

Abstract
Background and Purpose
Glutamate transmission is dysregulated in both multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), the animal model of MS. A characteristic of EAE is increased glutamate transmission associated with up-regulation of AMPA receptors. However, little is known about the role of NMDA receptors in the synaptic modifications induced by EAE.
Experimental Approach
The contribution of NMDA receptors to the alterations of glutamate transmission and disease severity in EAE mice was assessed by means of neurophysiological, morphological, Western blot, metabolic and clinical score assessments.
Key Results
In our EAE mice, there was an NMDA receptor-dependent increase of glutamate release, associated with marked activation of the astroglia. Presynaptic NMDA receptors became overactive during EAE, increasing synaptic glutamate release by a mechanism dependent on voltage-gated sodium channels. By means of NAD(P)H autofluorescence analysis, we also found that EAE has a glutamate and NMDA receptor-dependent dysfunction of mitochondrial activity, which is known to contribute to the neurodegenerative damage of MS and EAE. Furthermore, pharmacological blockade of NMDA receptors in vivo ameliorated both synaptic transmission defects and of the clinical disease course of EAE mice, while EAE induced in mice with a genetically enhanced NMDA receptor signalling had opposite effects.
Conclusions and Implications
Our data, showing both sensitization of NMDA receptors and their involvement in the progression of the EAE disease, supggest that pharmacological impairment of NMDA receptor signalling would be a component of a neuroprotection strategy in MS.
Keywords: excitotoxicity, glutamate, inflammation, multiple sclerosis, neurodegeneration
Introduction
Increased extracellular glutamate concentration, resulting in excessive synaptic excitation, has been convincingly associated with neuronal damage in many acute and chronic neurological disorders, through a process termed excitotoxicity (Olney, 1969; Forder and Tymianski, 2009). Glutamate toxicity has in fact been proposed as the basis of the pathogenesis of ischaemic brain damage (Arundine and Tymianski, 2004) and of several neurodegenerative diseases, including Alzheimer's disease, Huntington's disease, Parkinson's disease, amyotrophic lateral sclerosis (Albin and Greenamyre, 1992) and multiple sclerosis (MS) (Gonsette, 2008).
MS is a chronic, inflammatory and degenerative disease of the CNS, characterized by focal lesions with inflammation, demyelination, infiltration of immune cells, oligodendroglial death and axonal and neuronal degeneration (Compston and Coles, 2008). Several lines of evidence support the hypothesis of a dysregulation of the glutamate system in both MS and experimental autoimmune encephalomyelitis (EAE), the animal model of MS (Bolton and Paul, 1997; Stover et al., 1997; Smith et al., 2000; Matute et al., 2001; Pitt et al., 2003; Sarchielli et al., 2003; Killestein et al., 2005; Srinivasan et al., 2005; Cianfoni et al., 2007; Newcombe et al., 2008; Olechowski et al., 2010; Pampliega et al., 2011). In vivo observations have shown that imbalanced glutamate homeostasis contributes to neuronal and oligodendroglial pathology in MS (Werner et al., 2001; Pitt et al., 2003; Srinivasan et al., 2005), and that blockade of glutamate receptors ameliorates the clinical course of both MS (Plaut, 1987) and EAE (Pitt et al., 2000; Smith et al., 2000; Centonze et al., 2009). In EAE, neurodegeneration follows an inflammation-driven process leading to increased glutamate transmission and late excitotoxicity (Pitt et al., 2003; Srinivasan et al., 2005; Centonze et al., 2009). Our studies were the first to demonstrate synaptic alterations in the brain of EAE mice. We have observed that EAE causes a dramatic potentiation of excitatory transmission by altering both the pre- and postsynaptic sites of the glutamate synapses in the striatum (Centonze et al., 2009; Rossi et al., 2010; 2012; Musumeci et al., 2011). This synaptic alteration contributes to dendritic spine degeneration and severe motor deficits. Accordingly, blockade of glutamate AMPA receptors ameliorated the clinical and synaptic deficits in EAE (Centonze et al., 2009).
While the role of these receptors has been investigated in these studies, little is known about the potential involvement of NMDA receptors in EAE pathophysiology (receptor nomenclature follows Alexander et al., 2011). NMDA receptors are one of the main mediators of the intracellular effects caused by glutamate overload in the synaptic cleft, such as increased calcium entry, oxidative stress and mitochondrial dysfunction (Rosenstock et al., 2010), and recently found to be profoundly compromised in both MS and EAE (Haider et al., 2011; Sajad et al., 2011). In the present work, therefore, we have analysed the involvement of NMDA receptors in the pathogenic mechanisms of EAE. We have selected the nucleus striatum for our investigations because, among the grey matter areas, basal ganglia are vulnerable to the neurodegenerative process associated with MS (Bakshi et al., 2002; Tao et al., 2009; Audoin et al., 2010; Batista et al., 2012; Lebel et al., 2012), are involved in fatigue and cognitive dysfunction of MS patients (Calabrese et al., 2010; Loitfelder et al., 2011) and because striatal excitatory synaptic transmission has already been shown to be affected in EAE (Centonze et al., 2009; Rossi et al., 2011). This study provides evidence of increased NMDA receptor function in EAE mice, which leads to enhanced glutamate release in the synaptic cleft, eventually resulting in mitochondrial dysfunction and severe disease course.
Methods
Experimental animals
All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals and the European Communities Council Directive of 24 November, 1986 (86/609/EEC). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 85 animals were used in the experiments described here. Female C57BL/6 mice were used for immunization experiments, and female mutant mice lacking the d-aspartate oxidase (Ddo) gene and their wild-type (WT) littermates were used for the studies shown in Figure 6. As reported (Errico et al., 2006; 2008), WT (Ddo+/+) and knockout (KO, Ddo–/–) mice were derived from mating of heterozygous (Ddo+/−) mice, back-crossed to F5 generation to C57BL/6 strain. Mice were randomly assigned to standard cages, with four to five animals per cage, and kept at standard housing conditions with a light/dark cycle of 12 h and free access to food and water.
Figure 6.
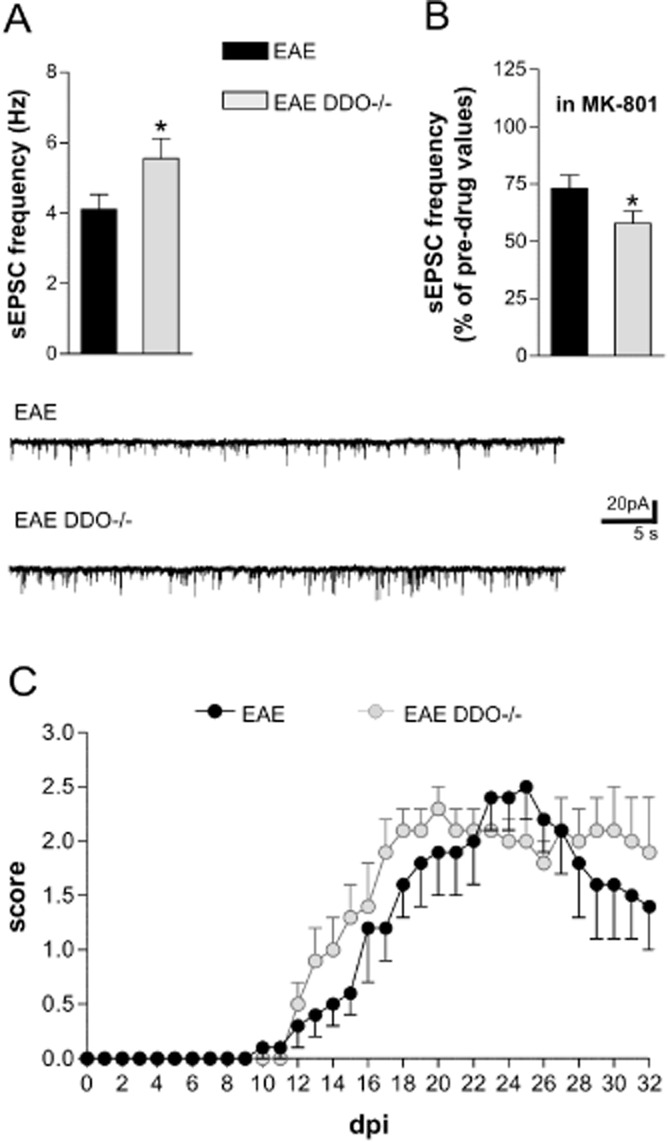
Effect of NMDA receptor potentiation on the synaptic and clinical defects of EAE. (A) The frequency of sEPSCs is exacerbated in DDO–/– EAE mice (#P < 0.05 vs. EAE). (B) The reduction of sEPSC frequency mediated by MK801 is potentiated in DDO–/– EAE mice. The electrophysiological traces below are example of sEPSC alterations in EAE and in DDO–/– EAE. (C) Clinical EAE scores (means ± SEM) over time in DDO–/– (n = 16) and WT mice (n = 17). Despite the fact that the median disease score each day was not significantly different between the groups, the disease course of EAE was faster in DDO−/− mice in both preclinical and acute phases of EAE (clinical onset: 17 ± 1 dpi in EAE mice and 14 ± 1 dpi in DDO–/– mice; day of maximum score: 23 ± 1 dpi in EAE mice and at 20 ± 1 dpi in DDO–/– mice; t-test P < 0.05). Mice with comparable scores (20–25 dpi; score > 2.0) were chosen for the recordings.
Induction and clinical assessment of EAE
Chronic relapsing EAE was induced in 6–8 weeks old mice as previously described (Centonze et al., 2009; Rossi et al., 2011). Briefly, mice were immunized by the subcutaneous injection of 200 μg myelin oligodendrocyte glycoprotein (MOG 35−55) (>85% purity; Espikem, Florence, Italy) in 300 μL of complete Freund's adjuvant (CFA) containing 8 mg·mL−1 Mycobacterium tuberculosis (strain H37Ra; Difco Laboratories Inc., Franklin Lakes, NJ, USA). Pertussis toxin (Sigma-Aldrich, Milan, Italy) (500 ng) was injected on the day of the immunization and again 2 days later. As controls, animals received no treatment (referred to as ‘naive’) or the same treatment as EAE mice without the immunogen, MOG peptide, including CFA and Pertussis toxin (referred to as ‘CFA’). Body weight and clinical score (0 = healthy; 1 = limp tail; 2 = ataxia and/or paresis of hind limbs; 3 = paralysis of hind limbs and/or paresis of forelimbs; 4 = tetraparalysis; 5 = moribund or death) were recorded daily by investigators blind to group identity. The experiment was repeated, and data were pooled. Median and interquartile range was calculated for each group per day to analyse the time course of EAE clinical scores. Mean and SEM were calculated for illustration in figures. For each animal, the onset day was recorded as the day post immunization (dpi) when the first clinical manifestations appeared (score > 0), while the peak day was defined as the day of highest score. The day of maximum score was defined as the middle time point between the first and the last day in which the animal maintained the highest score. Differences between groups were tested by Mann–Whitney test, for their score course, and Student's t-test for the day of onset and of maximum score. Significance level was established at P < 0.05. For the experiments performed during the symptomatic phase of the disease (20–25 dpi), animals with a representative score (always > 2.0) of each experimental group were killed.
