

Abstract
Large data sets on human genetic variation have been collected recently, but their usefulness for learning about history and natural selection has been limited by biases in the ways polymorphisms were chosen. We report large subsets of SNPs from the International HapMap Project1,2 that allow us to overcome these biases and to provide accurate measurement of a quantity of crucial importance for understanding genetic variation: the allele frequency spectrum. Our analysis shows that East Asian and northern European ancestors shared the same population bottleneck expanding out of Africa but that both also experienced more recent genetic drift, which was greater in East Asians.
According to the fossil record, anatomically modern humans first emerged in Africa ~200,000 years ago (200 kya) and then dispersed through Asia, Australia and Europe starting ~80–40 kya in an expansion known as the ‘out-of-Africa’ event. Although it is widely accepted that the non-African populations were founded by a relatively small group that experienced a founder event that we refer to as a bottleneck3–7, it is unknown how many bottlenecks there were or to what extent they were shared among populations. Patterns of genetic variation can provide information that complements the archaeological record. We focus on the frequencies of alleles of SNPs, the most common type of human variation. Demographic events affect the distribution of SNP allele frequencies3,4. For example, a large proportion of rare alleles indicates recent expansion, as mutations that have occurred since expansion will not have had time to spread through the population. Understanding neutral allele frequencies is also useful for identifying regions of the genome affected by natural selection.
A problem with past SNP-based studies is that they were biased by the way polymorphisms were discovered, meaning that the SNPs did not represent the true distribution of allele frequencies. One issue is that SNPs are generally discovered in a small set of samples, so those with rare alleles tend to be missed8,9. Statistical methods have been developed to correct for this bias4,9. However, every large data set thus far1,10 has involved ascertainment that has been too complex to model fully8, including SNPs discovered in different ways in samples from multiple populations. The only clean data sets for analysis of evolutionary history have been small5–7,11,12. A second bias is experimental: alleles that occur in only a few copies of the ascertainment sample are more likely to be missed; thus, there is a bias against discovering SNPs of low allele frequency13. This problem may have an effect even on very carefully collected data, and is of particular concern as it is impossible to estimate the magnitude of the bias.
To address these sources of bias fully, we identified SNPs by comparing two chromosomes within an individual of known ancestry (Fig. 1). As every SNP that is polymorphic between an individual’s two chromosomes has an equal likelihood of discovery, regardless of true allele frequency in the population, this eliminates experimental bias toward discovering alleles with higher population frequencies. We used the fact that HapMap Phase 2 attempted to genotype every SNP in public databases at the time of marker selection2. By focusing on subsets of SNPs discovered in specific individuals of West African, East Asian or northern European ancestry that were used in HapMap SNP discovery (Table 1), we obtained tens of thousands of SNPs that could be used to estimate the allele frequency spectrum in each population after computationally correcting for discovery in two chromosomes (see Methods).
Figure 1.
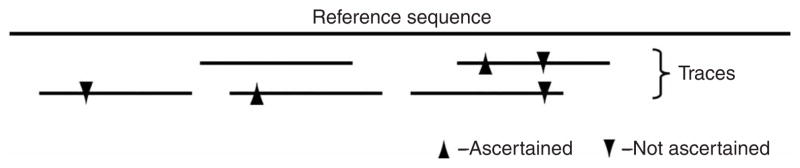
Discovery of SNPs by comparing two sequencing reads from an individual of known ancestry. SNPs useable for analysis are identified as sites where one read matches the reference sequence and the other does not (an arrowhead of either type indicates a mismatch compared with the reference sequence; see Methods). The leftmost SNP is not ascertained because there is only one read, and the rightmost SNP is not ascertained because both reads share the same allele.
Table 1.
Ascertainment libraries
| Individual | Ancestry | Ascertained SNPs | After all corrections | Chromosome 2p Phase 2 SNPs |
|---|---|---|---|---|
| Cor171191 | African American | 615,931 | 114,198 | 6,219 |
| Cor171091 | African American | 108,214 | 17,176 | |
| Cor73401 | European American (CEU) | 293,093 | 52,184 | |
| HuAA30 | European Americana | 115,445 | 18,006 | 643 |
| Cor113211 | East Asian | 261,605 | 45,721 | |
| HuFF30 | East Asiana | 32,338 | 5,250 | 186 |
| Cor104701 | Biaka Pygmy | 37,922 | 3,997 |
The table lists the seven ascertainment libraries for which sequence traces were available. The columns specify each individual’s ancestry, the number of ascertained SNPs, the number of SNPs remaining after all corrections were applied and the number of SNPs genotyped in the chromosome 2p data set. Chromosome 2p data are available only for libraries subjected to whole-genome shotgun sequencing (it is not available for the flow-sorted chromosome libraries).
