

Abstract
PML nuclear bodies (PML NBs), also called ND10, are matrix-bound nuclear structures that have been implicated in a variety of functions, including DNA repair, transcriptional regulation, protein degradation, and tumor suppression. These domains are also known for their potential to mediate an intracellular defense mechanism against many virus types. This is likely why they are targeted and subsequently manipulated by numerous viral proteins. Paradoxically, the genomes of various DNA viruses become associated with PML NBs, and initial sites of viral transcription/replication centers are often juxtaposed to these domains. The question is why viruses start their transcription and replication next to their supposed antagonists. Here, we report that PML NBs are targeted by the adenoviral (Ad) transactivator protein E1A-13S. Alternatively spliced E1A isoforms (E1A-12S and E1A-13S) are the first proteins expressed upon Ad infection. E1A-13S is essential for activating viral transcription in the early phase of infection. Coimmunoprecipitation assays showed that E1A-13S preferentially interacts with only one (PML-II) of at least six nuclear human PML isoforms. Deletion mapping located the interaction site within E1A conserved region 3 (CR3), which was previously described as the transcription factor binding region of E1A-13S. Indeed, cooperation with PML-II enhanced E1A-mediated transcriptional activation, while deleting the SUMO-interacting motif (SIM) of PML proved even more effective. Our results suggest that in contrast to PML NB-associated antiviral defense, PML-II may help transactivate viral gene expression and therefore play a novel role in activating Ad transcription during the early viral life cycle.
INTRODUCTION
Human adenovirus type 5 (Ad5) early region 1A (E1A) is the first protein expressed upon infection and plays an essential role in transcriptional activation and induction of cell cycle progression (1). At early times of infection, two major E1A proteins, E1A-12S (243R) and E1A-13S (289R), are synthesized from alternatively spliced mRNA transcripts of the E1A gene (2, 3). The proteins are identical except for a 46-amino-acid (aa) conserved region (CR3) that is unique to the larger E1A protein (3). Despite being very similar, these proteins show significant differences in their biological activities. The larger E1A isoform (E1A-13S) is considered to be primarily responsible for transactivating viral gene expression by interacting with a wide range of transcription factors via its CR3 (4–8).
The PML (promyelocytic leukemia) protein was first described in acute promyelocytic leukemia (APL), showing that PML was fused to retinoic acid receptor alpha (RARα) due to a chromosomal translocation (t15;17) (9–20). The PML gene encodes multiple isoforms, derived from alternative mRNA splicing, differing only in the C-terminal parts of the proteins (21). PML seems to be responsible for the assembly of nuclear bodies by recruiting other constitutive components such as Daxx (death domain-associated protein), Sp100 (speckled protein of 100 kDa), or SUMO (small ubiquitin-related modifier) (22–24). Other components, such as p53, pRb, and CBP/p300, are present only under certain circumstances (summarized in 25). Modification of PML by SUMO is critical for PML nuclear body (PML NB) formation (23, 26), and SUMOylated proteins, which are often found in PML NBs (27) are recruited to PML NBs by the SUMO-interacting motif (SIM) of PML (24).
Since various proteins localize to PML nuclear bodies, these domains have been associated with numerous functions, such as protein degradation (28), transcriptional regulation (29, 30), cellular senescence (31–34), tumor suppression (35, 36), DNA repair (37, 38), apoptosis (39, 40), and epigenetic regulation (41).
PML itself is a member of the RING finger family of proteins, including proteins involved mainly in transcriptional regulation. Although PML does not exhibit DNA binding activity, its RBCC domain displays RING finger-dependent transactivating activity (42). Indeed, PML has been found to act both as a transcriptional coactivator (43, 44) and as a corepressor (45, 46). It is thought to regulate transcription through targeting various DNA binding transcription factors/cofactors such as CBP/p300 and nuclear receptor cofactor TIF1 α (30, 44, 47, 48).
PML and PML-associated factors are suspected to mediate intracellular antiviral defense mechanisms and are therefore targeted by multiple viral proteins during infection (49–51). Interestingly, genomes of human DNA viruses such as adenoviruses, herpesviruses, polyomaviruses, and papillomaviruses associate with PML NB components at initial stages in their replication cycles (49, 52–56). Consequently, newly formed transcription and replication sites are often found juxtaposed to the PML nuclear domains (57).
In our present study, we further have clarified the relationship between adenoviral proteins and PML nuclear structures and have analyzed of the role of PML during productive adenoviral infection with respect to functional cooperation with specific E1A variants. We show that E1A-13S interacts with the PML-II isoform through conserved region 3 (CR3) of E1A, and this complex elevates transcription from the adenoviral E2 early promoter. Mutation of the SUMO-interacting motif in PML-II further enhanced this E1A-dependent transactivation. Furthermore, PML-II combined with E1A-13S stimulated cellular gene expression through interacting with the cellular transcriptional coactivator p300, suggesting that adenovirus takes advantage of PML and associated factors to regulate cellular as well as viral transcription.
MATERIALS AND METHODS
Cell lines.
HepaRG (58), HALP/HALP PML-II (59), and H1299 (60) cells and mouse embryonic fibroblasts (MEF) (61) were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 100 U of penicillin and 100 μg of streptomycin per ml in a 5% CO2 atmosphere at 37°C. For HepaRG cells, the medium was additionally supplemented with 5 μg/ml of bovine insulin and 0.5 μM hydrocortisone.
Plasmids and transient transfections.
Ad5 E1A-12S, Ad5 E1A-13S, and the Ad5 E1A mutant proteins were expressed from pcDNA3 vector. N-terminal flag-tagged human PML-isoforms I to VI were expressed from the pLKO.1-puro vector and were kindly provided by Roger Everett (Glasgow, United Kingdom). All PML isoforms are named according to the nomenclature of Jensen et al. (21): PML-I (AAG50180), PML-II (AF230410), PML-III (S50913), PML-IV (AAG50185), PML-V (AAG50181), and PML-VI (AAG50184). The PML-II SIM mutation was introduced by site-directed mutagenesis using forward primer 5′-GGAACGCGGTGGGGGGATCAGCAGC-3′ and reverse primer 5′-GCTGCTGATCCCCCCACCGCGTTCC-3′ (see also Fig. 8A). For transient transfection, subconfluent cells were treated with a mixture of DNA and 25-kDa linear polyethylenimine (Polysciences) as described previously (62).
Fig 8.

PML-II positively regulates adenovirus progeny production in human cells. Parental HepaRG, HALP, and HALP EYFP–PML-II cells were infected with wild-type H5pg4100 at a multiplicity of 50 FFU/cell. Viral particles were harvested at 48 h postinfection, and virus yield was determined by quantitative E2A-72K immunofluorescence staining on 293 cells.
Viruses.