Minipump implantation and continuous intracranial infusions
One week before immunization, some EAE mice were implanted with a minipump under ketamine (100 mg·kg−1) anaesthesia, in order to allow continuous i.c.v. infusion of either vehicle (n = 10) or MK801 (6.5 mM; Tocris, Bristol, UK) (n = 13) for 4 weeks. Alzet osmotic minipumps (model 1004; Durect Corporation, Cupertino, CA) connected via catheter tube to intracranial cannula (Alzet Brain Infusion Kits 3) delivered vehicle or MK801 into the right lateral ventricle at a continuous rate of 0.11 μL·h−1. The coordinates used for i.c.v. minipump implantation were as follows: antero-posterior = −0.4 mm from bregma; lateral = −1 mm; depth: 2.5 mm from the skull. After recovery, EAE-MK801 mice did not show any overt behavioural abnormalities.
Electrophysiology
Mice were killed by cervical dislocation under halothane anaesthesia during the preclinical phase (7–9 dpi) or the acute phase (20–25 dpi), depending on the experiment as specified in the results. Corticostriatal coronal slices (200 μm) were prepared from fresh tissue blocks of the brain with the use of a vibratome (Centonze et al., 2009; Rossi et al., 2011). A single slice was then transferred to a recording chamber and submerged in a continuously flowing artificial cerebrospinal fluid (ACSF) (34°C, 2–3 mL·min−1) gassed with 95% O2- 5% CO2. The composition of the control ACSF was (in mM) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, 25 NaHCO3.
Mouse striatum could be readily identified under low power magnification, whereas individual neurons were visualized in situ using a differential interference contrast (Nomarski) optical system. This employed an Olympus BX50WI (Japan) upright microscope with ×40 water immersion objective combined with an infra-red filter, a monochrome CCD camera (COHU 4912), and a personal computer compatible system for analysis of images and contrast enhancement (WinVision, 2000, Delta Sistemi, Verona, Italy). Recording pipettes were advanced towards individual striatal cells in the slice under positive pressure and, on contact, tight GΩ seals were made by applying negative pressure. The membrane patch was then ruptured by suction and membrane current and potential monitored using an Axopatch 1D patch clamp amplifier (Axon Instruments, Foster City, CA). Whole-cell access resistances measured in voltage clamp were in the range of 5–20 MΩ.
Whole-cell patch clamp recordings were made with borosilicate glass pipettes (1.8 mm o.d.; 2–3 MΩ), in voltage-clamp mode, at the holding potential (HP) of −80 mV. To study evoked (eEPSCs), spontaneous (sEPSCs) and miniature glutamate-mediated excitatory postsynaptic currents (mEPSCs), the recording pipettes were filled with internal solution of the following composition (mM): K+-gluconate (125), NaCl (10), CaCl2, (1.0), MgCl2 (2.0), 1,2-bis (2-aminophenoxy) ethane-N,N,N,N-tetraacetic acid (BAPTA; 0.5), HEPES (19), GTP; 0.3, Mg-ATP; 1.0), adjusted to pH 7.3 with KOH. Bicuculline (10 μM) was added to the perfusing solution to block GABAA-mediated transmission.
For synaptic stimulation, bipolar electrodes were used to activate corticostriatal glutamatergic fibres (0.1 Hz). Pulse interval was 40 ms for the experiments on paired pulse ratio (PPR). Amplitudes of eEPSCs were largely dependent on the intensity of stimulation and on the distance between the stimulating and recording sites, and normally ranged between 100 and 500 pA. Evoked, spontaneous, and miniature synaptic events were stored by using P-CLAMP 9 (Axon Instruments) and analysed off line on a personal computer with Mini Analysis 5.1 (Synaptosoft, Leonia, NJ) software. The detection threshold of spontaneous and miniature excitatory events was set at twice the baseline noise. Baseline noise was calculated with Mini Analysis 5.1 by analysing random raw traces (3 of about 400 ms for each cell) not containing synaptic events. Membrane noise was not significantly different between CFA (n = 20) and EAE (n = 22) neurons (1.6 ± 0.10 pA for CFA neurons, 1.4 ± 0.18 pA for 20–25 dpi EAE neurons). The absence of false positive events was confirmed by visual inspection for each experiment. Offline analysis was performed without knowledge of the treatments, on spontaneous and miniature synaptic events recorded during fixed time epochs (3–5 min, 3–6 samplings), sampled every 5 or 10 min. Only cells that exhibited stable frequencies in control (less than 20% changes during the control samplings) were taken into account.
Striatal neurons from EAE mice were compared with those obtained from mice treated with CFA alone. One to six cells per animal were recorded. For each type of experiment and time point, at least five distinct animals were employed for each experimental group. Throughout the text, ‘n’ refers to the number of animals, unless otherwise specified. Electrophysiological data are presented as the mean ± SEM, and statistical analysis was performed using a paired or unpaired Student's t-test or Wilcoxon's test. The significance level was established at P < 0.05.
Drugs were applied by dissolving them to the desired final concentration in the bathing ACSF; these were (in μM) CNQX (10), MK801 (30), tetrodotoxin (TTX, 1), 4-aminopyridine (4-AP, 100), bicuculline (10).
Western blotting
Total protein extracts from striatal microdissection performed at 25 dpi were lysed in standard lysis buffer containing sucrose (320 mM), HEPES (pH 7.4 1 mM) and MgCl2 (1 mM) supplemented with protease inhibitors cocktail (Sigma-Aldrich). Quantification was performed by using BCA protein assay (Thermo, Waltham, MA, USA). Samples (25μg) of each extract was analysed by electrophoresis on SDS-poly-acrylamide gel and subsequently blotted on nitrocellulose (Millipore, Billerica, MA, USA). The following primary antibodies were used: mouse anti-β-actin 1:5000 (Sigma-Aldrich), anti-GluN1 1:2000 (Sigma), anti-GluN2A 1:1000 (Sigma-Aldrich), anti-GluN2B 1:3000 (Millipore). Secondary antibodies (Biorad, Milan, Italy) were incubated for 2 h at room temperature and signals were revealed by using the ECL kit (Millipore).
Immunohistochemistry and confocal microscopy
EAE and CFA mice were deeply anaesthetized at 21 dpi with 2,2,2-tribromoethanol (3.6 mg kg-1, i.p.; Avertine from Sigma-Aldrich) and perfused through the aorta with ice-cold 4% paraformaldehyde. Brains were post-fixed for at least 4 h at 4°C and equilibrated with 30% sucrose overnight. Coronal sections (30μm) were permeabilized in PBS with Triton-X 0.25% (TPBS). All subsequent incubations were performed in TPBS. Sections were pre-incubated with 10% normal donkey serum solution for 1 h at room temperature and incubated with the primary antibody rabbit anti-glial fibrillary acidic protein (GFAP) (1:500) and anti-vimentin (1:100) overnight at 4°C. After being washed three times for 10 min each, sections were incubated with the secondary antibody Alexa-488-conjugated donkey anti-mouse (1:200, Invitrogen) for 2 h at room temperature, rinsed and DAPI counterstained. Sections were mounted with Vecta-shield (Vector Labs, Burlingame, CA, USA) on poly-l-lysine-coated slides, air-dried and coverslipped. Images were acquired using a LSM5 Zeiss confocal laser-scanner microscope (Zeiss, Göttingen, Germany). By using a 20× objective (zoom 0.5×, pixel resolution 1024 × 1024, 5 μm Z-step, pinhole of 1 airy units) immunostaining stacks of the striatum from coronal sections were z-projected and exported in TIFF file format by NIH ImageJ software (http://rsb.info.nih.gov/ij/). GFAP-positive cells were counted on the z-projections. Their number was divided by the surface area of striatal sections to obtain the astroglial density. Astroglial surface was measured as the area of GFAP-positive immunoreactivity, and analysed as percentage of the total striatal area and as a mean cellular surface, maintaining between the groups the same threshold of GFAP signal intensity defining the positivity. Measurements were performed without knowledge of the treatments, and repeated for seven to nine images from five sections per animal (n = 3 mice per group).
To assess astroglial proliferation, CFA and EAE mice (20 dpi) were injected with 5-iodo-2′-deoxyuridine (IdU) (100 mg·kg−1, Sigma) for 10 h before killing. by anaesthetic overdose. Mice were transcardially perfused with 4% paraformaldehyde in PBS pH 7.2. Brains were then fixed 4% paraformaldehyde in PBS pH 7.2 for 12 h at +4°C and cryoprotected for 24 h in 30% sucrose (Sigma) in PBS at +4°C. Brains were subsequently sectioned at 10 μm. One section every 300 μm (from the anterior bregma +1.4 to the posterior bregma −0.4) was used for GFAP/IdU detection and cell counting (Gobeske et al., 2009; Pohl et al., 2011). GFAP/IdU staining was performed as previously described (Centonze et al., 2009). Briefly, sections were incubated in HCl 2 M for 20 min and then in buffer borate 0.1 M, pH 8.5 for 10 min. Sections were then incubated in blocking solution as above described before receiving rabbit anti-GFAP (1:500) and mouse anti-IdU (1:100, BD) overnight at 4°C. After being washed three times for 10 min each, sections were incubated with the secondary antibody conjugated with Alexa-488 and Alexa-546 fluorochrome.
NAD(P)H fluorescence imaging
CFA and EAE mice (20–25 dpi) were deeply anaesthetized with a mixture of ketamine and xylazine (85 and 15 mg·kg−1, respectively) and decapitated. Brains were removed and placed in ice-cold cutting solution. Coronal sections (350 μm) were cut with a Vibratome (Leica VT1000S, Leica Microsystems AG, Wetzlar, Germany), and slices were kept in ACSF at room temperature for 90 min. Cutting and recording ACSF solutions were both 300–305 mOsm·L−1. Recording ACSF contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2 and 10 glucose equilibrated with 95% O2/5% CO2. Cutting solution contained: 3 mM KCl, 1.25 mM NaH2PO4, 6 mM MgSO4, 26 mM NaHCO3, 0.2 mM CaCl2, 10 mM glucose, 220 mM sucrose, and 0.43 mM ketamine.
