

Summary
The nuclear factor-κB (NF-κB) pathway is a critical regulator of innate and adaptive immunity. Noncanonical K63-linked polyubiquitination plays a key regulatory role in NF-κB signaling pathways by functioning as a scaffold to recruit kinase complexes containing ubiquitin-binding domains. Ubiquitination is balanced by deubiquitinases that cleave polyubiquitin chains and oppose the function of E3 ubiquitin ligases. Deubiquitinases therefore play an important role in the termination of NF-κB signaling and the resolution of inflammation. In this review, we focus on NF-κB regulation by deubiquitinases with an emphasis on A20 and CYLD. Deubiquitinases and the ubiquitin/proteasome components that regulate NF-κB may serve as novel therapeutic targets for inflammatory diseases and cancer.
Keywords: deubiquitinases, NF-κB, A20, CYLD, inflammation
Introduction
The nuclear factor κB (NF-κB) transcription factor has been intensely studied since its discovery by Sen and Baltimore in 1986 (1). Although NF-κB was first described as a factor binding to the κ light chain enhancer in B cells, its critical importance in not only adaptive immunity but also innate immune responses have been well established. Furthermore, NF-κB is also a critical regulator of anti-apoptotic genes and cell survival. The importance of NF-κB is not limited to the immune system, since it also plays diverse roles regulating development, metabolism and key aspects of the central nervous system (Reviewed in 2). Given these roles, it is not surprising that dysregulation of NF-κB plays an underlying role in chronic inflammation, septic shock, autoimmunity, and cancer (Reviewed in 3).
NF-κB is composed of homo- and heterodimeric protein complexes containing RelA, c-Rel, NF-κB1, NF-κB2 and RelB (Reviewed in 4). Each of the NF-κB proteins contains an approximately 300 amino acid domain located in the amino-terminus known as the Rel homology domain (RHD). The RHD is essential for DNA binding, nuclear localization, and dimerization of NF-κB proteins (Reviewed in Ghosh et al., this volume). Furthermore, NF-κB1 and NF-κB2 are precursor proteins that are processed by the proteasome to yield the mature subunits, p50 and p52, respectively. NF-κB is expressed in virtually all cell types and is activated by diverse stimuli ranging from stress, radiation, cytokines, bacterial and viral products, and antigen. NF-κB complexes are sequestered in the cytoplasm in unstimulated cells by inhibitory IκB proteins (IκBα plays a predominant role) that all share a series of ankyrin repeat domains (Reviewed in 5 and Hinz et al., this volume). NF-κB activating stimuli trigger the degradation of IκBα with the concomitant nuclear translocation of NF-κB where it activates target genes. The degradation of IκBα is preceded by its site-specific phosphorylation on two amino-terminal serine residues by a multi-subunit kinase complex IKK [inhibitor of NF-κB (IκB) kinase] consisting of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ or NEMO) (Reviewed in 6 and Liu et al., this volume). The multisubunit SCFβTRCP (Skp, Cullin, F-box containing complex, beta-transducin repeat containing protein) E3 ubiquitin ligase complex recognizes phosphorylated IκBα and ubiquitinates IκBα leading to its degradation by the proteasome (7, reviewed in Kanarek & Ben-Neriah, this volume). NF-κB is tightly regulated by multiple checks and balances in order to prevent persistent NF-κB activation that could have deleterious effects on the host. Indeed, IκBα is an NF-κB target gene and inhibits NF-κB activation in a negative feedback loop (8). Numerous other negative regulatory mechanisms, both in the cytoplasm and nucleus, ensure that NF-κB activation is transient.
The signaling events upstream of IKK have been largely elucidated for NF-κB activators such as the cytokines tumor necrosis factor (TNF) and interleukin-1β and bacterial lipopolysaccharide (LPS). A pervasive theme is the importance of ubiquitin in the activation of IKK and NF-κB (Reviewed in Chen, this volume). Ubiquitin is a 76 amino acid polypeptide that is covalently attached to lysine residues on protein substrates (9). Although ubiquitination is best known as a trigger for protein degradation by the proteasome, emerging studies in the past decade have revealed multi-faceted roles of ubiquitin in receptor trafficking, signal transduction, and the DNA damage response (10). Ubiquitination requires the coordinated activities of three classes of enzymes: E1 ubiquitin activating enzymes, E2 ubiquitin conjugating enzymes, and E3 ubiquitin ligases (Reviewed in 11). The E1 enzyme activates ubiquitin in an adenosine triphosphate (ATP)-dependent manner, forming a thioester linkage between the catalytic cysteine of the E1 and the carboxy terminal glycine of ubiquitin. The E1 transfers the activated ubiquitin to the active-site cysteine residue of an E2 ubiquitin conjugating enzyme forming an E2-ubiquitin thioester. Finally, an E3 ubiquitin ligase conjugates ubiquitin to a protein substrate by linking the C-terminal glycine of ubiquitin to the ε-amino group of a lysine residue. The E3 ligase provides specificity because it directly contacts the substrate, however the mechanisms of ubiquitin transfer differ based on the class of E3 enzyme. For example, HECT (homologous to E6-associated protein C-terminus) domain E3 ligases form a thioester intermediate with ubiquitin, and directly transfer the ubiquitin to a substrate (12). Conversely, RING (really interesting new gene) domain, U-box, and F-box E3 ligases lack the catalytic cysteine residue needed to form the thioester intermediate and instead function as scaffolds and transfer ubiquitin to substrates in conjunction with specific E2 enzymes (13). The human proteome consists of 2 E1 enzymes, ~40-50 E2 enzymes, and over 600 E3 enzymes thus highlighting the complexity and versatility of ubiquitination.
Proteins may be modified by ubiquitin in a variety of ways. Monoubiquitination occurs when a single ubiquitin molecule is conjugated to a lysine residue. Ubiquitin itself has seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63) that may serve as acceptor sites for ubiquitination to form polyubiquitin chains (Reviewed in 14). Alternatively, the amino-terminus of ubiquitin can be used to form linear polyubiquitin chains in a head-to-tail linkage. An E3 ligase complex known as LUBAC (linear ubiquitin chain assembly complex) containing two RING-type E3 ligases HOIL1 and HOIP specifically assembles linear polyubiquitin chains that play an important role in NF-κB activation by multiple stimuli (15). SHARPIN was recently identified as a third component of the LUBAC complex that is essential for linear ubiquitination of the IKK subunit NEMO and NF-κB activation (16-18). Although the functional significance of linear polyubiquitin chains is just now coming into focus, the importance of K48-linked and K63-linked polyubiquitin chains for a variety of biological processes are well established. K48-linked polyubiquitin chains are recognized by the 26S proteasome and generally trigger the degradation of the protein substrate (11). K63-linked polyubiquitin chains typically do not trigger protein degradation but instead regulate nonproteolytic functions including protein trafficking, kinase activation, the DNA damage response and signal transduction (Reviewed in 19). Other types of polyubiquitin chains are poorly understood although K11, K27, and K29-linked polyubiquitin chains have each been linked to protein degradation (20-22). The topology of the different polyubiquitin chains likely dictates the distinct functional outcomes.
The type of ubiquitin linkage formed during a ubiquitination reaction is dependent on the protein transferring the ubiquitin to the substrate (E2 or HECT E3), although RING E3 ligases may also play a role in linkage selection (10). For HECT domain E3 ligases, the chain type specificity has been mapped to the last 60 amino acids of the C lobe of the HECT domain (23). The E2 enzyme Ubc13, together with Uev1a, is specific for the synthesis of K63-linked polyubiquitin chains (24, 25). Other E2s such as UbcH5 are more promiscuous and may participate in the formation of multiple types of polyubiquitin chains (26, 27).
TNF binding to the TNF receptor (TNFR) leads to receptor trimerization and the recruitment of adapter molecules TRADD (TNFR-associated death domain) and RIP1(receptor-interacting protein 1) and the E3 ubiquitin ligases TRAF2 (TNFR-associated factor 2), TRAF5, cIAP1/2 (cellular inhibitor of apoptosis 1/2), and LUBAC (28, 29). Mass-spectrometric analysis of the TNFR1 complex has revealed multiple polyubiquitin linkages conjugated to proteins in the receptor complex including K48, K63, K11, and linear chains (18). In response to TNF stimulation, RIP1 undergoes K63-linked polyubiquitination at lysine 377 by cIAP1/2 and is also conjugated with linear ubiquitin chains (18, 30, 31). Although the precise role of TRAF2 in TNFR signaling is unclear, a recent study showed that the lipid sphingosine-1-phosphate (S1P) directly activates TRAF2 ligase activity (32). The sphingosine kinase 1 (Sphk1) and S1P are both essential for RIP1 K63-linked polyubiquitination and NF-κB activation (32). It has also been reported that TRAF2 is not essential for K63-linked polyubiquitination of RIP1 but rather that TRAF2 and TRAF5 may facilitate cIAP1/2-mediated RIP1 ubiquitination (33-35). RIP1 ubiquitination likely functions as a molecular scaffold to recruit proteins bearing ubiquitin-binding domains (UBDs). For example, the TAK1 kinase complex consisting of TAK1 and the ubiquitin binding adaptors TAB2 and TAB3 are recruited to RIP1 via TAB2/TAB3 polyubiquitin binding (36). Similarly, the IKK complex is recruited to RIP1 via NEMO binding to RIP1 polyubiquitin chains mediated through its UBD domains (31). Assembly of the ubiquitin-directed membrane signaling complex permits TAK1 to phosphorylate the IKKβ activation loop leading to NF-κB activation (37). Linear ubiquitination of NEMO by LUBAC enhances the interaction of NEMO with the TNFR signaling complex, thus stabilizing the complex and is required for efficient NF-κB activation (28).
NF-κB activation occurs by a similar mechanism in IL-1R/TLR4 signaling. The adapter molecule myeloid differentiation factor 88 (Myd88) recruits kinases of the IL-1 receptor-associated kinase (IRAK) family (IRAK1 and IRAK4) to the receptor complex, which triggers the oligomerization and activation of the E3 ubiquitin ligase TRAF6 (38). TRAF6 is dependent on the dimeric E2 enzyme complex Ubc13/Uev1a for activation and downstream signaling (25). TRAF6 undergoes K63-linked autoubiquitination, although it is unclear what role autoubiquitination plays in NF-κB signaling (39). Regardless, TRAF6 is essential for downstream TAK1 [TGFβ (transforming growth factor β)-activated protein kinase], IKK, MAPK (mitogen-associated protein kinase), and NF-κB activation (40).
