

Abstract
West Nile virus strain Kunjin (WNVKUN) is an enveloped, positive-sense RNA virus within the virus family Flaviviridae. Many flaviviruses have been shown to manipulate multiple signaling pathways, including autophagic, innate immune, and stress responses, in order to benefit replication. In particular, we have demonstrated that WNVKUN regulates the unfolded protein response (UPR), skewing the downstream effectors toward chaperone expression and Xbp-1 activation while preventing PERK-mediated translation attenuation and C/EBP homologous protein (CHOP) upregulation. WNVKUN-regulated UPR signaling can then be hijacked in order to affect type I interferon (IFN) responses, preventing IFN-mediated STAT1 phosphorylation and nuclear translocation. To extend our previous observations, we aimed to investigate the contribution of ATF6- and IRE1-mediated signaling during WNVKUN replication and how the two sensors contribute to the inhibition of IFN signaling. ATF6-deficient cells infected with WNVKUN showed decreased protein and virion production. These cells also demonstrated increased eIF2α phosphorylation and CHOP transcription, absent in infected matched control cells. Thus, we propose that in the absence of ATF6, WNVKUN is incapable of manipulating the PERK-mediated response to infection. In contrast, infection of IRE1−/− knockout cells showed no discernible differences compared to IRE1+/+ cells. However, both ATF6 and IRE1 were required for WNVKUN-induced inhibition of STAT1 phosphorylation. We suggest that the combination of abhorrent UPR signaling, promotion of cell death, and increased innate immune responses contributes to the replication defects in ATF6-deficient cells, thus demonstrating the dual importance of ATF6 in maintaining cell viability and modulating immune responses during WNVKUN infection.
INTRODUCTION
The endoplasmic reticulum (ER) is a multifunctional signaling organelle, controlling protein and lipid synthesis, calcium homeostasis, and components of redox signaling (1). Accumulation of misfolded or unfolded proteins in the lumen of the ER activates a complex signaling cascade termed the unfolded protein response (UPR), which acts to decrease ER loading and increase capacity and folding to restore homeostasis to the organelle (2). Briefly, dissociation of the chaperone BiP from the three transmembrane stress sensors PERK, ATF6, and IRE1 activates each one (3), by dimerization and autophosphorylation (PERK and IRE1) (4–6) or by inclusion into COPII vesicles and trafficking to the Golgi apparatus (ATF6) (7). Numerous transcription factors are upregulated downstream of these sensors, which, in turn, increase expression of chaperones (BiP, protein disulfide isomerase [PDI], calnexin) (8–10), degradative factors (EDEM-1, Erdj4) (8), redox recovery molecules (11), and proapoptotic proteins (C/EBP homologous protein [CHOP]) (12–15). These effectors combine to reduce input into the ER (16), refold or degrade misfolded proteins (17), increase ER capacity (18, 19), and in times of irreversible stress, activate cell death pathways (15).
Transcriptional activation of downstream UPR genes is controlled by a number of cis-acting different promoter elements. The ER stress response element (ERSE) is located upstream of many ER chaperones (20) and binds both cleaved ATF6 and Xbp-1(s) (10, 21), although ATF6 has a much higher affinity for ERSE. Additionally, Xbp-1 can bind to the UPR response element (UPRE), which controls expression of components of ER-associated degradation (9). Transcriptional effectors downstream of PERK are initially induced by binding of ATF4 to amino acid response elements (AARE), of which CHOP is a major target gene (22). As each UPR-induced transcription factor has different binding capacities to UPR target genes, this leads to extensive cross talk in the transcriptional upregulation and long-term potentiation of ER homeostasis. For example, CHOP expression is strongly induced by ATF4 (via PERK activation) but can also be stimulated by ATF6 and Xbp1 (23). BiP (and other chaperones) transcription is under the control of the ERSE and can thus be induced by ATF6 but also Xbp-1. However, Xbp-1 mRNA expression is regulated by ATF6 but splicing and, hence, translation of the active transcription factor are under the control of IRE1 (9, 24). To complicate matters further, UPR-induced mRNAs have been demonstrated to have differential stabilities in stressed cells, to allow adaptation, and to facilitate cell survival (25, 26).
It is believed that this cross talk between the arms of the UPR is essential for the coordinated response of the cell to differing levels of ER stress. For example, early events in UPR signaling activate eIF2α phosphorylation (16, 27), which immediately inhibits translation and protein input into the ER. Following this, negative feedback mechanisms via GADD34 (28) (driven by ATF4) and p58IPK (29, 30) (downstream of Xbp-1 and ATF6) restore translation, switching the response to chaperone expression, lipid biosynthesis, and ER-associated degradation (all requiring translational machinery). This long-term response allows the cell to adapt to the stress and promote survival. However, in acute ER stress, increased early signaling through PERK drives high expression of CHOP, which inhibits expression of the antiapoptotic Bcl-2 (31) while simultaneously upregulating proapoptotic proteins such as Ero1, Bim, and DR5 (13, 14, 32, 33). Thus, early regulation of PERK signaling, through either lower stimulation or negative feedback, can determine cell fate following ER stress.