Individual corticostriatal slices were transferred to the recording chamber and were perfused with warmed (37°C) oxygenated ACSF at 2 mL·min−1 for 10 min before recording. Drugs were applied by dissolving them to the desired final concentration in the bathing ACSF (10 min). Drugs used were (in μM) CNQX (10), MK801 (30) and bicuculline (10). Bicuculline was applied for the entire duration of the recording to inhibit GABA receptors.
NADH has broad excitation and emission spectra with peaks at ∼350 and 460 nm respectively. Excitation (360 nm) was delivered via an optic fibre/monochromator system (DeltaRAM VTM; Photon Technology International, Lawrenceville, NJ, USA) reflected onto the slice surface via a dichroic mirror (DMLP 400 nm, Chroma Technology, Brattleboro, VT, USA). Fluorescence emission was collected using a cooled interline transfer CCD camera (MicroMAX System, Princeton Instruments, Trenton, NJ, USA). All experiments used a 410 nm long-pass glass filter between the dichroic mirror and the camera to maximize light capture. Imaging was performed after focusing onto the surface of slices, using 10× water immersion objective (numerical aperture 0.3; Olympus), and collected after 2 × 2 binning of the 512 × 512 line image. For analysis, performed without knowledge of the treatments,, the image data were filtered by using 3 × 3 pixel averaging and presented as the change in fluorescence intensity/pre-stimulus fluorescence intensity (ΔF/Fo). Data were gathered over a 2500 μm2 circle surface tangent to the stimulus electrode.
Corticostriatal excitatory fibers were stimulated using a bipolar electrode (50 mm tip) in the presence of bicuculline (10 μm). Stimuli were delivered via a Master-8 controller, DC supply and constant current isolation unit (AMPI, Jerusalem, Israel). We used for all acquisitions the same protocol: the stimulus frequency was 100 Hz for 500 ms (interval 70 μs) with the intensity of 2 mA. Between successive tetanic stimuli 5 min intervals were maintained. The fluorescence excitation/imaging at 4 Hz (100 ms of exposition) began 4 s before onset of the electric stimulus and continued for a total of 25 s, so that the kinetics of initial NAD(P)H oxidation events (transient negative deflections) could be assessed.
In the study, n refers to number of animals, with five acquisitions from each slice, with a maximum of three slices obtained from each experimental animal used for each protocol (six animals each group).
Materials
CNQX, MK801 and tetrodotoxin (TTX) were obtained from Tocris Cookson; 4-aminopyridine (4-AP) and bicuculline from Sigma-RBI, St. Louis, MO.
Results
Role of NMDA receptors in EAE-induced alterations of sEPSCs
EAE causes increased frequency and duration of glutamate-mediated sEPSCs, reflecting enhanced glutamate release from presynaptic nerve terminals (Rossi et al., 2010) and enhanced postsynaptic glutamate AMPA receptor expression and sensitivity (Centonze et al., 2009) respectively.
NMDA receptors control glutamate transmission at both pre- and postsynaptic levels (MacDermott et al., 1999), and might therefore play many roles in the abnormalities of glutamate transmission in EAE mice. To address this possibility, we incubated corticostriatal brain slices from both control (CFA) and EAE mice in the presence of MK801 (10 min), a selective antagonist of NMDA receptors. According to previous findings (Centonze et al., 2009; Rossi et al., 2010), sEPSC frequency and duration (both decay time and half width) increased in EAE mice in the presymptomatic phase of the disease (7–10 days post immunization, cell number = 21) and were still abnormal in the symptomatic stage (20–25 dpi, cell number = 21) in comparison with CFA (cell number = 19) and naive mice (cell number = 18) (Figure 1A,B). MK801 reduced the frequency of sEPSCs in EAE mice (20–30 dpi, P < 0.05, n = 15; Figure 1C), while sEPSC decay time and half width were unaffected (P > 0.05; not shown). On the contrary, in slices from CFA (cell number = 11) and EAE mice in the presymptomatic phase (cell number = 9), MK801 did not have any effect on either sEPSC frequency (P > 0.05; Figure 1C) or duration (decay time and half width; P > 0.05; not shown). When we blocked the glutamate AMPA receptors with CNQX (10 min), the sEPSCs were fully abolished in both CFA (n = 8) and EAE mice (7–10 dpi: n = 5; 20–25 dpi: cell number = 6; Figure 1C).
Figure 1.
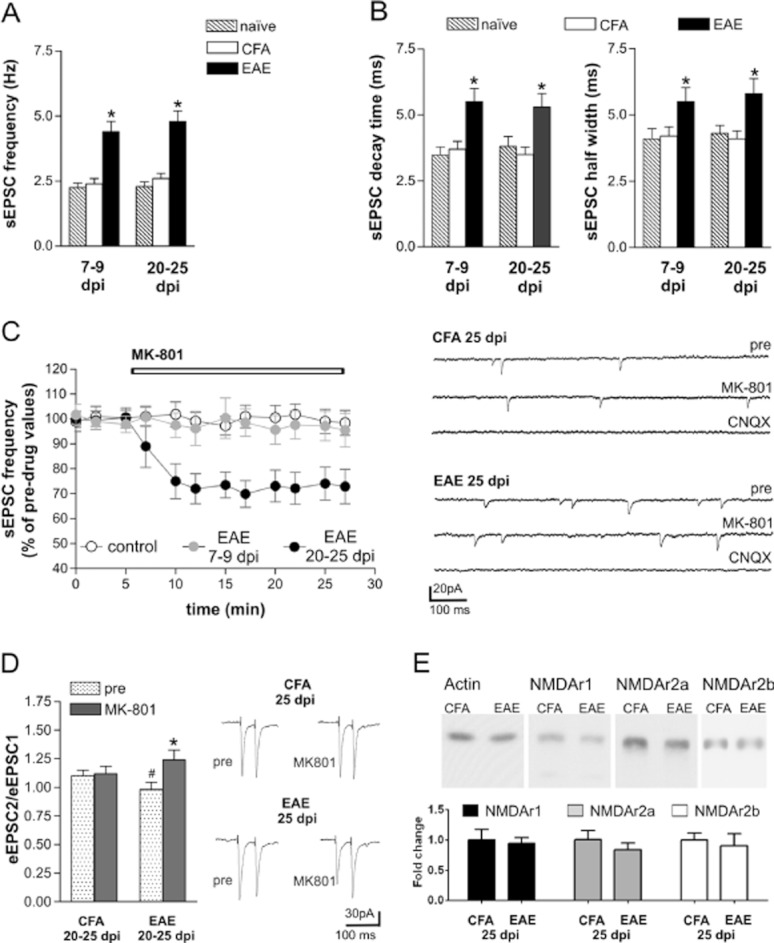
Role of NMDA receptors in EAE-induced alterations of sEPSC. (A) The frequency of glutamatergic sEPSCs recorded from striatal neurons increased in the preclinical (7–9 dpi) and in the acute (20–25 dpi) phase of EAE (*P < 0.01) in comparison with both control groups, CFA and naive mice. (B) Decay time and half width of sEPSCs were increased in both preclinical and clinical stages of EAE in comparison with both control, CFA and naive mice. (C) MK801, a selective NMDA receptor antagonist, reduced sEPSC frequency in EAE mice (20–25 dpi) but not in CFA mice and in EAE mice before the disease onset (7–9 dpi). The electrophysiological traces are examples of voltage-clamp recordings before and during the application of MK801, in CFA and in EAE mice. The sEPSCs were fully abolished in the presence of the AMPA receptor blocker CNQX (10 min). (D) Paired-pulse ratio (PPR) of eEPSCs was reduced in EAE mice (#P < 0.05 compared with CFA mice in basal conditions), and MK801 increased PPR in EAE mice but not in CFA mice (*P < 0.01 compared with pre-drug). The electrophysiological traces on the right are examples of eEPSCs evoked with a 70 ms interstimulus interval before (left) and during the application of MK801(right) in CFA and in EAE mice. (E) Western blot analysis of the expression of NMDA receptors subunits in striata of EAE and CFA mice at 25 dpi. Quantification of GluN1, GluN2A and GluN2B subunits shows no difference between CFA and EAE from three independent experiments, suggesting that NMDA receptor expression was not altered in EAE. Western blot data were normalized to actin and plotted as percentage of CFA mice. In all experiments performed during the symptomatic phase of the disease (20–25 dpi), animals with a representative score of the disease (>2.0) were killed.
Together these results suggest that an abnormal activation of the NMDA receptors localized at presynaptic level in EAE mice contributed to increased sEPSC frequency in EAE striatum during the symptomatic phase of the disease. On the contrary, presynaptic NMDA receptor signalling did not affect the glutamate transmission before onset of the disease.
Effects of MK801 on paired pulse ratio (PPR) of eEPSCs
The selective effect of MK801 in partially restoring sEPSC frequency in EAE suggests that this antagonist is mainly acting presynaptically to reduce glutamate release in EAE. We confirmed this idea by assessing the effect of MK801 on the PPR of electrically evoked EPSCs (eEPSCs), as pharmacological manipulations that reduce transmitter release from presynaptic terminals are well known to increase this neurophysiological parameter (Manabe et al., 1993; Nicoll and Malenka, 1999; Thomson, 2000). MK801 reduced eEPSCs and increased PPR in EAE mice (20–25 dpi, cell number = 6), while it did not change eEPSCs or PPR in CFA animals (cell number = 5) (Figure 1D). Notably, basal PPR was reduced in EAE compared with CFA mice (P < 0.05), a finding which is in agreement with the idea that presynaptic glutamatergic terminals are more prone to release glutamate in response to synaptic activation in EAE than in controls (Figure 1D). Thus, presynaptic NMDA receptors become overactive during EAE and play a regulatory role in promoting synaptic glutamate release.
Expression of NMDA receptors in EAE
Because the activity of NMDA receptors is significantly affected by the level of their expression (Yashiro and Philpot, 2008), we investigated whether the increased sensitivity of NMDA receptors in EAE (25 dpi) was associated with altered expression of such proteins. By Western blot assays, we found that the major NMDA receptor subunits GluN1, GluN2A and GluN2B (also known as NR1, NR2A and NR2B) did not show significant changes in EAE striatum compared with those from CFA striatum (Figure 1E).