Deubiquitinases (DUBs) are proteases that cleave ubiquitin from target proteins and therefore oppose the function of E3 ligases. There are approximately 100 DUBs encoded in the human genome (41), subdivided into five families based on specific structural domains: ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Josephins, and JAB1/MPN/MOV34 metalloenzymes (JAMMs) (42). The UCH, USP, OUT, and Josephin DUBs are cysteine proteases, whereas JAMMs are zinc metalloproteases (42, 43). The USPs comprise the largest subfamily of DUBs, containing more than 50 members (44). A common feature of DUBs are the presence of motifs predicted to interact with ubiquitin, including the zinc finger ubiquitin-specific protease (ZnF-UBP) domain, the ubiquitin-interacting motif (UIM) and the ubiquitin-associated domain (UBA) (45). It is likely that these domains regulate the recognition and recruitment of ubiquitinated substrates and therefore provide specificity.
DUBs play important functional roles that can be broadly categorized in three categories. Ubiquitin is encoded by four genes (UBC, UBB, UBA52, and UBA80) and is made as a precursor protein consisting of multiple ubiquitin proteins or ubiquitin fused to the amino terminus of ribosomal proteins (46). DUBs play an essential role in cleaving these precursor proteins to generate free ubiquitin (45). Second, DUBs cleave ubiquitin from proteins that are modified post-translationally to either rescue protein degradation by removal of K48-linked chains or modulate signaling or trafficking by removal of K63-linked chains (44). The cleaved ubiquitin molecules may be recycled for additional ubiquitination events, thus contributing to ubiquitin homeostasis (44). Third, DUBs may edit ubiquitin chains either by modifying the number of ubiquitins or by altering the type of linkage (i.e. K63 to K48) (47). The A20 deubiquitinase is an example of a ubiquitin-editing enzyme that modifies ubiquitin linkages and is a major focus of this review article.
Discovery of A20 and its mechanisms of action
A20 was first identified in 1990 as a gene rapidly induced by TNF stimulation in human umbilical vein endothelial cells and was also known as TNFα-induced protein 3 (TNFAIP3) (48). Subsequent studies identified two NF-κB binding sites in the promoter as the critical TNF-responsive cis elements (49). Shortly thereafter, additional NF-κB activating stimuli such as the viral oncoproteins LMP1 (encoded by Epstein Barr virus) and Tax (encoded by the human T-cell leukemia virus 1), and the phorbol ester phorbol myristate acetate (PMA) were found to induce expression of A20 (50). Although A20 was found to contain several repeats of a Cys2/Cys2 zinc finger motif, there was little clue regarding its biological function. The first indication for a function of A20 arose upon analysis of A20 expression in different isolates of the breast cancer cell line MCF-7 that were either sensitive or resistant to TNF killing. A20 was markedly upregulated in the cells resistant to TNF-induced cell death (51). Indeed, transfection of A20 into cells provided protection from TNF killing suggesting that A20 was a bona fide inhibitor of TNF-induced cytotoxicity (51). Thus, the first ascribed function of A20 was as an inhibitor of cell death.
In 1996, several groups showed that overexpression of A20 inhibited NF-κB activation in response to TNF or IL-1 stimulation (52-54). In one of these studies, a yeast two-hybrid screen identified A20 as an interacting protein of TRAF2, a key signaling mediator of the TNF signaling pathway (53). The N-terminus of A20 was shown to interact with TRAF2 whereas the C-terminal zinc fingers were critical for NF-κB inhibition (53). A20 also abrogated NF-κB activation in response to TRAF2 overexpression, suggesting that TRAF2 was the target of A20 in the TNFR pathway. The adapter molecule RIP1 was also a potential target for A20, because A20 inhibited RIP1-induced activation of NF-κB (55). A20 also inhibited IL-1 signaling at the level of TRAF6 and an interaction was also observed between A20 and TRAF6 (56). Collectively, these studies, while based on overexpression experiments, identified key targets for A20 in the TNFR and IL-1R pathways.
A breakthrough in our understanding of the physiological function of A20 came in 2000 when the Ma group reported the phenotype of A20-deficient mice (57). Mice lacking A20 succumbed shortly after birth due to multi-organ tissue inflammation and cachexia (57). A20-deficient mice were also exquisitely sensitive to inflammatory stimuli and rapidly perished when exposed to sub-lethal doses of TNF, IL-1, or LPS (57). The spontaneous inflammation and perinatal death was likely due to uncontrolled activation of NF-κB which was persistently activated in TNF-stimulated A20-deficient MEFs. This study clearly established that A20 was a critical negative feedback regulator of NF-κB essential for homeostasis of the immune system.
In the early 2000s, the mechanism of how A20 inhibited NF-κB was still poorly understood. However, in 2004 two independent reports (47, 58) demonstrated that A20 contains a DUB domain from the ovarian tumor (OTU) family in its N-terminus. Remarkably, A20 was found to inhibit NF-κB via its DUB domain by hydrolyzing K63-linked polyubiquitin chains on key NF-κB signaling molecules (47). Furthermore, one of the C-terminal zinc finger domains (ZnF4) was found to harbor intrinsic E3 ligase activity (47). Rabex-5 also contains an A20-like ZnF with E3 ligase activity suggesting a new class of E3 ligases (59, 60). Therefore, A20 is a novel ubiquitin-editing enzyme with both DUB and E3 ligase activity. Although paradoxical that a protein would contain domains with opposing activities, it is likely that the DUB and E3 ligase activities of A20 are tightly regulated and function in a cooperative and sequential manner. The ubiquitin-editing function of A20 has been mainly described in the TNF signaling pathway. Upon TNF stimulation, A20 expression is induced by NF-κB, A20 is recruited to RIP1 and cleaves K63-linked polyubiquitin chains on RIP1 (47). At later times after TNF stimulation (i.e., 3-6 h), A20 conjugates K48-linked polyubiquitin chains on RIP1 to trigger its degradation by the proteasome (47). Therefore, A20 inactivates RIP1 via sequential deubiquitinase and E3 ligase activities (Fig. 1). A20 may also target substrates for degradation via the lysosomal pathway, since A20 localizes to lysosomes and triggers the degradation of TRAF2 in lysosomes (61, 62).
Fig. 1. Mechanisms of A20 inhibition of NF-κB.
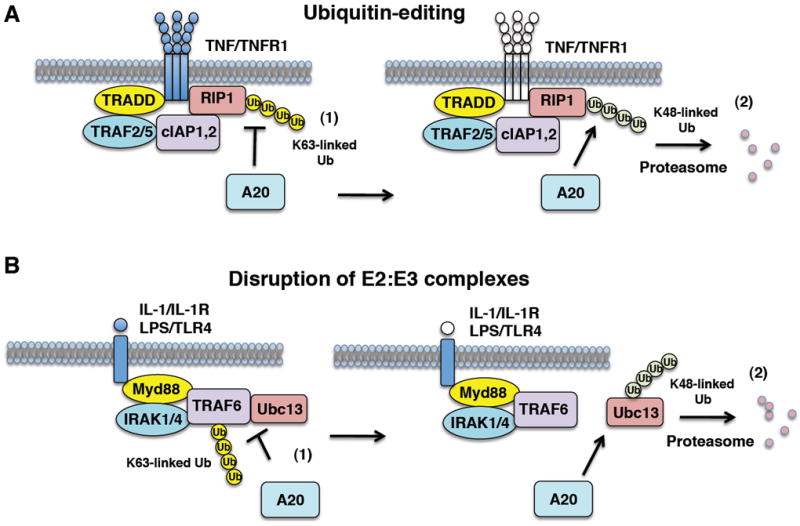
(A) The ubiquitin-editing function of A20. In response to TNF stimulation, A20 expression is induced and inhibits NF-κB in a negative feedback loop in a two-step manner. (1) A20 first hydrolyzes K63-linked polyubiquitin chains on RIP1 in an OTU-dependent manner to inhibit IKK and NF-κB signaling. (2) A20 then conjugates K48-linked polyubiquitin chains onto RIP1 to trigger its proteasomal degradation. (B) Disruption of E2:E3 ubiquitin enzyme complexes by A20. The E3 ligase TRAF6 inducibly interacts with the E2 enzymes Ubc13 and UbcH5c upon IL-1R/TLR4 stimulation. (1) A20 interacts with Ubc13, UbcH5c and TRAF6 and disrupts the binding between TRAF6 and the E2 enzymes. (2) A20 then conjugates K48-linked polyubiquitin chains on Ubc13 (and UbcH5c) to trigger its proteasomal degradation.
The functional effects of A20 on TRAF6 activation have been described in a number of studies. Overexpression of A20, but not a catalytically inactive DUB mutant, inhibits TRAF6 ubiquitination, and genetic ablation of A20 triggers persistent LPS-induced TRAF6 ubiquitination (63). Although A20 clearly inhibits K63-linked TRAF6 ubiquitination, it does not cause its degradation as observed with RIP1, suggesting a distinct mechanism of inhibition. Indeed, A20 was recently shown to downregulate the activity of TRAF6 and other E3 ligases including TRAF2 by a novel mechanism involving the disruption of E2:E3 ubiquitin enzyme complexes (64). In the IL-1R/TLR4 pathway, A20 disrupts the IL-1-inducible binding of TRAF6 with the E2 enzymes Ubc13 and UbcH5c (Figure 1) (64). Similarly, A20 antagonizes TNF-dependent interactions between TRAF2, cIAP1/2, and Ubc13 in the TNFR pathway (64). A20 then targets Ubc13 and UbcH5c for proteasome-dependent degradation (64). The catalytic cysteine residue (C103) of A20 in the OTU domain as well as zinc finger domain 4 (ZnF4) are critical for A20 to disrupt E2:E3 complexes and trigger E2 degradation (64). Interestingly, the OTUB1 deubiquitinase uses a similar mechanism to inhibit DNA damage-induced chromatin ubiquitination (65). OTUB1 interacts with and inhibits Ubc13 independently of its DUB domain (65).
