

Holomycin is a member of the N-acylated dithiolopyrrolone class of natural product antibiotics reported to interfere with bacterial RNA metabolism.[1] The antibiotic scaffold contains an unusual bicyclic core (1, Scheme 1) comprised of a cyclic ene-disulfide fused to an aminopyrrolone ring. While a much more compact framework than the mycotoxin gliotoxin or the FDA-approved histone deacetylase inhibitor FK228 (Scheme 1), holomycin joins these molecules in that it possesses a disulfide bridged heterocycle which is essential to its biologic activity. Disulfides in these molecules are likely to be precursors, reduced in cells to their active dithiol forms.[2] In Streptomyces clavuligerus, holomycin biosynthesis proceeds via formation of a tethered Cys-Cys-S-enzyme intermediate on a nonribosomal peptide synthetase (NRPS) template. Subsequent oxidation by four flavoproteins[3] likely contributes to the formation of a pair of enethiols (one endocyclic, the other exocyclic) that undergo enzymatic oxidation to close the disulfide imbedded in the bicyclic scaffold (1).
Scheme 1.

Compounds containing biologically active disulfides. (A) The conversion of the dihydro-forms of antibiotics holomycin (1) and gliotoxin to their disulfide forms by dithiol oxidases HlmI and GliT, respectively. (B) The structure of the drug FK228, the biological activity of which also depends on an intramolecular disulfide.
For gliotoxin it has been established that the enzyme GliT, which acts late to convert dithio-gliotoxin (Scheme 1) to its isolated disulfide, plays a self-protective role in the producing Aspergilli.[4] In Aspergillus fumigatus, for example, the deletion of gliT resulted in mutants that are much more sensitive to gliotoxin when compared to the wild type. This heightened sensitivity is presumably due to decreased ability of the producer to sequester gliotoxin as the disulfide, resulting in consequent build up of the active dithiol form. Toxicity of the dithiol form of gliotoxin may result from modification of host proteins or redox-cycling, to generate reactive oxygen species.[5]
In analogy, we have recently shown that HlmI, part of the holomycin (hlm) biosynthetic gene cluster in S. clavuligerus, encodes a comparable flavin-dependent dihydroholomycin (2, Scheme 1) oxidase that converts 2 to the bicyclic oxidized form (1, Scheme 1).[6] In this previous study we took advantage of the significant upregulation of the hlm pathway in an S. clavuligerus mutant with a deletion of the ORF15 gene. ORF15 is involved in the biosynthesis of another antibiotic, clavulanate, but the ΔORF15 strain overproduces holomycin 10–100 fold relative to wild type.[7] In both S. clavuligerus wild type and ΔORF15 background, deletion of the hlmI gene resulted in 100–1000 fold decrease in production of holomycin and a significantly heightened sensitivity to exogenously added holomycin.[6] Unresolved was the fate of any presumptively toxic dihydroholomycin accumulating in the ΔhlmI and ΔhlmI/ΔORF15 strains and how these mutants might deal with such a dithiol load. Herein, we describe the use of HPLC-ESI+-MS- and NMR-based comparative metabolomics to identify dihydroholomycin and dihydroholothin derivatives that accumulate in the ΔhlmI and ΔhlmI/ΔORF15 strains and to investigate the related bacterial self-protection mechanisms.
Detection of S-Methylated Forms of Dihydroholomycin
To gain insight into whether dihydroholomycin intermediates build up in the hlmI deletion mutants, we began by comparing the metabolomes of the ΔhlmI/ΔORF15 and ΔORF15 strains via high-resolution HPLC-ESI+-MS. Peaks with masses corresponding to dihydroholomycin (calcd for C7H9N2O2S2, m/z 217.0100) and dihydroholothin (calcd for C5H7N2OS2, m/z 174.9994) were not observed. Instead, two strong peaks (obsd m/z 231.0244 [M + H]+, calcd for C8H11N2O2S2, m/z 231.0256) were present in ΔhlmI/ΔORF15 but not ΔORF15 (Figure 1A top trace). The mass difference of 14.0157 amu relative to dihydroholomycin indicated the addition of a CH2 unit, suggesting either S-methylation (3, Figure 1B regio and/or cis/trans-stereoisomers, and Figure 1A top trace) or addition of a methyl group to the N-3 amide nitrogen of the dithiolopyrrolone scaffold, as observed in the structure of the related natural product thiolutin.[8]
Figure 1.