Thus, our results argue against marked changes in the expression level in the whole striatum of NMDA receptors themselves, as the primary mechanism leading to synaptic hyperactivity in EAE, suggesting the possibility of a finer modulation, perhaps confined to the presynaptic site.
Role of voltage-dependent Na+ channels in abnormal NMDA receptor function in EAE
Increased expression of voltage-gated Na+ channels results in intra-axonal accumulation of Na+ ions in MS and in EAE (Saab et al., 2004; Craner et al., 2004b; Waxman, 2005). Previous work has established the critical role of intracellular Na+ ions in the potentiation of the activity of NMDA receptors in neurons (Xin et al., 2005; Yu, 2006). We asked therefore if Na+ ions flowing through voltage-dependent Na+ channels in axons could be responsible for the observed facilitation of presynaptic NMDA receptor function in EAE. In the presence of the voltage-dependent Na+ channel blocker TTX, MK801 was no longer able to reduce the frequency of synaptic events (mEPSCs) in acute EAE mice (cell number = 5; P > 0.05), indicating that the abnormal sensitivity of presynaptic NMDA receptors in this disease was indeed caused by intra-axonal Na+ accumulation (Figure 2A).
Figure 2.
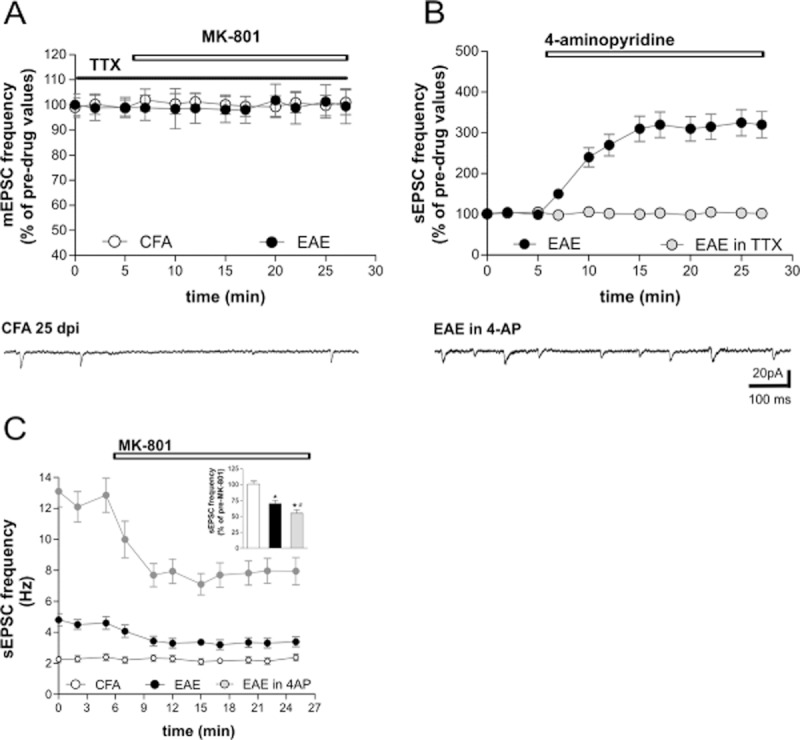
Role of voltage-dependent Na+ channels in abnormal NMDA receptor function in EAE. (A) Pre-incubation with TTX prevented the depressant action of MK801 on sEPSC frequencies in EAE mice. (B) The graph shows that 4-AP, a K+ channel blocker, further increased the frequency of sEPSCs in EAE mice and co-incubation with TTX completely abolished this effect, indicating that it was dependent on voltage-dependent Na+ channels. (C) The graph shows the effect of MK801 incubation on the sEPSC frequencies of 4-AP-treated slices. As shown in the inset, the treatment with 4-AP enhanced the depressant effect of MK801 in EAE mice (*P < 0.01 compared with CFA mice and #P < 0.05 compared with EAE untreated mice). Recordings were performed on EAE mice (20–25 dpi) with a representative score of the disease > 2.0.
We confirmed this conclusion in a further set of complementary experiments, in which action potentials and the resulting Na+ accumulation were enhanced in axons with 4-AP, a K+ channel blocker recently approved for the treatment of some disabling symptoms of MS (Hayes, 2011). By incubating EAE striatal slices (20–25 dpi, cell number = 8) in the presence of 4-AP, we observed a further increase (about three times; P < 0.01) of the sEPSC frequency relative to pre-drug recordings or EAE frequency values (Figure 2B and C). Co-incubation with TTX blocked the effect (cell number = 8, P > 0.05), indicating that it was entirely mediated by the promotion of action potentials (Figure 2B). When we blocked NMDA receptors in EAE slices incubated with 4-AP (20–25 dpi, cell number = 6), we observed a more pronounced inhibition of sEPSC frequency (P < 0.05) compared with the effects of MK801 in EAE slices not treated with 4-AP (Figure 2C), indicating the cumulative sensitization of the NMDA receptors dependent on endogenous (mediated by EAE) and exogenous (mediated by 4-AP) intra-axonal Na+ accumulation.
Altogether these experiments confirm the crucial role of Na+ ion influx through voltage-gated Na+ channels in sensitization of presynaptic NMDA receptor in EAE.
Astroglial activation in EAE
While providing a possible mechanism for sensitization of presynaptic NMDA receptors, the results presented above do not explain how these receptors are preferentially activated in EAE. Presynaptic NMDA receptors are a rather selective target of glutamate released from astrocytes (Jourdain et al., 2007; Parri and Crunelli, 2007), indicating that enhanced stimulation of these receptors could be mediated by astroglial cell activation in EAE. We assessed astroglial activation by astrocyte immunostaining in the striatum of EAE mice (21 dpi). Coronal striatal slices from CFA and EAE mice were stained with GFAP and vimentin antibodies (Figure 3A and E). Virtually all cells expressing vimentin at robust levels, co-expressed GFAP (Figure 3E). We found an increased number of astroglial cells in striatum from EAE mice, compared with CFA (n = 3 each group; P < 0.01) (Figure 3B). Accordingly, in EAE striatum the total surface covered by astroglial cells, measured as area of the GFAP-positive cells, was almost double that measured on CFA slices (P < 0.05; Figure 3C). Marked morphological differences were not obvious between the two groups, in terms of the calculated mean astroglial area (Figure 3D). We also injected CFA and EAE mice with the S-phase tracer IdU for 10 h on the day of killing (20 dpi, n = 3 for each group, P < 0.0001) to label proliferating cells within the striatum. Although the great majority of proliferating cells detected within striatum belonged to the microglia/macrophage lineage (Centonze et al., 2009), approximately 17% of GFAP-expressing cells in EAE brains incorporated the IdU tracer, suggesting that reactive astrocytes increase their number during EAE-induced acute inflammation (Figure 3E).
Figure 3.
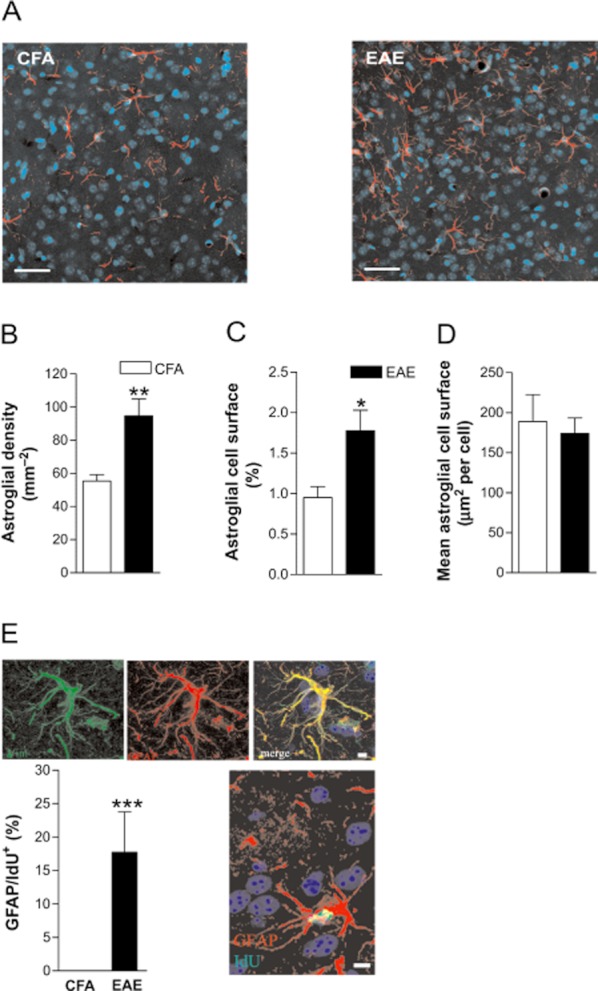
Astroglial activation in the symptomatic phase of EAE. Coronal striatal sections from control and EAE mice (25 dpi) were stained with anti-GFAP antibody and counterstained with DAPI. (A) Representative images from CFA and EAE striata (magnification 20×). Scale bar, 100 μm. (B) EAE striatal slices showed a highly increased number of GFAP-positive cells, counted on confocal microscopy images and expressed as density (mm–2) in comparison with CFA (n = 3; **P < 0.01). (C) The area of GFAP immunopositivity, expressed as percentage of the total striatal area, was greater in EAE striatal slices compared with CFA (*P < 0.05). (D) The mean cell area was not significantly different between EAE and CFA (P > 0.05). (E) Reactive GFAP expressing cells proliferate within the EAE striatum. Sections were stained for GFAP and vimentin a specific marker of astroglia activation; virtually all astrocytes from EAE striatum co-expressed both marker. CFA and EAE mice (20 dpi, n = 3 for each group) were injected with IdU for 10 h and then killed. IdU+ cells were distributed within the striatum of EAE mice, but very few, if any, proliferating cells were scored into the striatum of CFA mice. Double labelling for GFAP and IdU revealed that 17.7 ± 6.1% of IdU-positive cells co-expressed the GFAP marker. ***P < 0.001. Scale bar, 10 μm. The experiment was performed on EAE mice (20 dpi) with a representative score of the disease > 2.0.
