed equal expression of WT and mutant nsp2-3 (Fig. 4B). These results demonstrated that, at least in the context of the nsp2-3 expression system, the selective removal of PLP2 DUB activity significantly disrupted its ability to suppress IFN-β promoter activity.
Arteriviruses Lacking PLP2 DUB Activity Elicit an Enhanced Host Innate Immune Response.
Having identified mutations in PLP2 that reduce its ability to suppress Ub-mediated innate immune signaling (Fig. 4A) without adversely affecting nsp2|nsp3 cleavage (Fig. 3A), we were in a position to directly evaluate the importance of PLP2 DUB activity for immune evasion during arterivirus infection. The six (combinations of) mutations used in Figs. 3B and 4 were introduced into an EAV full-length cDNA clone, and mutant viruses were launched by electroporation of in vitro transcribed RNA into BHK-21 cells. Immunofluorescence microscopy subsequently confirmed expression of both nsp2 and the nucleocapsid (N) protein, followed by virus spread to initially untransfected cells. These findings indicated that viruses carrying these (combinations of) mutations were replication competent.
We next focused on the two mutants showing the greatest decrease in inhibitory activity in the IFN-β promoter activity assay: I353R and T312A/I313V/I353R (Fig. 4A). We first characterized the replication kinetics of these mutants in a time course experiment in primary equine lung fibroblasts (ELFs), which are derived from the natural host species of EAV and are likely to maintain an intact innate immune response. ELFs were infected with WT or mutant EAV at a multiplicity of infection (m.o.i.) of 0.5 or 5. The first-cycle replication kinetics as determined by real-time quantitative reverse transcriptase PCR (qRT-PCR) measurement of viral genome RNA levels did not notably differ between WT and mutant EAV (Fig. 5 A and B). In addition, viral titers in cell culture supernatants harvested from infected ELFs at 24 h postinfection (hpi) revealed no significant difference between WT and mutant EAV (Fig. 5C). Finally, immunofluorescence microscopy of ELFs infected with m.o.i. 5 revealed expression of nsp2 from 6 hpi onward, and by 9 hpi, expression of N protein could be seen in all cells for both WT and mutant EAV (Fig. S2). These results demonstrated that the replication kinetics of the PLP2 Ub-binding surface mutants and WT control were essentially the same, in line with our previous conclusion that cleavage of the nsp2|nsp3 site is hardly affected by these mutations. At the same time, Western blot analysis showed that, compared with WT EAV, the DUB activity of both mutants was severely impaired during infection (Fig. 5D).
Fig. 5.

EAV PLP2 mutants display similar replication kinetics as WT virus. Equine lung fibroblasts were infected with WT or mutant EAV at m.o.i. 0.5 (A) or 5 (B–D). (A and B) At the indicated time points, total RNA was isolated for qRT-PCR measurement of EAV genomic RNA levels. Results, which were obtained in three independent experiments, were analyzed using the standard curve method and normalized against the relative quantities of GAPDH and β-actin mRNA. Error bars represent SDs. (C) At 24 hpi, cell culture supernatants were harvested, and virus titers were determined by plaque assay on ELFs. Results were obtained in three independent experiments, and error bars represent SDs. (D) Cells were lysed at 10 hpi, and total ubiquitination was assessed by Western blot analysis. AU, arbitrary units; hpi, hours postinfection; N, nucleocapsid; pfu, plaque-forming units; Ub, ubiquitin.
Subsequently, we used real-time qRT-PCR to measure the levels of mRNAs encoding IFN-β, the IFN-stimulated protein MX1, and the proinflammatory cytokine IL8 and investigate the effect of mutations I353R and T312A/I313V/I353R on innate immune signaling during infection of ELFs. Initially, we infected ELFs with WT or mutant EAV at m.o.i. 5, but in this setup, IFN-β mRNA levels remained below the detection limit at all time points analyzed (from 3 to 12 hpi), suggesting that the innate immune response triggered by EAV on initial infection is very limited. Therefore, we next infected ELFs with WT or mutant EAV at m.o.i. 0.25, resulting in infection of ∼20% of cells. We hypothesized that this would allow for IFN-β–mediated priming of uninfected cells as a result of paracrine signaling from cells infected during the first round. The expression of IFN-stimulated genes would then result in a more potent response during the second cycle of infection. Indeed, following such a low m.o.i. infection with WT EAV, low but detectable levels of IFN-β mRNA were induced by 20 hpi (Fig. 6A). Interestingly, at both 20 and 24 hpi, cells infected with either mutant showed significantly increased levels of IFN-β mRNA compared with WT virus-infected cells (P < 0.05), with mutant T312A/I313V/I353R showing the most pronounced difference. In addition, the levels of MX1 mRNA differed significantly between WT and mutant EAV-infected cells at 24 hpi (Fig. 6B). In contrast, at 20 and 24 hpi, no significant difference in the levels of IL8 mRNA was observed on infection with WT or mutant EAV (Fig. 6C). Equally efficient replication of WT and mutant EAV was corroborated by comparing viral genome RNA levels, for which no significant differences were measured (Fig. 6D). In summary, these data show that the PLP2 DUB function is indeed involved in the inhibition of innate immune signaling during EAV infection in primary equine cells and that it is possible to specifically inactivate this function to suppress immune evasion of otherwise fully replication-competent viruses.