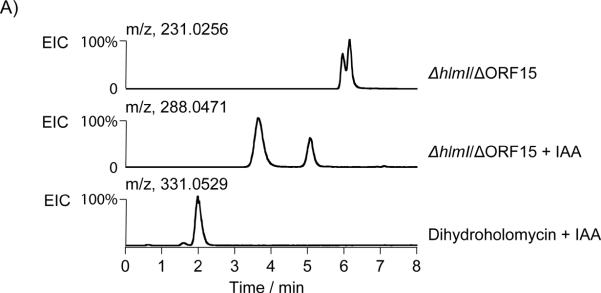

Identification and characterization of metabolites related to the hlm pathway in the ΔhlmI /ΔORF15 mutant. (A) Characterization of the holomycin plus 14.0157 amu metabolite (m/z of 231.0256) in ΔhlmI /ΔORF15 by HPLC-MS and chemical derivatization with iodoacetamide (IAA). Shown are the extracted ion chromatograms (EIC) of ΔhlmI/ΔORF15 (top, [M + H]+, 231.0256), ΔhlmI/ΔORF15 treated with IAA (middle, [M + H]+, 288.0471), and dihydroholomycin standard treated with IAA (bottom, [M + H]+, 331.0529). (B) The structures of hlm biosynthetic pathway intermediates, semi-synthetic derivative, and select disulfide containing metabolites. Compounds 3, 5, 6, and 8–11 are all novel compounds upregulated in the ΔhlmI/ΔORF15 mutant. Compounds, or portions of compounds, colored in red represent structural elements that were supported by HR-MS(MS) analysis, but could not fully be characterized by NMR spectroscopy. Blue arrows represent HMBC correlations that support the structural assignments in these compounds.
As a first pass to distinguish between S- or N-methylation, an excess of thiolalkylating agent iodoacetamide (IAA) was added to the culture supernatant of ΔhlmI/ΔORF15. After incubation at RT for one hour, the m/z 231.0256 peak disappeared and two well separated new peaks emerged, both of which had molecular ions (obsd m/z 288.0466 [M + H]+, calcd for C9H11N3O3S2, m/z 288.0471) suggestive of single IAA adducts (Figure 1A middle trace). Additionally, both new peaks retained the signature UV absorption at 370 nm, suggesting that they are related to, and retain the chromophore of, holomycin[9] (dihydroholomycin has a unique UV absorption at 330 nm, different from that of holomycin or alkylated dihydroholomycin, see Supporting Figure 2). However, no bis-IAA adduct was detected, whereas a control reaction with authentic dihydroholomycin led exclusively to formation of the bis-IAA adduct (Figure 1A bottom trace, obsd m/z 331.0553 [M + H]+, calcd for C11H15N4O4S2, m/z 331.0529). The proposed structure for the bis-IAA adduct (4) is shown in Figure 1B. These findings indicate that ΔhlmI/ΔORF15 does not yield detectable quantities of dihydroholomycin and that at least two mono-S-methylated dihydroholomycin isomers (Figure 1A, top trace) are present in its stead.