H5pg4100 served as wild-type virus (63). H5dl347 and H5dl348 carry cloned segments corresponding to E1A-12S and E1A-13S mRNAs, respectively, in place of the E1A gene. H5dl312 lacks a large segment of the E1A gene and therefore does not produce E1A products (64). H5pm4150 carries a frameshift mutation in the E4orf3 open reading frame and H5pm4149 carries stop codons in the E1B-55K open reading frame, so they do not express E4orf3 and E1B-55K, respectively (65, 66). All viruses were propagated, and titers were determined as fluorescence-forming units (FFU), in HEK293 monolayer cultures (63). To measure virus growth, infected cells were harvested at 48 h postinfection (p.i.) and lysed by three cycles of freeze-thawing. The cell lysates were serially diluted in DMEM for infection of HEK293 cells, and virus yield was determined by quantitative E2A immunofluorescence staining at 24 h after infection.
Luciferase reporter assay.
For dual-luciferase assays, subconfluent H1299 cells were transfected as described above, using 1 μg of reporter (pGL-C3G5-luc or pGL-E2-early promoter), 1 μg of pRL-TK (Promega), which expresses Renilla luciferase under the control of the herpes simplex virus (HSV) thymidine kinase promoter, and 1 μg of effector plasmids (E1A-12S, E1A-13S, PML-II, and pG4-p300). Cell extracts were prepared, measured, and normalized as described recently (67).
Antibodies and protein analysis.
Primary antibodies specific for Ad proteins included E1A mouse monoclonal antibody (MAb) M58 and E1A mouse MAb M73 (68) and E2A mouse MAb B6-8 (69). E1A rabbit MAb 610 was a kind gift of Roger Grand. Primary antibodies specific for cellular and epitopically expressed proteins included PML rabbit polyclonal antibody (PAb) NB100-59787 (Novus Biologicals, Inc.), PML mouse MAb clone 36.1-104 (Millipore), flag mouse MAb flag-M2 (Sigma-Aldrich, Inc.), and β-actin mouse MAb AC-15 (Sigma-Aldrich, Inc.).
All protein extracts were prepared in radioimmunoprecipitation assay (RIPA) lysis buffer as published recently (70). For immunoprecipitation, flag-M2-coupled protein A-Sepharose beads (Sigma-Aldrich, Inc.) were used or protein A-Sepharose (3 mg/immunoprecipitation) was coupled with 1 μg of MAb for 1 h at 4°C. The Ab-coupled protein A-Sepharose was added to pansorbin-Sepharose (50 μl/lysate; Calbiochem)-precleared extracts and rotated for 2 h at 4°C. Proteins bound to the Ab-coupled protein A-Sepharose were precipitated by centrifugation, washed three times, boiled for 3 min at 95°C in 2× Laemmli buffer, and analyzed by immunoblotting exactly as described recently (62).
Indirect immunofluorescence.
For indirect immunofluorescence, cells were grown on glass coverslips as already published (71). To visualize intranuclear E1A staining, cells were extracted with 0.5% Triton X-100 in CSK buffer as described by Carvalho and coworkers (72). Cells were fixed in 4% paraformaldehyde (PFA) at 4°C for 20 min and permeabilized in phosphate-buffered saline (PBS)–0.5% Triton X-100 for 30 min at room temperature. After 1 h of blocking in Tris-buffered saline–bovine serum albumin-glycine (TBS-BG) buffer, coverslips were treated for 1 h with the primary antibody diluted in PBS, washed three times in PBS–0.1% Tween 20, and then incubated with the corresponding Alexa 488 (Invitrogen)- or Cy3 (Dianova)-conjugated secondary antibodies. Coverslips were washed three times in PBS–0.1% Tween 20 and mounted in Glow medium (Energene), and digital images were acquired with a confocal laser scanning microscope (Zeiss-CLSM-510). Images were cropped using Adobe Photoshop CS4 and assembled with Adobe Illustrator CS4.
Quantitative RT-PCR analysis.
Subconfluent HepaRG cells were infected with wild-type virus at a multiplicity of 50 FFU/cell and harvested 24 h p.i. Total RNA was isolated with TRIzol reagent (Invitrogen) as described by the manufacturer. The amount of total RNA was measured, and 1 μg of RNA was reverse transcribed using the reverse transcription system from Promega. To amplify specific viral genes, primers were designed as follows: 18S rRNA forward primer, 5′-CGGCTACCACATCCAAGGAA-3′; 18S rRNA reverse primer, 5′-GCTGGAATTACCGCGGCT-3′; E2A forward primer 5′-GAAATTACGGTGATGAACCCG-3′; and E2A reverse primer, 5′-CAGCCTCCATGCCCTTCTCC-3′. Quantitative reverse transcription-PCR (RT-PCR) was performed with a first-strand method in a Rotor-Gene 6000 (Corbett Life Sciences, Sydney, Australia) in 0.5-ml reaction tubes containing a 1/50 dilution of the cDNA template, 10 pmol/μl of each synthetic oligonucleotide primer, and 5 μl/sample SensiMix SYBR (Bioline). The PCR conditions were as follows: 10 min at 95°C, followed by 40 cycles of 30 s at 95°C, 30 s at 62°C, and 30 s at 72°C. The average threshold cycle (CT) value was determined from triplicate reactions, and levels of viral mRNA relative to cellular 18S rRNA were calculated as described recently. The identities of the products obtained were confirmed by melting curve analysis.
RESULTS
Ad5 E1A-13S localizes to endogenous PML in transiently transfected cells.
Although reports have proposed different functions of E1A-12S and E1A-13S during adenoviral infection (73), E1A has been described to localize to PML nuclear bodies (72). In this context, we decided to express both E1A isoforms separately and visualize their localization with respect to endogenous PML bodies. Both isoforms localized to the nucleus in large amounts (Fig. 1A, panels g and k), whereas PML exhibited the characteristic punctuate nuclear staining (Fig. 1A, panels f and j). Consistent with published results, E1A-12S as well as E1A-13S relocalized PML bodies, causing them to appear larger and fewer in number (74).
Fig 1.

E1A-13S colocalizes with endogenous PML in transiently transfected cells. (A) HepaRG cells were transfected with 3 μg of pE1A-12S or pE1A-13S, fixed with 4% PFA 48 h posttransfection, and double labeled with MAb M-58 (anti-E1A [α-E1A]) and PAb NB 100-59787 (α-PML). (B) HepaRG cells were extracted with cytoskeleton buffer prior to fixation with 4% PFA. For both panels, primary Abs were detected with Cy3 (α-PML; green)- and Alexa 488 (α-E1A; red)-conjugated secondary Ab. For nuclear staining, the DNA-intercalating dye DRAQ5 (Biostatus) was used. Representative α-E1A (panels c, g, and k) and α-PML (panels b, f, and j) staining patterns of at least 50 analyzed cells are shown. Overlays of single images (merge) are shown in panels d, h, and l. Magnification, ×7,600.
Since the large amounts of intranuclear E1A make it very difficult to observe any distinct staining patterns, the cells were treated as described in Materials and Methods (“Indirect immunofluorescence”) prior to fixation (Fig. 1B) (72). After preextraction, E1A-13S accumulates in several brightly stained nuclear aggregates juxtaposed to endogenous PML in ∼30% of the transfected cells (Fig. 1B, panels i to l). For the smaller E1A-12S isoform, no staining juxtaposed to PML could be observed (Fig. 1B, panels e to h). Our observations are consistent with previous reports showing that transcriptionally active proteins often tend to associate with the nuclear matrix (75).