The ancestry of HuAA and HuFF was determined by using ancestry informative markers (Supplementary Methods).
In practice, we had to deal with some complications. First, HapMap Phase 2 did not design genotyping assays for SNPs attempted in Phase 1 (ref. 1). As genotyping in the two phases had different success rates and characteristically different allele frequencies1,2, we randomly dropped SNPs from Phase 1 and Phase 2 to balance the success rate, repeating this procedure chromosome by chromosome (Supplementary Methods online). The second complication was that the choice of SNPs for Phase 2 HapMap was influenced by a published data set of ~1.6 million SNPs in European Americans, African Americans and Chinese individuals10. SNPs that were observed to have <5% minor allele frequency in all three populations in that study—or that were in complete linkage disequilibrium (LD) with another SNP targeted for genotyping in HapMap—were not attempted in HapMap Phase 2. For these SNPs, we substituted the allele frequencies from ref. 10 (1.5% of SNPs) or from HapMap SNPs in complete LD (5% of SNPs) (see Methods). To test the validity of these corrections, we used the fact that genotyping of all SNPs on the p arm of chromosome 2 had been attempted in Phase 2 of HapMap, irrespective of previous genotyping1. The chromosome 2p data (Table 1) are an excellent match to the whole-genome data (Supplementary Note online).
We obtained allele frequency spectra by identifying SNPs within an individual of known ancestry and studying their allele frequencies in many samples of the same ancestry (Fig. 2). We focused on derived alleles (the new mutations that have arisen in the population), which we identified by comparing to both chimpanzee and orangutan (see Methods). Two qualitative patterns were evident. First, there was a deficiency of rare alleles in Europeans (CEU) and East Asians (CHB+JPT) compared with expectation for a constant-sized population (Fig. 2). This is consistent with a contraction in non-African history (P ≪ 10−12; Supplementary Table 1 and Supplementary Note online) and has been attributed to an out-of-Africa bottleneck3,4,7. By contrast, West Africans (YRI) had more rare derived alleles than expected (Fig. 2), consistent with an expansion (P ≪ 10−12; Supplementary Table 1 and Supplementary Note). An African expansion has been indicated by previous genetic data, although there has been a controversy about whether data have supported it conclusively3,4,7.
Figure 2.

Derived allele frequency spectra in each population. The derived allele frequency spectrum (the proportion of SNPs of each possible derived allele frequency) is shown for each of the HapMap populations, after discovery of SNPs in two reads of the same ancestry. The YRI spectrum is based on SNPs ascertained in both the Cor17109 and the Cor17119 libraries; the CEU spectrum is based on the Cor7340 and the HuAA libraries; and the CHB+JPT spectrum is based on the Cor11321 and the HuFF libraries. SNPs ascertained in individuals of the same ancestry are pooled together (Supplementary Fig. 1 online), as are allele frequency data from the two East Asian populations, CHB and JPT (Supplementary Fig. 2 online). Also shown is the expected derived allele frequency spectrum for a population of constant size throughout history and the same ascertainment scheme. Although all spectra are biased by discovery in two chromosomes, they are comparable because the bias is identical for all spectra (we account for this bias in our analyses; see Methods).
The second qualitative pattern was that East Asians had fewer rare derived alleles than Europeans (Fig. 2). This difference was subtle compared to the difference between Africans and non-Africans, but was highly significant (P ≪ 10−12) and suggests increased genetic drift in East Asian history. Previous analyses have offered hints of greater East Asian genetic drift compared with European genetic drift—a pattern that has been supported by lower diversity in microsatellite data14,15, fewer distinct haplotypes in SNP data16 and multiple aspects of genetic variation7—but these analyses have not been fully corrected for ascertainment biases, and there has been no proof that the signal is significant. In addition, previous studies have not ruled out the alternative explanation of continued migration between Europeans and Africans since the Asian-European split17.