Structural studies of the A20 DUB domain and ZnF4 have provided important new mechanistic insight regarding A20 ubiquitin-editing function. A20 DUB activity does not appear to be specific for K63-linked polyubiquitin chains, but rather preferentially deubiquitinates K48-linked chains in vitro (66). However in vivo, A20 deubiquitinates K63-linked polyubiquitin chains indicating that other factors may determine the specificity of A20 for K63-linked polyubiquitin chains. Indeed, as discussed later, A20 functions in the context of a multi-protein ubiquitin-editing complex. The catalytic mechanism of the A20 OTU domain is unique compared to other cysteine proteases, however A20 shares a minimal catalytic triad with a conserved cysteine (Cys103), histidine (His256,) and possibly aspartic acid (Asp70) (67, 68). The DUB domain also contains two highly conserved surface sites adjacent to the active site that likely comprise the ubiquitin binding elements (67). Surprisingly, instead of disassembling K63-linked polyubiquitin chains from a substrate in a processive manner, A20 cleaves at the junction of the substrate and the polyubiquitin chain (68).
A20 ZnF4 is essential for NF-κB inhibition and also is responsible for the E3 ligase activity of A20 (47, 64). The crystal structure of A20 ZnF4 together with ubiquitin and UbcH5a has revealed that A20 ZnF4 does not directly bind to E2 enzymes or substrates, but rather interacts with monoubiquitin and K63-linked polyubiquitin chains (69). Distinct regions of A20, predominantly ZnF1 and surrounding regions mediate binding to RIP1, whereas ZnF5-7 interact with E2 enzymes (69). Thus, distinct regions of A20 contribute to its ubiquitin-editing function and downregulation of NF-κB signaling.
A20 regulation of innate and adaptive immunity
In most cell types, A20 is inducible by proinflammatory cytokines or mitogens and inhibits NF-κB in a negative feedback loop. However, T lymphocytes express high basal levels of A20 that are diminished upon stimulation with T-cell receptor agonists (70), suggesting that A20 is regulated differently in lymphocytes compared to other cell types. The distinct regulation of A20 expression in different tissues may occur, in part, due to post-transcriptional control by microRNAs. A recent study identified the microRNA miR-29c as a negative regulator of A20 expression in hepatocytes (71). Interestingly, NF-κB directly binds to the miR-29c promoter and suppresses its transcription which presumably relieves the negative control of A20 expression (72). In addition, A20 is regulated at the post-translational level by phosphorylation of Ser381 by IKKβ (73). Although the mechanism is still unclear, A20 phosphorylation enhances its ability to suppress NF-κB signaling.
The spontaneous inflammation in A20-deficient mice is not triggered by uncontrolled TNF signaling since crossing A20-deficient mice with mice deficient in TNFα or the TNFR1 does not rescue the spontaneous inflammation and premature lethality (63). Furthermore, the phenotype is unaltered on a Rag1−/− background, indicating that lymphocytes are dispensable for the spontaneous inflammation (57). However, genetic deletion of the TLR adapter molecule Myd88 rescued A20-deficient mice from uncontrolled inflammation and lethality, suggesting that Myd88-dependent TLR signaling pathways were dysregulated (74). Indeed, it was revealed that TLR signaling was constitutively activated in A20-deficient mice due to inappropriate and exaggerated responses to commensal intestinal bacteria (74). Taken together, these findings underscore the importance of A20 in controlling immune homeostasis, particularly in the intestine.
Due to the limitations of studying conventional A20-deficient mice as a result of the premature lethality, A20 conditional knockout mice have provided important new insight in the role of A20 in innate and adaptive immunity as well as other biological functions. Deletion of A20 in B lymphocytes, dendritic cells (DCs), or myeloid cells does not recapitulate the severe inflammatory phenotype observed in A20-deficient mice, although autoimmunity does occur in each case. Genetic ablation of A20 in B cells triggers the production of autoantibodies and the development of autoimmune disease resembling systemic lupus erythematosus (SLE) (75-77). Interestingly, polymorphisms in the human A20 gene locus have been linked to development of SLE (78). B cells lacking A20 exhibit increased NF-κB activation and proliferation in response to anti-CD40, LPS, and CpG DNA (75). The increased cytokine production by A20-deficient B cells causes a sustained inflammatory response which leads to a breakdown in B-cell tolerance together with an expansion of myeloid cells and effector T cells (75, 76). Since these mice do not develop lymphomas, it is clear that loss of A20 alone is not sufficient for the development of B-cell lymphomas.
Deletion of A20 in DCs causes a more severe form of systemic autoimmunity also similar to SLE characterized by autoantibody production, nephritis, splenomegaly, and lymphadenopathy (79). DCs lacking A20 spontaneously undergo maturation and are hyperresponsive to TLR ligands and cytokines (79). These hyperactive DCs directly stimulate B cells and also provoke the activation of T cells (79). Thus, loss of A20 in DCs leads to autoimmunity and spontaneous activation of both B and T cells. These results are consistent with an earlier siRNA study showing that knockdown of A20 in conventional DCs leads to enhanced maturation, cytokine production and antigen presentation (80).
Myeloid-specific deletion of A20 does not result in an SLE-like disease but rather leads to an autoimmune disease resembling rheumatoid arthritis. Although serum levels of inflammatory cytokines TNF, IL-1β and IL-6 were elevated in the knockout mice, inflammation was primarily restricted to the joints (81). As expected, macrophages lacking A20 exhibit enhanced NF-κB activation and proinflammatory cytokine production (81). Interestingly, the arthritis was not dependent on T and B lymphocytes or the TNF pathway but rather was triggered by the TLR4-Myd88 pathway (81). This particular aspect is shared with the conventional A20-deficient mice, which undergo spontaneous inflammation via a Myd88-dependent pathway. Nevertheless, it is remarkable that uncontrolled inflammation in myeloid cells results in joint-specific pathology.
Epidermis-specific A20 knockout mice have also been generated, and surprisingly these mice do not develop spontaneous skin inflammation (82). Instead, lack of A20 in the epidermis leads to keratinocyte hyperplasia and developmental abnormalities such as disheveled hair and abnormal ectodermal appendages also observed with mice overexpressing ectodysplasin-A1 (EDA-A1) or ectodysplasin receptor (EDAR) (82, 83). Indeed, A20 was shown to function as a negative feedback regulator of NF-κB in the EDAR pathway in the skin, independent of its DUB domain (82). Taken together, A20 is important for skin homeostasis and appendage development by inhibiting the EDAR pathway.
Because A20 has been identified as a susceptibility locus for inflammatory bowel disease (IBD), A20 was also deleted in intestinal epithelial cells (IECs) by crossing with Cre transgenic mice driven by the IEC-specific promoter villin (84). Although A20IEC-KO mice do not develop spontaneous inflammation or colitis, they exhibit enhanced susceptibility to experimental colitis as characterized by increased colon shortening, crypt loss, and immune cell infiltration (84). Interestingly, this phenotype is not TLR-dependent but is instead triggered by increased TNF-dependent apoptosis of IECs that compromises intestinal barrier function and promotes inflammation (84). Thus, A20 is a key anti-apoptotic protein in IECs that is essential to maintain epithelial barrier integrity and homeostasis under proinflammatory conditions (Reviewed in Pasparakis, this volume). As mentioned previously, the two main functions of A20 are to inhibit NF-κB and inflammation and to prevent apoptosis. The collective A20 conditional gene targeting studies reveal that the main function of A20 in B cells, DCs, myeloid cells and keratinocytes is to restrict NF-κB signaling, whereas in the intestinal epithelium A20 is mainly an anti-apoptotic protein. Further studies are needed to determine if the anti-apoptotic function of A20 plays physiological roles in other tissues or organs.
In addition to regulating TLR signaling, A20 also inhibits signaling through other pattern recognition receptors including NOD2 (nucleotide-binding oligomerization domain containing 2) and RIG-I/MDA5. NOD2 (also known as CARD15) recognizes muramyl dipeptide (MDP), a derivative of peptidoglycan (85). Upon sensing of MDP, NOD2 engages the adaptor molecule RIP2 (also known as RICK) which becomes modified by K63-linked polyubiquitin chains in a cIAP1/cIAP2-dependent manner (86). RIP2 polyubiquitination is critical for NF-κB and MAPK activation by recruiting the TAK1 kinase complex leading to IKK and JNK/p38 activation and the induction of key cytokines and chemokines important for host defense (87). A20 inhibits NOD2 signaling by deubiquitinating RIP2 to block downstream NF-κB signaling (88, 89) (Fig. 2). Consistently, A20-deficient bone marrow-derived macrophages exhibit enhanced MDP-dependent RIP2 ubiquitination and NF-κB activation (89). Further in vivo studies showed that MDP induced greater levels of serum IL-6 in A20-deficient mice (89). Together, these results suggest that A20 is an important negative regulator of NOD2 signaling by inhibiting RIP2 ubiquitination. It is unclear if ubiquitin editing and the E3 ligase activity of A20 is important to inactivate RIP2 as is necessary for RIP1 in the TNFR pathway.
Fig. 2. Signaling pathways regulated by A20, CYLD, and other DUBs.
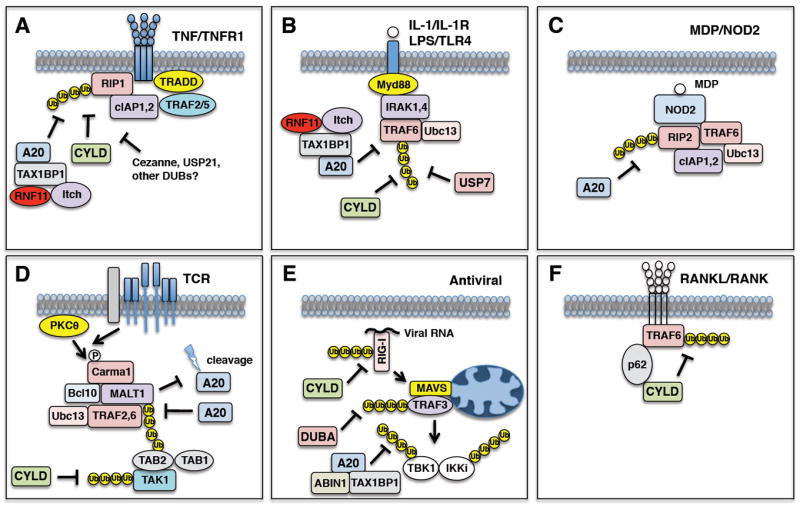
(A) TNF binding to TNFR1 triggers the K63-linked polyubiquitination of RIP1 which is negatively regulated by A20, CYLD and possibly other DUBs (Cezanne, USP21, etc.). (B) IL-1 binding to the IL-1R or LPS binding to TLR4 triggers the activation and K63-linked polyubiquitination of TRAF6 which is negatively regulated by A20, CYLD and USP7. (C) Sensing of MDP by NOD2 facilitates the K63-linked polyubiquitination of RIP2 which is counteracted by A20. (D) T-cell receptor engagement by antigen and costimulation activates PKCθ and the CBM complex leading to MALT1 K63-linked polyubiquitination. A20 inhibits MALT1 ubiquitination, however MALT1 cleaves A20 to inactivate its function. CYLD also hydrolyzes K63-linked polyubiquitin chains from TAK1 in peripheral T cells. (E) RIG-I/MDA5 sense viral nucleic acid and inducibly interact with the mitochondrial adaptor molecule MAVS which assembles a signaling complex containing the E3 ligase TRAF3 and the kinases TBK1/IKKi. CYLD inhibits antiviral signaling by removing K63-linked polyubiquitin chains from RIG-I. DUBA removes K63-linked polyubiquitin chains from TRAF3. A20, together with ABIN1 and TAX1BP1, remove K63-linked polyubiquitin chains from TBK1/IKKi. (F) RANK ligand/RANK ligation in osteoclasts triggers TRAF6 activation and polyubiquitination. CYLD cleaves K63-linked polyubiquitin chains from TRAF6 and requires the adaptor molecule p62 to interact with TRAF6.