Ad5 E1A-13S localizes to PML nuclear bodies during adenoviral infection.
Detailed studies have revealed an astonishing amount of cross talk between adenoviral proteins in the modulation of host cell factors, e.g., PML and PML-associated proteins. Previously published data illustrate that the viral protein E4orf3 is necessary and sufficient to disrupt the structure of cellular PML NBs during Ad5 infection (72, 76–78). In addition, viral early E1B-55K protein has been shown to interact and functionally cooperate with PML bodies (79–83).
To evaluate whether other adenoviral proteins interfere with or alter the cooperation between E1A and PML, we further analyzed localization during productive infection (Fig. 2). Cells were infected with wild-type virus (H5pg4100) and virus mutants expressing E1A-12S (H5dl347) or E1A-13S (H5dl348). To exclude expression of other adenoviral proteins mediating PML interaction, viruses lacking E1B-55K (H5pm4149) or E4orf3 (H5pm4150) were included as controls.
Fig 2.

E1A-13S colocalizes with endogenous PML in infected cells. Human hepatocytes (HepaRG) were infected with wild-type (H5pg4100) or mutant (H5dl347, H5dl348, H5pm4149, and H5pm4150) viruses at a multiplicity of 50 FFU/cell and fixed with 4% PFA at 24 h p.i. The cells were extracted with cytoskeleton buffer prior to fixation and double labeled with MAb M-58 (α-E1A) and PAb NB 100-59787 (α-PML). Detection of primary Abs and nuclear staining were performed as for Fig. 1. Representative α-E1A (red) (c, g, k, o, s, and w) and α-PML (green) (b, f, j, n, r, and v) staining patterns of at least 50 analyzed cells are shown. Overlays of the single images (merge) are shown in panels d, h, l, p, t, and x. Magnification, ×7,600.
As expected, PML was relocalized (Fig. 2b, f, j, n, and r) into so-called track-like structures in infected cells. The virus mutant lacking E4orf3 protein (H5pm4150) was no longer able to relocalize PML bodies, although their shape was different from the punctate staining in mock-infected cells (Fig. 2b and v). We detected E1A colocalization with PML track-like structures in wild-type virus (H5pg4100)- as well as H5dl348-infected cells expressing only E1A-13S (Fig. 2g and o), whereas E1A-12S (H5dl347) is distributed much more diffusely in the host cell nucleus (Fig. 2k). E1A also localized to PML after infection with virus lacking E4orf3 or E1B-55K (Fig. 2t and x). Based on these observations, we conclude that E1A-13S localizes to PML nuclear bodies independently of E4orf3 or E1B-55K expression.
Ad5 E1A-13S specifically interacts with the human PML-II isoform in transiently transfected cells.
Human cells express at least seven distinguishable PML isoforms (21), of which PML-I to -VI possess a nuclear localization sequence. To ascertain whether E1A specifically interacts with different PML isoforms, human cells were transfected with E1A-13S or E1A-12S and plasmids encoding human PML isoforms I to VI. By precipitating flag-tagged PML and subsequently staining for E1A, we detected a highly specific interaction between E1A-13S and PML isoform II (Fig. 3D, lane 4). E1A-12S could not be coprecipitated with any PML isoform (Fig. 3B), although the steady-state levels showed equal amounts of transfected PML and E1A isoforms (Fig. 3A and C).
Fig 3.
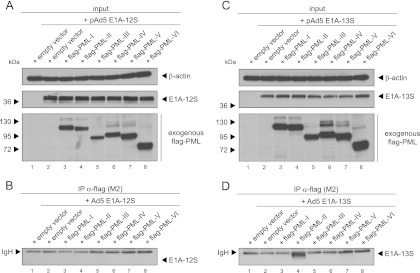
E1A-13S binds a specific PML isoform in transiently transfected H1299 cells. Subconfluent H1299 cells were transfected with 3 μg of wild-type pE1A-12S or pE1A-13S and 5 μg of different lentiviral constructs encoding N-terminal flag-tagged human PML isoforms I to VI and harvested after 48 h to prepare total cell extracts. Immunoprecipitation of flag-PML was performed using MAb flag-M2 (α-flag), and proteins resolved by 10% SDS-PAGE were visualized by immunoblotting. Input levels in total cell lysates (A and C) and coprecipitated proteins (B and D) were detected using MAb 610 (α-E1A), MAb flag-M2 (α-flag), and MAb AC-15 (α-β-actin). Molecular masses in kDa are indicated on the left, while the relevant proteins are labeled on the right. Note that heavy chains (IgH) are detected at 55 kDa in panels B and C.
Mutational screening of E1A-13S reveals PML-II isoform interaction within CR3.
Since E1A-13S differs from E1A-12S only in CR3, composed of 46 aa, we assumed that PML-II binding depends on this or adjacent regions in the E1A protein. To more precisely identify the binding domain required for E1A-13S/PML-II interaction, we constructed E1A-13S mutants with overlapping 14- to 16-aa deletions spanning the proposed region (Fig. 4A). Although the E1A-13S mutants had slightly altered gel migration properties, input levels of E1A-13S and PML-II were equal in our coimmunoprecipitation assays (Fig. 4B). These experiments revealed that deletion mutant 7 (E1A-13SΔ176-191) (Fig. 4A) exhibits strongly impaired PML-II binding properties (Fig. 4C, lane 11).
Fig 4.

Mapping experiments identify an E1A mutant defective in binding PML-II. (A) Overlapping deletions of 14 to 16 aa spanning CR3 and adjacent regions are shown below the schematic representation of E1A-12S and E1A-13S protein structures. Detailed amino acid deletions of each mutant are listed on the right. (B and C) Subconfluent H1299 cells were transfected with 3 μg of wild type pE1A-13S or pE1A-13S deletion mutants and 3 μg of pPML-II and harvested after 48 h before preparation of total cell extracts. Coimmunoprecipitation assays were performed as described for Fig. 3. Note that the different E1A-13S deletion mutants show different mobilities. Molecular masses in kDa are indicated on the left, appropriate proteins on the right.
To further narrow down the interaction region, a shorter deletion mutant, E1A-13SΔ182-191, was constructed and tested for PML-II binding (Fig. 5A). We confirmed binding deficiency of deletion mutant E1A-13SΔ182-191 with the PML-II isoform by immunofluorescence analysis and coimmunoprecipitation experiments (data not shown).
Fig 5.