We also generated a complementary data set that supports greater East Asian genetic drift. We aligned hundreds of millions of base pairs from DNA from individuals of known ancestry for whom shotgun genome sequence was available, and we counted the differences per base pair between chromosomes (Supplementary Methods and Supplementary Table 2 online). Because differences accumulate in a clocklike manner, they provide an estimate of the average time since genetic divergence. Excluding hypermutable CpG dinucleotides, West African diversity was 0.8359 ± 0.0048 differences per kilobase, European 0.6044 ± 0.0038 and East Asian 0.5741 ± 0.0051. The most recent common genetic ancestor was more recent in East Asians than Europeans (P ≪ 10−6), as expected for a population with greater genetic drift.
An East Asian population that has undergone greater genetic drift is expected to have allele frequencies with greater divergence (on average) from Africans. To test this, we examined FST18 between each of the non-African populations and the African population, using SNPs ascertained in African Americans (Table 1). As African Americans have some European ancestry, we also repeated the analysis using SNPs identified from a Biaka Pygmy and using SNPs identified from sections of the genome of an African American in which we were confident there no European ancestry (Supplementary Note). In all analyses, FST was significantly larger between East Asians and West Africans (0.178–0.187) than between Europeans and West Africans (0.143–0.158) (P ≪ 10−12; Supplementary Table 3 and Supplementary Note online).
We note that the lower FST between Europeans and Africans could be due to more migration between these populations since European-Asian divergence rather than to greater East Asian drift17. To explore this possibility, we carried out two analyses. First, we studied the sequence divergence data, noting that whereas migration could affect sequence divergence between Africans and non-Africans, genetic drift should affect only allele frequency differentiation. West African–European divergence (0.8345 ± 0.003) was indistinguishable from West African–Asian divergence (0.8312 ± 0.004), providing no evidence for asymmetric migration (P = 0.74; Supplementary Note). Second, we measured the allele frequency difference at 111,604 SNPs between Europeans and East Asians and then tested for a correlation with the allele frequency difference of the same SNPs between West Africans and Mbuti Pygmies19 (Supplementary Note). We did not observe any correlation (r = −0.004; P = 0.21), contrary to what would be expected if there were increased migration between Europeans and Africans since the European-Asian divergence.
Thus far, our analyses were model free. We next searched for models of history that could approximate important features in the data. We began with a model of a single bottleneck, searching independently in Europeans and East Asians by varying the time of occurrence and bottleneck intensity, defined as F = T/2N (the number of generations it lasted divided by twice the effective population size6). A bottleneck fit the data better than a model of a constant population size (P ≪ 10−12; Supplementary Note and Fig. 3). We estimated F = 0.151 ± 0.009 for the Europeans and F = 0.201 ± 0.009 for the East Asians (P < 10−4 for the difference; Supplementary Note), supporting greater East Asian genetic drift. However, the 80- to 40-kya date usually ascribed to the out-of-Africa expansion based on the archaeological record20 is older than the estimated bottleneck dates: 23 ± 2 kya for East Asians and 32 ± 3 kya for Europeans. Thus, this model does not provide inferences about human history that are plausible in the context of the archaeological record.
Figure 3.

Modeling provides an excellent fit to the observed allele frequency spectra. (a,b) CEU (a) and CHB+JPT (b) data compared with that predicted by the models. Model 1 allows for one bottleneck in the history of each population, and model 2 allows for two bottlenecks (Supplementary Methods). For presentation (not actual analysis), the spectra are divided into 15 bins. Model 2 provides a better fit to the data, with mean squared error (averaged over bins) reduced to 62% of the model 1 value in CEU and 47% of the model 1 value in CHB+JPT.
To identify a model that provides a better fit to both the data and the archaeological record, we explored a model with two bottlenecks and found that it fit both the European and East Asian data sets significantly better (P < 10−6; Fig. 3 and Supplementary Note). We estimated that the ancient bottleneck did not have any significant difference in time or intensity between the two populations (P = 0.71; Supplementary Note). We estimated that the recent bottleneck took place 18 ± 3 kya in Europeans and 16 ± 2 kya in East Asians, with estimated intensities of F = 0.091 ± 0.016 and F = 0.123 ± 0.015 (Supplementary Note). Jointly modeling the allele frequencies in Europeans and East Asians (Supplementary Methods), we estimated that the populations diverged 17 ± 3 kya, suggesting that the divergence and bottlenecks may have been associated with the same demographic upheavals, perhaps the last glacial maximum21 (Supplementary Note).