RIG-I/MDA5 constitute a class of pattern recognition receptors that recognize viral nucleic acid and trigger the production of type I interferons (90). RIG-I specifically binds to uncapped 5′-triphosphate RNA derived from viral genomes, whereas MDA5 detects double-stranded RNA (91, 92). RIG-I/MDA5 sensing of viral nucleic acid leads to a conformational change and inducible binding with the mitochondrial adaptor molecule MAVS (also known as IPS-1, Cardif, or VISA), which then triggers the activation of TRAF3 and the noncanonical IκB kinases TBK1/IKKi (93). IRF3 and IRF7 are phosphorylated by TBK1/IKKi triggering their dimerization and nuclear translocation and subsequent induction of type I interferons (94). A20 is induced by virus infection and blocks the phosphorylation and dimerization of IRF3 in a negative feedback loop (95-97). A20 also inhibits TLR3-induced activation of NF-κB and IFN-β (97). The main target of A20 in the RIG-I pathway appears to be TBK-1/IKKi, since A20 interacts with these kinases and inhibits their K63-linked polyubiquitination (95, 98) (Fig. 2). Interestingly, the OTU domain of A20 is dispensable for A20 to inhibit antiviral signaling (96, 98). Additional studies are needed to examine responses of A20 conditional knockout mice to virus infection. Other DUBs that target the RIG-I/MDA5 pathway include CYLD and DUBA which target RIG-I and TRAF3 ubiquitination respectively (99, 100) (Fig. 2).
A20 is also a key negative regulator of NF-κB signaling downstream of the T-cell receptor (TCR) and B-cell receptor (BCR) in T and B lymphocytes, respectively. As mentioned earlier, A20 is regulated uniquely in lymphocytes compared to other cell types because of its high basal levels and mechanism of inactivation (70). MALT1 is a key signaling molecule in the TCR and BCR pathways and assembles a protein complex termed ‘CBM’ with Carma1 and Bcl10 (101). The CBM complex links protein kinase C (PKCθ in T cells and PKCβ in B cells) with the IKK complex to trigger NF-κB activation (101). A20 targets MALT1 in the TCR and BCR pathways by removing K63-linked polyubiquitin chains in an OTU domain-dependent manner (102) (Fig. 2). However, MALT1 inactivates A20 by cleavage after arginine 439 to disrupt its inhibitory effect on NF-κB (103) (Fig. 2). Therefore, MALT1 proteolytic activity regulates the threshold of NF-κB activation and ‘fine tunes’ TCR and BCR signaling by cleaving A20 and possible other substrates such as RelB (104). MALT1 also cleaves the NF-κB inhibitor CYLD in the TCR pathway, however CYLD cleavage is required for JNK, but not NF-κB activation (105). In B-cell lymphomas, the delicate balance between MALT1 and A20 is disrupted leading to constitutive NF-κB activation and enhanced cell survival. MALT1 may serve as a therapeutic target for lymphoma since inhibitors of MALT1 catalytic activity suppress NF-κB activation and rescue A20 cleavage in diffuse large B-cell lymphomas (DLBCL) of the activated B-cell (ABC) type (106). However, because A20 is frequently mutated or deleted in lymphoid malignancies it is unclear if MALT inhibitors will trigger cell death in the absence of functional A20.
Regulation of A20 by interacting proteins
A20 preferentially cleaves K48-linked polyubiquitin chains in vitro suggesting that it may rely on cofactors to target K63-linked polyubiquitin chains and provide target specificity in vivo (66). Indeed, A20 functions in the context of a multi-protein complex referred to as the ‘A20 ubiquitin-editing complex’ consisting of A20, TAX1BP1 and the E3 ubiquitin ligases Itch and RNF11. The A20 ubiquitin-editing complex is assembled in response to TNF, IL-1 or LPS stimulation and loss of either subunit of the complex (TAX1BP1, Itch, or RNF11) impairs A20 function and the negative feedback of NF-κB. It is also possible that there are additional yet-to-be identified subunits and further the composition of the A20 complex may vary depending on the cell type or specific stimulus. For instance, A20, TAX1BP1 and ABIN1 form an inducible complex that restricts antiviral signaling and IFN-β production in response to virus infection (98, 107) (Fig. 2).
TAX1BP1 was initially isolated in yeast two-hybrid screens using the HTLV-I Tax oncoprotein, A20, and TRAF6 as bait (108-110). TAX1BP1 was first shown to regulate cell death by mediating the anti-apoptotic function of A20 (109). Subsequent gene targeting studies in mice established TAX1BP1 as a key regulator of A20 and NF-κB signaling. Mice lacking TAX1BP1 are hyperresponsive to inflammatory stimuli and also exhibit spontaneous inflammatory infiltrates in the heart and skin (111, 112). The spontaneous inflammation in TAX1BP1-deficient mice is dependent on hematopoietic cells although the precise cell type that mediates the inflammation has not been identified (112). Both macrophages and fibroblasts lacking TAX1BP1 have enhanced and persistent NF-κB activation in response to TNF, IL-1, or LPS stimulation (111, 112). TAX1BP1-deficient cells also have enhanced K63-linked ubiquitination of TRAF6 and RIP1 upon LPS or TNF stimulation, respectively (111, 112). Mechanistically, TAX1BP1 functions as an adaptor molecule that links A20 with its substrates RIP1 and TRAF6 in the TNFR and IL-1R/TLR4 pathways respectively (111, 112). Since TAX1BP1 contains a ubiquitin-binding domain (UBD) within its zinc finger domain, TAX1BP1 probably senses ubiquitinated substrates via the UBD initially and then recruits A20 to inactivate the substrates.
TAX1BP1 has been shown to interact with the HECT E3 ligase Itch via two ‘PPXY’ (where P=Proline, X=any amino acid and Y=tyrosine) motifs located within the C2H2 zinc finger motifs in TAX1BP1 (113). PPXY motifs interact with ‘WW’ (where W=tryptophan) domains and indeed Itch contains several WW domains (114). Itch was further shown to regulate the targeting of A20 to substrates, and A20 was unable to inhibit NF-κB in the absence of Itch (113). As observed with A20-deficient cells, Itch-deficient MEFs also exhibit elevated and persistent NF-κB signaling in response to TNF or IL-1 (113). Mice lacking Itch (Itchy mice) succumb to inflammation, predominantly in the lungs and skin, although the phenotype is not as severe as A20-deficient mice (115). Itch was previously demonstrated to interact with a RING E3 ligase RNF11 (RING Finger Protein 11) via a conserved PPXY motif in RNF11 (116). RNF11 regulates tumor growth factor β (TGF-β) signaling by interactions with Smurf2 and SMAD4 (117, 118). In addition, a high-throughput yeast two-hybrid screen identified numerous interacting proteins of RNF11 including A20, TAX1BP1, Itch, NEMO and ABIN1 suggesting that RNF11 may regulate NF-κB (119). Indeed, RNF11 inducibly interacts with TAX1BP1, Itch and RIP1 upon TNF stimulation, and is required for A20 to suppress NF-κB (120). Consistently, siRNA-mediated knockdown of RNF11 leads to enhanced NF-κB activation in monocytes and impairs A20-mediated inhibition of NF-κB (120). Taken together, the E3 ligases Itch and RNF11 are both essential subunits of the A20 ubiquitin-editing complex although their precise roles remain to be determined.
ABIN1 (A20 binding inhibitor of NF-κB 1) was identified in a yeast two-hybrid screen as an A20-binding protein and subsequently shown to inhibit NF-κB upon overexpression (55, 121). ABIN1 may also serve as an adaptor molecule for A20 and certain substrates such as NEMO (122). ABIN1 contains a UBD termed UBAN (UBD in ABIN proteins and NEMO) that interacts with both linear and K63-linked polyubiquitin chains (123, 124). ABIN1-deficient mice are embryonic lethal due to fetal liver apoptosis, anemia and hypoplasia (124). ABIN1 also plays a critical role in inhibiting TNF-induced cell death, although ABIN1-deficient MEFs are largely normal for NF-κB signaling (124). Recently, knockin mice were generated that harbor a mutation in the ABIN1 UBAN domain that disrupts the ubiquitin binding of ABIN1 (125). These mice develop an autoimmune disease similar to lupus characterized by spontaneous formation of germinal centers, isotype switching and production of autoreactive antibodies (125). Therefore, ABIN1 sensing of ubiquitin appears to be important for the suppression of autoimmunity. The ABIN1-related proteins ABIN2 and ABIN3 also contain UBAN domains, interact with A20 and inhibit NF-κB, however genetic studies have revealed distinct functions for these molecules (123, 126-128). ABIN2 is essential for the stabilization of the Tpl2 kinase which ensures efficient TLR4-induced ERK activation in macrophages (129, 130). Although ABIN3 is inducible by LPS, the NF-κB inhibitory function appears to be selective for human versus mouse ABIN3 (128). Further studies are necessary to determine the role of ABIN3 in NF-κB signaling.
Other proteins found to interact with A20 in yeast two-hybrid screens include 14-3-3 and YMER. Several isoforms of 14-3-3 interact with A20 and may modulate the localization of A20 by acting as a chaperone (131, 132). However, it does not appear that 14-3-3 proteins regulate the NF-κB inhibitory function of A20 (132). YMER (also known as CCDC50) also interacts with A20 and harbors a UBD important for inhibition of NF-κB (133). YMER may act as an adaptor molecule for A20 although additional studies are needed.