Comparative Metabolome Analysis via NMR Spectroscopy
To further characterize biosynthetic intermediates or shunt metabolites that accumulate as a result of the hlmI deletion, we employed NMR spectroscopy-based comparison of the ΔhlmI/ΔORF15 and ΔORF15 metabolomes. Differential analyses of 1D and 2D NMR-spectra (DANS) enables largely unbiased comparisons of related metabolome samples and has previously been employed in microbial[10] and animal systems[11] to identify metabolites associated with genetic or environmental perturbations.[12]
Initial 1H spectra acquired for crude extracts of ΔhlmIΔORF15 and ΔORF15 revealed at least nine ΔhlmIΔORF15-unique singlets between 5.5 and 6.7 ppm (Figure 2A, for full spectra see Supporting Figures 5 and 6). DANS comparison of dqfCOSY spectra acquired for extracts of ΔhlmIΔORF15 and ΔORF15 revealed no additional differential proton signals. To identify the ΔhlmI/ΔORF15-upregulated metabolites via 1H,13C-HMQC and 1H,13C-HMBC spectra, partial fractionation of the ΔhlmIΔORF15 and ΔORF15 samples was required as the compounds of interest accounted for less than 2% of the total ΔhlmI/ΔORF15 metabolome (extracted from 500 mL of a two day culture at an OD600nm ~16). Reverse-phase flash chromatography of the ΔhlmI/ΔORF15 and ΔORF15 extracts yielded three metabolite pools, of which pool 1 contained the most polar metabolites and pool 3 the least polar metabolites. 1H spectra were acquired for these six pools (Supporting Information, Section 3).
Figure 2.

Comparative 1H NMR and 2D HPLC-ESI+-MS analysis of crude extracts of ΔhlmI/ΔORF15 and ΔORF15. (A) Region of 1H spectra containing proton singlets diagnostic for H-1 or H-1' of compounds 3, 5, 6, and 8–11. Peaks marked with red X remain uncharacterized, but may belong to compounds 8 and/or 9. (B) Comparison of mass spectra (175–470 m/z) versus retention time (3–12 min.) plots corresponding to extracts of the two mutants ΔhlmI/ΔORF15 and ΔORF15 revealed a number of cross peaks diagnostic for compounds 3, 5, 6, and 8–11 that are exclusively found, or dramatically upregulated, in ΔΔhlmI/ΔORF15.
Identification of a Methylated Asymmetric Dimeric Holomycin Derivative
Comparison of 1H spectra corresponding to pool 3 ΔhlmI/ΔORF15 and pool 3 ΔORF15 samples showed two groups of singlets (three peaks between 6.3–5.7 ppm and 4 peaks between 2.5–2.0 ppm) dramatically upregulated in the ΔhlmI/ΔORF15 spectrum (Supporting Figure 11). 1H,13C-HMBC and 1H,13CHMQC spectra acquired for ΔhlmI/ΔORF15 pool 3 revealed that the ΔhlmI/ΔORF15-upregulated singlets belong to two new compounds with correlation patterns and chemical shifts similar to those observed for holomycin (1) and dihydroholomycin (2). Based on 1H,13C-HMBC and an additional ROESY spectra, one of the two unknown compounds was identified as bis-S-methylated dihydroholothin (5) (Figure 1B). The remaining ΔhlmI/ΔORF15-dependent analyte in pool 3, like compound 5, appeared to contain a primary non-acylated amine substituent that showed 3-bond 1H,13C-HMBC correlations from its NH2-protons to an amide carbonyl carbon and a methylsulfanyl-substituted olefin carbon. Notably, an exocyclic olefinic proton, analogous to H-1 of 5, coupled with carbons of the enamine ring system and a second N-7-acetylated pyrrolone ring system suggesting a thioether linkage. ROESY correlations between H-1 and H-1' further supported these structural assignments and determined the stereochemistry of this compound, 6 (Figure 1B). High-resolution UPLC-ESI+-MS confirmed the molecular formulae for both 5 and 6. The pool 3 1H,13C-HMBC spectrum further indicated the presence of at least four minor stereoisomers of compound 6.