Role of NMDA receptors in mitochondrial dysfunction in EAE
Abnormal stimulation of NMDA receptors leads to excitotoxic neuronal damage by altering mitochondrial activity (Rosenstock et al., 2010; Stanika et al., 2010; Kambe et al., 2011). Mitochondrial dysfunction has been recently proposed to contribute significantly to neurodegenerative damage in MS (Kalman et al., 2007; Mahad et al., 2008; Witte et al., 2010; Campbell et al., 2011) and in EAE (Qi et al., 2006; Das et al., 2008; Sajad et al., 2011). Our hypothesis is that NMDA receptor hyperactivity in EAE mice may contribute to an altered mitochondrial activity. Thus, we measured NAD(P)H autofluorescence signals in striatal slices from EAE (20–25 dpi) and CFA mice following electrical activation of corticostriatal glutamatergic nerve terminals (Figure 4A). Under UV excitation the NADP(H) autofluorescence signal, reported as the ratio of the fluorescence intensity over the pre-stimulus fluorescence intensity (ΔF/Fo), presents two components: the early negative component (initial) reflects consumption of NAD(P)H while the late positive component (overshoot) corresponds to the regeneration of NAD(P)H (Loizzo et al., 2010). The detected signal was derived from electrically responsive cells, neurons and glial cells (Shuttleworth et al., 2003). As shown in Figure 4B, we observed a significant difference of the NAD(P)H fluorescent profiles between the two groups: both the early and late components of NAD(P)H signals were clearly reduced in EAE striatum (EAE vs. CFA P < 0.01) in response to glutamate synaptic stimulation. Of note, the presence of an increased number of glia cells and of glutamate responsive infiltrating cells (Miglio et al., 2005; Lindblad et al., 2012) in EAE slices (Centonze et al., 2009) should provide an additional contribute to the NAD(P)H response in comparison to CFA. However, the marked reduction of the NAD(P)H signal in EAE indicates a minor contribution from these cells, demonstrating a severe impairment of mitochondrial respiratory chain activity in neuronal cells of EAE mice.
Figure 4.
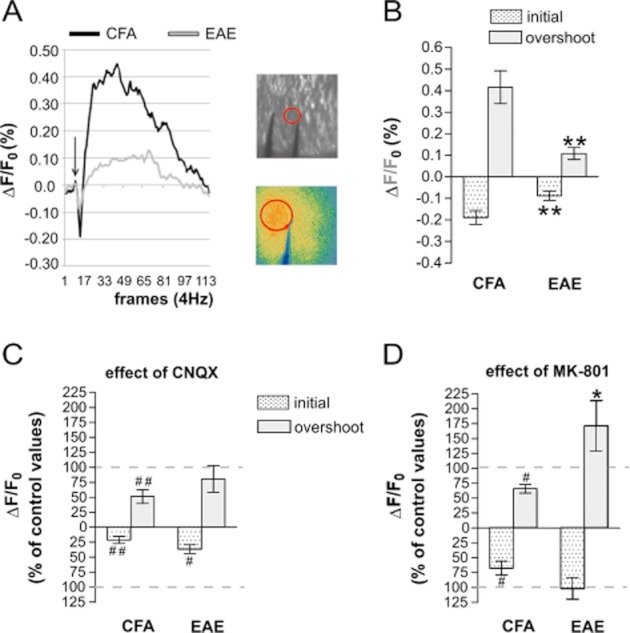
Role of NMDA receptors in mitochondrial dysfunction in EAE. (A) NAD(P)H fluorescence changes evoked by electrical stimuli (50 pulses,100 Hz) applied in corticostriatal slices from CFA and EAE mice. Fluorescence levels are expressed by pseudo-colors as ΔF/Fo (means ± SEM; n = 6). Data were gathered over a 2500 μm2 circle surface tangent to the stimulus electrode. (B) Peak values of the biphasic curves induced by synaptic stimulation are means ± SEM (CFA, n = 6; EAE, n = 6; **P < 0.01 for both components). (C) CNQX reduced NAD(P)H fluorescence in CFA (##P < 0.01 CFA + CNQX vs. CFA untreated slices) and in EAE mice (#P < 0.05 EAE + CNQX vs. EAE untreated slices). (D) MK801 reduced NAD(P)H fluorescence only in CFA mice (#P < 0.05 CFA + MK801 vs. CFA untreated slices; *P < 0.05 EAE + MK801 vs. CFA + MK801). Recordings were performed on EAE mice (20–25 dpi) with a representative score of the disease > 2.0.
In order to identify the role of glutamatergic receptors in the defective mitochondrial activity in EAE, we incubated the striatal slices either with CNQX or MK801, for 10 min and then performed NADP(H) imaging experiments. As shown in Figure 4C, in the presence of CNQX, the NAD(P)H autofluorescence signal was markedly reduced in CFA mice, as expected following blockade of one of the main postsynaptic components (AMPA receptors) of glutamate transmission (CFA + CNQX vs. CFA P < 0.01). In EAE slices, in which the mitochondrial respiratory chain activity was compromised, the effect of CNQX was less prominent; only the initial component was partially reduced (Figure 4C; EAE + CNQX vs. EAE P < 0.05 only for the initial component). When the experiments were performed in the presence of MK801, we observed a significant reduction of the NAD(P)H response in CFA slices as expected by blocking NMDA receptors (Figure 4D; both early component and overshoot: CFA-MK801 vs. CFA P < 0.05). Interestingly, in EAE slices MK801 did not exert any significant effect on the NAD(P)H signal (Figure 4D; both early component and overshoot: EAE-MK801 vs. EAE P > 0.05). Notably, the MK801-mediated reduction in CFA was significantly different from the MK801 effect observed in EAE (Figure 4D, P < 0.05). Together, these results suggest that the pharmacological treatments exerted a different effect on the mitochondrial activity of CFA and EAE mice, ameliorating the NAD(P)H response in EAE mice. In fact, under glutamate stimulation, despite the presence of glutamate receptor blockers, we observed in EAE mice a ‘restoration’ of the NADPH response, consistent with a role of NMDA receptors in mitochondrial dysfunction in EAE.
Effects of NMDA receptor inhibition on the synaptic and clinical defects of EAE
To further investigate the role of NMDA receptors on EAE glutamate transmission we blocked thesereceptors in vivo by chronic i.c.v. infusion of MK-801 (EAE-MK801) starting 1 week before the immunization. We killed the mice during the symptomatic phase of the disease (20–25 dpi) and recorded sEPSC in EAE-MK801 mice and in their relative controls. We observed a significant inhibition of sEPSC frequency (cells number = 14, P < 0.05), while EAE-induced alterations of sEPSC decay time and duration were unaffected by the treatment (P > 0.05; Figure 5A,B). Such results support the previous in vitro observations and provide an in vivo demonstration of the involvement of the NMDA receptors in the perturbations of glutamate release in EAE mice.
Figure 5.
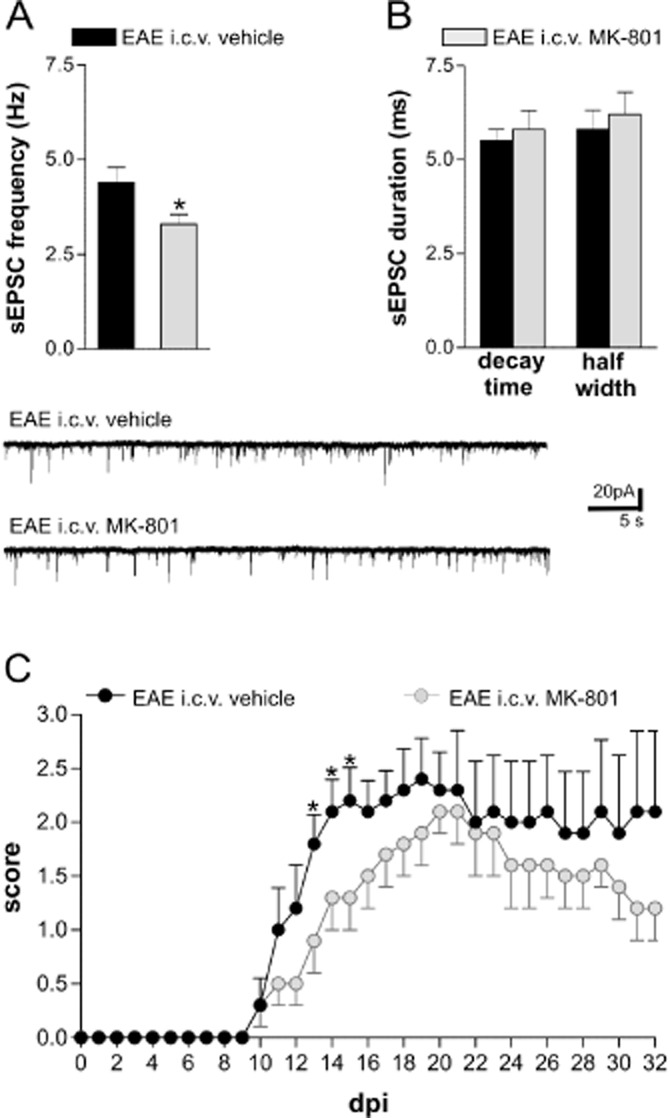
In vivo blockade of NMDA receptors ameliorates synaptic alterations and clinical deficits in EAE. (A) In vivo treatment with MK801 was able to reduce the sEPSC frequency in EAE mice compared with EAE mice receiving vehicle (*P < 0.05). (B) Both sEPSC half width and decay time were similar in mice treated with MK801 or vehicle. The electrophysiological traces on the bottom are examples of voltage-clamp recordings showing the reduction of sEPSC frequency in a neuron from EAE-MK801 mice compared with those from EAE-vehicle mice. Mice with comparable scores (20–25 dpi; score > 2.0) were chosen for the recordings. (C) Clinical EAE scores (means ± SEM) over time in mice treated i.c.v. with MK801 (n = 13) and vehicle (n = 13) 1 week before immunization. The severity of EAE was milder in EAE-MK801 mice compared with EAE-vehicle due to an improvement of the early clinical symptoms (13, 14 and 15 dpi; Mann–Whitney test, *P < 0.05) and a delay of the acute phase of the disease (18 ± 1 dpi in EAE-MK801 mice and 15 ± 1 in EAE-vehicle mice; t-test, P < 0.05).