Fig. 6.

EAV lacking PLP2 DUB activity elicits an enhanced innate immune response. ELFs were infected with EAV encoding WT or mutant PLP2 at m.o.i. 0.25 and at 20 and 24 hpi RNA was isolated for real-time qRT-PCR measurement of the levels of (A) IFN-β mRNA, (B) MX1 mRNA, (C) IL8 mRNA, or (D) EAV genomic RNA. Results, which were obtained in three independent experiments, were analyzed using the standard curve method and normalized against the relative quantities of GAPDH and β-actin mRNA. Error bars represent SDs, and asterisks indicate a significant difference relative to WT at the same time point. *P < 0.05; **P < 0.01. AU, arbitrary units; hpi, hours postinfection.
Discussion
Because of the widespread adoption of polyprotein synthesis and cleavage as a genome expression strategy in +RNA viruses and retroviruses, proteases have become key regulatory enzymes of the replication of many important human and animal pathogens. Moreover, virus–host coevolution has offered ample opportunity to develop additional protease functions, which promote virus replication by targeting cellular rather than viral substrates. Because both functions depend on the same protease active site, it has been intrinsically difficult to study them independently during virus infection, mainly because protease inactivation per se is generally incompatible with virus viability. The independent assessment of the cleavage of host cell targets requires its decoupling from viral polyprotein processing, which to date has only been achieved for the poliovirus 2A protease (53–56). Our present work on EAV PLP2 illustrates how structure-guided mutagenesis can be applied to specifically disrupt the interaction of a viral protease with its cellular substrate. The identification of a Ub-binding surface that is distant from the PLP2 active site allowed us to specifically inactivate the DUB function of the enzyme and ultimately probe its relevance in the context of the infected cell. Compared with WT EAV, viruses that lack PLP2 DUB activity induced a significantly enhanced innate immune response in primary equine cells while displaying essentially identical replication kinetics, thus demonstrating the importance of this activity in the evasion of innate immune signaling by arteriviruses.
Analysis of the EAV PLP2 fold using the DALI server identified members of the OTU superfamily as its closest structural relatives, supporting its classification as a member of this superfamily. However, the limited sequence similarity between arterivirus PLP2 and other OTU proteases was previously rated as statistically insignificant (29), and our structure reveals topological features that deviate considerably from a typical OTU fold. Domain I of known OTU domain structures contains a pair of solvent-exposed α-helices that pack perpendicularly against two internal helices (Fig. 1 E, G, and H). Although the internal helices are conserved in PLP2 (α1 and α2), the solvent exposed helices have been replaced by a single helix (α4) that packs parallel to helices α1 and α2 to form a three-helix bundle that comprises most of domain I (Fig. 1 E and F). This topology subtly resembles the L domain of papain (57) and markedly reduces the size of domain I and its contribution to Ub binding relative to other OTU DUBs that have been determined in complex with Ub (38, 43–45, 48). The compact fold of arterivirus PLP2 may be required for its function in replicase polyprotein maturation, an activity that is not shared by cellular OTU DUBs or the OTU protease of CCHFV (58).
A more striking deviation from known OTU domain structures is the presence of the zinc finger in PLP2 domain II (Fig. 1C) and its critical importance for catalytic activity (16). Ub-binding domains that contain zinc fingers exist within a number of OTU DUBs, but none comprise part of the distal Ub-binding site within the OTU fold. Instead, they exist as accessory domains that are connected to the OTU domain through flexible linkers (42), where, in the case of A20, they appear to provide additional polyubiquitin linkage specificity and possibly target the protein to specific signaling complexes (59). For PLP2, the zinc finger is an integral part of the OTU fold, and it plays a central role in binding and positioning the distal Ub molecule on the protease surface (Fig. 2A). Currently, OTU DUBs are grouped into three subclasses based on their structural characteristics: the Otubains, the A20-like OTUs, and the OTUs (42). Given the unique features of EAV PLP2, we propose that arterivirus PLP2 enzymes represent a fourth subclass of zinc-dependent OTUs.