S-Methylated Dihydroholomycin Derivatives
Comparison of the pool 1 1H spectra revealed three 1H singlets (5.97, 5.90, and ~2.3 ppm) in the ΔhlmI/ΔORF15 spectrum that were absent from the ΔORF15 spectrum. 1H,13C-gHMBC spectra indicated that the two protons at 5.97 and 5.90 ppm belong to similar 1H-13C spin systems. Both protons couple with carbons belonging to a methylsulfanyl moiety (~17 ppm) and appear to be attached to carbons that are part of a holomycin-like olefinic system. The proton at 5.97 ppm showed an additional correlation to another olefinic carbon that does not couple with any other protons. Additionally, the pool 1 ΔhlmI/ΔORF15 1H,13C-gHMBC spectrum showed two major correlations from methyl protons (~1.8 ppm) to carbons at ~ 170 ppm, suggesting the presence of acetylated enamines. The absence of any additional methylsulfanyl correlations in the 1H,13C-gHMBC spectrum further supported the hypothesis that these compounds are a pair of monomethyl dihydroholomycin stereoisomers (3) and its trans-stereoisomer, (Figure 1B).
Because of poor NMR-spectroscopic line shapes of the proton signals in the 1H,13C-HMBC spectrum, not all carbons in the proposed structures 3 could be detected. Derivatization with 2-iodo-N-(1-phenylethyl)acetamide under basic conditions led to isolation of a compound whose 1H, 1H,13C-HMQC and 1H,13CHMBC and high resolution UPLC-ESI+-TOF MS(MS) spectra were consistent with the structure 7, confirming the presence of 3 in the ΔhlmI/ΔORF15 pool 1 sample. The trans-stereoisomer of 7 was not observed. High-resolution UPLCESI+-MS analysis of pool 1 and the 2-iodo-N-(1-phenylethyl)acetamide derivative further supported these structural assignments. In addition, high- and unit-resolution HPLC-ESI+-MS analysis of ΔhlmI/ΔORF15 extracts showed several chromatographic peaks whose mass spectra suggested that they represent disulfide-bridged dimers of 3 such as 8 and 9, which likely formed as a result of non-enzymatic oxidation during processing of the samples (see Supporting Information, Section 8d).
Analysis of 1H and 1H,13C-gHMBC spectra of ΔhlmI/ΔORF15 pool 2 suggested the presence of a bis-methylated dihydroholomycin and a corresponding sulfoxide, both of which were absent in the corresponding ΔORF15 sample. As in the case of 3, poor NMR line shapes prevented complete NMR-spectroscopic characterization. However, high resolution HPLC-ESI+-MS of enriched 10 and 11 showed peaks whose mass spectra are consistent with the proposed structures (obsd m/z 245.0401 [M + H]+, calcd for C9H13N2O2S2, m/z 245.0413 for 10, and obsd m/z 219.0240 [M + H]+, calcd for C7H11N2O2S2, m/z 219.0256 for 11).
Targeted metabolite profiling of wild type, ΔORF15, and ΔhlmI S. clavuligerus strains
Next we asked whether the compounds we identified by comparing the ΔhlmI/ΔORF15 and ΔORF15 metabolomes occur in the wild type strain and what the relative quantities were. For this purpose, we used targeted HPLC-ESI+-MS-based profiling using the diagnostic mass spectral fragmentation patterns and retention times established for the ΔhlmI/ΔORF15-dependent metabolites 3, 5, 6, and 8–11 (Figure 2B). Of compounds 3, 5, 6, and 8–11, only 1 and 6 are found in all the strains profiled (Supporting Figure 1). The ΔORF15 mutation increases the amount of compound 6 by approximately tenfold relative to wild type, whereas the ΔhlmI/ΔORF15 double mutations increase the amount of 6 by two and three orders of magnitude, respectively. The primary enamine-containing compounds 5, 9, and 11 were only observed in extracts of the ΔhlmI/ΔORF15 mutant.