Interestingly, many viruses also preferentially activate ATF6 and/or IRE1 pathways over PERK in order to benefit replication. Human cytomegalovirus (HCMV) upregulates BiP and Xbp-1 splicing in infected cells (34). Hepatitis C virus (HCV) replication activates ATF6 signaling (35, 36), while other flaviviruses, such as Japanese encephalitis virus (JEV) and dengue virus (DENV), upregulate Xbp-1 splicing and downstream transcription of endoplasmic reticulum-associated degradation (ERAD) components (37). Our previous studies have shown that IRE1 and ATF6 signaling is upregulated in West Nile virus strain Kunjin (WNVKUN)-infected cells and that PERK depletion does not affect replication (38). Additionally, this differential signaling can inhibit STAT1 activation in response to interferon (IFN) stimulation, allowing cross-regulation of two essential cellular processes during WNV replication (38).
Thus, we were interested to investigate the individual contributions of ATF6 and IRE1 during WNVKUN replication and examine whether dysregulation of the canonical UPR would disrupt (i) virus replication and/or (ii) the cellular response to infection.
MATERIALS AND METHODS
Cells and virus stocks.
Cell lines consisting of immortalized ATF6 and IRE1 knockout mouse embryonic fibroblasts (MEFs) were a kind gift from Randal Kaufman (University of Michigan) (24, 39) and were maintained in high-glucose Dulbecco's modified Eagle medium (DMEM; Life Technologies) supplemented with sodium pyruvate, 10% (vol/vol) heat-inactivated fetal calf serum (FCS; Life Technologies), penicillin/streptomycin (100 IU/ml and 100 μg/ml, respectively; Life Technologies), 200 μM GlutaMAX (Glx; Life Technologies), 1% (vol/vol) nonessential amino acids (NEAA; Life Technologies), and 1 μM β-mercaptoethanol (Life Technologies). Vero cells were maintained in 5% FCS (Lonza), GlutaMAX, and penicillin/streptomycin as indicated above. All cells were grown at 37°C in a 5% CO2 incubator. WNVKUN stocks were propagated from an existing MRM61C secondary stock in Vero C1008 cells at 37°C in DMEM supplemented with 0.2% (wt/vol) bovine serum albumin (BSA) for 32 h. Following infection, the virus-containing supernatant was collected and centrifuged at 4,800 rpm for 5 min to remove cell debris and then aliquots were stored at −80°C. Viral titer was determined by plaque assay as described below.
Antibodies and reagents.
Tunicamycin (containing homologues A, B, C, and D) (Sigma) was dissolved in dimethyl sulfoxide (DMSO) (Sigma) to a concentration of 5 mM. Mouse alpha interferon (IFN-α) was sourced from PBL Interferon and diluted to 100 U/μl in phosphate-buffered saline (PBS) and 0.1% bovine serum albumin (BSA). Mouse anti-NS1, anti-NS5, and envelope monoclonal antibodies were a kind gift from Roy Hall (University of Queensland). WNVKUN-specific rabbit anti-NS3 polyclonal antiserum has been described previously (40). Rabbit anti-eIF2a and rabbit anti-p-eIF2a (Ser51) polyclonal antibodies were purchased from Life Technologies, and rabbit anti-BiP and rabbit anti-actin polyclonal antibodies were sourced from Sigma. Mouse anti-CHOP antibody (Santa Cruz) was a kind gift from Julie Atkins (La Trobe University), and mouse anti-phospho-STAT1 (Tyr701) was purchased from BD Transduction Laboratories.
Plaque assay.
Vero C1008 cells were seeded in DMEM complete medium in 6-well plates and incubated at 37°C overnight. Virus stocks or supernatant samples were diluted 10-fold in 0.2% BSA/DMEM, and cells were infected with 300 μl of stock dilutions (in duplicate) and incubated at 37°C for 60 min. Two milliliters of a semisolid overlay containing 0.3% (wt/vol) low-melting-point agarose, 2.5% (wt/vol) FCS, P/S, Glx, HEPES, and NaCO3 was added to cells and solidified at 4°C for 30 min. Cells were incubated at 37°C for 3 days, fixed in 4% (vol/vol) formaldehyde (in PBS) for 1 h, and stained in 0.4% crystal violet (with 20% [vol/vol] methanol and PBS) at room temperature for 1 h. Plaques were manually counted, and numbers of PFU per ml were calculated as a measure of infectious virion secretion.
Virus infections and time course assays.
Knockout and control MEFs were seeded into 6-well or 24-well plates in infection medium (6% FCS, 200 μM Glx, and 1% [vol/vol] NEAA) and cultured overnight. Prior to infection, all cells were counted using a hemocytometer, and parallel lines were infected with quaternary WNVKUN stocks at a multiplicity of infection (MOI) of 10 in cold infection medium. Infections were synchronized on ice for 1 h, with rocking to ensure complete coverage, and then virus uptake was induced by the addition of prewarmed infection medium to a final volume of 1.5 or 0.4 ml (in 6-well or 24-well plates, respectively). Cells were incubated at 37°C for the time specified before collection of RNA, protein, or infected supernatant samples as described below.
Western blotting.