We believe that the structure-guided inactivation of the PLP2 DUB function may contribute to the engineering of improved modified live vaccines (MLVs) for arteriviruses. Although generally effective strategies for the prevention and control of equine viral arteritis have been developed, several field strains are poorly neutralized by antibodies from horses vaccinated with the ARVAC vaccine strain (60), and EAV outbreaks continue to cause significant disruptions of the horse breeding industry (61). Although our current work focuses on EAV PLP2, the corresponding protease of PRRSV has been shown to have very similar immune evasive properties in a variety of experimental settings (18, 30, 31). Since its discovery in the late 1980s, PRRSV has spread around the globe and now ranks among the most important swine pathogens. PRRSV infection was estimated to cause annual losses in the order of $500 million in the United States alone (62), and the emergence of highly virulent strains in China is of particular concern (63–65). Moreover, the virus has proven difficult to control, and it has been suggested to counteract innate immunity, thus undermining the overall immune response and viral clearance in infected animals (66). Consequently, inactivation of the PLP2 DUB function may be an important step in the design of improved vaccine candidates. Virus lacking PLP2 DUB activity should induce a more robust innate response and therefore also a more potent adaptive immune reaction than that achieved with the currently available MLVs. Multiple studies have suggested that additional arterivirus nsps may contribute to innate immune evasion (67, 68), and consequently, vaccine efficacy may be further bolstered by targeting a combination of such functions. As in this study, detailed insight into the molecular interactions of these other arterivirus proteins and their host ligands will likely be required to achieve this goal without crippling the basic replication capacity of the virus. In cell culture, our most promising EAV PLP2 mutant, carrying mutations T312A/I313V/I353R, induced an approximately eightfold increase in IFN-β mRNA levels compared with WT virus. Although we were unable to investigate this effect in vivo due to the lack of a small animal model for EAV infection, the disabling of immune evasion mechanisms can result in significant virus attenuation. One striking example is the modification of the immune evasive influenza virus NS1 protein, which yielded attenuated viruses that are promising vaccine candidates (69).
In addition to opening possibilities for vaccine development, the mutants described in this paper should provide excellent tools for identifying the cellular targets of PLP2, which to date remain unknown. Although Western blot analysis suggests that PLP2 acts in a very promiscuous fashion, causing a general decrease in the levels of ubiquitinated host proteins (Fig. 5D), our qRT-PCR results support the idea that there is at least some degree of specificity in the inhibition of innate immune signaling, as evidenced by the PLP2-mediated inhibition of IFN-β mRNA transcription but not IL8 mRNA transcription (Fig. 6 A and C). Because the expression of IFN-β depends largely on the activation of the transcription factor IFN regulatory factor 3 (IRF3) and the expression of proinflammatory cytokines like IL8 does not (27), our findings suggest that PLP2-mediated inhibition of innate immunity is primarily directed at IRF3-dependent signaling. Future experiments will aim at elucidating the target specificity of arterivirus PLP2.
Arteriviruses are not the only virus family harboring proteases with multiple substrate specificities. Especially interesting in this respect are the distantly related coronaviruses (CoVs), which infect a wide variety of species, including livestock, companion animals, bats, and humans. Six CoV species have now been found to infect humans, causing symptoms ranging from mild respiratory illness to acute respiratory syndromes, as in the case of severe acute respiratory syndrome (SARS) CoV (70) and the recently emerged human CoV EMC/2012 (71, 72). The replicase polyproteins of coronaviruses harbor one or two papain-like proteases (PLpros) that participate in polyprotein maturation and presumably promote evasion of innate immunity by means of their DUB activity (32, 34, 37, 73). Although the CoV PLpros belong to the Ub-specific protease (USP) (74, 75) rather than the OTU superfamily of DUBs, it is likely that coronavirus PLpro substrate specificities can be decoupled using a similar structure-guided approach. Here we have illustrated how structure-guided mutagenesis of such a viral protease may be used to enhance the innate immune response to infection, a strategy that may be applied in the design of MLVs targeting arteriviruses and other virus families encoding similar dual-specificity proteases.
Materials and Methods
PLP2 Plasmids.