Interaction with PML-II depends on a highly conserved transcription factor binding region within CR3. (A) Schematic representation of E1A-12S and E1A-13S, with the enlarged interaction region depicted at the bottom. Mapped is the newly constructed PML-II/E1A-binding mutant 13SΔ182-191 with a shorter deletion than the previously identified 13SΔ176-191. (B and C) Subconfluent H1299 cells were transfected with 3 μg of wild-type pE1A-12S, pE1A-13S, and pE1A-13SΔ182-191 or infected with wild-type virus (H5pg4100) or virus mutants (H5dl312, H5dl347, and H5dl348). The cells were harvested at 48 h after transfection or 24 h p.i., and total cell extracts were prepared. Immunoprecipitation of endogenous PML was performed using PAb NB100-59787 (α-PML), and proteins were resolved by 10% SDS-PAGE and visualized by immunoblotting. Steady-state expression levels of total cell lysates (input) and coprecipitated proteins (IP) were detected using MAb 610 (α-E1A), PAb NB 100-59787 (α-PML), and MAb AC-15 (α-β-actin). Molecular masses in kDa are on the left, and corresponding proteins are on the right.
Ad5 E1A-13S interacts with endogenous PML in plasmid-transfected and infected cells.
Next, we verified our observations by coimmunoprecipitation experiments with endogenous PML. We transfected human cells with E1A-12S, E1A-13S, or E1A-13SΔ182-191 or infected them with wild-type virus (H5pg4100) and virus mutants expressing only E1A-12S (H5dl347), only E1A-13S (H5dl348), or no E1A protein (H5dl312) as a negative control (Fig. 5B). After precipitation of endogenous PML and subsequent staining for E1A, we could detect specific interaction of endogenous PML with transfected E1A-13S as well as viral E1A in H5pg4100- and H5dl348-infected cells (Fig. 5C, lanes 3, 5, and 6). In contrast, E1A-12S, deletion mutant E1A-13SΔ182-191, and viral E1A encoded by the virus mutant lacking E1A-13S (H5dl347) could not be precipitated with endogenous PML (Fig. 5C).
In sum, interaction of E1A-13S with PML could be mapped to aa 182 to 191 at the end of the CR3 of E1A-13S. This binding region (aa 182 to 191) is highly conserved in different Ad types and has previously been identified as the variable transcription factor binding domain or promoter-targeting domain of E1A-13S (84–86).
Ad5 E1A-13S and PML cooperation impacts viral and cellular transcription.
PML bodies have been implicated in regulating transcription (30, 86). Furthermore, it is known that several viral genomes, including that of adenovirus, localize close to PML bodies in order to start transcription and replication (53, 87). Therefore, E1A likely affects transcriptional regulation from adenoviral promoters in cooperation with PML-II.
To address this idea, we used luciferase reporter assays and transfected luciferase-dependent plasmids carrying adenoviral promoters (67) together with E1A-13S and PML-II (Fig. 6). PML-II alone was not able to stimulate transcription from the Ad5 E2 early promoter, whereas E1A-13S activated luciferase activity from the viral promoter 10-fold. Furthermore, PML-II efficiently stimulated transcription from the Ad5 promoter when coexpressed with E1A-13S. This was further verified by the positive correlation between increasing levels of expressed PML-II and activated transcription at constant E1A-13S levels (Fig. 6B). As a control we also included E1A-12S and E1A-13SΔ182-191 in this reporter gene assay. We observed that neither E1A-12S nor E1A-13SΔ182-191 stimulated transcription from the adenoviral promoter in combination with equal amounts of PML-II (Fig. 6B).
Fig 6.

PML stimulates E1A-13S-dependent transcriptional activation of the adenoviral E2 early promoter. (A) Schematic representation of the luciferase reporter assay. The luciferase gene is under the control of the Ad5 promoter, which is bound and activated by a complex of E1A-13S and PML-II. (B) H1299 cells were transfected with 0.2 μg of pRenilla-Luc, 0.5 μg pGL-E2 early promoter, 0.2 μg pE1A (E1A-12S, E1A-13S, and E1A-13SΔ182-191), and increasing amounts of pPML-II in the combinations indicated (+). At 24 h posttransfection, total cell extracts were prepared and luciferase activity was determined. The means and standard deviations for three independent experiments are presented. (C) H1299 cells were transfected with 0.2 μg of pRenilla-Luc, 0.5 μg pGL-E2-early promoter, 0.2 μg pE1A-13S, and 1.5 μg pPML-I-VI in the combinations indicated. At 24 h posttransfection, total cell extracts were prepared and luciferase activity was determined. The means and standard deviations for three independent experiments are presented.
Recently, Pelka et al. reported that cellular transcription factor p300 binds E1A-13S within the CR3 region and is recruited to Ad promoters during infection (88). p300 possesses intrinsic acetyltransferase activity and is therefore an important transcriptional coactivator; it is recruited to gene promoters via its association with numerous DNA binding transcription factors (89). PML was also found to interact with p300/CBP and regulate transcription (90, 91). In this context, we tested whether E1A-13S and PML-II affect p300 coactivator function in transcriptional regulation. We performed luciferase reporter assays with a GAL4-responsive promoter and p300 fused to a GAL4 DNA binding domain, in combination with E1A-13S and PML-II (Fig. 7A). PML-II alone only slightly stimulated transcription from the GAL4 promoter, whereas E1A-13S enhanced p300 transcriptional activity 2-fold. Consistent with our observations described above, coexpression of E1A-13S with PML-II strongly stimulated the luciferase activity from the promoter. Cotransfection of increasing PML-II amounts further stimulated p300-dependent transcriptional activation (Fig. 7B). On the other hand, coexpression of PML-II together with E1A-13SΔ182-191 did not activate transcription from the GAL4 promoter (Fig. 7B).
Fig 7.

E1A-13S and PML cooperate to stimulate p300 transcriptional coactivation. (A) Schematic presentation of the luciferase reporter assay. The luciferase gene is under the control of a promoter containing 5 GAL4 sequences, which is bound and activated by a complex of GAL-p300, E1A-13S, and PML-II. (B) H1299 cells were transfected with 0.2 μg of pRenilla-Luc, 0.5 μg pGL-C3G5-luc, 0.5 μg pG4-p300, 1 μg pE1A (E1A-13S or 13SΔ182-191), and increasing amounts of pPML-II in the combinations indicated (+). At 24 h posttransfection, total cell extracts were prepared and luciferase activity was determined. The means and standard deviations for three independent experiments are presented. (C) H1299 cells were transfected with 0.2 μg of pRenilla-Luc, 0.5 μg pGL-C3G5-luc, 0.5 μg pG4-p300, 1 μg pE1A-13S, and 2 μg pPML-I to -VI in the combinations indicated. At 24 h posttransfection, total cell extracts were prepared and luciferase activity was determined. The means and standard deviations for three independent experiments are presented.
Additionally, we performed the reporter gene assay shown in Fig. 6 and 7 with the other nuclear PML isoforms. Our results showed that other PML isoforms do positively affect E1A-dependent transactivation, although to a lesser extent than PML-II (Fig. 6C and 7C).
Taken together, our results on transcriptional regulation suggest that PML-II is a dose-dependent coactivator of E1A-13S-dependent transactivation. PML-II not only is able to modulate transcription from Ad5 promoters in combination with E1A-13S but also stimulates cellular gene expression by affecting key cellular coactivators such as p300.