WNVKUN-infected cells were aspirated in PBS and then lysed in SDS lysis buffer (0.1% SDS, 1 mM EDTA, 50 mM Tris-HCl [pH 8.0]) containing a protease inhibitor cocktail (Astral Scientific) and the phosphatase inhibitors sodium orthovanadate (25 mM), sodium fluoride (25 mM), and β-glycerophosphate (25 mM) (Sigma). Lysates were diluted in SDS loading buffer (Bio-Rad), boiled at 95°C for 5 min, and separated on a Tris-glycine polyacrylamide gel. Proteins were then transferred to Hi-Bond ECL nitrocellulose membrane (Amersham Biosciences), and the membrane was blocked with 5% (wt/vol) skim milk (Diploma) in Tris-buffered saline (TBS) with 0.05% Tween (TBS-T) or 5% (wt/vol) BSA (Sigma) in TBS-T. Primary antibodies were incubated at 4°C with membrane overnight in blocking solution as described above. Following primary incubation, the membrane was washed in TBS-T and then incubated with secondary antibodies conjugated to Alexa Fluor 647 or Alexa Fluor 488 (Life Technologies) in TBS-T at room temperature for 2 h. The membrane was washed twice in TBS-T and then TBS, and proteins were visualized on the Storm fluorescence scanner (Amersham Biosciences) on either the 635-nm or 430-nm emission channel.
Immunofluorescence and CHOP nuclear scoring.
Cells were infected with WNVKUN at an MOI of 10 and at 36 h postinfection (p.i.) were fixed and permeabilized on coverslips with either acetone/methanol (1:1) at −20°C for 5 min or 4% (vol/vol) paraformaldehyde followed by 0.1% (vol/vol) Triton X-100. Cells were incubated with primary antibodies diluted in 1% BSA/PBS, washed twice in 0.2% BSA/PBS, and subsequently incubated with secondary antibodies conjugated to either Alexa Fluor 488 or 594 (Molecular Probes) in 1% BSA/DMEM at room temperature for 45 min. Cells were washed in PBS and then mounted onto glass slides using Ultramount mounting medium (Fronine). Immunofluorescent staining was visualized on a Zeiss confocal microscope, and pictures were assembled using Adobe Photoshop. For CHOP nuclear translocation quantitation, 150 infected cells for each cell type were scored for nuclear staining by comparison with cytoplasmic CHOP labeling. This numeration was performed in duplicate in replicate experiments.
RNA extraction and qRT-PCR.
RNA was extracted from WNVKUN-infected cells with TRIzol reagent (Invitrogen) as indicated by the manufacturers. Total RNA was then treated with RQ1 DNase (Promega) at 37°C for 30 min to remove any contaminating DNA. cDNA was synthesized with Superscript III reverse transcriptase (RT) (Invitrogen) with gene-specific reverse primers at 50°C for 50 min. Following heat inactivation at 65°C, RT reactions were diluted 10-fold in diethyl pyrocarbonate (DEPC)-treated deionized water. cDNA levels were quantified using quantitative PCR (qPCR) with Sybr GreenER (Bio-Rad) on an ICycler PCR cycling machine (Bio-Rad). Primers were designed for the internal control GAPDH (glyceraldehyde-3-phosphate dehydrogenase), CHOP, and IFN-β as well as the WNVKUN genome (primer sequences are available from the corresponding author upon request). Fold induction of each gene was calculated by comparing threshold cycle values (CT) to those for the internal control GAPDH and mock-infected cells at each time point.
Cell cytotoxicity assays.
Cells were lysed by treatment with 5% Triton X-100 at 37°C for 1 h. Cell viability was then measured by lactate dehydrogenase activity using a cell cytotoxicity kit (Promega) as recommended by the manufacturers. Briefly, lysed cells were transferred to a new 96-well microtiter plate and incubated with the substrate and reagent for 45 min. The reaction was halted by the addition of a stop solution, and enzyme activity was assessed by colorimetric analysis at 490-nm absorbance on a microplate reader (Anthos Lucy 2 luminometer). Each sample was assayed in triplicate.
RESULTS
WNVKUN production is decreased in ATF6−/− cells.
We have previously observed that UPR effectors downstream of ATF6 and IRE1 are upregulated during WNVKUN replication while PERK signaling appears dispensable (38). To investigate the contributions of each ER stress sensor during virus replication, ATF6−/− and IRE1−/− knockout MEFs and matched controls were infected with WNVKUN and RNA, protein, and infected supernatants were collected at 1, 12, 24, 36, and 48 h postinfection for analysis. As shown in Fig. 1A, viral RNA levels in the ATF6 cell lines peaked at 36 h p.i., with no significant differences between the knockout and control cells. However, viral protein expression was reduced at 12 and 24 h p.i. in the ATF6−/− cells (Fig. 1C), resulting in a modest but significant reduction in virions secreted at 36 h p.i. (Fig. 1E) (P < 0.05). In contrast, no significant differences were observed between the IRE1 knockout and control cells in RNA, protein, or virion production (Fig. 1B, D, and F), suggesting that IRE1 may be dispensable for efficient WNVKUN replication. Interestingly, total protein levels (as assessed by cellular actin and BiP content) were decreased in all infected cell lines from 24 h p.i. onwards, although this was more pronounced in ATF6−/− cells (Fig. 1C and D).
Fig 1.