We caution that the two-bottleneck hypothesis is an idealization of a family of models with features that could fit the data. For example, geographic dispersion models14,15 suggest that instead of sudden bottleneck, non-African populations might have experienced a long, drawn-out period of genetic drift, comprised of many mild bottlenecks (the bottleneck intensity measure we use in our modeling provides a reasonable approximation to either a sudden or extended period of genetic drift; Supplementary Note), and further analyses will be necessary to distinguish among these hypotheses. We also did not explore the possibility of gene flow between East Asian and North European ancestors after their initial population separation, which might result in an underestimation of the population divergence time. However, our modeling places an important constraint on the ordering of demographic events. If Europeans and East Asians diverged after the ancient bottleneck, we would expect FST > 0.21, and if after the recent bottleneck, FST < 0.04. The observed value was intermediate (0.10–0.11; P ≪ 10−12; Supplementary Table 4 online), indicating that divergence occurred around the time or shortly before the more recent bottleneck.
The main contributions of this work are the generation of large data sets of simply identified SNPs from HapMap that are useful for population genetic studies, and the use of these data to obtain new insights about human history. Our historical results demand further exploration; in particular, our two-bottleneck model implies a time of ~140–80 kya for the first bottleneck (Supplementary Note), older than the conventionally estimated dates of the out-of-Africa expansion. Possible explanations are that our time estimates may reflect an averaging of an out-of-Africa bottleneck with earlier events11,22 or that the ‘out-of-Africa’ bottleneck may have coincided with the migrations of anatomically modern humans to the Middle East 135–90 kya20,23–25 rather than with the subsequent European and Asian dispersal. We emphasize that there are also other demographic events in human history that our models are not capturing: for example, the explosive population expansion that occurred in the last ten thousand years (Supplementary Note). The data sets we have generated are publicly available, and further study should elucidate the background pattern of human variation and assist in screens for disease genes and regions affected by natural selection.
METHODS
SNP ascertainment
For Phase 2 of the International Haplotype Map project, SNPs were chosen from the list of all available in dbSNP build 121. We focused on the subset that had been discovered by comparing genomic DNA sequencing reads from an individual of known ancestry1,2. These were obtained by whole-genome shotgun approaches or, for some individuals, targeted studies of specific chromosomes. The sequences in the trace archive have ancillary information that indicates which sequences came from a specific library. The SNP discovery tool ssahaSNP26 was used to identify variant alleles relative to the public reference sequence (NCBI build 34). Using two or more overlapping sequencing reads from an individual, we called a SNP as heterozygous in a given individual if the sequencing reads met the neighborhood quality standard threshold and if the reads showed both alleles at the SNP position (Fig. 1 and Table 1).
The key strategy for obtaining an unbiased data set in this study is based on the observation that with some exceptions, genotyping of all SNPs in dbSNP—including all those identified by comparing an individual’s two chromosomes—was attempted in the HapMap1,2. Genotyping of every SNP was attempted in HapMap Phase 2 genotyping with four categories of exceptions that we dealt with as follows. (i) A SNP had already been attempted in Phase 1 (ref. 1). For these, we used Phase 1 genotyping data and corrected for the differential success of SNPs in Phase 1 and Phase 2 (Supplementary Methods). (ii) A SNP design or genotyping failed for Phase 2. We ignored these SNPs in our analysis, which is appropriate because this class of SNPs is not expected to be frequency biased. (iii) A SNP was present in the study of ~1.6 million SNPs10 and showed complete LD (r2 = 1) with another SNP that was attempted in either Phase 1 or Phase 2. For such SNPs, we substituted the allele frequency of a SNP in complete LD, if one existed in HapMap (5% of SNPs in our final data; Supplementary Note); otherwise, we substituted the allele frequency information from the ref. 10 data set (1.1% of SNPs; see below). (iv) A SNP was of minor allele frequency <5% in all three samples of the ref. 10 data set. For these SNPs, we used the available allele frequency information from the ref. 10 data set (0.4% of SNPs). All of these corrections are important, as SNPs of type (i) are subject to the complex ascertainment of the first phase of HapMap1, which is characteristically different from that of Phase 2; SNPs of type (iii) tend to have higher minor allele frequencies than average; and SNPs of type (iv) have lower minor allele frequencies.