PML-II positively regulates adenovirus progeny production in human cells.
To reveal the effect of PML-II on the production of adenoviral progeny, we determined virus growth in HepaRG cell lines in which endogenous PML was knocked down (HALP cells) and the short hairpin RNA (shRNA)-resistant PML-II isoform was reconstituted (HALP PML-II) (59). In accordance with recent reports, PML knockdown showed a modest effect on adenovirus replication compared to that in the parental cell line (80, 92). However, reconstitution of the single isoform PML-II enhanced virus yield severalfold compared to that in the parental cell line (Fig. 8). Consistent with the data obtained from the reporter assays, this strongly indicates that PML-II is a positive regulator of Ad5 replication in human cells. To further test the positive effect of PML isoforms, we measured early gene expression in an abortive infection in PML-null mouse embryonic fibroblasts (MEF) to confirm the data obtained from the reporter gene experiments. In this context, we performed time course experiments with PML−/− MEF compared to PML+/+ MEF cells. We observed a delay in the protein expression of E2A (Fig. 9A) as well as reduced E2A mRNA expression in MEF cells without PML (Fig. 9B).
Fig 9.

Ad5 E2A is delayed in an abortive infection in PML-null mouse embryonic fibroblasts. (A) Subconfluent parental MEF (PML+/+) or PML knockout MEF (PML−/−) cells were infected with wild-type virus H5pg4100 at a multiplicity of 50 FFU/cell. The cells were harvested 24 h p.i., and total RNA was extracted, reverse transcribed, and quantified by RT-PCR analysis using primers specific for E2A. The data were normalized to 18S rRNA levels. (B) Subconfluent parental MEF (PML+/+) or PML knockout MEF (PML−/−) cells were infected with wild-type virus H5pg4100 at a multiplicity of 50 FFU/cell. The cells were harvested at 24, 48, and 72 h. p.i., and total cell extracts were prepared. Steady-state expression levels of total cell lysates were detected using MAb 610 (α-E1A), MAb B6 (α-E2A), MAb clone 36.1-104 (α-PML), and MAb AC-15 (α-β-actin). Molecular masses in kDa are on the left, and corresponding proteins are on the right.
Transcriptional coactivation by PML does not depend on its functional SIM.
PML formation of PML NBs, its dynamic behavior, and its mode of function are based mainly on SUMOylation of the PML protein itself (24, 26, 93). The PML SIM is required to form PML NBs when exogenously expressed in PML−/− cells and enables PML to interact noncovalently with SUMOylated proteins, including PML itself (24). In line with these observations, we were interested in whether the PML SIM, which is necessary for interacting with other SUMOylated factors in the PML NBs, plays a role in transcriptional activation of Ad promoters by PML-II together with E1A-13S (Fig. 10A). Therefore, we generated a PML-II mutant with a nonfunctional SIM. Interestingly, this mutant stimulated E1A-13S-dependent transcription from the Ad5 E2 early promoter more efficiently than wild type PML-II (Fig. 10B). We also tested whether the PML-II SIM mutant affected E1A-13S binding properties. The PML-II SIM mutant and wild-type PML-II were expressed at equal amounts (Fig. 10C), and coimmunoprecipitation experiments showed no differences in binding capabilities between wild-type PML-II and the PML-II SIM mutant (Fig. 10D, lanes 3 and 5).
Fig 10.
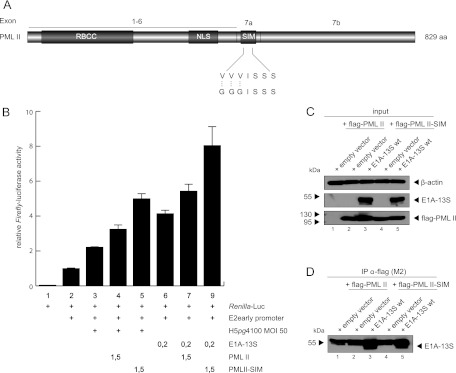
SIM of PML is not important for stimulating E1A-13S-dependent transcriptional activation. (A) Schematic representation of the PML-II isoform showing the SIM amino acid sequence. Shown is the newly constructed PML-II SIM construct with introduced mutations. (B) H1299 cells were transfected with 0.2 μg pRenilla-Luc, 0.5 μg pGL-E2-early promoter, 0.2 μg pE1A-13S, and 1.5 μg pPML-II and pPML-II SIM in the combinations indicated (+). At 24 h posttransfection, total cell extracts were prepared and luciferase activity was determined. The means and standard deviations for three independent experiments are presented. (C and D) Subconfluent H1299 cells were transfected with 3 μg of pE1A-13S and 5 μg of pPML-II or pPML-II SIM and harvested after 48 h, and total cell extracts were prepared. Coimmunoprecipitation assays were performed as described for Fig. 3. (C and D) Input levels in total cell lysates (C) and coprecipitated proteins (D). Molecular masses in kDa are on the left, while corresponding proteins are indicated on the right.
DISCUSSION
PML bodies are considered to mainly be part of an intracellular antiviral defense mechanism, which is activated by interferon upon viral infection (50, 51, 94, 95). Nevertheless, there is also evidence for a proviral role of PML bodies (49). Here, we show for the first time an adenoviral protein taking direct advantage of PML: E1A-13S apparently physically interacts with the PML-II isoform to positively affect E1A-dependent transcriptional activation. Since neither E1A nor PML possesses DNA binding activity, our data support the idea that PML activates transcription through stabilizing coactivator/transcription factor complexes. Based on our observations that PML-II enhances p300 coactivator function, it is also likely that PML acts as a platform for E1A-13S to interact with other coactivators or transcription factors.
PML isoforms that do not interact with E1A-13S in coimmunoprecipitation experiments do activate E1A-13S-dependent transcription to some extend (Fig. 6C and 7C). So far, it is known that PML isoforms form homo- and heterodimers via their RBCC/TRIM motif (21). As the reporter gene assays were not performed with PML knockout cells, there is endogenous PML-II present, which could heterodimerize with the other PML isoforms. On the other hand it is also possible that the interaction of E1A and PML-II is more stable than for the other PML isoforms and that those weak interactions could not be monitored in our coimmunoprecipitation assays due to use of a stringent lysis buffer.
It was already known that human papillomavirus (HPV) infection is positively influenced by PML expression (96). Human cytomegalovirus (HCMV) and herpes simplex virus 1 (HSV-1) genome transcription takes place exclusively at PML bodies (53, 56, 97). During lytic infection, HSV-1 recruits several cellular DNA repair factors from PML bodies into viral replication centers to support viral replication (98–100). Simian virus 40 (SV40) transcription and replication occur preferentially in the vicinity of PML NBs (101), and similar observations were made for human polyomaviruses (54, 102). These reports suggest that PML NBs might be a “host cell depot” for transcription or DNA repair factors.