WNVKUN production is decreased in ATF6−/− cells. ATF6 and IRE1 knockout MEFs and their matched controls were infected with WNVKUN (MOI, 10) for 1, 12, 24, 36, and 48 h, and supernatants, protein, and RNA samples were collected at each time point. (A and B) Viral genomic RNA was quantified by qRT-PCR using gene-specific primers and compared to that for the internal control GAPDH. Error bars indicate 1 standard deviation of three independent experiments. (C and D) Protein lysates collected at each time point were analyzed for viral protein expression (Env) and chaperone upregulation (BiP) using Western blotting, with actin used as a loading control. (E and F) Infectious virion secretion (from supernatant samples) was analyzed by plaque assay on Vero cells. Error bars indicate 1 standard deviation of three independent experiments, and asterisks show statistically significant differences in samples, as determined by a Student t test (P < 0.05).
Infected knockout and control cells were also fixed and stained with the WNVKUN viral protein NS1 and the ER chaperone BiP to assess viral replication complex formation and infection efficiency (Fig. 2). All cell lines had a similar infection rate (data not shown) over 12 to 48 h, negating the possibility that the defects observed in ATF6−/− cells were due simply to an initially lower percentage of infected cells. NS1 subcellular labeling was comparable, and distinct replication complexes were observed in all samples, although a slight decrease in intensity was detected in the ATF6−/− cells. In addition, we observed no significant differences in the distribution and localization of the WNVKUN replication complex formation in each of the cell types (Fig. 2), indicating that intracellular replication of WNVKUN occurs unimpeded in the presence or absence of ATF6 and IRE1.
Fig 2.

WNVKUN replication complex formation is not altered in knockout cell lines. ATF6−/−, IRE1−/−, and their matched control cells were infected with WNVKUN as indicated above, fixed at 36 h p.i., and then labeled with anti-NS1 (green) and anti-BiP (red) antibodies. Cells were viewed on an Olympus epifluorescence microscope.
eIF2α phosphorylation and downstream CHOP activity are upregulated in WNVKUN-infected ATF6−/− cells.
Various studies have shown that the three arms of the UPR are interdependent, with cross talk demonstrated between the acute-phase (PERK- and eIF2α-based responses) and the long-term (ATF6 and IRE1 signaling) effectors. Additionally, some of these effectors (such as p58IPK) are able to directly dephosphorylate eIF2α (29, 41), thus switching the UPR from the initial response of translation inhibition to stress adaptation, inducing the production of chaperones and degradative enzymes (42). We have observed that WNVKUN skews the UPR toward ATF6 and IRE1, with negligible eIF2α phosphorylation, as cell lines deficient in PERK permit moderate increases in WNVKUN viral protein and virion production (38). Thus, we hypothesized that WNVKUN-induced increases in ATF6 and/or IRE1 signaling downregulate PERK signaling, which, in the absence of either of these sensors, would result in the resumption of eIF2α phosphorylation and global attenuation of transcription.
To investigate this further, ATF6- and IRE1-deficient cells were infected with WNVKUN for 36 h or stimulated with tunicamycin (an ER stress inducer) for 12 h and assessed for eIF2α phosphorylation. As shown in Fig. 3, phospho-eIF2α was increased by tunicamycin treatment in all cell lines. However, in WNVKUN-infected cells, the ratio of phospho-eIF2α to total eIF2α was higher in the ATF6−/− MEFs than in the control cells (Fig. 3A, compare lanes 3 and 6). This was quantified by densitometric analyses, which showed that the proportion of phospho-eIF2α (as standardized to total eIF2α levels) was almost 2-fold higher in the ATF6−/− cells than in the wild-type (WT) controls. This was not observed in the IRE1 cell lines (Fig. 3B). Interestingly, higher baseline levels of phospho-eIF2α in tunicamycin-treated cells were also evident in the ATF6 and IRE1 knockout cells, suggesting an increased propensity for PERK activation despite infection/drug treatment. As phospho-eIF2α is a potent inhibitor of translation, this may also explain the decreased viral protein levels in ATF6−/− MEFs during early replication (Fig. 1).
Fig 3.
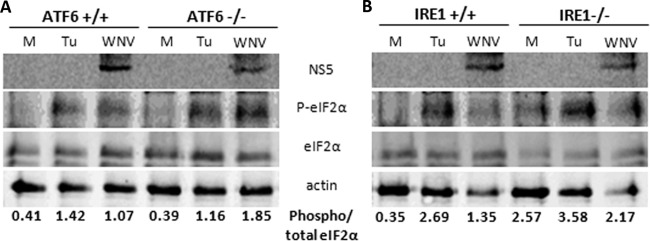
eIF2α phosphorylation is elevated in ATF6−/− MEFs. ATF6−/−, IRE1−/−, and their matched control cell lines were infected with WNVKUN (MOI, 10) over the time course indicated or treated with 2 μM tunicamycin for 12 h. (A) At 36 h p.i., protein lysates were collected and probed for levels of phospho-eIF2α (Ser51), total eIF2α, NS5 (as an infection control), and actin by Western blotting. The ratio of phospho to total eIF2α levels was determined by densitometry analysis (Quantity One; Bio-Rad) using the Western blot analysis shown.