Inhibition of NMDA receptors ameliorates the clinical course of a form of EAE in rats (Wallström et al., 1996). However, more recently, systemic treatment with 0.1 mg·kg−1 of MK801, starting from the symptomatic phase (dpi 21), did not have any effect on the clinical course of EAE (Matute, 2010). Therefore, we explored the effect of NMDA receptor inhibition on EAE severity in our experimental conditions. Mice receiving chronic i.c.v. infusions of MK801 (EAE-MK801) (cell number = 13) showed a less severe disease course compared with EAE-vehicle mice (cell number = 10). In particular, at the beginning of the symptomatic phase of the disease (13, 14 and 15 dpi), EAE-MK801 mice had a significantly lower clinical score than EAE-vehicle mice (P < 0.05) (Figure 5A). In addition, by analysing the temporal course of the disease, we detected a significant difference in the day of maximum score, which was 3 days later in the EAE-MK801 than in the EAE-vehicle mice (18 ± 1 dpi in EAE-MK 801 mice and 15 ± 1 in EAE-vehicle mice; P < 0.05). No significant difference was observed for the onset day (14 ± 1 dpi in EAE-MK 801 mice and 12 ± 1 dpi in EAE-vehicle mice; P > 0.05).
Such results suggest that pretreatment with antagonists of the NMDA receptors given i.c.v. can improve the early EAE clinical symptoms and delay the acute phase of the disease.
Effects of NMDA receptor potentiation on the synaptic and clinical defects of EAE
Based on the previous observations, we investigated the effect of EAE on mice with targeted deletion of the DDO gene. DDO is the limiting enzyme in the degradation of d-aspartate, the main endogenous agonist of NMDA receptors and precursor of NMDA, and DDO–/– mice express abnormally high concentrations of d-aspartate and NMDA in the brain (Errico et al., 2006; 2008). As expected, genetic disruption of DDO exacerbated the abnormalities of sEPSC frequency induced by EAE in comparison with WT mice (Figure 6A; cell number = 12, P < 0.05), while sEPSC decay time and half width were unaffected (data not shown). Furthermore, incubation of EAE-DDO–/– striatal slices with MK801 induced a more pronounced reduction in sEPSC frequency, relative to EAE-WT mice (cell number = 12 each group; Figure 6B). Such a result is in good agreement with the idea that NMDA receptor signalling is enhanced in these mice and with previous neurophysiological data in naïve DDO–/– mice (Errico et al., 2008).
Regarding the clinical symptoms, although the median disease score of each day was not significantly different between DDO–/– (n = 16) and WT mice (EAE n = 17; P > 0.05), the time course of EAE was faster in DDO–/– mice compared with their WT counterparts. Disease onset was in fact 3 days earlier in these mutants (17 ± 1 dpi in EAE mice and 14 ± 1 dpi in DDO–/– mice; P < 0.05), as was the day of maximum score (23 ± 1 dpi in EAE mice and at 20 ± 1 dpi in DDO–/– mice; P < 0.05) (Figure 6C). Such results showed that, although the EAE severity did not change in the presence of high levels of NMDA, progression of the disease was faster.
Discussion and Conclusions
Excessive synaptic excitation by the neurotransmitter glutamate triggers neuronal damage in acute and chronic neurological disorders, including EAE and MS (Forder and Tymianski, 2009; Rossi et al., 2011). High levels of glutamate have been measured in the cerebrospinal fluid (Stover et al., 1997; Sarchielli et al., 2003) and in the brains of MS patients (Srinivasan et al., 2005; Cianfoni et al., 2007), and over-activation of glutamate receptors causes MS-like lesions (Matute et al., 2001) and inflammatory neuronal death in MS (Rossi et al., 2011). Furthermore, glutamate transporter expression is altered in MS (Vallejo-Illarramendi et al., 2006; Vercellino et al., 2007) and in EAE (Hardin-Pouzet et al., 1997; Ohgoh et al., 2002), and both AMPA and NMDA receptors are up-regulated (Newcombe et al., 2008).
How the myelin-targeting autoimmune reaction results in abnormal glutamate transmission has not been fully elucidated, but it is now well recognized that activated immune cells infiltrating the brain and resident immune cells, such as microglia and astroglia, interact extensively with synaptic transmission. Proinflammatory cytokines, such as TNF-α and IL1-β, released from activated microglia in the grey matter are crucial molecular players involved in the enhanced glutamate transmission observed both in EAE and MS (Centonze et al., 2009; Musumeci et al., 2011; Rossi et al., 2012). In particular, we observed that during EAE, TNF-α affected sEPSC decay time and duration in the striatum (Centonze et al., 2009). This postsynaptic effect did not depend on NMDA activity but involved changes in expression and phosphorylation of AMPA receptors (Centonze et al., 2009; Musumeci et al., 2011). Recently, we demonstrated IL1-β in cerebrospinal fluid from MS patients increased sEPSC frequency in vitro and provided evidence that this effect was not due to a sensitization of presynaptic NMDA receptors but to an involvement of TRPV1 channels (Rossi et al., 2012).
In the present study, we provide a further presynaptic molecular mechanism as the basis for the enhanced glutamate release in EAE striatum, which involves an increased presynaptic NMDA receptor function in EAE mice following abnormal intra-axonal Na+ accumulation (Saab et al., 2004; Craner et al., 2004a; Waxman, 2005) and is likely to be mediated by astrocyte activation (Giraud et al., 2010; Ziehn et al., 2010; Guo et al., 2011; present study). By Western blot, we could not detect any changes in the expression levels of the major NMDA receptor subunits (GluN1, GluN2A, GluN2B) during EAE. We cannot however exclude the possibility that an increase in their expression at presynaptic sites, sufficient to influence glutamate release, may be below the detection limit of the method used. Alternatively, a sensitization of these receptors may occur through different mechanisms, such as posttranslational modifications (Mu et al., 2003), or by an increase of agonist acting on such receptors at several possible levels. Presynaptic NMDA receptors may in fact respond to glutamate spillover from neighbouring synapses, to retrograde release of glutamate from the same post-synaptic site, to glutamate released from the same pre-synaptic terminal autoreceptors, from surrounding astroglia or from axo-axonic glutamatergic synapses (Duguid and Smart, 2009). An increase of glutamate release from one of these sources may thus be also implied in increased presynaptic NMDA receptor function during EAE. One possibility may be that the increase of glutamate transmission observed in EAE independently of NMDA receptors (Centonze et al., 2009; Rossi et al., 2010) may act in a positive feedback loop on presynaptic NMDA autoreceptors (Starke et al., 1989), further enhancing glutamate release. Furthermore, in hippocampus presynaptic NMDA receptors positively regulated synaptic vesicle release in response to astroglial release of glutamate (Yang et al., 2006; Jourdain et al., 2007; Parri and Crunelli, 2007), suggesting that astroglial activation found in our EAE model may result in enhanced activation of presynaptic NMDA receptors.
Not only presynaptic NMDA receptor activity but also the reverse mode of action of the Na+/Ca2+ exchanger contributes to increase the frequency of sEPSCs in the striatum (Rossi et al., 2010). Both alterations are favoured by intra-axonal Na+ accumulation known to occur in EAE (Craner et al., 2003; 2004b; Saab et al., 2004; Waxman, 2005; Rossi et al., 2010; present study), and are likely to interact with each other in controlling glutamate release from presynaptic nerve terminals. In agreement with these results, a recent study demonstrated that pharmacological blockade of reversed Na+/Ca2+ exchanger function suppresses, in parallel, sensitivity of NMDA receptors, as expected for crosstalk between these two critical determinants of glutamate release (Brustovetsky et al., 2011).
We have shown here that the blockade of voltage-dependent Na+ channels abolished NMDA receptor sensitization specifically in EAE, while the inhibition of K+ channels increased sensitivity. Together these results indicate that the influx of Na+ through voltage-gated Na+ channels plays a crucial role in sensitization of presynaptic NMDA receptors in EAE and suggest that this may act at least in part through the release of the voltage-dependent block of NMDA receptors.
Interestingly, the effect of the enhanced NMDA receptor sensitization seems to be only relevant during the symptomatic phase of the disease because we observed that blockade of NMDA receptors did not influence the sEPSC frequency in presymptomatic mice. This observation is in accordance with the occurrence of Na+ accumulation through altered voltage-dependent Na+ channels in the symptomatic phase of the disease and with a prominent astroglial activation. It is likely that NMDA receptor-independent mechanisms, such as those induced by IL1-β, are involved in glutamate enhancement before the onset of clinical signs.
Excessive synaptic signalling through NMDA receptors causes excitotoxic neuronal damage by impairing mitochondrial activity (Rosenstock et al., 2010; Stanika et al., 2010; Kambe et al., 2011), whose relationship with excitatory transmission can be explored by measuring NAD(P)H autofluorescence changes induced by synaptic activation in brain slices (Loizzo et al., 2010). Changes in NAD(P)H fluorescence have long been used as a measure of oxidative phosphorylation changes (Chance et al., 1962; Connor et al., 1976; Mayevsky et al., 1988; Schuchmann et al., 2001).
In brain slices, excitatory synaptic stimulation results typically in initial transient signal decreases in NAD(P)H fluorescence, followed by longer-lasting NAD(P)H increases, that overshoot pre-stimulus NAD(P)H levels, before returning slowly to baseline.
Mitochondrial NADH dynamics are predominant contributors to both phases of evoked NAD(P)H transient signals (Schuchmann et al., 2001; Shuttleworth et al., 2003; Brennan et al., 2006; 2007) and were reduced in our EAE model, confirming severe impairment of mitochondrial activity in this disorder (Qi et al., 2006; Das et al., 2008; Sajad et al., 2011). Such effect might be explained by the higher tonic excitatory transmission typical of EAE mice (Centonze et al., 2009), which may cause lower basal levels of NAD(P)H and a consequent lower quantity of NAD(P)H oxidation driven by evoked synaptic transmission. The lack of a reduction of NAD(P)H signal in EAE in the presence of glutamate receptor inhibitors might be explained by the low basal levels of NAD(P)H, making undetectable a change triggered by the evoked synaptic transmission. However, this is unlikely because in the presence of CNQX, which was more effective than MK801 in CFA mice, a small but significant effect on the NAD(P)H initial component in EAE mice was still detectable. An alternative interpretation may be that the inhibition of AMPA and NMDA receptors before synaptic stimulation was sufficient to induce a ‘recovery’ of the mitochondrial activity by counteracting the abnormal EAE glutamate release. Consequently, the evoked NAD(P)H signal was not reduced as expected under stimulation. In support of this hypothesis, MK801 showed a tendency to ameliorate NAD(P)H autofluorescence rebound in EAE mice, consistent with a role of these receptors in mitochondrial dysfunction of EAE mice. Accordingly, the alteration of mitochondrial activity by NMDA receptor activation has already been demonstrated in other models of neurodegenerative damage (Rosenstock et al., 2010; Stanika et al., 2010; Kambe et al., 2011). Finally, we corroborated the electrophysiological results obtained in vitro by investigating the EAE-associated synaptic alterations in two sets of complementary in vivo experiments in which NMDA receptor-mediated neurotransmission was inhibited or physiologically potentiated. In particular, we observed that the glutamate signalling was reduced following chronic NMDA inhibition or enhanced partially in DDO–/– mice characterized by an increased NMDA activation. In addition, although local electrophysiological alterations in the EAE brain cannot be strictly correlated with the clinical outcome of the disease, we explored the time course of EAE in these two groups of mice and we observed concordant effects; a systemic enhancement of NMDA receptor signalling accelerated the course of the disease, while a central inhibition of NMDA signalling delayed it. However, we did not observe a strong effect on EAE severity. Several reasons could explain such observations, including the route of drug administration or the use of KO mice in which compensatory mechanisms could occur, together with the quite complex role played by the NMDA receptors in EAE pathophysiology (Paul and Bolton, 2002; Basso et al., 2008; Reijerkerk et al., 2010; Guo et al., 2012).