We currently favor the model that PML NBs possess antiviral properties due to the large number of repressive proteins localizing to these compartments, such as Daxx or p53 (29, 34, 103). Therefore, during evolution adenoviruses have apparently acquired a mechanism to counteract this PML NB function, disrupting the integrity of PML NBs by expressing early viral E4orf3, which causes PML NB components to redistribute into so-called track-like structures (52, 72). Consequently, repressive factors would become accessible for targeting by further viral factors, such as Ad5 E1B-55K, that localize to PML tracks (52, 79). Recently, our group showed that the constitutive PML NB protein Daxx represses Ad5 gene expression and productive infection (80). This is counteracted by capsid protein pVI-dependent Daxx relocalization from the nuclear bodies at immediate-early times of infection, followed by E1B-55K-dependent proteasomal degradation (67, 80).
In addition to Daxx (81, 104), E1B-55K physically interacts with two different PML isoforms, PML-IV and -V (82). Since PML-IV is the only isoform described to recruit or modulate p53 (105), the interaction between E1B-55K and PML-IV might be a key regulatory event to modify or repress p53 functions. Together, these observations imply that adenoviral proteins specifically target and counteract repressive proteins from PML bodies, or take advantage of PML and associated factors as discussed above, in order to regulate cellular processes to benefit viral transcription/replication.
So far, E4orf3 was the only adenoviral protein known to physically interact with PML-II (76). Although our data show that E1A-13S interacts with PML-II independently of E4orf3, the fact that both adenoviral proteins target the same PML isoform provides evidence for cooperation between E1A, E4orf3, and PML-II during productive adenoviral infection, although we cannot observe an effect of adding E4orf3 expression vector to the luciferase assays shown in Fig. 6 and 7 (data not shown).
Our observation that mutation of the SIM in PML-II further enhanced E1A-dependent transactivation supports our model that Ad5 selectively benefits from PML NB functions. The PML SIM has been proposed to mediate noncovalent interactions with other PML isoforms and SUMOylated proteins (24, 106). Our data imply that the coactivator properties of PML do not necessarily depend on its localization to PML bodies. Thus, it is tempting to speculate that protein interactions within PML bodies, mediated by the SIM, may inhibit transcriptional stimulation by PML itself.
However, the enormous amount of PML-associated proteins may offer additional mechanisms to further understand E1A-13S/PML-II functional interaction. Nevertheless, our data support the idea that viral factors targeting PML not only represents a means of counteracting host mechanisms but also appear to be required and beneficial for productive viral infection.
ACKNOWLEDGMENTS
We thank Roger Everett, Philippe Gripon, Roger Grand, and Thomas Shenk for providing reagents, and we greatly appreciate critical comments and advice from Andrew Turnell, Thomas Sternsdorf, Hans Will, and Michael Nevels.
The Heinrich Pette Institute, Leibniz Institute for Experimental Virology, is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit (BMG). Part of this work was supported by grants from the Erich und Gertrud Roggenbuck Stiftung, the Horst Müggenburg Stiftung, and the Wilhelm Sander-Stiftung to S.S. and T.D.
Footnotes
Published ahead of print 7 November 2012
REFERENCES
- 1. Flint J, Shenk T. 1989. Adenovirus E1A protein paradigm viral transactivator. Annu. Rev. Genet. 23:141–161 [DOI] [PubMed] [Google Scholar]
- 2. Chow LT, Broker TR, Lewis JB. 1979. Complex splicing patterns of RNAs from the early regions of adenovirus-2. J. Mol. Biol. 134:265–303 [DOI] [PubMed] [Google Scholar]
- 3. Perricaudet M, Akusjarvi G, Virtanen A, Pettersson U. 1979. Structure of two spliced mRNAs from the transforming region of human subgroup C adenoviruses. Nature 281:694–696 [DOI] [PubMed] [Google Scholar]
- 4. Hiebert SW, Blake M, Azizkhan J, Nevins JR. 1991. Role of E2F transcription factor in E1A-mediated trans activation of cellular genes. J. Virol. 65:3547–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kovesdi I, Reichel R, Nevins JR. 1986. Identification of a cellular transcription factor involved in E1A trans-activation. Cell 45:219–228 [DOI] [PubMed] [Google Scholar]
- 6. Liu F, Green MR. 1990. A specific member of the ATF transcription factor family can mediate transcription activation by the adenovirus E1a protein. Cell 61:1217–1224 [DOI] [PubMed] [Google Scholar]
- 7. Nevins JR. 1990. Adenovirus E1A-dependent trans-activation of transcription. Semin. Cancer Biol. 1:59–68 [PubMed] [Google Scholar]
- 8. Stevens JL, Cantin GT, Wang G, Shevchenko A, Berk AJ. 2002. Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 296:755–758 [DOI] [PubMed] [Google Scholar]
- 9. Ascoli CA, Maul GG. 1991. Identification of a novel nuclear domain. J. Cell Biol. 112:785–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang KS, Stass SA, Chu DT, Deaven LL, Trujillo JM, Freireich EJ. 1992. Characterization of a fusion cDNA (RARA/myl) transcribed from the t(15;17) translocation breakpoint in acute promyelocytic leukemia. Mol. Cell. Biol. 12:800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 66:675–684 [DOI] [PubMed] [Google Scholar]
- 12. Dyck JA, Maul GG, Miller WH, Jr, Chen JD, Kakizuka A, Evans RM. 1994. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 76:333–343 [DOI] [PubMed] [Google Scholar]
- 13. Goddard AD, Borrow J, Solomon E. 1992. A previously uncharacterized gene, PML, is fused to the retinoic acid receptor alpha gene in acute promyelocytic leukaemia. Leukemia. 6(Suppl 3):117S–119S [PubMed] [Google Scholar]
- 14. Kakizuka A, Miller WH, Jr, Umesono K, Warrell RP, Jr, Frankel SR, Murty VV, Dmitrovsky E, Evans RM. 1991. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 66:663–674 [DOI] [PubMed] [Google Scholar]
- 15. Kastner P, Perez A, Lutz Y, Rochette-Egly C, Gaub MP, Durand B, Lanotte M, Berger R, Chambon P. 1992. Structure, localization and transcriptional properties of two classes of retinoic acid receptor alpha fusion proteins in acute promyelocytic leukemia (APL): structural similarities with a new family of oncoproteins. EMBO J. 11:629–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koken MH, Puvion-Dutilleul F, Guillemin MC, Viron A, Linares-Cruz G, Stuurman N, de Jong L, Szostecki C, Calvo F, Chomienne C. 1994. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 13:1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melnick A, Fruchtman S, Zelent A, Liu M, Huang Q, Boczkowska B, Calasanz M, Fernandez A, Licht JD, Najfeld V. 1999. Identification of novel chromosomal rearrangements in acute myelogenous leukemia involving loci on chromosome 2p23, 15q22 and 17q21. Leukemia 13:1534–1538 [DOI] [PubMed] [Google Scholar]
- 18. Melnick A, Licht JD. 1999. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93:3167–3215 [PubMed] [Google Scholar]
- 19. Pandolfi PP, Alcalay M, Fagioli M, Zangrilli D, Mencarelli A, Diverio D, Biondi A, Lo Coco F, Rambaldi A, Grignani F. 1992. Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J. 11:1397–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weis K, Rambaud S, Lavau C, Jansen J, Carvalho T, Carmo-Fonseca M, Lamond A, Dejean A. 1994. Retinoic acid regulates aberrant nuclear localization of PML-RAR alpha in acute promyelocytic leukemia cells. Cell 76:345–356 [DOI] [PubMed] [Google Scholar]