To confirm the increased PERK activation in the ATF6−/− cells, CHOP transcription was investigated in ATF6 wild-type and knockout cell lines. CHOP is potently induced by ATF4, which is translationally activated by phospho-eIF2α. Correlating with increased phospho-eIF2α, a modest increase in CHOP mRNA levels was evident at 36 h p.i. in WNVKUN-infected ATF6−/− cells (Fig. 4A). To confirm downstream transcriptional activity, we assessed CHOP subcellular localization in WNVKUN-infected cells at 36 h p.i. by immunofluorescence. As shown in Fig. 4B, CHOP translocated to the nucleus in most infected cell types, although with different efficacies compared to that of the tunicamycin-treated control. The percentage of cells with nuclear CHOP was quantified and showed that significantly more ATF6−/− cells accumulated CHOP in the nucleus than did matched controls (Fig. 4C) (P < 0.05). Additionally, CHOP labeling was much higher in ATF6−/− cells, a trend which correlated with the increased transcription also observed at this time point.
Fig 4.

Increased CHOP activity in WNVKUN-infected ATF6−/− MEFs leads to cell death. (A) Total RNA extracted from infected and mock cells at each time point was analyzed for CHOP transcription using qRT-PCR, and fold induction was calculated by comparison to mock cells at the same time point. Error bars indicate 1 standard deviation of three independent experiments. (B) Knockout and wild-type MEFs were infected with WNVKUN for 36 h (MOI, 10) or treated with 2 μM tunicamycin for 12 h. Cells were fixed and subsequently labeled with anti-CHOP (red) and anti-NS3 (green) antibodies before analysis by immunofluorescence. Cells were also counterstained with the nuclear dye DRAQ5 and viewed on a Zeiss confocal microscope. The right-hand-side panels indicate the efficiency of CHOP nuclear translocation in the presence of tunicamycin alone. (C) CHOP nuclear translocation was quantified as a percentage of infected cells, and error bars indicate 1 standard deviation of two independent quantitations (of ∼150 cells each). Asterisks denote statistically significant differences between samples, as determined by a Student t test (P < 0.05). (D) Knockout and wild-type MEFs were infected or mock infected with WNVKUN in a 96-well microtiter plate for 48 h. Cells were lysed in 5% (vol/vol) Triton X-100, and then lactate dehydrogenase activity was assessed as a measure of cell viability using a cytotoxicity kit. Error bars indicate 1 standard deviation of 3 independent experiments with triplicates, and asterisks denote P values of <0.05.
These observations suggest that CHOP activity is enhanced in ATF6−/− cells in response to infection, which is presumably regulated by WNVKUN in wild-type cells to maintain efficient replication. We hypothesized that this increased activity would also lead to premature cell death in infected cells via increased transcription of proapoptotic molecules. To investigate this possibility further, virus-induced cytotoxicity was compared in the ATF6-deficient cells and the matched controls after 48 h of infection. Corresponding with increased CHOP transcription and nuclear localization, ATF6−/− cells also had a significantly lower percentage survival rate after infection with WNVKUN (Fig. 4D) (P < 0.05). This result may also explain the lower overall protein levels observed in the time course assays (Fig. 1B), as global translation would likely have been inhibited by increased eIF2α phosphorylation as well as reduced cell viability. Thus, we suggest that CHOP-induced increases in proapoptotic effectors in the WNVKUN-infected ATF6−/− cells contributed to the increased cell death observed.
Late-phase IFN signaling is partially restored in ATF6−/− and IRE1−/− MEFs.
We have previously shown that chemical or viral induction of the UPR can inhibit type I IFN-mediated STAT1 phosphorylation and nuclear translocation and that this effect is partly mediated by the expression of hydrophobic nonstructural proteins (particularly NS4A) (38). Given that ATF6 and IRE1 downstream signaling is potently activated by WNVKUN and that ATF6-deficient MEFs show decreased replication, we were interested to investigate if either one of these arms is also required for the UPR-dependent, viral-induced inhibition of IFN signaling. Knockout or control cells were infected with WNVKUN for 24 h and then stimulated with mouse IFN-α for 1 h, and STAT1 phosphorylation was detected by a Western blot in infected and uninfected cells. As expected, a decrease in phospho-STAT1 was observed in infected control cells after stimulation with IFN-α (Fig. 5A and B, compare columns 2 and 4). However, in both ATF6−/− and IRE1−/− WNVKUN-infected cells, we observed a greater amount of phospho-STAT1 (Fig. 5A and B, compare columns 6 and 8), with the phospho-STAT1 levels being similar in mock and infected cells. Interestingly, higher phospho-STAT1 levels were observed in unstimulated, infected ATF6+/+ cells than in the knockouts; whether this is due to the increased replication and, hence, immune detection was not able to be ascertained. As MEFs are immunocompetent, we were also interested in determining if this restoration may be due to differences in IFN-β transcription between cell lines. However, as shown in Fig. 5C, no significant variations in IFN-β transcription were observed.
Fig 5.