Assigning an allele to be ancestral or derived
The power to study demographic history can be increased by examining the unfolded allele frequency spectrum (that is, by knowing which is the ancestral allele and which is the new mutation). We studied only SNPs for which we could determine this with high reliability. We removed all SNPs in hypermutable CpG dinucleotides from the analysis. In addition, we required that both the chimpanzee and orangutan allele agree in their determination of the derived allele state: We aligned sequence traces from the chimpanzee27 and from the orangutan (see URLs section below; used with permission from Washington University Genome Sequencing Center) to the human reference sequence using ssahaSNP26. If both chimpanzee and orangutan sequence traces aligned across the base position of human SNPs, the nonhuman bases were recorded. If the chimpanzee and orangutan alleles further agreed and coincided with one of the two human alleles, then the ancestral allele was determined as the shared human-chimpanzee-orangutan allele; otherwise, we discarded the SNP from the data set.
Determination of SNP allele frequencies
We obtained the genotypes of 60 unrelated Yoruba individuals from Ibadan, Nigeria (YRI), 60 unrelated European American individuals from Utah, USA (CEU), 45 unrelated Han Chinese individuals from Beijing (CHB) and 45 unrelated Japanese individuals from Tokyo (JPT) from Phase 1 and Phase 2 of the International HapMap Project (HapMap public release #19)1,2. The YRI and CEU individuals are unrelated, as we considered only the parents from each of the 30 parent-offspring trios1. We substituted the allele frequencies of 1.5% of SNPs from the ref. 10 data set owing to ascertainment filters described above. For these SNPs, we mapped the allele frequencies of the European American sample to the CEU sample, of the African American sample to the YRI sample (ignoring one sample, as it is one of the ascertainment libraries we considered, Cor17109) and of the Han Chinese sample to the combined CHB+JPT sample, as validated in the Supplementary Note. Although substitution of alleles from the African Americans is, in principle, problematic because African Americans have some European ancestry, this does not have a substantial quantitative effect on our inferences. The correlation of the allele frequencies of the HapMap YRI and the ref. 10 African American samples was 0.98 for the SNPs of low minor allele frequency and 0.93 for the SNPs in complete LD (based on all SNPs on chromosomes 2p; Supplementary Note).
Allele frequency modeling and demographic inference
For each model of demographic history of a single population, we used a maximum likelihood formulation to capture the probability of the data under any possible demography and conditioned on ascertainment. The data analyzed consist of the exact derived and ancestral allele counts for each SNP. For each set of parameter values, we obtained the expected proportion of SNPs of any derived allele frequency by the multi-epoch model as described in ref. 4. To correct for ascertainment in two chromosomes, we then multiplied by the probability 2f (1 − f) of an individual being heterozygous for a SNP of allele frequency f. We obtained maximum likelihood estimates by evaluating the likelihood based on these corrected expected frequency spectra over a grid of each model’s parameters, further refining the grid around the maximal values, using a few hundred values for each parameter. To verify that this obtains the maximum likelihood, we also maximized the likelihood numerically using the Nedler-Mead simplex method28. Standard errors of the maximum likelihood estimates and statistical tests are based on bootstrapping 1,000 random data sets using the moving block bootstrap (MBB)29, randomly resampling contiguous runs of SNPs from the data to take into account the effect of correlation between SNPs in the analysis (Supplementary Table 5 online).
We normalized the maximum likelihood estimates to fit the observed sequence heterozygosity in each population analyzed (Supplementary Methods). We estimated the autosomal mutation rate after removing CpG dinucleotides as 7.94 × 10−10 per year (1.99 × 10−8 per generation for 25 years per generation), assuming 7 million years for human-chimpanzee genetic divergence. We note that errors in this estimate of human-chimpanzee divergence will result in proportionate errors in all date estimates. However, we emphasize that the inferences about population expansion and contraction are independent of this normalization (Supplementary Note).
By using the counts of ancestral and derived alleles at each SNP in our analysis (instead of using allele frequency estimates), we aimed to avoid bias in our likelihood-based estimates of demographic parameters. To verify this property, we randomly chose 60 individuals out of the sample of 90 CHB+JPT individuals, to match the CEU and YRI sample size. Results were similar to what we obtained with the full set of data (data not shown).