For bacterial expression of EAV PLP2, a cDNA fragment encoding residues 261–392 of EAV pp1a and an in-frame C-terminal His6 purification tag were inserted downstream of a Ub fusion partner in the pASK3 vector (76), yielding plasmid pASK3-ePLP2. A mammalian expression construct encoding an EAV nsp2-3 polyprotein was made by cloning residues 261–1,064 of EAV pp1a in-frame with an N-terminal HA tag in the pcDNA3.1 vector (Invitrogen). All mutants were engineered by site-directed mutagenesis using Pfu DNA polymerase (Fermentas). Primer sequences are available on request. All constructs were verified by sequence analysis.
Purification and Crystallization of EAV PLP2 Bound to Ub.
E. coli BL21-Gold(DE3) cells were transformed with pASK3-ePLP2 and cultured to an optical density (OD600) of 0.7 in lysogeny broth (LB) medium at 37 °C. The culture was then supplemented with 200 ng/mL of anhydrous tetracycline and incubated for 3 h at 28 °C with shaking to induce expression of the Ub-PLP2-His6 fusion protein. The cells were pelleted and resuspended in ice-cold lysis buffer [20 mM 2-(N-Morpholino)ethanesulfonic acid (MES), pH 7, 500 mM NaCl, 10% (vol/vol) glycerol, 5 mM imidazole, pH 7.4, 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP)] and lysed using a French pressure cell [American Instrument Company (AMINCO)]. The lysate was clarified by centrifugation and loaded onto a Ni-NTA column (Qiagen) preequilibrated with lysis buffer. After washing with lysis buffer supplemented with 15 mM imidazole, recombinant PLP2 was eluted from the column using an equilibration buffer supplemented with 150 mM imidazole and exchanged into 50 mM Tris, pH 8.0, 300 mM NaCl and 5 mM DTT before storing at 4 °C. The endogenous DUB activity of PLP2 resulted in the efficient removal of the N-terminal Ub-tag from the fusion protein during expression in E. coli; therefore, affinity chromatography yielded highly pure PLP2 carrying a C-terminal His6 purification tag only.
The mechanism-based suicide inhibitor Ub–3Br was prepared according to Messick et al. (38) and Borodovsky et al. (39), as described by James et al. (43). Ub-3Br was covalently bound to purified PLP2 by gently mixing the proteins in a 3:2 molar ratio for 1 h at 37 °C. The resulting PLP2-Ub complex was purified by gel filtration (Superdex 75) followed by anion exchange (Source 15Q) chromatography and then exchanged into 20 mM Tris, pH 8.0, 50 mM NaCl before concentrating to 10 mg/mL and storing at 4 °C.
The PLP2-Ub complex was crystallized by hanging-drop vapor diffusion at 10 mg/mL in mother liquor consisting of 100 mM MES, pH 6.2, 18% (wt/vol) polyethylene glycol (PEG) 20,000. Crystals were flash-cooled and stored in liquid nitrogen after sweeping them through mother liquor supplemented with 20% (vol/vol) glycerol.
X-Ray Data Collection and Crystal Structure Determination.
X-ray diffraction data for a multiwavelength anomalous dispersion (MAD) experiment were collected at the Canadian Light Source (beam line 08ID-1). Data were collected at three different wavelengths over the absorption edge of zinc from a single crystal of the PLP2-Ub complex held at 100 K in an N2 (g) stream. The data were processed using MOSFLM and SCALA (77), and structure factor phases were determined using phenix.autosol (78). Initial phases generated by SOLVE were improved by density modification using RESOLVE within the PHENIX package. After reserving a random subset of reflections for cross-validation using the free R-factor (79), a model was built using phenix.autobuild (78) and manually completed and refined using COOT (80) and phenix.refine (78). Crystallographic data and model refinement statistics are summarized in Table S1.
In Vitro Enzymatic Assays.