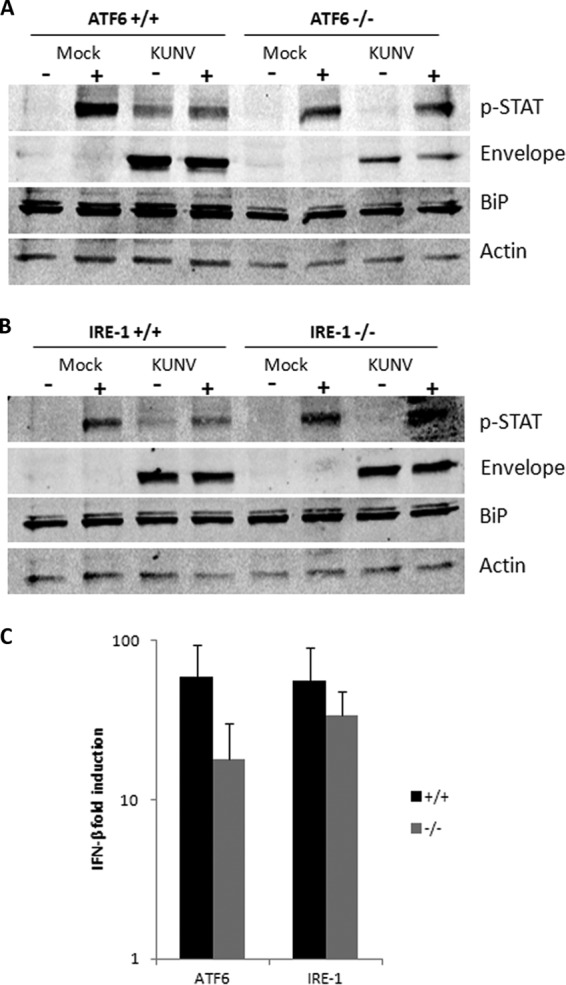
Late-phase IFN signaling is partially restored in ATF6−/− and IRE1−/− MEFs. (A and B) Knockout ATF6 and IRE1 MEFs and their matched controls were infected with WNVKUN (MOI, 10) for 36 h and then stimulated with 100 U/ml of mouse IFN-α for 1 h. Protein lysates were collected and probed for phospho-STAT1 (Y701), envelope, BiP, and actin using Western blotting. Results are indicative of at least two independent experiments. (C) RNA collected from infected cells was analyzed for IFN-β transcription by qRT-PCR using gene-specific primers. Error bars indicate 1 standard deviation of three independent experiments.
Thus, we suggest that the WNVKUN-induced defects imposed on IFN-mediated signaling are dependent on the downstream signaling of the ER stress response, particularly those events triggered by ATF6 and IRE1.
DISCUSSION
The UPR is a complex signaling pathway with myriad downstream effectors, many of which are still being discovered. These effectors, such as transcription factors, stimulatory or inhibitory proteins, or translational blocks, maintain homeostasis and function in the ER under stressful conditions. The contributions of the initial stress sensors (PERK, ATF6, and IRE1) to downstream signaling can also differ according to the type and extent of the stimulus. We investigated the roles of the ER stress sensors ATF6 and IRE1 during WNVKUN replication, as we had previously shown that PERK appears dispensable for WNVKUN replication and that ATF6 and IRE1 are preferentially activated in infected cells (38). To this end, we infected MEFs deficient in either ATF6 or IRE1 (and matched WT controls) with WNVKUN and assessed the contribution of these effectors to viral replication and immune evasion. While IRE1−/− cells had no discernible differences in viral RNA, protein, or virion production, we found that cells deficient in ATF6 had significant defects in viral protein and infectious virion production. However, viral RNA levels were only slightly attenuated, suggesting that the effect on replication was mediated at a translational level rather than affecting replication complex function and RNA synthesis. Interestingly, this observation coincided with an increase in eIF2α phosphorylation in the ATF6 knockout cells, which leads to global translational inhibition. Thus, we concluded that the decreases in WNVKUN protein production may be partially attributed to the inhibition of translation due to immoderate eIF2α phosphorylation.
We also investigated the extent of CHOP signaling in ATF6-deficient cells and showed that the increased eIF2α phosphorylation levels also resulted in elevated CHOP transcription and nuclear protein accumulation in infected cells. Additionally, this caused a decrease in cell viability compared to that of the wild-type controls, a result which we hypothesized was due to the transcriptional upregulation of proapoptotic molecules (such as Bim and Ero1α) by nuclear-bound CHOP. Thus, it is reasonable to suggest that the premature induction of apoptosis in the absence of ATF6 may have a limiting effect on WNVKUN replication, in particular, protein and virion production. Additionally, various studies have reported sustained IRE1 signaling in ATF6−/− cells which may also have contributed to the increased cell death; both regulated IRE1-dependent decay (RIDD) and Jun N-terminal protein kinase (JNK) activation have significant proapoptotic effects which would have only increased the cytotoxicity of virus replication. Interestingly, CHOP mRNA has been shown to be selectively degraded by IRE1 during ER stress (26); as this was not observed in ATF6−/− cells, it suggests that the overstimulation of CHOP transcription may also be due to an overall dysregulation of the entire UPR pathway due to the absence of ATF6.
The effects of the ablation of ATF6 signaling on WNVKUN replication can be attributed to a number of reasons. Fundamentally, the loss of chaperone production in response to the accumulation of viral proteins in the ER would have a significant outcome; not only would viral protein folding and function be perturbed, but they also could potentiate further UPR signaling through the remaining functional sensors IRE1 and PERK, resulting in aberrant activation of downstream effectors, such as CHOP and JNK. Multiple studies have shown that cross talk between the stress sensors is vital for correct responses to ER stress, in particular, the dephosphorylation of eIF2α (and, thus, resumption of translation) by ATF6/IRE1-mediated signaling (29, 30, 41). We have previously observed that WNVKUN preferentially activates ATF6 and IRE1 signaling while simultaneously inhibiting PERK and downstream effectors. We now propose a mechanism by which viral replication activates ATF6, not only to induce chaperone production and potentially assist with membrane proliferation but also to prevent PERK signaling, thus maintaining cell viability for efficient replication (Fig. 6A). This is supported by our data, which show that loss of ATF6 results in increased signaling through PERK and associated downstream effectors during WNVKUN replication (Fig. 6B), which has the dual effect of inhibiting viral protein and virion production and inducing premature cell death.