URLs
Data are available at http://genepath.med.harvard.edu/~reich/Clean_Asc_HapMap_Data.html. NCBI build history: http://www.ncbi.nlm.nih.gov/SNP/buildhistory.cgi. NCBI human genome assembly release notes: http://www.ncbi.nlm.nih.gov/genome/guide/human/release_notes.html#Top. NCBI trace archive: http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?. Orangutan sequence traces: http://www.ncbi.nlm.nih.gov/Traces/trace.cgi?cmd=retrieve&val=+species_code%3D%22pongo+pygmaeus+abelii%22 (used with permission from Washington University Genome Sequencing Center). HapMap: http://www.hapmap.org.
Supplementary Material
Acknowledgments
We thank D. Altshuler, M. Bernstein, A. Keinan, E. Lander, M. Mandel, M. Mirazon Lahr, A. Price, M. Przeworski, S. Schaffner, C. Stringer and B. Weir for discussions, comments and assistance with stages of this study. We are grateful to G. Marth for sharing the multi-epoch model source code with us. Orangutan sequence traces were produced by the Genome Sequencing Center at Washington University School of Medicine (ftp://ftp.ncbi.nih.gov/pub/TraceDB/pongo_pygmaeus_abelii); we thank R. Wilson for permission to use these data. Sequence traces for the human ABC libraries used in this study were produced by Agencourt Biosciences Corporation and were obtained from ftp://ftp.ncbi.nih.gov/pub/TraceDB/homo_sapiens; we thank D. Smith and E. Eichler for permission to use these data. A.K. was supported by the Rothschild fellowship from Yad Hanadiv foundation. J.C.M. was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (NIH). N.P. was supported by a career transition award from the NIH. D.R. was supported by NIH grant U01 HG004168 and a Burroughs Wellcome Career Development Award in the Biomedical Sciences.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The International HapMap Consortium. The Phase II HapMap. in the press. [Google Scholar]
- 3.Adams AM, Hudson RR. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics. 2004;168:1699–1712. doi: 10.1534/genetics.104.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pluzhnikov A, Di Rienzo A, Hudson RR. Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics. 2002;161:1209–1218. doi: 10.1093/genetics/161.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reich DE, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 7.Voight BF, et al. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci USA. 2005;102:18508–18513. doi: 10.1073/pnas.0507325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen R, Hubisz MJ, Clark AG. Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics. 2004;168:2373–2382. doi: 10.1534/genetics.104.031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinds DA, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 11.Garrigan D, Mobasher Z, Kingan SB, Wilder JA, Hammer MF. Deep haplotype divergence and long-range linkage disequilibrium at xp21.1 provide evidence that humans descend from a structured ancestral population. Genetics. 2005;170:1849–1856. doi: 10.1534/genetics.105.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williamson SH, et al. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc Natl Acad Sci USA. 2005;102:7882–7887. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet. 2006;38:375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- 14.Prugnolle F, Manica A, Balloux F. Geography predicts neutral genetic diversity of human populations. Curr Biol. 2005;15:R159–R160. doi: 10.1016/j.cub.2005.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad DF, et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 17.Bowcock AM, et al. Drift, admixture, and selection in human evolution: a study with DNA polymorphisms. Proc Natl Acad Sci USA. 1991;88:839–843. doi: 10.1073/pnas.88.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution Int J Org Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 19.Becquet C, Patterson N, Stone AC, Przeworski M, Reich D. Genetic structure of chimpanzee populations. PLoS Genet. 2007;3:e66. doi: 10.1371/journal. pgen.0030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellars P. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science. 2006;313:796–800. doi: 10.1126/science.1128402. [DOI] [PubMed] [Google Scholar]
- 21.Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- 22.Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahr MM, Foley R. Multiple dispersals and modern human origins. Evol Anthropol. 2005;3:48–60. [Google Scholar]
- 24.Stringer CB, Grun R, Schwarcz HP, Goldberg P. ESR dates for the hominid burial site of Es Skhul in Israel. Nature. 1989;338:756–758. doi: 10.1038/338756a0. [DOI] [PubMed] [Google Scholar]
- 25.Vanhaereny M, et al. Middle Paleolithic shell beads in Israel and Algeria. Science. 2006;312:1785–1788. doi: 10.1126/science.1128139. [DOI] [PubMed] [Google Scholar]
- 26.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 28.Lagarias JC, Reeds JA, Wright MH, Wright PE. Convergence properties of the Nelder-Mead simplex method in low dimensions. SIAM J Optim. 1998;9:112–147. [Google Scholar]
- 29.Lahiri SN. Resampling Methods for Dependent Data. Springer; New York: 2003. [Google Scholar]
- 30.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.