- 21. Jensen K, Shiels C, Freemont PS. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene 20:7223–7233 [DOI] [PubMed] [Google Scholar]
- 22. Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006–1016 [DOI] [PubMed] [Google Scholar]
- 23. Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF, III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. 2006. The mechanisms of PML-nuclear body formation. Mol. Cell 24:331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Negorev D, Maul GG. 2001. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 20:7234–7242 [DOI] [PubMed] [Google Scholar]
- 26. Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752 [PubMed] [Google Scholar]
- 27. Seeler JS, Dejean A. 2003. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 4:690–699 [DOI] [PubMed] [Google Scholar]
- 28. Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P, de The H. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 193:1361–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Leo C, Zhu J, Wu X, O'Neil J, Park EJ, Chen JD. 2000. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 20:1784–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhong S, Salomoni P, Pandolfi PP. 2000. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2:E85–E90 [DOI] [PubMed] [Google Scholar]
- 31. Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. 2002. Deconstructing PML-induced premature senescence. EMBO J. 21:3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. 2000. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 14:2015–2027 [PMC free article] [PubMed] [Google Scholar]
- 33. Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. 2002. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21:2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406:207–210 [DOI] [PubMed] [Google Scholar]
- 35. Salomoni P, Ferguson BJ, Wyllie AH, Rich T. 2008. New insights into the role of PML in tumour suppression. Cell Res. 18:622–640 [DOI] [PubMed] [Google Scholar]
- 36. Salomoni P, Pandolfi PP. 2002. The role of PML in tumor suppression. Cell 108:165–170 [DOI] [PubMed] [Google Scholar]
- 37. Bischof O, Kim SH, Irving J, Beresten S, Ellis NA, Campisi J. 2001. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell Biol. 153:367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carbone R, Pearson M, Minucci S, Pelicci PG. 2002. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 21:1633–1640 [DOI] [PubMed] [Google Scholar]
- 39. Hofmann TG, Will H. 2003. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ. 10:1290–1299 [DOI] [PubMed] [Google Scholar]
- 40. Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H. 2004. PML nuclear bodies and apoptosis. Oncogene 23:2819–2824 [DOI] [PubMed] [Google Scholar]
- 41. Torok D, Ching RW, Bazett-Jones DP. 2009. PML nuclear bodies as sites of epigenetic regulation. Front. Biosci. 14:1325–1336 [DOI] [PubMed] [Google Scholar]
- 42. Ahn JH, Brignole EJ, III, Hayward GS. 1998. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol. Cell. Biol. 18:4899–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guiochon-Mantel A, Savouret JF, Quignon F, Delabre K, Milgrom E, De The H. 1995. Effect of PML and PML-RAR on the transactivation properties and subcellular distribution of steroid hormone receptors. Mol. Endocrinol. 9:1791–1803 [DOI] [PubMed] [Google Scholar]
- 44. Vallian S, Gaken JA, Gingold EB, Kouzarides T, Chang KS, Farzaneh F. 1998. Modulation of Fos-mediated AP-1 transcription by the promyelocytic leukemia protein. Oncogene 16:2843–2853 [DOI] [PubMed] [Google Scholar]
- 45. Alcalay M, Tomassoni L, Colombo E, Stoldt S, Grignani F, Fagioli M, Szekely L, Helin K, Pelicci PG. 1998. The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol. Cell. Biol. 18:1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vallian S, Chin KV, Chang KS. 1998. The promyelocytic leukemia protein interacts with Sp1 and inhibits its transactivation of the epidermal growth factor receptor promoter. Mol. Cell. Biol. 18:7147–7156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Doucas V, Tini M, Egan DA, Evans RM. 1999. Modulation of CREB binding protein function by the promyelocytic (PML) oncoprotein suggests a role for nuclear bodies in hormone signaling. Proc. Natl. Acad. Sci. U. S. A. 96:2627–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhong S, Delva L, Rachez C, Cenciarelli C, Gandini D, Zhang H, Kalantry S, Freedman LP, Pandolfi PP. 1999. A RA-dependent, tumour-growth suppressive transcription complex is the target of the PML-RARalpha and T18 oncoproteins. Nat. Genet. 23:287–295 [DOI] [PubMed] [Google Scholar]
- 49. Everett RD. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20:7266–7273 [DOI] [PubMed] [Google Scholar]
- 50. Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 51. Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207–2221 [DOI] [PubMed] [Google Scholar]
- 52. Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10:196–207 [DOI] [PubMed] [Google Scholar]
- 53. Ishov AM, Maul GG. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134:815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jul-Larsen A, Visted T, Karlsen BO, Rinaldo CH, Bjerkvig R, Lonning PE, Boe SO. 2004. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp. Cell Res. 298:58–73 [DOI] [PubMed] [Google Scholar]
- 55. Maul GG. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20:660–667 [DOI] [PubMed] [Google Scholar]
- 56. Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75 [DOI] [PubMed] [Google Scholar]
- 57. Sourvinos G, Everett RD. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 21:4989–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655–15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, Everett RD. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 124:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mitsudomi T, Oyama T, Gazdar AF, Minna JD, Okabayashi K, Shirakusa T. 1992. Mutations of ras and p53 genes in human non-small cell lung cancer cell lines and their clinical significance. Nihon Geka Gakkai Zasshi 93:944–947 [PubMed] [Google Scholar]
- 61. Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP. 1998. Role of PML in cell growth and the retinoic acid pathway. Science 279:1547–1551 [DOI] [PubMed] [Google Scholar]
- 62. Muller D, Schreiner S, Schmid M, Groitl P, Winkler M, Dobner T. 2012. Functional cooperation between human adenovirus type 5 early region 4, open reading frame 6 protein, and cellular homeobox protein HoxB7. J. Virol. 86:8296–8308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Groitl P, Dobner T. 2007. Construction of adenovirus type 5 early region 1 and 4 virus mutants. Methods Mol. Med. 130:29–39 [DOI] [PubMed] [Google Scholar]
- 64. Winberg G, Shenk T. 1984. Dissection of overlapping functions within the adenovirus type 5 E1A gene. EMBO J. 3:1907–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, Seed RI, Catnaigh PO, McCabe CJ, Stewart GS, Dobner T, Grand RJ, Martin A, Turnell AS. 2012. Adenovirus E4orf3 targets transcriptional intermediary factor 1gamma for proteasome-dependent degradation during infection. J. Virol. 86:3167–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kindsmuller K, Schreiner S, Leinenkugel F, Groitl P, Kremmer E, Dobner T. 2009. A 49-kilodalton isoform of the adenovirus type 5 early region 1B 55-kilodalton protein is sufficient to support virus replication. J. Virol. 83:9045–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schreiner S, Martinez R, Groitl P, Rayne F, Vaillant R, Wimmer P, Bossis G, Sternsdorf T, Ruszsics Z, Dobner T, Wodrich H. 2012. Transcriptional activation of the adenoviral genome is mediated by capsid protein. PLoS Pathog. 8 (2):e1002549 doi:10.1371/journal.ppat.1002549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Harlow E, Franza BR, Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 55:533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484 [DOI] [PubMed] [Google Scholar]