It could be argued that treatment with i.c.v. MK801 and the increased level of d-aspartate had a generalized effect not only on NMDA receptors located on synapses but also on those expressed by lymphocytes and on the blood–brain barrier (BBB) (Paul and Bolton, 2002; Miglio et al., 2005; Lindblad et al., 2012). However, if MK801 was really able to act on immune cells, dampening their response or affecting the migration of these cells into the CNS, it would have prevented all the synaptic abnormalities of EAE mice (Centonze et al., 2009; Musumeci et al., 2011; Rossi et al., 2012), and their scores as well. Instead, we have observed a mild effect on the score and a selective effect of MK801 treatment on sEPSC frequency both in vitro and in vivo. Similarly, we have observed that the lack of DDO affected only the frequency and not the kinetics of sEPSCs. Our results, thus, indicate NMDA receptors in the presynaptic compartment as the most likely to have a major role in the pathophysiology of striatal neurons studied here.
In conclusion, our data show that during EAE, a sensitization of NMDA receptors enhanced glutamate transmission presynaptically in striatum. Our in vitro pharmacological data and in vivo experiments suggest that neuronal pre-synaptic NMDA receptors contribute to this synaptic effect. Moreover, we showed in vivo that a systemic enhancement of NMDA receptor signalling accelerated the time course of EAE and that a central inhibition of NMDA signalling postponed the acute phase. Thus, although this role of NMDA receptors in the disease course may be due to a synergy of several factors, our data show that they play a role in both the clinical symptoms and the synaptic alterations. Together with previous findings (Centonze et al., 2009; Rossi et al., 2010; 2011; 2012; Musumeci et al., 2011), the current data further support a role for synaptic impairment in the pathophysiology of EAE and elucidate the molecular mechanisms responsible for dysregulation of excitatory synapses associated with central inflammatory events. Although additional studies are needed to provide mechanistic insights into the role of NMDA receptors at the many levels of EAE pathogenesis and about their relationship with the many cellular players involved, our work supports the view that pharmacological inhibition of NMDA receptors might be important when considering neuroprotective strategies in MS.
Acknowledgments
We wish to thank Massimo Tolu and Vladimiro Batocchi for helpful technical assistance. This investigation was supported by the Italian National Ministero della Salute and by Fondazione TERCAS to DC, by Fondazione Italiana Sclerosi Multipla (FISM) to SR and by a grant from the European Community (AXREGEN: Axonal regeneration, plasticity & stem cells- Grant agreement 21 4003) founding PhD fellowship of NH. LM was supported by grant FISM 2009/R/18.
Glossary
- ACSF
artificial cerebrospinal fluid
- CFA
Freund's adjuvant
- DDO
d-aspartate oxidase
- dpi
day post immunization
- EAE
experimental autoimmune encephalomyelitis
- EPSC
excitatory postsynaptic currents
- GFAP
glial fibrillary acidic protein
- MS
multiple sclerosis
- PPR
paired-pulse ratio
- WT
wild-type
Conflicts of interest
Silvia Rossi received honoraria for writing from Bayer Schering and funding for traveling from Novartis, Teva, Merck Serono. She is involved as sub-investigator in clinical trials for Novartis, Merck Serono, Teva, Bayer Schering, Sanofi-aventis, Biogen Idec.
Valentina De Chiara received funding for traveling by Teva. She is involved as study coordinator in clinical trials for Novartis, Merck Serono, Teva, Bayer Schering, Sanofi-aventis, Biogen Idec.
Giorgio Bernardi is the principal investigator in clinical trials for Merck Serono and Teva.
Diego Centonze is an Advisory Board member of Merck-Serono, Teva, Bayer Schering, and received funding for traveling and honoraria for speaking or consultation fees from Merck Serono, Teva, Novartis, Bayer Schering, Sanofi-aventis, Biogen Idec. He is also an external expert consultant of the European Medicine Agency (EMA), and the principal investigator in clinical trials for Novartis, Merck Serono, Teva, Bayer Schering, Sanofi-aventis, Biogen Idec.
The other authors have no conflict of interests to declare.
References
- Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733–738. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition (2011) Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoin B, Zaaraoui W, Reuter F, Rico A, Malikova I, Confort-Gouny S, et al. Atrophy mainly affects the limbic system and the deep grey matter at the first stage of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2010;81:690–695. doi: 10.1136/jnnp.2009.188748. [DOI] [PubMed] [Google Scholar]
- Bakshi R, Benedict RH, Bermel RA, Caruthers SD, Puli SR, Tjoa CW, et al. T2 hypointensity in the deep gray matter of patients with multiple sclerosis: a quantitative magnetic resonance imaging study. Arch Neurol. 2002;59:62–68. doi: 10.1001/archneur.59.1.62. [DOI] [PubMed] [Google Scholar]
- Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, et al. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;2008:1532–1543. doi: 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista S, Zivadinov R, Hoogs M, Bergsland N, Heininen-Brown M, Dwyer MG, et al. Basal ganglia, thalamus and neocortical atrophy predicting slowed cognitive processing in multiple sclerosis. J Neurol. 2012;259:139–146. doi: 10.1007/s00415-011-6147-1. [DOI] [PubMed] [Google Scholar]
- Bolton C, Paul C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther. 1997;282:397–402. [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. Modulation of the amplitude of NAD(P)H fluorescence transients after synaptic stimulation. J Neurosci Res. 2007;85:3233–3243. doi: 10.1002/jnr.21288. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T, Brittain MK, Sheets PL, Cummins TR, Pinelis V, Brustovetsky N. KB-R7943, an inhibitor of the reverse Na+ /Ca2+ exchanger, blocks N-methyl-D-aspartate receptor and inhibits mitochondrial complex I. Br J Pharmacol. 2011;162:255–270. doi: 10.1111/j.1476-5381.2010.01054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M, Rinaldi F, Grossi P, Mattisi I, Bernardi V, Favaretto A, et al. Basal ganglia and frontal/parietal cortical atrophy is associated with fatigue in relapsing-remitting multiple sclerosis. Mult Scler. 2010;16:1220–1228. doi: 10.1177/1352458510376405. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69:481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:3442–3452. doi: 10.1523/JNEUROSCI.5804-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation-reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- Cianfoni A, Niku S, Imbesi SG. Metabolite findings in tumefactive demyelinating lesions utilizing short echo time proton magnetic resonance spectroscopy. AJNR Am J Neuroradiol. 2007;28:272–277. [PMC free article] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Connor JA, Kreulen DL, Prosser CL. Relation between oxidative metabolism and slow rhythmic potentials in mammalian intestinal muscle. Proc Natl Acad Sci U S A. 1976;73:4239–4243. doi: 10.1073/pnas.73.11.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Kataoka Y, Lo AC, Black JA, Baker D, Waxman SG. Temporal course of upregulation of Na(v)1.8 in Purkinje neurons parallels the progression of clinical deficit in experimental allergic encephalomyelitis. J Neuropathol Exp Neurol. 2003;62:968–975. doi: 10.1093/jnen/62.9.968. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004a;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004b;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res. 2008;86:2992–3001. doi: 10.1002/jnr.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Presynaptic NMDA Receptors. In: Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton, FL: CRC Press; 2009. pp. 313–328. [PubMed] [Google Scholar]
- Errico F, Pirro MT, Affuso A, Spinelli P, De Felice M, D'Aniello A, et al. A physiological mechanism to regulate d-aspartic acid and NMDA levels in mammals revealed by d-aspartate oxidase deficient mice. Gene. 2006;374:50–57. doi: 10.1016/j.gene.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Errico F, Rossi S, Napolitano F, Catuogno V, Topo E, Fisone G, et al. d-aspartate prevents corticostriatal long-term depression and attenuates schizophrenia-like symptoms induced by amphetamine and MK-801. J Neurosci. 2008;28:10404–10414. doi: 10.1523/JNEUROSCI.1618-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Giraud SN, Caron CM, Pham-Dinh D, Kitabgi P, Nicot AB. Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB-dependent CCL2 expression in reactive astrocytes. Proc Natl Acad Sci U S A. 2010;107:8416–8421. doi: 10.1073/pnas.0910627107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeske KT, Das S, Bonaguidi MA, Weiss C, Radulovic J, Disterhoft JF, et al. BMP signaling mediates effects of exercise on hippocampal neurogenesis and cognition in mice. Plos ONE. 2009;4:e7506. doi: 10.1371/journal.pone.0007506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsette RE. Neurodegeneration in multiple sclerosis: the role of oxidative stress and excitotoxicity. J Neurol Sci. 2008;274:48–53. doi: 10.1016/j.jns.2008.06.029. [DOI] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Delgado M, Sohn J, Miers L, et al. Macroglial plasticity and the origins of reactive astroglia in experimental autoimmune encephalomyelitis. J Neurosci. 2011;31:11914–11928. doi: 10.1523/JNEUROSCI.1759-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ko EM, Delgado M, Horiuchi M, Soulika A, et al. Disruption of NMDA receptors in oligodendroglial lineage cells does not alter their susceptibility to experimental autoimmune encephalomyelitis or their normal development. J Neurosci. 2012;32:639–645. doi: 10.1523/JNEUROSCI.4073-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin-Pouzet H, Krakowski M, Bourbonniere L, Didier-Bazes M, Tran E, Owens T. Glutamate metabolism is down-regulated in astrocytes during experimental allergic encephalomyelitis. Glia. 1997;20:79–85. doi: 10.1002/(sici)1098-1136(199705)20:1<79::aid-glia8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Hayes KC. Impact of extended-release dalfampridine on walking ability in patients with multiple sclerosis. Neuropsychiatr Dis Treat. 2011;7:229–239. doi: 10.2147/NDT.S10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, et al. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kalman B, Laitinen K, Komoly S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2007;188:1–12. doi: 10.1016/j.jneuroim.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Kambe Y, Nakamichi N, Takarada T, Fukumori R, Nakazato R, Hinoi E, et al. A possible pivotal role of mitochondrial free calcium in neurotoxicity mediated by N-methyl-d-aspartate receptors in cultured rat hippocampal neurons. Neurochem Int. 2011;59:10–20. doi: 10.1016/j.neuint.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Killestein J, Kalkers NF, Polman CH. Glutamate inhibition in MS: the neuroprotective properties of riluzole. J Neurol Sci. 2005;233:113–115. doi: 10.1016/j.jns.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Lebel RM, Eissa A, Seres P, Blevins G, Wilman AH. Quantitative high-field imaging of sub-cortical gray matter in multiple sclerosis. Mult Scler. 2012;18:433–441. doi: 10.1177/1352458511428464. [DOI] [PubMed] [Google Scholar]