To ensure complete removal of the N-terminal Ub-tag from recombinant PLP2, E. coli C2523 containing (WT or mutant) pASK3-ePLP2 were cotransformed with plasmid pCG1, which encodes the DUB ubiquitin-specific processing protease 1 (Ubp1) (76). The DUB activity of PLP2 (WT and mutants) was assayed using 7-amino-4-methylcoumarin (AMC)–labeled versions of Ub (Ub-AMC) (Boston Biochem) and the C-terminal peptide motif of Ub: RLRGG-AMC (Enzo Life Sciences). The enzymes cleave the AMC label causing a significant increase in the fluorescence quantum yield of the dye. All reactions were performed in 50 mM Tris-Cl buffer at pH 8 and 100 mM NaCl. Time-dependent fluorescence traces were collected by a Fluorolog-3 Horiba Jobin Yvon fluorimeter. The monochromators were set to 360 nm (excitation) and 460 nm (emission). The slits were set between 1 and 3 nm bandpass depending on substrate concentration. Enzyme activities in all mutants were characterized by the specificity constant . At substrate concentrations significantly smaller than Km, the formation of product follows pseudo–first-order kinetics; therefore, the temporal evolution of product fluorescence F follows the equation
. At substrate concentrations significantly smaller than Km, the formation of product follows pseudo–first-order kinetics; therefore, the temporal evolution of product fluorescence F follows the equation
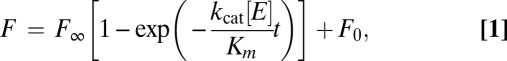 |
where 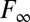 is the fluorescence when all AMC is liberated,
is the fluorescence when all AMC is liberated,  is the background fluorescence, [E] is the total enzyme concentration, and t is the time elapsed. From fitting the fluorescence time traces to Eq. 1, all parameters including the specificity constant
is the background fluorescence, [E] is the total enzyme concentration, and t is the time elapsed. From fitting the fluorescence time traces to Eq. 1, all parameters including the specificity constant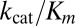 are obtained. Our assays indicate that PLP2 exhibits pseudo–first-order kinetics for Ub-AMC at concentrations less than 0.2 µM and for RLRGG-AMC at concentrations less than 150 µM.
are obtained. Our assays indicate that PLP2 exhibits pseudo–first-order kinetics for Ub-AMC at concentrations less than 0.2 µM and for RLRGG-AMC at concentrations less than 150 µM.
Reverse Genetics.
Mutations in the EAV PLP2 coding sequence were engineered in an appropriate shuttle vector and subsequently transferred to pEAN551/AB, a derivative of EAV full-length cDNA clone pEAN551 carrying additional (translationally silent) AflII and BspEI restriction sites (17, 81). The virus derived from pEAN551/AB was used as a WT control in all experiments. All constructs were verified by sequence analysis.
In vitro RNA transcription from XhoI-linearized WT or mutant EAV full-length cDNA clones was performed using the mMESSAGE mMACHINE T7 Kit (Ambion). Five micrograms of full-length EAV RNA was electroporated into 5.0 × 106 BHK-21 cells using the Amaxa Cell Line Nucleofector Kit T and the program T-020 of the Amaxa Nucleofector (Lonza) according to the manufacturer’s instructions. Cells were incubated at 39.5 °C, and virus-containing supernatants were harvested at 24 h after transfection. Titers were determined by plaque assay on primary ELFs essentially as described before (82).
To verify the presence of the correct mutations, RNA was isolated from virus-containing supernatants using the QIAamp Viral RNA Mini Kit (Qiagen) and converted to cDNA using RevertAid H Minus reverse transcriptase (Fermentas) and random hexameric primers. The region of PLP2 encoding the mutations was subsequently PCR amplified using Pfu DNA polymerase (Fermentas) and sequenced.
Real-Time qRT-PCR.
Confluent ELFs were infected with WT or mutant EAV at m.o.i. 5, 0.5, or 0.25 and incubated at 37 °C. At the indicated time points, cell lysates were harvested in TriPure Isolation Reagent (Roche). After the addition of chloroform, the aqueous phase was mixed in a 1:1 ratio with buffer RA1 of the Nucleospin RNA II kit (Macherey-Nagel). RNA was isolated as per the manufacturer’s instructions and reverse transcribed using RevertAid H Minus RT (Fermentas) and the oligo(dT)20 primer. Finally, samples were assayed by real-time qRT-PCR on a CFX384 Touch Real-Time PCR detection system (BioRad) using iTaq SYBR Green Supermix with ROX (BioRad). Primers (Table S2) targeting mRNAs encoding equine GAPDH, β-actin, IFN-β, MX1, and the EAV genome were designed using Primer3 (83) or sequences were kindly provided by Udeni Balasuriya (University of Kentucky, Lexington, KY) in the case of IL8. The real-time PCR was followed by a melting-curve analysis to verify the specificity of the reaction. Results were quantified using the standard curve method and normalized against the geometric mean of the relative quantities of GAPDH and β-actin mRNA. Data from three independent experiments were analyzed with SPSS Statistics software using a one-sample t test or unpaired Student t test, where appropriate. P < 0.05 was considered statistically significant.