Activity of dimethyldihydroholomycin against S. clavuligerus strains
To evaluate the effect of S-methylation on the activity of dihydroholomycin, we chemically synthesized an S,S'-dimethyldihydroholomycin standard, which contained both isomeric forms. Our attempt to synthesize the S-monomethyldihydroholomycin was unsuccessful due to apparent rapid formation of the dimethyl species. Therefore we were unable to obtain and to test the inhibitory effect of monomethylated compounds. The 1H and 13C NMR spectroscopic data of the synthetic S,S'-dimethyldihydroholomycin were consistent with those of the isolated sample of compound 10, confirming our structural assignments (Supporting Figure 13 and 14).[13] The antimicrobial activity of S,S'-dimethyldihydroholomycin was tested in a bioassay against S. clavuligerus wild type, ΔORF15, ΔhlmI, and ΔORF15/ΔhlmI strains in parallel with the same concentrations of holomycin. S,S'-dimethyldihydroholomycin did not inhibit the growth of any of the tested strains, whereas holomycin displayed inhibitory effect against all four strains, especially for the ΔhlmI mutants (Supporting Figure 3) as observed in our previous work.[6] These results suggest that the dithiol groups of reduced holomycin are relevant to the biological activity of this antibiotic and S-methylation of the dithiols in the producing bacteria significantly decrease that antimicrobial activity as a self-protection mechanism. However, we could not directly test the activity of dihydroholomycin as it is oxidized in minutes to holomycin in air.[6]
Discussion
These studies were undertaken with the specific question of how S. clavuligerus protects itself during holomycin production when it has insufficient capacity (e.g. in the hlmI null mutant) to oxidize the presumed proximal toxin, dihydroholomycin, to the disulfide that is normally excreted into the medium. More generally the study probes the question of what chemical strategies producer microbes utilize to blunt the reactivity of unwanted thiol groups when disulfide formation is suppressed. Our findings indicate that S-methylation in combination with dimerization plays a major role.
Building upon previous studies that utilized NMR- and HPLC-MS-based comparative metabolomics to connect metabolic outputs to specific biosynthetic pathway gene mutations,[10b] we have profiled the chemical species upregulated in the ΔhlmI/ΔORF15 mutant relative to ΔORF15. The three ΔhlmI/ΔORF15-upregulated metabolites fully characterized in this study 3, 5 and 6, all represent novel secondary metabolites, one of which, the dimeric 6, is also consistently produced by the wild type strain. The structures of several additional ΔhlmI/ΔORF15-upregulated compounds were proposed with high confidence, although their NMR-spectroscopic characterization remained incomplete due to the characteristically poor line shapes of these compounds.
We show that a mix of mono- and dimethyl thioethers, with and without N-acetylation, accumulate in the mutant extracts (Scheme 2A). From prior in vitro studies we assigned HlmA and HlmI as the two enzymes catalyzing the last two steps in holomycin assembly.[3, 6] Results of the current study are consistent with these roles, although either step could be the ultimate step. The unusual dimeric adduct 6 detected at higher levels in the ΔhlmI/ΔORF15 mutant, relative to all the other S. clavuligerus strains probed, has a thioether bridge from the exocyclic ene-sulfur of one pyrrolone unit to the C-6 position of the N-acylaminopyrrolone moiety of a second unit (Figure 1B). In this adduct, two of the sulfurs are S-methylated, as found in the monomeric adducts noted above (3). One might have expected an intermolecular disulfide from the two remaining thiols (the disulfide can be seen but only after oxidation during workup of crude extracts) but this adduct instead contains a thioether linkage, and thus one of the anticipated sulfur atoms appears to have been lost. One possible path to its formation would be for a monomethylated dihydroholomycin, with a free exocyclic enethiol to add into the C-5-6 double bond of a second molecule via conjugate addition (Scheme 2B). The resultant tetrahedral adduct could then eliminate −SH or −SCH3, depending on the state of the sulfur that is departing, to yield the dimeric thioether.[14] This route illustrates the nucleophilic character of the exocyclic enethiol of reduced holomycin, and demonstrates why the producer organism would go to substantial lengths to avoid its accumulation. It also reveals the potential electrophilicity of the cyclic enamide. The reactivity of this enamide is presumably enhanced by acylation of the exocyclic amine by HlmA; however, it may also be further augmented by oxidation of one or other of the sulfur atoms, such as in compound 11.[14]
Scheme 2.