Fig 6.
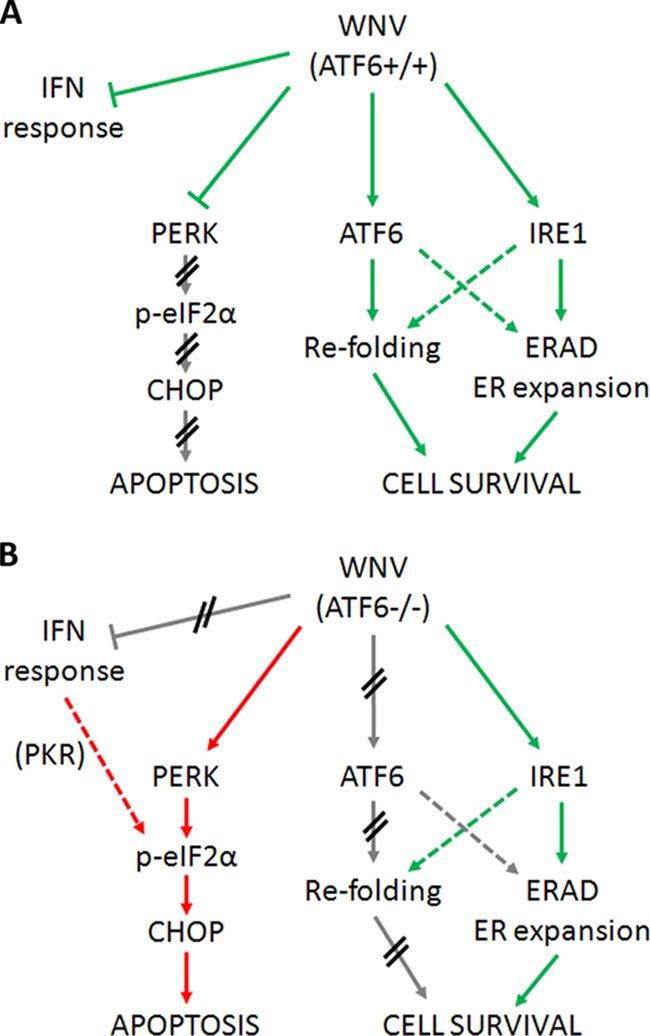
ATF6 is required during WNV replication to maintain cell viability. (A) During WNVKUN replication in WT cells, PERK and IFN signaling is actively inhibited to prevent cell death (red arrows) while ATF6- and IRE1-mediated effectors are induced (green arrows) to assist with viral protein and virion production (via chaperone expression, expansion of ER membranes). (B) However, in ATF6-deficient cells, the modulation of UPR signaling is lost, resulting in uncontrolled signaling through PERK (red arrows) as well as resumption of IFN signaling. Although WNVKUN can still signal via IRE1 (green arrows), the cells must override this manipulation to aid in viral clearance. The proviral effects of ATF6-induced chaperone expression are also ablated, resulting in translational defects and premature cell death in WNVKUN-infected cells. We propose that ATF6 (via a downstream effector) is required to modulate PERK and IFN responses.
During ER stress conditions, multiple basic cellular functions, such as translation, protein folding, membrane proliferation, secretion, and trafficking, not to mention effects on redox processes, calcium signaling, and immunity, are perturbed. Thus, the multiple crossovers that exist between UPR effectors and these pathways are logical, as optimal functioning of the ER is vital for the majority of all cellular processes. We have previously shown that virus-induced UPR signaling is required for the inhibition of type I IFN responses and that this coregulation is, in part, mediated by the hydrophobic membrane proteins of WNV (38). Thus, we investigated if either ATF6 or IRE1 signaling was necessary for the virus-induced inhibition of type I IFN signaling (Fig. 5). In the absence of either IRE1 or ATF6, STAT1 phosphorylation was partially restored in WNVKUN-infected cells, implying that both are required for IFN inhibition (Fig. 6). This effect is most likely not dependent on endogenous IFN production, as IFN-β transcription was only marginally decreased in knockout cells (Fig. 5C). It is interesting to note that protein kinase R (PKR), which is induced by phospho-STAT1, also feeds into the eIF2α/ATF4 signaling pathway. Restoration of IFN signaling in ATF6 or IRE1 knockouts may thus also contribute to the increased translational and proapoptotic effects in these cells via PKR and downstream signaling, thus inducing an antiviral state detrimental to replication.