A key feature of the A20 ubiquitin-editing complex is the inducible formation of the complex in response to cytokine stimulation. We have recently demonstrated that TAX1BP1 is phosphorylated by IKKα on Ser593 and Ser624 upon TNF or IL-1 stimulation (134). Phosphorylation of TAX1BP1 is essential for assembly of the A20 ubiquitin-editing complex and the termination of NF-κB signaling (134). TAX1BP1 phosphorylation likely triggers a conformational change which promotes binding to other subunits of the A20 complex. Previous studies have demonstrated that macrophages lacking IKKα or expressing a catalytically inactive IKKα have enhanced canonical NF-κB activation and produce more inflammatory cytokines (135, 136). Thus, IKKα phosphorylation of TAX1BP1 plays a central role in the assembly and function of the A20 ubiquitin-editing complex (Fig. 3).
Fig. 3. Activation and assembly of the A20 ubiquitin-editing complex.
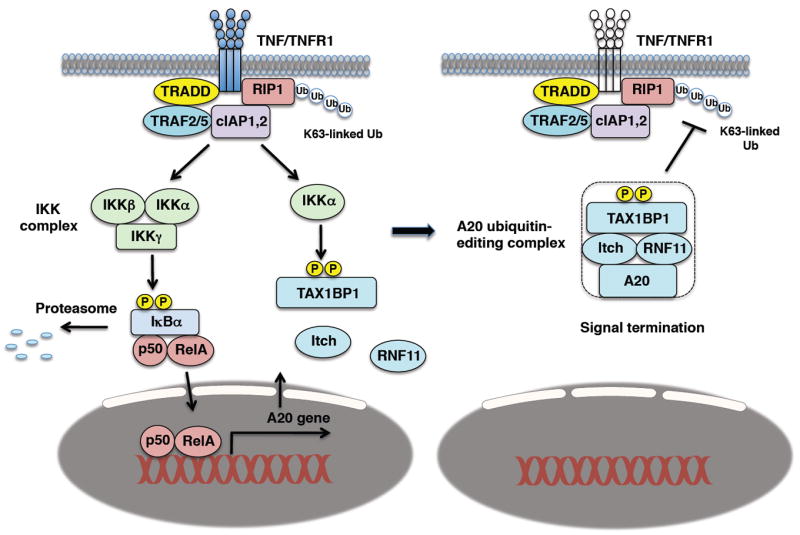
TNF stimulation activates NF-κB and induces A20 expression as part of a negative feedback loop. IKKα phosphorylates TAX1BP1 on Ser593 and Ser624 which nucleates the A20 ubiquitin-editing complex and is required for interactions between TAX1BP1, Itch, RNF11 and A20. The A20 ubiquitin-editing complex inhibits RIP1 K63-linked polyubiquitination to terminate NF-κB signaling downstream of TNFR1.
A20, inflammatory disease, and cancer
Since A20 is of central importance as a negative regulator of innate and adaptive immune pathways, it is not surprising that is has been implicated in the development of human inflammatory and autoimmune diseases as well as lymphoid malignancies. A20 has been identified as a susceptibility locus for rheumatoid arthritis, SLE, type 1 diabetes, inflammatory bowel disease (IBD), celiac disease, psoriasis, and coronary artery disease (137-143). Mucosal biopsies from Crohn’s disease patients revealed decreased A20 expression (144), whereas an SLE-associated A20 variant with a SNP within the DUB domain of A20 (Phe127Cys) was less effective than wildtype A20 in NF-κB inhibition (138). An African-derived polymorphism has also been found in the DUB domain of A20 (Ala125Val) that increases the risk of IBD (145). This A20 variant reduces A20 DUB activity, possibly by impairing the structure of the DUB domain. Taken together, certain polymorphisms within the A20 locus predispose to autoimmunity, either due to reduced expression or impaired function of A20.
Deletion of chromosome band 6q is a common event in non-Hodgkin’s lymphomas (146). Indeed, the A20 gene which maps to this region is commonly deleted in several subtypes of B-cell lymphomas including marginal zone lymphoma, DLBCL, follicular lymphoma, MALT lymphoma, and Hodgkin’s lymphoma (147-151). Loss of A20 may also occur by inactivating point mutations or epigenetic inactivation of A20 by promoter methylation (150). A20 deletions have also been observed in Sezary syndrome, a cutaneous T-cell lymphoma (152). Genetic lesions in the NF-κB pathway occur frequently in B-cell lymphomas (approximately 40% of lymphomas have NF-κB pathway mutations), and target both canonical and noncanonical NF-κB pathways (153). Interestingly, few mutations were observed in A20 in Epstein Barr virus (EBV) positive Hodgkin’s lymphomas, suggesting that EBV may potentially inactivate A20 (151). However, these observations did not extend to EBV-associated acquired immunodeficiency syndrome-related lymphomas where A20 mutations were identified in both EBV+ and EBV− cases (154). However, EBV latent membrane protein 1 (LMP1), which is a strong activator of NF-κB, was not expressed in the majority of EBV cases lacking A20 (154). Besides A20, mutations have been found in other negative regulators such as ABIN1, although these occur less frequently compared to A20 mutations (155).
Although A20 clearly functions as a tumor suppressor in lymphoid malignancies owing to its potent NF-κB inhibiting function, A20 may exert oncogenic activity in certain solid tumors. This can be explained by the anti-apoptotic role of A20 which may predominate over NF-κB inhibition in certain tissues. Large-scale cancer genome sequencing efforts have identified numerous A20 mutations in lymphoid malignancies, but very few thus far in solid tumors. A20 is highly expressed in aggressive breast carcinomas lacking expression of the estrogen receptor (ER), progesterone receptor (PR), or in tumor samples with high histological grade (156). Stable transfection of A20 in the breast cancer line MCF-7 conferred protection against cell death upon tamoxifen treatment (156). A20 is also overexpressed in glioma cells and may exert oncogenicity in glioblastoma. Knockdown of A20 in glioma cells reduces proliferation, promotes cell cycle arrest and apoptosis (157). A20 is also overexpressed in glioma stem cells and is important for cell survival, self-renewal, and tumorigenesis (158). Taken together, it appears that A20 can function as an oncogene or tumor suppressor depending on the context and tissue.
CYLD and the regulation of NF-κB
The cylindromatosis gene Cyld encodes a tumor suppressor commonly mutated in familial cylindromatosis, a genetic condition characterized by benign tumors of skin appendages (159). CYLD is a deubiquitinase of the USP family with a C-terminal catalytic domain where mutations and truncations frequently occur in cylindromatosis patients and lead to impaired DUB activity (160). CYLD was found to be a NEMO-interacting protein that inhibits IKK and NF-κB by removing K63-linked polyubiquitin chains from TRAF2, TRAF6, and NEMO (160-162). Other CYLD substrates important for NF-κB regulation to be discussed later include TAK1, Bcl3, and RIP1 (163-165) (Fig. 2). In addition to regulating NF-κB, CYLD has also been implicated in a number of other pathways including antiviral signaling, MAPK pathways, proximal TCR signaling, cell cycle, and calcium signaling (99, 166-169).
In vitro studies have confirmed that CYLD preferentially cleaves K63-linked and linear polyubiquitin chains compared to K48-linked chains (66). Similar to A20, CYLD may also depend on ubiquitin-binding adaptor molecules in certain pathways to ensure specificity. For example, p62 (also known as sequestosome 1) links CYLD with TRAF6 downstream of RANK and neurotrophin receptors in osteoclasts and neurons, respectively (170, 171). Optineurin may be a ubiquitin-binding adaptor for CYLD in the TNFR pathway, since knockdown of optineurin impaired CYLD binding to RIP1 and NF-κB inhibition in TNF stimulated cells (172). However, it is unclear if CYLD is dependent on either p62 or optineurin to interact with other known CYLD substrates and also if CYLD relies on additional adapter molecules.
Several Cyld genetic mouse models have been generated that have elucidated novel functions of CYLD in diverse biological processes. CYLD plays essential roles in T-cell development in addition to regulating immune homeostasis and inflammation. Cyld−/− mice have fewer mature CD4+ and CD8+ T cells in the thymus and periphery owing to a defect in proximal TCR signaling (167, 173). CYLD cleaves K48-linked and K63-linked polyubiquitin chains from the lck kinase and links lck with its substrate ZAP70 (167). The development of natural killer T (NKT) cells is also impaired in Cyld−/− mice due to impaired expression of the costimulatory molecule ICOS (inducible costimulator) that is essential for NKT cell development (174). CYLD deficiency leads to spontaneous activation of peripheral T lymphocytes, leading to an autoimmune disease resembling inflammatory bowel disease (163). Consistently, Cyld−/− mice were more prone to inflammation and tumor formation in an experimentally induced colitis model (175). In mature T cells, CYLD restrains IKK and NF-κB activation in the TCR pathway by removing K63-linked polyubiquitin chains from TAK1 (163) (Fig. 2). CYLD also downregulates Streptococcus pneumoniae-induced NFAT (nuclear factor for activated T cells) activation and inflammation by inhibiting TAK1 ubiquitination (176). CYLD deficiency also triggers spontaneous B-cell activation and hyperplasia as a result of constitutive NF-κB activation (177, 178). Furthermore, macrophages from Cyld−/− mice are hyperresponsive to TLR stimuli, anti-CD40, and TNF and exhibit enhanced NF-κB activation and proinflammatory cytokine production (175). Finally, DCs from mice expressing the short splice variant of CYLD lacking exons 7 and 8 exhibit a hyperactive phenotype accompanied by enhanced NF-κB activation (179). Together, these findings underscore the importance of CYLD in both the development and homeostasis of specific immune cell subsets.
Studies with Cyld−/− mice have also revealed critical roles for CYLD in the regulation of bone homeostasis and germ cell apoptosis. Receptor activator of NF-κB (RANK) is a member of the TNFR family and together with its ligand RANKL are critical for the activation and differentiation of osteoclasts, cells involved in bone resorption (180). Cyld−/− mice exhibit aberrant osteoclast differentiation and develop severe osteoporosis (170). CYLD inhibits RANK-mediated signaling in osteoclasts in a negative feedback loop by deubiquitinating TRAF6 (170) (Fig. 2). As mentioned earlier, CYLD requires the p62 adapter molecule to engage TRAF6 for its inactivation. Cyld−/− male mice are sterile due to testicular atrophy caused by an impairment of the early wave of germ cell apoptosis, a hallmark of spermatogenesis (165). CYLD deficiency promotes the activation of NF-κB and induction of anti-apoptotic genes in germ cells. RIP1 is a key target of CYLD in testicular cells and is persistently ubiquitinated in the absence of CYLD (165).