Proposed pathway in ΔhlmI/ΔORF15 mutant strain to generate (A) mono- and bis-methylated intermediates (including 3, 5, and 10) that have been characterized by MS and/or NMR, and (B) heterodimeric compound 6. SAM: S-adenosyl methionine.
In particular, these results summarize the biochemical consequence of upregulating holomycin production in S. clavuligerus (due to the deletion of the ORF15 gene) when the last step oxidation is disabled. In absence of the dithiol oxidase HlmI S. clavuligerus is subjected to build-up of the active (reduced) form of its own toxin. One way to blunt this activity is to remove the nucleophilicity of the enethiol groups. While S-acetylation possibly provides an alternate detoxification strategy, the resultant thioesters would no doubt be readily hydrolysable and rather create a reactive acylating agent. Instead S-methylation, likely the result of methyl transfer from S-adenosylmethionine, is observed. This process could be sequential with monomethyl species as free intermediates (Scheme 2A). The inactivity of the dimethyldihydroholomycin standard observed in the bioassay supports the role of S-methylation as a back-up plan for self-protection. Another possible route of detoxification is for dihydroholomycin to react with mycothiol or mycothione, an abundant cellular thiol in actinomycete,[15] via disulfide exchange or nucleophilic addition. However, we were not able to detect such mycothiol-holomycin adducts.
The identity of the putative SAM-dependent methyltransferase in S. clavuligerus producers is as yet unknown. One might expect an analogous S-methylation self-protection strategy in the ΔgliT mutants of Aspergillus fumigatus to blunt the nucleophilicity and consequent toxicity of the dithiol form of gliotoxin. In fact, a wild type gliotoxin producer, Gliocladium deliquescens adopts this thiol methylation strategy, and has been reported to irreversibly methylate gliotoxin to yield bisdethiobis(methylthio)gliotoxin.[16] Similarly, bismethyl thiol forms of gliotoxin have been isolated from other fungal strains.[17] These are exactly the metabolites that should arise from bis methylation of the dihydro form of gliotoxin in detoxification routes. The bis thioether versions of gliotoxin have been shown to be substantially less toxic than gliotoxin itself.[17a, 18] Additionally, in our earlier DANS study on intermediates in the gliotoxin biosynthetic pathway, we detected both mono and bis methylation on incompletely processed scaffolds.[10b] The S-methylation may be catalyzed by the methyltransferase GliN encoded as a backup plan to protect the producer from the toxicity of the biosynthetic intermediates.
S-methylation of dithiols in bacterial and fungal strains, using the readily available SAM as electrophilic methyl donor, may thus be the preferred protection mechanism against endogenous and exogenous dithiol forms of toxic metabolites when the native dithiol oxidase is absent. Indirectly, this study strengthens our hypothesis that the molecular species with antibiotic properties is the penultimate metabolite in the holomycin pathway, dihydroholomycin. In the producing organism, this metabolite is rapidly converted to the bicyclic disulfide via HlmI action and then exported to the extracellular matrix. When taken up by a susceptible neighboring microbe, the disulfide will be in a microenvironment rich in thiols. Reduction to the mixed disulfide and/or the dithiol in a target cell may be nonenzymatic or enzymatic and generate the reactive nucleophilic enethiol forms of the antibiotic. It is possible that dihydroholomycin, like the dihydro form of gliotoxin, exerts some of its toxicity through redox cycling to generate reactive oxygen species, in which case S-methylation can also prevent that toxicity. Although inhibition of RNA synthesis has been proposed as an end effect of the holomycin class of antibiotics[1b, 1c] the molecular targets of this compact yet latently reactive framework remain to be fully characterized.