Deubiquitination During Infection.
Confluent ELFs were infected with WT or mutant EAV at m.o.i. 5 and incubated for 10 h at 37 °C. Cells were then lysed in 500 µL 2× Laemmli sample buffer [250 mM Tris, 2% (wt/vol) SDS, 20% (vol/vol) glycerol, 0.01% bromophenol blue, 2 mM DTT, pH 6.8], and total ubiquitination was assessed by Western blot analysis as described in SI Materials and Methods.
Ectopic Expression Experiments.
Ectopic expression experiments were performed essentially as described before (31) but are described in detail in SI Materials and Methods, together with a description of the additional plasmids, cells, and antibodies used.
Supplementary Material
Acknowledgments
We thank Adolfo García-Sastre, John-Paul Bacik, John Hiscott, Udeni B. Balasuriya, Alexander E. Gorbalenya, Aartjan J. W. te Velthuis, Adriaan H. de Wilde, Kathleen C. Lehman, and Diede Oudshoorn for helpful discussions. We thank V. Larmour for technical assistance and S. Labiuk and the staff of the Canadian Light Source (CLS) beamline 08ID-1 for assistance with data collection. We kindly thank the following people for providing us with reagents: Erwin van den Born, Craig E. Cameron, Natalia Frias-Staheli, Michaela U. Gack, Paul N. Moynagh, Adolfo García-Sastre, and Gijs A. Versteeg. The CLS is supported by Natural Sciences and Engineering Research Council of Canada (NSERC), the National Research Council, the Canadian Institutes of Health Research, and the University of Saskatchewan. This research was supported in part by the Division of Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW) through ECHO Grant 700.59.008 (to M. Kikkert and E.J.S.) and by NSERC Grant 311775-2010 (to B.L.M.). B.L.M. holds a Manitoba Research Chair award. The research was also supported in part by the European Union Seventh Framework Programme (FP7/2007-2013) under SILVER Grant 260644.
Footnotes
Conflict of interest statement: The authors have filed a provisional patent application that relates to some aspects of this work.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4IUM).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218464110/-/DCSupplemental.
References
- 1.Firth AE, Brierley I. Non-canonical translation in RNA viruses. J Gen Virol. 2012;93(Pt 7):1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dougherty WG, Semler BL. Expression of virus-encoded proteinases: Functional and structural similarities with cellular enzymes. Microbiol Rev. 1993;57(4):781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya AE, Donchenko AP, Blinov VM, Koonin EV. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989;243(2):103–114. doi: 10.1016/0014-5793(89)80109-7. [DOI] [PubMed] [Google Scholar]
- 4.Gorbalenya AE, Koonin EV, Lai MM. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991;288(1-2):201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hellen CU, Kräusslich HG, Wimmer E. Proteolytic processing of polyproteins in the replication of RNA viruses. Biochemistry. 1989;28(26):9881–9890. doi: 10.1021/bi00452a001. [DOI] [PubMed] [Google Scholar]
- 6.Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257(24):14806–14810. [PubMed] [Google Scholar]
- 7.Kräusslich HG, Nicklin MJ, Toyoda H, Etchison D, Wimmer E. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J Virol. 1987;61(9):2711–2718. doi: 10.1128/jvi.61.9.2711-2718.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102(49):17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 10.Ventoso I, MacMillan SE, Hershey JW, Carrasco L. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett. 1998;435(1):79–83. doi: 10.1016/s0014-5793(98)01027-8. [DOI] [PubMed] [Google Scholar]
- 11.Balasuriya UB, MacLachlan NJ. The immune response to equine arteritis virus: Potential lessons for other arteriviruses. Vet Immunol Immunopathol. 2004;102(3):107–129. doi: 10.1016/j.vetimm.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Huang YW, Meng XJ. Novel strategies and approaches to develop the next generation of vaccines against porcine reproductive and respiratory syndrome virus (PRRSV) Virus Res. 2010;154(1-2):141–149. doi: 10.1016/j.virusres.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang Y, Snijder EJ. The PRRSV replicase: Exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010;154(1–2):61–76. doi: 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(Pt 4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 15.Han J, Rutherford MS, Faaberg KS. The porcine reproductive and respiratory syndrome virus nsp2 cysteine protease domain possesses both trans- and cis-cleavage activities. J Virol. 2009;83(18):9449–9463. doi: 10.1128/JVI.00834-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snijder EJ, Wassenaar AL, Spaan WJ, Gorbalenya AE. The arterivirus Nsp2 protease. An unusual cysteine protease with primary structure similarities to both papain-like and chymotrypsin-like proteases. J Biol Chem. 1995;270(28):16671–16676. doi: 10.1074/jbc.270.28.16671. [DOI] [PubMed] [Google Scholar]
- 17.Posthuma CC, et al. Formation of the arterivirus replication/transcription complex: A key role for nonstructural protein 3 in the remodeling of intracellular membranes. J Virol. 2008;82(9):4480–4491. doi: 10.1128/JVI.02756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frias-Staheli N, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2(6):404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18(5):520–528. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 20.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37(Pt 5):937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 21.Enesa K, et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. 2008;283(11):7036–7045. doi: 10.1074/jbc.M708690200. [DOI] [PubMed] [Google Scholar]