- Lindblad SS, Mydel P, Hellvard A, Jonsson IM, Bokarewa MI. The N-methyl-d-aspartic acid receptor antagonist memantine ameliorates and delays the development of arthritis by enhancing regulatory T cells. Neurosignals. 2012;20:61–71. doi: 10.1159/000329551. [DOI] [PubMed] [Google Scholar]
- Loitfelder M, Fazekas F, Petrovic K, Fuchs S, Ropele S, Wallner-Blazek M, et al. Reorganization in cognitive networks with progression of multiple sclerosis: insights from fMRI. Neurology. 2011;76:526–533. doi: 10.1212/WNL.0b013e31820b75cf. [DOI] [PubMed] [Google Scholar]
- Loizzo S, Pieri M, Ferri A, Carri MT, Zona C, Fortuna A, et al. Dynamic NAD(P)H post-synaptic autofluorescence signals for the assessment of mitochondrial function in a neurodegenerative disease: monitoring the primary motor cortex of G93A mice, an amyotrophic lateral sclerosis model. Mitochondrion. 2010;10:108–114. doi: 10.1016/j.mito.2009.11.001. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Matute C. Calcium dyshomeostasis in white matter pathology. Cell Calcium. 2010;47:150–157. doi: 10.1016/j.ceca.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Perez-Cerda F, Perez-Samartin A, Sanchez-Gomez MV. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci. 2001;24:224–230. doi: 10.1016/s0166-2236(00)01746-x. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Nioka S, Chance B. Fiber optic surface fluorometry/reflectometry and 31-p-NMR for monitoring the intracellular energy state in vivo. Adv Exp Med Biol. 1988;222:365–374. doi: 10.1007/978-1-4615-9510-6_43. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglio G, Varsaldi F, Lombardi G. Human T lymphocytes express N-methyl-D-aspartate receptors functionally active in controlling T cell activation. Biochem Biophys Res Commun. 2005;338:1875–1883. doi: 10.1016/j.bbrc.2005.10.164. [DOI] [PubMed] [Google Scholar]
- Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD. Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron. 2003;40:581–594. doi: 10.1016/s0896-6273(03)00676-7. [DOI] [PubMed] [Google Scholar]
- Musumeci G, Grasselli G, Rossi S, De Chiara V, Musella A, Motta C, et al. Transient receptor potential vanilloid 1 channels modulate the synaptic effects of TNF-α and of IL-1β in experimental autoimmune encephalomyelitis. Neurobiol Dis. 2011;43:669–677. doi: 10.1016/j.nbd.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Newcombe J, Uddin A, Dove R, Patel B, Turski L, Nishizawa Y, et al. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol. 2008;18:52–61. doi: 10.1111/j.1750-3639.2007.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, et al. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;125:170–178. doi: 10.1016/s0165-5728(02)00029-2. [DOI] [PubMed] [Google Scholar]
- Olechowski CJ, Parmar A, Miller B, Stephan J, Tenorio G, Tran K, et al. A diminished response to formalin stimulation reveals a role for the glutamate transporters in the altered pain sensitivity of mice with experimental autoimmune encephalomyelitis (EAE) Pain. 2010;149:565–572. doi: 10.1016/j.pain.2010.03.037. [DOI] [PubMed] [Google Scholar]
- Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- Pampliega O, Domercq M, Soria FN, Villoslada P, Rodriguez-Antiguedad A, Matute C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflammation. 2011;8:63. doi: 10.1186/1742-2094-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri R, Crunelli V. Astrocytes target presynaptic NMDA receptors to give synapses a boost. Nat Neurosci. 2007;10:271–273. doi: 10.1038/nn0307-271. [DOI] [PubMed] [Google Scholar]
- Paul C, Bolton C. Modulation of blood-brain barrier dysfunction and neurological deficits during acute experimental allergic encephalomyelitis by the N-methyl-D-aspartate receptor antagonist memantine. J Pharmacol Exp Ther. 2002;302:50–57. doi: 10.1124/jpet.302.1.50. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Pitt D, Nagelmeier IE, Wilson HC, Raine CS. Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology. 2003;61:1113–1120. doi: 10.1212/01.wnl.0000090564.88719.37. [DOI] [PubMed] [Google Scholar]
- Plaut GS. Effectiveness of amantadine in reducing relapses in multiple sclerosis. J R Soc Med. 1987;80:91–93. doi: 10.1177/014107688708000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, Riethmacher D, et al. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci. 2011;31:1069–1080. doi: 10.1523/JNEUROSCI.5035-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J Biol Chem. 2006;281:31950–31962. doi: 10.1074/jbc.M603717200. [DOI] [PubMed] [Google Scholar]
- Reijerkerk A, Kooij G, van der Pol SM, Leyen T, Lakeman K, Hof B, et al. The NR1 subunit of NMDA receptor regulates monocyte transmigration through the brain endothelial cell barrier. J Neurochem. 2010;113:447–453. doi: 10.1111/j.1471-4159.2010.06598.x. [DOI] [PubMed] [Google Scholar]
- Rosenstock TR, Bertoncini CR, Teles AV, Hirata H, Fernandes MJ, Smaili SS. Glutamate-induced alterations in Ca2+ signaling are modulated by mitochondrial Ca2+ handling capacity in brain slices of R6/1 transgenic mice. Eur J Neurosci. 2010;32:60–70. doi: 10.1111/j.1460-9568.2010.07268.x. [DOI] [PubMed] [Google Scholar]
- Rossi S, De Chiara V, Furlan R, Musella A, Cavasinni F, Muzio L, et al. Abnormal activity of the Na/Ca exchanger enhances glutamate transmission in experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:1379–1385. doi: 10.1016/j.bbi.2010.07.241. [DOI] [PubMed] [Google Scholar]
- Rossi S, Muzio L, De Chiara V, Grasselli G, Musella A, Musumeci G, et al. Impaired striatal GABA transmission in experimental autoimmune encephalomyelitis. Brain Behav Immun. 2011;25:947–956. doi: 10.1016/j.bbi.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, et al. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol. 2012;71:76–83. doi: 10.1002/ana.22512. [DOI] [PubMed] [Google Scholar]
- Saab CY, Craner MJ, Kataoka Y, Waxman SG. Abnormal Purkinje cell activity in vivo in experimental allergic encephalomyelitis. Exp Brain Res. 2004;158:1–8. doi: 10.1007/s00221-004-1867-4. [DOI] [PubMed] [Google Scholar]
- Sajad M, Zargan J, Sharma J, Chawla R, Arora R, Umar S, et al. Increased spontaneous apoptosis of rat primary neurospheres in vitro after experimental autoimmune encephalomyelitis. Neurochem Res. 2011;36:1017–1026. doi: 10.1007/s11064-011-0441-2. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Greco L, Floridi A, Gallai V. Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol. 2003;60:1082–1088. doi: 10.1001/archneur.60.8.1082. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Kovacs R, Kann O, Heinemann U, Buchheim K. Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res Brain Res Protoc. 2001;7:267–276. doi: 10.1016/s1385-299x(01)00080-0. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Brennan AM, Connor JA. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J Neurosci. 2003;23:3196–3208. doi: 10.1523/JNEUROSCI.23-08-03196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128:1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- Stanika RI, Winters CA, Pivovarova NB, Andrews SB. Differential NMDA receptor-dependent calcium loading and mitochondrial dysfunction in CA1 vs. CA3 hippocampal neurons. Neurobiol Dis. 2010;37:403–411. doi: 10.1016/j.nbd.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke K, Gothert M, Kilbinger H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- Stover JF, Lowitzsch K, Kempski OS. Cerebrospinal fluid hypoxanthine, xanthine and uric acid levels may reflect glutamate-mediated excitotoxicity in different neurological diseases. Neurosci Lett. 1997;238:25–28. doi: 10.1016/s0304-3940(97)00840-9. [DOI] [PubMed] [Google Scholar]
- Tao G, Datta S, He R, Nelson F, Wolinsky JS, Narayana PA. Deep gray matter atrophy in multiple sclerosis: a tensor based morphometry. J Neurol Sci. 2009;282:39–46. doi: 10.1016/j.jns.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Vallejo-Illarramendi A, Domercq M, Perez-Cerda F, Ravid R, Matute C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21:154–164. doi: 10.1016/j.nbd.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol. 2007;66:732–739. doi: 10.1097/nen.0b013e31812571b0. [DOI] [PubMed] [Google Scholar]
- Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137:89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Cerebellar dysfunction in multiple sclerosis: evidence for an acquired channelopathy. Prog Brain Res. 2005;148:353–365. doi: 10.1016/S0079-6123(04)48028-5. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;2:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Witte ME, Geurts JJ, de Vries HE, van der Valk P, van Horssen J. Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion. 2010;10:411–418. doi: 10.1016/j.mito.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Xin WK, Kwan CL, Zhao XH, Xu J, Ellen RP, McCulloch CA, et al. A functional interaction of sodium and calcium in the regulation of NMDA receptor activity by remote NMDA receptors. J Neurosci. 2005;25:139–148. doi: 10.1523/JNEUROSCI.3791-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XM. The role of intracellular sodium in the regulation of NMDA-receptor-mediated channel activity and toxicity. Mol Neurobiol. 2006;33:63–80. doi: 10.1385/MN:33:1:063. [DOI] [PubMed] [Google Scholar]
- Ziehn MO, Avedisian AA, Tiwari-Woodruff S, Voskuhl RR. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis, EAE. Lab Invest. 2010;90:774–786. doi: 10.1038/labinvest.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]