Cyld−/− mice are more susceptible to chemically induced skin tumors thus underscoring the important tumor suppressor function of CYLD in the skin. CYLD functions as an inhibitor of atypical NF-κB activation by controlling nuclear translocation of the NF-κB coactivator Bcl3. Treatment of keratinocytes with TPA (12-O-tetradecanoylphorbol-13-acetate) and/or ultraviolet (UV) radiation triggered the K63-linked polyubiquitination and nuclear translocation of Bcl3 where it activated expression of the cyclin D1 gene (164). CYLD inhibits Bcl3 by antagonizing the K63-linked polyubiquitination of Bcl3 thus preventing its nuclear translocation. Keratinocytes from Cyld−/− mice stimulated with UV and TPA exhibited elevated cyclin D1 expression and higher proliferation rates (164). CYLD regulation of Bcl3 nuclear translocation may extend to other cell types including B cells, DCs and smooth muscle cells (178, 179, 181).
Loss of the tumor suppressor function of CYLD in the skin not only predisposes to cylindromas but also to other skin tumors such as basal cell carcinoma and melanoma. CYLD expression is downregulated by the transcriptional repressor Snail in both basal cell carcinoma and melanoma (182, 183). Similarly, CYLD is downregulated by Notch/Hes1 in T-cell acute lymphoblastic lymphoma (T-ALL), thus leading to constitutive NF-κB activation (184). CYLD is also downregulated in colon and hepatocellular carcinomas (185). Finally, mutations in components of the canonical and noncanonical NF-κB pathways have been found in patients with multiple myeloma (186, 187). CYLD is among the most frequently mutated genes in multiple myeloma and likely contributes to the persistent NF-κB activation and enhanced cell survival in these tumors. Unlike A20, mutations in CYLD are a rare event in B-cell lymphomas (188). It should be stressed that the tumor suppressor activity of CYLD is not entirely dependent on NF-κB, since CYLD also regulates the cell cycle and microtubule polymerization (168, 189, 190).
Although CYLD and A20 substrates in NF-κB pathways overlap to a large degree (i.e. RIP1 and TRAF6) there is little functional redundancy between these proteins. This is due to temporal differences in inhibition by A20 and CYLD. Whereas CYLD is important for dampening basal NF-κB activation, A20 is more critical to inhibit activated NF-κB in a negative feedback loop. CYLD interacts with NEMO and is phosphorylated by IKK as a mechanism to inactivate CYLD DUB activity (191). Transient CYLD phosphorylation thus allows for NF-κB activation to proceed in a stimulus-dependent and transient manner prior to inhibition by the A20 ubiquitin-editing complex. However, constitutive phosphorylation of CYLD by IKK inactivates CYLD in HTLV-1-induced leukemia to promote heightened NF-κB activation (192). A similar mechanism is thought to occur in breast carcinogenesis where IKKε phosphorylates and inactivates CYLD (193). CYLD may potentially be inactivated by post-translational modifications such as phosphorylation in other malignancies since persistent NF-κB activation is common in many cancers.
Additional deubiquitinases regulating NF-κB
In addition to A20 and CYLD, a number of other DUBs have been implicated as regulators of NF-κB signaling (Fig. 2). Cezanne (Cellular zinc finger anti-NF-κB) is a deubiquitinase of the ovarian tumor superfamily with sequence similarity to A20 (194). Cezanne is induced by TNF stimulation and inhibits NF-κB in a negative feedback loop (195). Cezanne requires its catalytic activity to inhibit NF-κB and is recruited to the TNFR where it suppresses RIP1 ubiquitination (195). Interestingly, Cezanne has been recently identified as the first deubiquitinase with specificity for K11-linked chains (196). Because K11-linked polyubiquitin chains have been identified in the activated TNFR1 complex, these results imply that Cezanne may inhibit TNF-induced NF-κB activation by hydrolyzing K11-linked polyubiquitin chains on substrates present in the TNFR1 complex. Future genetic studies will be essential to confirm the importance of Cezanne in the inhibition of NF-κB and inflammation.
The deubiquitinase USP21 (ubiquitin-specific peptidase 21) has also been implicated as an inhibitor of NF-κB in the TNF pathway. USP21 interacts with RIP1 and deubiquitinates RIP1 in a DUB-dependent manner (197). Knockdown of USP21 with siRNA enhances RIP1 ubiquitination and TNF-induced NF-κB activation (197). However, it is unclear if USP21 exhibits specificity for a particular type of polyubiquitin chain. USP31 deubiquitinates K63-linked polyubiquitin chains and may also be another regulator of TNF-induced NF-κB signaling (198). USP7 was shown to negatively regulate NF-κB in TLR pathways by deubiquitinating TRAF6 and NEMO (199) (Fig. 2). Interestingly, the herpes simplex virus ICP0 protein exploits USP7 to inhibit innate responses to HSV (199). USP2 was recently identified as a DUB that acts as a positive regulator of TNF-induced NF-κB activation and induction of proinflammatory cytokines (200). However, the target of USP2 in the TNFR pathway is unknown and requires further study.
Recently, a novel deubiquitinase, MCP-induced protein 1 (MCPIP1) (also known as Zc3h12a), has been identified for TRAF2 and TRAF6 that is essential for the termination of JNK and NF-κB in inflammatory signaling pathways (201). Mcpip1−/− mice exhibited stunted growth after weaning, splenomegaly, lymphadenopathy and died prematurely (201). Bone marrow-derived macrophages (BMDMs) from Mcpip1−/− mice produced more proinflammatory cytokines including TNF, IL-1β and IL-6 compared to wild-type BMDMs (201). Apparently, MCPIP1 functions as a deubiquitinase despite little sequence homology with members from all known DUB families. Although MCPIP1 deubiquitinates K63-linked polyubiquitin chains from TRAF2, TRAF6 and RIP1, it is unclear how MCPIP1 functions in relation to other DUBs such as A20, CYLD, Cezanne, or USP21.
Most DUBs that regulate NF-κB operate upstream of the IKK complex and target TRAFs or RIP1, however there is evidence that certain DUBs target IKK or downstream of IKK. USP11 has been shown to deubiquitinate IKKα and IκBα in the TNFR pathway (202, 203). USP11 presumably removes K48-linked chains from IκBα and thus protects IκBα from degradation by the proteasome. Consistently, knockdown of USP11 leads to enhanced TNF-induced IκBα degradation and NF-κB activation (203). USP15, a COP9 signalosome-associated DUB, also removes K48-linked polyubiquitin chains from IκBα (204). It is possible that USP11 and USP15 may cooperate to remove degradative polyubiquitin chains from IκBα to dampen NF-κB activation.
Perspectives: conclusions and outstanding questions
A20, CYLD, and a number of other DUBs are key regulators of NF-κB signaling. The multiple NF-κB inhibitory DUBs and apparent lack of redundancy among these DUBs raises obvious questions regarding their individual mechanistic roles. Distinct adapter molecules for A20, CYLD, and potentially other DUBs likely confers specificity for each DUB; however, multiple DUBs may potentially cooperate to inhibit identical targets. One possibility is that each DUB may hydrolyze distinct polyubiquitin chains (i.e. K11, K63) on the same targets. For example, A20 and CYLD may hydrolyze K63-linked polyubiquitin chains on RIP1, whereas Cezanne may remove K11-linked polyubiquitin chains from RIP1 or another molecule in the TNFR complex. It is also important to consider that the negative regulation of NF-κB is dynamic and is governed by temporal and spatial differences in how each DUB functions to inhibit NF-κB.
Given the importance of ubiquitination/deubiquitination in the regulatory control of inflammatory and cell survival pathways, components of either ubiquitination or deubiquitination pathways may serve as attractive targets for autoimmune diseases or cancer (205). Indeed, the proteasome inhibitor Bortezomib is a Food and Drug Administration approved front-line treatment for multiple myeloma and is in clinical trials for other malignancies (206). Although deubiquitinase-specific drugs have not yet entered clinical trials, the USP7 DUB may prove to be a suitable target for certain malignancies that lack genetic lesions in p53. USP7 is a key regulator of the p53 E3 ligase MDM2 by stabilizing MDM2 which subsequently degrades p53 (207). Small-molecule inhibitors of USP7 have been developed that promote MDM2 ubiquitination and degradation with concomitant stabilization of p53. The E2 enzyme Ubc13 may also serve as an attractive target because of its importance in activating NF-κB while inhibiting p53 activation (208). A small-molecule inhibitor of Ubc13 has been identified that disrupts the binding of Ubc13 and Uev1a (209). This inhibitor blocks TNF-induced NF-κB activation and also sensitizes tumor cells to chemotherapeutic agents (209). Finally, it will be interesting to identify small molecules that mimic A20 function by disrupting the binding of Ubc13 with E3 ligases such as TRAF6. An A20 ‘mimic’ or Ubc13 antagonist may have therapeutic value for the treatment of lymphoid malignancies that lack A20 expression and have persistent NF-κB activation.