- 70. Wimmer P, Blanchette P, Schreiner S, Ching W, Groitl P, Berscheminski J, Branton PE, Will H, Dobner T. 21 May 2012. Cross-talk between phosphorylation and SUMOylation regulates transforming activities of an adenoviral oncoprotein. Oncogene, in press. doi:10.1038/onc.2012.187 [DOI] [PubMed] [Google Scholar]
- 71. Endter C, Hartl B, Spruss T, Hauber J, Dobner T. 2005. Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24:55–64 [DOI] [PubMed] [Google Scholar]
- 72. Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjarvi G, Carmo-Fonseca M, Dejean A. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shenk T, Flint J. 1991. Transcriptional and transforming activities of the adenovirus E1A proteins. Adv. Cancer Res. 57:47–85 [DOI] [PubMed] [Google Scholar]
- 74. Eskiw CH, Dellaire G, Mymryk JS, Bazett-Jones DP. 2003. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 116:4455–4466 [DOI] [PubMed] [Google Scholar]
- 75. Nardozza TA, Quigley MM, Getzenberg RH. 1996. Association of transcription factors with the nuclear matrix. J. Cell. Biochem. 61:467–477 [DOI] [PubMed] [Google Scholar]
- 76. Hoppe A, Beech SJ, Dimmock J, Leppard KN. 2006. Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J. Virol. 80:3042–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Leppard KN, Emmott E, Cortese MS, Rich T. 2009. Adenovirus type 5 E4 Orf3 protein targets promyelocytic leukaemia (PML) protein nuclear domains for disruption via a sequence in PML isoform II that is predicted as a protein interaction site by bioinformatic analysis. J. Gen. Virol. 90:95–104 [DOI] [PubMed] [Google Scholar]
- 78. Puvion-Dutilleul F, Chelbi-Alix MK, Koken M, Quignon F, Puvion E, de The H. 1995. Adenovirus infection induces rearrangements in the intranuclear distribution of the nuclear body-associated PML protein. Exp. Cell Res. 218:9–16 [DOI] [PubMed] [Google Scholar]
- 79. Leppard KN, Everett RD. 1999. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 80:997–1008 [DOI] [PubMed] [Google Scholar]
- 80. Schreiner S, Wimmer P, Sirma H, Everett RD, Blanchette P, Groitl P, Dobner T. 2010. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 84:7029–7038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sieber T, Dobner T. 2007. Adenovirus type 5 early region 1B 156R protein promotes cell transformation independently of repression of p53-stimulated transcription. J. Virol. 81:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wimmer P, Schreiner S, Everett RD, Sirma H, Groitl P, Dobner T. 2010. SUMO modification of E1B-55K oncoprotein regulates isoform-specific binding to the tumour suppressor protein PML. Oncogene 29:5511–5522 [DOI] [PubMed] [Google Scholar]
- 83. Zhao LY, Colosimo AL, Liu Y, Wan Y, Liao D. 2003. Adenovirus E1B 55-kilodalton oncoprotein binds to Daxx and eliminates enhancement of p53-dependent transcription by Daxx. J. Virol. 77:11809–11821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu F, Green MR. 1994. Promoter targeting by adenovirus E1a through interaction with different cellular DNA-binding domains. Nature 368:520–525 [DOI] [PubMed] [Google Scholar]
- 85. Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. 2008. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J. Virol. 82:7252–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Webster LC, Ricciardi RP. 1991. trans-dominant mutants of E1A provide genetic evidence that the zinc finger of the trans-activating domain binds a transcription factor. Mol. Cell. Biol. 11:4287–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Regad T, Chelbi-Alix MK. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274–7286 [DOI] [PubMed] [Google Scholar]
- 88. Pelka P, Ablack JN, Torchia J, Turnell AS, Grand RJ, Mymryk JS. 2009. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 37:1095–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goodman RH, Smolik S. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 14:1553–1577 [PubMed] [Google Scholar]
- 90. LaMorte VJ, Dyck JA, Ochs RL, Evans RM. 1998. Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc. Natl. Acad. Sci. U. S. A. 95:4991–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. von Mikecz A, Zhang S, Montminy M, Tan EM, Hemmerich P. 2000. CREB-binding protein (CBP)/p300 and RNA polymerase II colocalize in transcriptionally active domains in the nucleus. J. Cell Biol. 150:265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ullman AJ, Hearing P. 2008. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J. Virol. 82:7325–7335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, Solomon E, de The H, Hay RT, Freemont PS. 1999. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J. Cell Sci. 112:381–393 [DOI] [PubMed] [Google Scholar]
- 94. Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31:145–158 [DOI] [PubMed] [Google Scholar]
- 95. Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, Grosveld F, Pandolfi PP, Pelicci PG, Dejean A. 1995. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 11:871–876 [PubMed] [Google Scholar]
- 96. Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. U. S. A. 101:14252–14257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ishov AM, Stenberg RM, Maul GG. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 138:5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 102:5844–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Weitzman MD, Carson CT, Schwartz RA, Lilley CE. 2004. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst.) 3:1165–1173 [DOI] [PubMed] [Google Scholar]
- 100. Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 78:4783–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tang Q, Bell P, Tegtmeyer P, Maul GG. 2000. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J. Virol. 74:9694–9700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shishido-Hara Y, Higuchi K, Ohara S, Duyckaerts C, Hauw JJ, Uchihara T. 2008. Promyelocytic leukemia nuclear bodies provide a scaffold for human polyomavirus JC replication and are disrupted after development of viral inclusions in progressive multifocal leukoencephalopathy. J. Neuropathol Exp. Neurol. 67:299–308 [DOI] [PubMed] [Google Scholar]
- 103. Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. 2000. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2:730–736 [DOI] [PubMed] [Google Scholar]
- 104. Schreiner S, Wimmer P, Groitl P, Chen SY, Blanchette P, Branton PE, Dobner T. 2011. Adenovirus type 5 early region 1B 55K oncoprotein-dependent degradation of cellular factor Daxx is required for efficient transformation of primary rodent cells. J. Virol. 85:8752–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19:6185–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin DY, Huang YS, Jeng JC, Kuo HY, Chang CC, Chao TT, Ho CC, Chen YC, Lin TP, Fang HI, Hung CC, Suen CS, Hwang MJ, Chang KS, Maul GG, Shih HM. 2006. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24:341–354 [DOI] [PubMed] [Google Scholar]