- 22.Kayagaki N, et al. DUBA: A deubiquitinase that regulates type I interferon production. Science. 2007;318(5856):1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 23.Li S, et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem. 2010;285(7):4291–4297. doi: 10.1074/jbc.M109.074971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 25.Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol. 2012;12(1):35–48. doi: 10.1038/nri3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oudshoorn D, Versteeg GA, Kikkert M. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev. 2012;23(6):273–282. doi: 10.1016/j.cytogfr.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen S, Thomsen AR. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86(6):2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20(7):R328–R333. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 29.Makarova KS, Aravind L, Koonin EV. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci. 2000;25(2):50–52. doi: 10.1016/s0968-0004(99)01530-3. [DOI] [PubMed] [Google Scholar]
- 30.Sun Z, Chen Z, Lawson SR, Fang Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol. 2010;84(15):7832–7846. doi: 10.1128/JVI.00217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Kasteren PB, et al. Arterivirus and nairovirus ovarian tumor domain-containing Deubiquitinases target activated RIG-I to control innate immune signaling. J Virol. 2012;86(2):773–785. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devaraj SG, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282(44):32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inn KS, et al. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol. 2011;85(20):10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol. 2009;83(13):6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang J, Tang H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell. 2010;1(12):1106–1117. doi: 10.1007/s13238-010-0141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D, et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol. 2011;85(8):3758–3766. doi: 10.1128/JVI.02589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng D, Chen G, Guo B, Cheng G, Tang H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008;18(11):1105–1113. doi: 10.1038/cr.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messick TE, et al. Structural basis for ubiquitin recognition by the Otu1 ovarian tumor domain protein. J Biol Chem. 2008;283(16):11038–11049. doi: 10.1074/jbc.M704398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borodovsky A, et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9(10):1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- 40.Andreini C, Bertini I, Cavallaro G. Minimal functional sites allow a classification of zinc sites in proteins. PLoS ONE. 2011;6(10):e26325. doi: 10.1371/journal.pone.0026325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue, Suppl 2):W545-9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komander D, Clague MJ, Urbé S. Breaking the chains: Structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 43.James TW, et al. Structural basis for the removal of ubiquitin and interferon-stimulated gene 15 by a viral ovarian tumor domain-containing protease. Proc Natl Acad Sci USA. 2011;108(6):2222–2227. doi: 10.1073/pnas.1013388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akutsu M, Ye Y, Virdee S, Chin JW, Komander D. Molecular basis for ubiquitin and ISG15 cross-reactivity in viral ovarian tumor domains. Proc Natl Acad Sci USA. 2011;108(6):2228–2233. doi: 10.1073/pnas.1015287108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capodagli GC, et al. Structural analysis of a viral ovarian tumor domain protease from the Crimean-Congo hemorrhagic fever virus in complex with covalently bonded ubiquitin. J Virol. 2011;85(7):3621–3630. doi: 10.1128/JVI.02496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juang Y-C, et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol Cell. 2012;45(3):384–397. doi: 10.1016/j.molcel.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiener R, Zhang X, Wang T, Wolberger C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature. 2012;483(7391):618–622. doi: 10.1038/nature10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang OW, et al. Phosphorylation-dependent activity of the deubiquitinase DUBA. Nat Struct Mol Biol. 2012;19(2):171–175. doi: 10.1038/nsmb.2206. [DOI] [PubMed] [Google Scholar]
- 49.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol. 2012;86(7):3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol Cell. 2010;38(5):722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10(10):659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446(7138):916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 53.Morrison JM, Racaniello VR. Proteinase 2Apro is essential for enterovirus replication in type I interferon-treated cells. J Virol. 2009;83(9):4412–4422. doi: 10.1128/JVI.02177-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ventoso I, Carrasco L. A poliovirus 2A(pro) mutant unable to cleave 3CD shows inefficient viral protein synthesis and transactivation defects. J Virol. 1995;69(10):6280–6288. doi: 10.1128/jvi.69.10.6280-6288.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu SF, Lloyd RE. Identification of essential amino acid residues in the functional activity of poliovirus 2A protease. Virology. 1991;182(2):615–625. doi: 10.1016/0042-6822(91)90602-8. [DOI] [PubMed] [Google Scholar]