Acknowledgments
The laboratory of E.W.H. is supported by NIH grants P01CA128115, R01CA135362, and R01GM083143.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol. 2009;1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 4.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 6.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 7.Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–94. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–5. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson KD, Urban MK, Haas AL. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J Biol Chem. 1980;255:7529–32. [PubMed] [Google Scholar]
- 10.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–86. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 12.Kee Y, Huibregtse JM. Regulation of catalytic activities of HECT ubiquitin ligases. Biochem Biophys Res Commun. 2007;354:329–33. doi: 10.1016/j.bbrc.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–42. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokunaga F, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–32. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda F, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature. 471:637–41. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tokunaga F, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature. 471:633–6. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 18.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 471:591–6. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 19.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–5. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto ML, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 39:477–84. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda H, Kerppola TK. Lysosomal localization of ubiquitinated Jun requires multiple determinants in a lysine-27-linked polyubiquitin conjugate. Mol Biol Cell. 2008;19:4588–601. doi: 10.1091/mbc.E08-05-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chastagner P, Israel A, Brou C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE. 2008;3:e2735. doi: 10.1371/journal.pone.0002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HC, Huibregtse JM. Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol Cell Biol. 2009;29:3307–18. doi: 10.1128/MCB.00240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–53. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 25.Deng L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 26.Dynek JN, et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. Embo J. 29:4198–209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HT, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–86. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 28.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–44. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Wertz IE, Dixit VM. Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev. 2008;19:313–24. doi: 10.1016/j.cytogfr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 30.O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17:418–24. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–57. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez SE, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Tada K, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–4. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 35.Mahoney DJ, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:11778–83. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanayama A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–48. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–51. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Chen ZJ. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res. 21:6–21. doi: 10.1038/cr.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS ONE. 2008;3:e4064. doi: 10.1371/journal.pone.0004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–96. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 42.Nijman SM, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–86. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Cope GA, et al. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–11. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 44.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 46.Finley D, Bartel B, Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394–401. doi: 10.1038/338394a0. [DOI] [PubMed] [Google Scholar]
- 47.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 48.Opipari AW, Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–8. [PubMed] [Google Scholar]
- 49.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–6. [PubMed] [Google Scholar]
- 50.Laherty CD, Perkins ND, Dixit VM. Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor kappa B. J Biol Chem. 1993;268:5032–9. [PubMed] [Google Scholar]
- 51.Opipari AW, Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424–7. [PubMed] [Google Scholar]
- 52.Jaattela M, Mouritzen H, Elling F, Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol. 1996;156:1166–73. [PubMed] [Google Scholar]
- 53.Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A. 1996;93:6721–5. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996;271:18068–73. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 55.Heyninck K, et al. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol. 1999;145:1471–82. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heyninck K, Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-kappaB activation at the level of TRAF6. FEBS Lett. 1999;442:147–50. doi: 10.1016/s0014-5793(98)01645-7. [DOI] [PubMed] [Google Scholar]
- 57.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evans PC, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378:727–34. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mattera R, Tsai YC, Weissman AM, Bonifacino JS. The Rab5 guanine nucleotide exchange factor Rabex-5 binds ubiquitin (Ub) and functions as a Ub ligase through an atypical Ub-interacting motif and a zinc finger domain. J Biol Chem. 2006;281:6874–83. doi: 10.1074/jbc.M509939200. [DOI] [PubMed] [Google Scholar]
- 60.Lee S, et al. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat Struct Mol Biol. 2006;13:264–71. doi: 10.1038/nsmb1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li L, et al. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochim Biophys Acta. 2008;1783:1140–9. doi: 10.1016/j.bbamcr.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li L, Soetandyo N, Wang Q, Ye Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim Biophys Acta. 2009;1793:346–53. doi: 10.1016/j.bbamcr.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 64.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 327:1135–9. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakada S, et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 466:941–6. doi: 10.1038/nature09297. [DOI] [PubMed] [Google Scholar]
- 66.Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009;10:466–73. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85. doi: 10.1042/BJ20071399. [DOI] [PubMed] [Google Scholar]
- 68.Lin SC, et al. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol. 2008;376:526–40. doi: 10.1016/j.jmb.2007.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bosanac I, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell. 40:548–57. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Tewari M, Wolf FW, Seldin MF, O’Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol. 1995;154:1699–706. [PubMed] [Google Scholar]
- 71.Wang CM, et al. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun. 411:586–92. doi: 10.1016/j.bbrc.2011.06.191. [DOI] [PubMed] [Google Scholar]
- 72.Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem. 110:1155–64. doi: 10.1002/jcb.22630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. I{kappa}B Kinase {beta} Phosphorylates the K63 Deubiquitinase A20 To Cause Feedback Inhibition of the NF-{kappa}B Pathway. Mol Cell Biol. 2007;27:7451–61. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med. 2008;205:451–64. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tavares RM, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 33:181–91. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chu Y, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 117:2227–36. doi: 10.1182/blood-2010-09-306019. [DOI] [PubMed] [Google Scholar]
- 77.Hovelmeyer N, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol. 41:595–601. doi: 10.1002/eji.201041313. [DOI] [PubMed] [Google Scholar]
- 78.Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–61. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kool M, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 35:82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 80.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14:258–65. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 43:908–12. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 82.Lippens S, et al. Keratinocyte-specific ablation of the NF-kappaB regulatory protein A20 (TNFAIP3) reveals a role in the control of epidermal homeostasis. Cell Death Differ. doi: 10.1038/cdd.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Newton K, French DM, Yan M, Frantz GD, Dixit VM. Myodegeneration in EDA-A2 transgenic mice is prevented by XEDAR deficiency. Mol Cell Biol. 2004;24:1608–13. doi: 10.1128/MCB.24.4.1608-1613.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vereecke L, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. 207:1513–23. doi: 10.1084/jem.20092474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 86.Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 87.Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol. 2009;19:1255–63. doi: 10.1016/j.cub.2009.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hasegawa M, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. Embo J. 2008;27:373–83. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hitotsumatsu O, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–90. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brennan K, Bowie AG. Activation of host pattern recognition receptors by viruses. Curr Opin Microbiol. 13:503–7. doi: 10.1016/j.mib.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 91.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 92.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 93.Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev. 243:91–8. doi: 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- 94.Servant MJ, et al. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J Biol Chem. 2001;276:355–63. doi: 10.1074/jbc.M007790200. [DOI] [PubMed] [Google Scholar]
- 95.Saitoh T, et al. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol. 2005;174:1507–12. doi: 10.4049/jimmunol.174.3.1507. [DOI] [PubMed] [Google Scholar]
- 96.Lin R, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281:2095–103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- 97.Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-kappaB and ISRE and IFN-beta promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- 98.Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 285:14999–5009. doi: 10.1074/jbc.M110.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang M, et al. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283:18621–6. doi: 10.1074/jbc.M801451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kayagaki N, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–32. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 101.Blonska M, Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duwel M, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. 2009;182:7718–28. doi: 10.4049/jimmunol.0803313. [DOI] [PubMed] [Google Scholar]
- 103.Coornaert B, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. 2008;9:263–71. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- 104.Hailfinger S, et al. Malt1-dependent RelB cleavage promotes canonical NF-{kappa}B activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci U S A. 108:14596–601. doi: 10.1073/pnas.1105020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Staal J, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. Embo J. 30:1742–52. doi: 10.1038/emboj.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ferch U, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2009;206:2313–20. doi: 10.1084/jem.20091167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao L, Coope H, Grant S, Ma A, Ley SC, Harhaj EW. ABIN1 cooperates with TAX1BP1 and A20 to inhibit antiviral signaling. J Biol Chem. doi: 10.1074/jbc.M111.283762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gachon F, et al. CREB-2, a cellular CRE-dependent transcription repressor, functions in association with Tax as an activator of the human T-cell leukemia virus type 1 promoter. J Virol. 1998;72:8332–7. doi: 10.1128/jvi.72.10.8332-8337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.De Valck D, et al. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene. 1999;18:4182–90. doi: 10.1038/sj.onc.1202787. [DOI] [PubMed] [Google Scholar]
- 110.Ling L, Goeddel DV. T6BP, a TRAF6-interacting protein involved in IL-1 signaling. Proc Natl Acad Sci U S A. 2000;97:9567–72. doi: 10.1073/pnas.170279097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. Embo J. 2007;26:3910–22. doi: 10.1038/sj.emboj.7601823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Iha H, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. Embo J. 2008;27:629–41. doi: 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shembade N, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9:254–62. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- 114.Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet. 1998;18:143–6. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 115.Matesic LE, Copeland NG, Jenkins NA. Itchy mice: the identification of a new pathway for the development of autoimmunity. Curr Top Microbiol Immunol. 2008;321:185–200. doi: 10.1007/978-3-540-75203-5_9. [DOI] [PubMed] [Google Scholar]
- 116.Kitching R, et al. The RING-H2 protein RNF11 is differentially expressed in breast tumours and interacts with HECT-type E3 ligases. Biochim Biophys Acta. 2003;1639:104–12. doi: 10.1016/j.bbadis.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 117.Subramaniam V, et al. The RING-H2 protein RNF11 is overexpressed in breast cancer and is a target of Smurf2 E3 ligase. Br J Cancer. 2003;89:1538–44. doi: 10.1038/sj.bjc.6601301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Azmi P, Seth A. RNF11 is a multifunctional modulator of growth factor receptor signalling and transcriptional regulation. Eur J Cancer. 2005;41:2549–60. doi: 10.1016/j.ejca.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 119.Colland F, et al. Functional proteomics mapping of a human signaling pathway. Genome Res. 2004;14:1324–32. doi: 10.1101/gr.2334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shembade N, Parvatiyar K, Harhaj NS, Harhaj EW. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. Embo J. 2009;28:513–22. doi: 10.1038/emboj.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heyninck K, Kreike MM, Beyaert R. Structure-function analysis of the A20-binding inhibitor of NF-kappa B activation, ABIN-1. FEBS Lett. 2003;536:135–40. doi: 10.1016/s0014-5793(03)00041-3. [DOI] [PubMed] [Google Scholar]