Overall, we suggest that the balance of prosurvival and proapoptotic effectors of the UPR is of vital importance to WNVKUN replication (Fig. 6). Virus-mediated manipulation of the signaling pathways toward chaperone production, membrane biogenesis, and misfolded protein degradation benefits replication, not only in the formation of replication compartments and folding of viral proteins but also in the maintenance of cell viability during infection. This is clearly demonstrated by the various defects in WNVKUN-infected ATF6 knockout cells, implying that virus-induced stimulation of ATF6 signaling is a prosurvival mechanism required for replication. This may also be partly due to ATF6-mediated inhibition of PERK signaling; both translation attenuation and induction of apoptosis in ATF6 knockouts resulted in inefficient replication. In contrast, while IRE1 downstream effectors may also contribute to WNVKUN replication, loss of IRE1 and associated signaling does not adversely affect RNA, protein, or virion production. However, both IRE1 and ATF6 were shown to be required for the UPR-mediated inhibition of IFN responses, demonstrating the importance of regulated UPR signaling in WNVKUN replication. These results further underscore the central role that the UPR plays in viral manipulation of host cells.
ACKNOWLEDGMENTS
We thank Roy Hall and Julie Atkin for generously supplying antibodies for this study. We are also grateful to Randal Kaufman for the ATF and IRE1 MEFs utilized during the study. We also acknowledge the assistance of Jose Pena with cell lines and advice.
This research was supported by a project grant (no. 1004619) to J.M.M. from the National Health and Medical Research Council of Australia.
Footnotes
Published ahead of print 5 December 2012
REFERENCES
- 1. Berridge MJ. 2002. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32:235–249 [DOI] [PubMed] [Google Scholar]
- 2. Bernales S, Papa FR, Walter P. 2006. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 22:487–508 [DOI] [PubMed] [Google Scholar]
- 3. Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. 2004. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell Biol. 167:445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cui WJ, Li JZ, Ron D, Sha BD. 2011. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 67:423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu CY, Schroder M, Kaufman RJ. 2000. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 275:24881–24885 [DOI] [PubMed] [Google Scholar]
- 6. Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE. 2008. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J. Biol. Chem. 283:469–475 [DOI] [PubMed] [Google Scholar]
- 7. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. 2000. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6:1355–1364 [DOI] [PubMed] [Google Scholar]
- 8. Lee AH, Iwakoshi NN, Glimcher LH. 2003. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23:7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891 [DOI] [PubMed] [Google Scholar]
- 10. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. 2000. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20:6755–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harding HP, Zhang YH, Zeng HQ, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11:619–633 [DOI] [PubMed] [Google Scholar]
- 12. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang YH, Jungreis R, Nagata K, Harding HP, Ron D. 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18:3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. 2004. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 279:50375–50381 [DOI] [PubMed] [Google Scholar]
- 14. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129:1337–1349 [DOI] [PubMed] [Google Scholar]
- 15. Tabas I, Ron D. 2011. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13:184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez J, Yaman I, Sarnow P, Snider MD, Hatzoglou M. 2002. Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2α. J. Biol. Chem. 277:19198–19205 [DOI] [PubMed] [Google Scholar]
- 17. Romisch K. 2005. Endoplasmic reticulum-associated degradation. Annu. Rev. Cell Dev. Biol. 21:435–456 [DOI] [PubMed] [Google Scholar]
- 18. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang LM, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. 2004. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21:81–93 [DOI] [PubMed] [Google Scholar]
- 19. Sriburi R, Jackowski S, Mori K, Brewer JW. 2004. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 167:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roy B, Lee AS. 1999. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 27:1437–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. 2004. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136:343–350 [DOI] [PubMed] [Google Scholar]
- 22. Cherasse Y, Maurin AC, Chaveroux C, Jousse C, Carraro V, Parry L, Deval C, Chambon C, Fafournoux P, Bruhat A. 2007. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 35:5954–5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma YJ, Brewer JW, Diehl JA, Hendershot LM. 2002. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318:1351–1365 [DOI] [PubMed] [Google Scholar]
- 24. Lee K, Tirasophon W, Shen XH, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. 2002. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16:452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. 2009. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186:323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Akha AAS, Raden D, Kaufman RJ. 2006. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4:e374 doi:10.1371/journal.pbio.0040374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. 2002. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 22:7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brush MH, Weiser DC, Shenolikar S. 2003. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23:1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. 2003. P58(IPK), a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α signaling. J. Biol. Chem. 278:15558–15564 [DOI] [PubMed] [Google Scholar]
- 30. Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG. 2002. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58(IPK). Proc. Natl. Acad. Sci. U. S. A. 99:15920–15925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Mol. Cell. Biol. 21:1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I. 2009. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186:783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshida T, Shiraishi T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. 2005. Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 65:5662–5667 [DOI] [PubMed] [Google Scholar]
- 34. Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tardif KD, Mori K, Kaufman RJ, Siddiqui A. 2004. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279:17158–17164 [DOI] [PubMed] [Google Scholar]
- 36. Tardif KD, Mori K, Siddiqui A. 2002. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76:7453–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu CY, Hsu YW, Liao CL, Lin YL. 2006. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 80:11868–11880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ambrose RL, Mackenzie JM. 2011. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 85:2723–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GDY, Kaufman RJ. 2007. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13:351–364 [DOI] [PubMed] [Google Scholar]
- 40. Westaway EG, MacKenzie JM, Kenney MT, Jones MK, Khromykh AA. 1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 71:6650–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, Hegde RS. 2007. The role of p58(IPK) in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18:3681–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rutkowski DT, Kaufman RJ. 2007. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32:469–476 [DOI] [PubMed] [Google Scholar]