- 56.Yu SF, Benton P, Bovee M, Sessions J, Lloyd RE. Defective RNA replication by poliovirus mutants deficient in 2A protease cleavage activity. J Virol. 1995;69(1):247–252. doi: 10.1128/jvi.69.1.247-252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamphuis IG, Kalk KH, Swarte MBA, Drenth J. Structure of papain refined at 1.65 A resolution. J Mol Biol. 1984;179(2):233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 58.Bergeron E, Albariño CG, Khristova ML, Nichol ST. Crimean-Congo hemorrhagic fever virus-encoded ovarian tumor protease activity is dispensable for virus RNA polymerase function. J Virol. 2010;84(1):216–226. doi: 10.1128/JVI.01859-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bosanac I, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-κB signaling. Mol Cell. 2010;40(4):548–557. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 60.Zhang J, et al. Molecular epidemiology and genetic characterization of equine arteritis virus isolates associated with the 2006-2007 multi-state disease occurrence in the USA. J Gen Virol. 2010;91(Pt 9):2286–2301. doi: 10.1099/vir.0.019737-0. [DOI] [PubMed] [Google Scholar]
- 61.Holyoak GR, Balasuriya UB, Broaddus CC, Timoney PJ. Equine viral arteritis: Current status and prevention. Theriogenology. 2008;70(3):403–414. doi: 10.1016/j.theriogenology.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 62.Neumann EJ, et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc. 2005;227(3):385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- 63.Tong GZ, et al. Highly pathogenic porcine reproductive and respiratory syndrome, China. Emerg Infect Dis. 2007;13(9):1434–1436. doi: 10.3201/eid1309.070399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, et al. Emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus in the Mid-Eastern region of China. Vet J. 2007;174(3):577–584. doi: 10.1016/j.tvjl.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 65.Tian K, et al. Emergence of fatal PRRSV variants: Unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE. 2007;2(6):e526. doi: 10.1371/journal.pone.0000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimman TG, Cornelissen LA, Moormann RJ, Rebel JM, Stockhofe-Zurwieden N. Challenges for porcine reproductive and respiratory syndrome virus (PRRSV) vaccinology. Vaccine. 2009;27(28):3704–3718. doi: 10.1016/j.vaccine.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 67.Chen Z, et al. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 function as interferon antagonist. Virology. 2010;398(1):87–97. doi: 10.1016/j.virol.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beura LK, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol. 2010;84(3):1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richt JA, García-Sastre A. Attenuated influenza virus vaccines with modified NS1 proteins. Curr Top Microbiol Immunol. 2009;333:177–195. doi: 10.1007/978-3-540-92165-3_9. [DOI] [PubMed] [Google Scholar]
- 70.Perlman S, Netland J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat Rev Microbiol. 2009;7(6):439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 72.van Boheemen S, et al. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6)
- 73.Clementz MA, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol. 2010;84(9):4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wojdyla JA, et al. Papain-like protease 1 from transmissible gastroenteritis virus: Crystal structure and enzymatic activity toward viral and cellular substrates. J Virol. 2010;84(19):10063–10073. doi: 10.1128/JVI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ratia K, et al. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA. 2006;103(15):5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gohara DW, et al. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr Purif. 1999;17(1):128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 77.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 78.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brünger AT. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355(6359):472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 80.Emsley P, Cowtan K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2126–2132.
- 81.van Dinten LC, den Boon JA, Wassenaar AL, Spaan WJ, Snijder EJ. An infectious arterivirus cDNA clone: Identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc Natl Acad Sci USA. 1997;94(3):991–996. doi: 10.1073/pnas.94.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nedialkova DD, Gorbalenya AE, Snijder EJ. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog. 2010;6(2):e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rozen S, Skaletsky HJ. 2000. Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols: Methods in Molecular Biology, eds Krawetz S, Misener S (Humana Press, Totowa, NJ), pp 365–386.
- 84.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.