- 122.Mauro C, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–8. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 123.Wagner S, et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene. 2008;27:3739–45. doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 124.Oshima S, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. 2009;457:906–9. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nanda SK, et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med. 208:1215–28. doi: 10.1084/jem.20102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Van Huffel S, Delaei F, Heyninck K, De Valck D, Beyaert R. Identification of a novel A20-binding inhibitor of nuclear factor-kappa B activation termed ABIN-2. J Biol Chem. 2001;276:30216–23. doi: 10.1074/jbc.M100048200. [DOI] [PubMed] [Google Scholar]
- 127.Wullaert A, et al. LIND/ABIN-3 is a novel lipopolysaccharide-inducible inhibitor of NF-kappaB activation. J Biol Chem. 2007;282:81–90. doi: 10.1074/jbc.M607481200. [DOI] [PubMed] [Google Scholar]
- 128.Weaver BK, Bohn E, Judd BA, Gil MP, Schreiber RD. ABIN-3: a molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol Cell Biol. 2007;27:4603–16. doi: 10.1128/MCB.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lang V, et al. ABIN-2 forms a ternary complex with TPL-2 and NF-kappa B1 p105 and is essential for TPL-2 protein stability. Mol Cell Biol. 2004;24:5235–48. doi: 10.1128/MCB.24.12.5235-5248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Papoutsopoulou S, et al. ABIN-2 is required for optimal activation of Erk MAP kinase in innate immune responses. Nat Immunol. 2006;7:606–15. doi: 10.1038/ni1334. [DOI] [PubMed] [Google Scholar]
- 131.Vincenz C, Dixit VM. 14-3-3 proteins associate with A20 in an isoform-specific manner and function both as chaperone and adapter molecules. J Biol Chem. 1996;271:20029–34. doi: 10.1074/jbc.271.33.20029. [DOI] [PubMed] [Google Scholar]
- 132.De Valck D, Heyninck K, Van Criekinge W, Vandenabeele P, Fiers W, Beyaert R. A20 inhibits NF-kappaB activation independently of binding to 14-3-3 proteins. Biochem Biophys Res Commun. 1997;238:590–4. doi: 10.1006/bbrc.1997.7343. [DOI] [PubMed] [Google Scholar]
- 133.Bohgaki M, et al. Involvement of Ymer in suppression of NF-kappaB activation by regulated interaction with lysine-63-linked polyubiquitin chain. Biochim Biophys Acta. 2008;1783:826–37. doi: 10.1016/j.bbamcr.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 134.Shembade N, Pujari R, Harhaj NS, Abbott DW, Harhaj EW. The kinase IKKalpha inhibits activation of the transcription factor NF-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nat Immunol. 12:834–43. doi: 10.1038/ni.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 136.Li Q, et al. Enhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1) Proc Natl Acad Sci U S A. 2005;102:12425–30. doi: 10.1073/pnas.0505997102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thomson W, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–3. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Musone SL, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–4. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fung EY, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009;10:188–91. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- 140.Trynka G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–83. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 141.Wang K, et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet. 19:2059–67. doi: 10.1093/hmg/ddq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-kappaB target genes. Proc Natl Acad Sci U S A. 2007;104:18601–6. doi: 10.1073/pnas.0709011104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nair RP, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Arsenescu R, et al. Signature biomarkers in Crohn’s disease: toward a molecular classification. Mucosal Immunol. 2008;1:399–411. doi: 10.1038/mi.2008.32. [DOI] [PubMed] [Google Scholar]
- 145.Lodolce JP, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol. 184:7001–9. doi: 10.4049/jimmunol.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thelander EF, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49:477–87. doi: 10.1080/10428190701817282. [DOI] [PubMed] [Google Scholar]
- 147.Compagno M, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kato M, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–6. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 149.Honma K, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009 doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 150.Chanudet E, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 24:483–7. doi: 10.1038/leu.2009.234. [DOI] [PubMed] [Google Scholar]
- 151.Schmitz R, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–9. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Braun FC, et al. Tumor suppressor TNFAIP3 (A20) is frequently deleted in Sezary syndrome. Leukemia. 25:1494–501. doi: 10.1038/leu.2011.101. [DOI] [PubMed] [Google Scholar]
- 153.Rossi D, et al. Alteration of BIRC3 and multiple other NF-{kappa}B pathway genes in splenic marginal zone lymphoma. Blood. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- 154.Giulino L, et al. A20 (TNFAIP3) genetic alterations in EBV-associated AIDS-related lymphoma. Blood. 117:4852–4. doi: 10.1182/blood-2010-10-310995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dong G, et al. A20, ABIN-1/2, and CARD11 mutations and their prognostic value in gastrointestinal diffuse large B-cell lymphoma. Clin Cancer Res. 17:1440–51. doi: 10.1158/1078-0432.CCR-10-1859. [DOI] [PubMed] [Google Scholar]
- 156.Vendrell JA, Ghayad S, Ben-Larbi S, Dumontet C, Mechti N, Cohen PA. A20/TNFAIP3, a new estrogen-regulated gene that confers tamoxifen resistance in breast cancer cells. Oncogene. 2007;26:4656–67. doi: 10.1038/sj.onc.1210269. [DOI] [PubMed] [Google Scholar]
- 157.Guo Q, et al. A20 is overexpressed in glioma cells and may serve as a potential therapeutic target. Expert Opin Ther Targets. 2009;13:733–41. doi: 10.1517/14728220903045018. [DOI] [PubMed] [Google Scholar]
- 158.Hjelmeland AB, et al. Targeting a20 decreases glioma stem cell survival and tumor growth. PLoS Biol. 8:e1000319. doi: 10.1371/journal.pbio.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bignell GR, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160–5. doi: 10.1038/76006. [DOI] [PubMed] [Google Scholar]
- 160.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–5. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 161.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 162.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 163.Reiley WW, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–85. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–77. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 165.Wright A, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–16. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 166.Reiley W, Zhang M, Sun SC. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279:55161–7. doi: 10.1074/jbc.M411049200. [DOI] [PubMed] [Google Scholar]
- 167.Reiley WW, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006;7:411–7. doi: 10.1038/ni1315. [DOI] [PubMed] [Google Scholar]
- 168.Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci U S A. 2007;104:8869–74. doi: 10.1073/pnas.0703268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stokes A, Wakano C, Koblan-Huberson M, Adra CN, Fleig A, Turner H. TRPA1 is a substrate for de-ubiquitination by the tumor suppressor CYLD. Cell Signal. 2006;18:1584–94. doi: 10.1016/j.cellsig.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 170.Jin W, et al. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–66. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wooten MW, et al. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–9. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- 172.Nagabhushana A, Bansal M, Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFalpha-induced NF-kappaB activation. PLoS ONE. 6:e17477. doi: 10.1371/journal.pone.0017477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tsagaratou A, Trompouki E, Grammenoudi S, Kontoyiannis DL, Mosialos G. Thymocyte-specific truncation of the deubiquitinating domain of CYLD impairs positive selection in a NF-kappaB essential modulator-dependent manner. J Immunol. 185:2032–43. doi: 10.4049/jimmunol.0903919. [DOI] [PubMed] [Google Scholar]
- 174.Lee AJ, et al. Regulation of natural killer T-cell development by deubiquitinase CYLD. Embo J. 29:1600–12. doi: 10.1038/emboj.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang J, et al. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042–9. doi: 10.1172/JCI28746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Koga T, et al. Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J Biol Chem. 2008;283:12546–54. doi: 10.1074/jbc.M710518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jin W, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem. 2007;282:15884–93. doi: 10.1074/jbc.M609952200. [DOI] [PubMed] [Google Scholar]
- 178.Hovelmeyer N, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med. 2007;204:2615–27. doi: 10.1084/jem.20070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Srokowski CC, et al. Naturally occurring short splice variant of CYLD positively regulates dendritic cell function. Blood. 2009;113:5891–5. doi: 10.1182/blood-2008-08-175489. [DOI] [PubMed] [Google Scholar]
- 180.Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. 2008;1143:123–50. doi: 10.1196/annals.1443.016. [DOI] [PubMed] [Google Scholar]
- 181.Takami Y, et al. Potential role of CYLD (Cylindromatosis) as a deubiquitinating enzyme in vascular cells. Am J Pathol. 2008;172:818–29. doi: 10.2353/ajpath.2008.070312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kuphal S, et al. GLI1-dependent transcriptional repression of CYLD in basal cell carcinoma. Oncogene. doi: 10.1038/onc.2011.163. [DOI] [PubMed] [Google Scholar]
- 183.Massoumi R, et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med. 2009;206:221–32. doi: 10.1084/jem.20082044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Espinosa L, et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 18:268–81. doi: 10.1016/j.ccr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis. 2007;28:21–7. doi: 10.1093/carcin/bgl081. [DOI] [PubMed] [Google Scholar]
- 186.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Schmidt A, et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 49:803–9. doi: 10.1002/gcc.20789. [DOI] [PubMed] [Google Scholar]
- 189.Gao J, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283:8802–9. doi: 10.1074/jbc.M708470200. [DOI] [PubMed] [Google Scholar]
- 190.Wickstrom SA, Masoumi KC, Khochbin S, Fassler R, Massoumi R. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. Embo J. 29:131–44. doi: 10.1038/emboj.2009.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Reiley W, Zhang M, Wu X, Granger E, Sun SC. Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol Cell Biol. 2005;25:3886–95. doi: 10.1128/MCB.25.10.3886-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wu X, Zhang M, Sun SC. Mutual regulation between deubiquitinase CYLD and retroviral oncoprotein Tax. Cell Biosci. 1:27. doi: 10.1186/2045-3701-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hutti JE, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell. 2009;34:461–72. doi: 10.1016/j.molcel.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Evans PC, et al. A novel type of deubiquitinating enzyme. J Biol Chem. 2003;278:23180–6. doi: 10.1074/jbc.M301863200. [DOI] [PubMed] [Google Scholar]
- 195.Enesa K, et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. 2008;283:7036–45. doi: 10.1074/jbc.M708690200. [DOI] [PubMed] [Google Scholar]
- 196.Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 17:939–47. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xu G, et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor alpha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J Biol Chem. 285:969–78. doi: 10.1074/jbc.M109.042689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tzimas C, Michailidou G, Arsenakis M, Kieff E, Mosialos G, Hatzivassiliou EG. Human ubiquitin specific protease 31 is a deubiquitinating enzyme implicated in activation of nuclear factor-kappaB. Cell Signal. 2006;18:83–92. doi: 10.1016/j.cellsig.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 199.Daubeuf S, et al. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood. 2009;113:3264–75. doi: 10.1182/blood-2008-07-168203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Metzig M, et al. An RNAi screen identifies USP2 as a factor required for TNF-alpha-induced NF-kappaB signaling. Int J Cancer. 129:607–18. doi: 10.1002/ijc.26124. [DOI] [PubMed] [Google Scholar]
- 201.Liang J, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. 207:2959–73. doi: 10.1084/jem.20092641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yamaguchi T, Kimura J, Miki Y, Yoshida K. The deubiquitinating enzyme USP11 controls an IkappaB kinase alpha (IKKalpha)-p53 signaling pathway in response to tumor necrosis factor alpha (TNFalpha) J Biol Chem. 2007;282:33943–8. doi: 10.1074/jbc.M706282200. [DOI] [PubMed] [Google Scholar]
- 203.Sun W, et al. USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal. 22:386–94. doi: 10.1016/j.cellsig.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. Embo J. 2007;26:1532–41. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell. 143:686–93. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 206.Mitsiades CS, Mitsiades N, Hideshima T, Richardson PG, Anderson KC. Proteasome inhibition as a new therapeutic principle in hematological malignancies. Curr Drug Targets. 2006;7:1341–7. doi: 10.2174/138945006778559247. [DOI] [PubMed] [Google Scholar]
- 207.Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–86. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 208.Laine A, Topisirovic I, Zhai D, Reed JC, Borden KL, Ronai Z. Regulation of p53 localization and activity by Ubc13. Mol Cell Biol. 2006;26:8901–13. doi: 10.1128/MCB.01156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Scheper J, et al. Protein-protein interaction antagonists as novel inhibitors of non-canonical polyubiquitylation. PLoS ONE. 5:e11403. doi: 10.1371/journal.pone.0011403. [DOI] [PMC free article] [PubMed] [Google Scholar]