Supplementary Material
Acknowledgement
This work is supported in part by grants: NIH GM49338 (CTW), GM099904-01 (BL), NIH GM008500 (Cornell University). The authors thank Steven Malcolmson for review of the manuscript.
References
- [1].a) Jimenez A, Tipper DJ, Davies J. Antimicrob. Agents Chemother. 1973;3:729–738. doi: 10.1128/aac.3.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tipper DJ. J. Bacteriol. 1973;116:245–256. doi: 10.1128/jb.116.1.245-256.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]; Oliva cB., O'Neill A, Wilson JM, O'Hanlon PJ, Chopra I. Antimicrob. Agents Chemother. 2001;45:532–539. doi: 10.1128/AAC.45.2.532-539.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]; b) Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- [3].Li B, Walsh CT. Proc. Natl. Acad. Sci. U. S. A. 2010;107:19731–19735. doi: 10.1073/pnas.1014140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O'Brien J, Nolan A, Stephens J, Fenelon O, Doyle S. PLoS Pathog. 2010;6:e1000952. doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Waring P, Sjaarda A, Lin QH. Biochem Pharmacol. 1995;49:1195–1201. doi: 10.1016/0006-2952(95)00039-3. [DOI] [PubMed] [Google Scholar]
- [6].Li B, Walsh CT. Biochemistry. 2011;50:4615–4622. doi: 10.1021/bi200321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].de la Fuente A, Lorenzana LM, Martin JF, Liras P. J Bacteriol. 2002;184:6559–6565. doi: 10.1128/JB.184.23.6559-6565.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Celmer WD, Solomons IA. J. Am. Chem. Soc. 1955;77:2861–2865. [Google Scholar]
- [9].Okamura K, Soga K, Shimauchi Y, Ishikura T, Lein J. J Antibiot (Tokyo) 1977;30:334–336. doi: 10.7164/antibiotics.30.334. [DOI] [PubMed] [Google Scholar]
- [10].a) Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. Proc Natl Acad Sci U S A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Forseth RR, Fox EM, Chung D, Howlett BJ, Keller NP, Schroeder FC. J Am Chem Soc. 2011;133:9678–9681. doi: 10.1021/ja2029987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pungaliya C, Srinivasan J, Fox BW, Malik RU, Ludewig AH, Sternberg PW, Schroeder FC. Proc Natl Acad Sci U S A. 2009;106:7708–7713. doi: 10.1073/pnas.0811918106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Davies J. Curr Opin Chem Biol. 2011;15:5–10. doi: 10.1016/j.cbpa.2010.11.001. [DOI] [PubMed] [Google Scholar]
- [13].Hjelmgaard T, Givskov M, Nielsen J. Org Biomol Chem. 2007;5:344–348. doi: 10.1039/b616411k. [DOI] [PubMed] [Google Scholar]
- [14].Schachtner JE, Nienaber J, Stachel HD, Waisser K. Pharmazie. 1999;54:335–340. [PubMed] [Google Scholar]
- [15].Newton GL, Buchmeier N, Fahey RC. Microbiol Mol Biol Rev. 2008;72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kirby GW, Robins DJ, Sefton MA, Talekar RR. J. Chem. Soc. Perkin Trans. 1980;1:119–121. [Google Scholar]
- [17].a) Li X, Kim SK, Nam KW, Kang JS, Choi HD, Son BW. J Antibiot (Tokyo) 2006;59:248–250. doi: 10.1038/ja.2006.35. [DOI] [PubMed] [Google Scholar]; b) Lee HJ, Lee JH, Hwang BY, Kim HS, Lee JJ. Arch Pharm Res. 2001;24:397–401. doi: 10.1007/BF02975182. [DOI] [PubMed] [Google Scholar]
- [18].Sun Y, Takada K, Takemoto Y, Yoshida M, Nogi Y, Okada S, Matsunaga S. J Nat Prod. 2012;75:111–114. doi: 10.1021/np200740e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.